

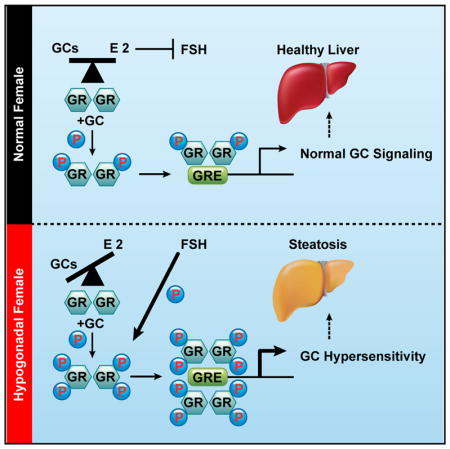
SUMMARY
Glucocorticoids (GCs) are master regulators of systemic metabolism. Intriguingly, Cushing’s syndrome, a disorder of excessive GCs, phenocopies several menopause-induced metabolic pathologies. Here, we show that the glucocorticoid receptor (GR) drives steatosis in hypogonadal female mice because hepatocyte-specific GR knockout mice are refractory to developing ovariectomy-induced steatosis. Intriguingly, transcriptional profiling revealed that ovariectomy elicits hepatic GC hypersensitivity globally. Hypogonadism-induced GC hypersensitivity results from a loss of systemic but not hepatic estrogen (E2) signaling, given that hepatocyte-specific E2 receptor deletion does not confer GC hypersensitivity. Mechanistically, enhanced chromatin recruitment and ligand-dependent hyperphosphorylation of GR underlie ovariectomy-induced glucocorticoid hypersensitivity. The dysregulated glucocorticoid-mediated signaling present in hypogonadal females is a product of increased follicle-stimulating hormone (FSH) production because FSH treatment in ovary-intact mice recapitulates glucocorticoid hypersensitivity similar to hypogonadal female mice. Our findings uncover a regulatory axis between estradiol, FSH, and hepatic glucocorticoid receptor signaling that, when disrupted, as in menopause, promotes hepatic steatosis.
In Brief
Menopause is well known to promote metabolic dysfunction in females. Loss of systemic estrogen has long been thought to be the primary mediator of these metabolic defects in post-menopausal women. Quinn et al. reveal that stress hormones are, in fact, the primary contributor to metabolic disturbances following ovariectomy in mice.
INTRODUCTION
Menopause occurs during the aging process in women and is characterized by a steady decline in ovarian function. Several pathologies accompany the onset of menopause, such as metabolic syndrome and fatty liver disease (Lobo et al., 2014). Although the molecular events giving rise to menopause-associated pathologies are largely unknown, loss of ovarian function is thought to be the underlying mechanism. This notion has led to hormone replacement therapy (HRT) being the mainstay clinical treatment for menopausal symptoms (Kaunitz and Manson, 2015). Although HRT is efficacious at alleviating the symptoms of menopause, it increases the risk for breast cancer (Chlebowski et al., 2003; Li et al., 2003). Furthermore, recent studies suggest that lack of female sex hormones, particularly estradiol, cannot fully explain the metabolic manifestations of menopause (Bingol et al., 2010; Fenkci et al., 2003; Mittelstrass et al., 2011; Song et al., 2011; Stampfer and Colditz, 1991; Turner et al., 2011; Wang et al., 2011).
Glucocorticoids (GCs) are primary stress hormones secreted in response to either physiological or psychological stressors. GCs mediate their physiological effects through the GC receptor (GR), a ligand-activated transcription factor. Because GCs are potent regulators of a variety of biological processes, their synthesis and secretion are tightly regulated to avoid insufficient or excessive production, leading to Addison’s disease or Cushing’s syndrome respectively. GCs are well known modulators of metabolism and have been linked pathogenically to obesity and steatosis (Arnaldi et al., 2010; Lemke et al., 2008; Macfarlane et al., 2014; Patel et al., 2011; Sun et al., 2013). Intriguingly, menopause phenocopies a spectrum of pathologies observed in Cushing’s syndrome patients such as osteoporosis, insulin resistance, and steatosis. However, the clinical link between GCs and menopause has not been studied empirically, despite human studies indicating trends toward higher cortisol levels during the menopausal transition (Woods et al., 2009). We show here that circulating GCs are elevated in a mouse surgical model of menopause via ovariectomy (OVX). Moreover, the rise in systemic GCs directly promotes metabolic syndrome and steatosis in ovariectomized mice because adrenalectomy (ADX) or genetic deletion of GR from hepatocytes blocks these metabolic abnormalities. Intriguingly, we discovered that hypogonadism in female mice promotes hepatic GC hypersensitivity. Furthermore, we have uncovered a mode of estradiol antagonism of hepatic GR signaling independent of liver-expressed estrogen receptor alpha (ERα), involving estradiol inhibition of follicle-stimulating hormone (FSH) production, leading to decreased GC-dependent phosphorylation of GR and diminished recruitment of GR to chromatin. Collectively, our data illuminate a role for GR signaling in the pathogenesis of metabolic syndrome/steatosis in estradiol-depleted mice and indicate
RESULTS
Loss of Ovarian Function Results in Cushing-like Syndrome in Mice
Given the clinical similarities between menopause and Cushing’s syndrome, we wanted to determine whether GCs drive metabolic syndrome/steatosis in a murine model of menopause. Consistent with Cushing’s syndrome, we observed significantly elevated corticosterone levels in ovariectomized mice compared with sham controls (Figure 1A). Furthermore, OVX promotes increased body weight coupled with a higher fat mass and lower lean mass, a phenomenon driven by adrenal hormones (Figures 1B and 1C).
Figure 1. GCs Drive Metabolic Dysfunction and Steatosis in Ovariectomized Mice.

(A) Serum corticosterone levels in sham-operated, ovariectomized, and OVX+ADX mice for 3 months. n = 4 per group.
(B) Body weight in sham, ovariectomized, and OVX+ADX mice. n = 5 per group.
(C) Body composition measured by dual-energy X-ray absorptiometry (DEXA)-scan of sham, ovariectomized, and OVX+ADX mice. n = 3 per group.
(D) Liver weight of sham, ovariectomized, and OVX+ADX mice. n = 4 per group.
(E) Glucose levels in sham, ovariectomized, and OVX+ADX mice fasted overnight. n = 3 per group.
(F) TG levels measured from sham, ovariectomized, and OVX+ADX mice. n = 3 per group.
(G) Representative H&E staining of livers from sham, ovariectomized, and OVX+ADX mice. Scale bars represent 50 μm.
(H) Representative images of oil red O-stained livers from sham, ovariectomized, and OVX+ADX mice. Scale bars represent 50 μm. that this pathway may be an alternative therapeutic target for the treatment of menopause-induced metabolic dysfunction.
(I) Immunoblot for GR in GRfl/fl and H-GRKO mice.
(J) Body weight of sham and ovariectomized GRflox/flox and H-GRKO mice. n = 3–8 per group.
(K) Representative oil red O staining of livers from ovariectomized GRflox/flox and H-GRKO mice 3 months after OVX. Scale bars represent 100 μm.
(L) Schematic of the experimental design for ADX rescue of ovariectomized mice.
(M) Final body weight at the end of the rescue experiment. n = 6–8 mice per group.
(N) Percent weight gain at the end of the rescue experiment. n = 6–8 mice per group.
(O) Representative oil red O staining of livers from ovariectomized mice receiving either sham or ADX surgery 3 months after OVX. Scale bars represent 100 μm.
Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
The metabolic abnormalities associated with menopause can manifest in the liver as steatosis and insulin resistance (Brady, 2015; Stefanska et al., 2015). We found that ovariectomized mice have increased liver weights compared with both sham and ovariectomized/adrenalectomized mice (Figure 1D). Furthermore, hyperglycemia was observed in fasted ovariectomized mice but not ovariectomized/adrenalectomized mice (Figure 1E). Triglyceride (TG) levels were measured, revealing the presence of steatosis selectively in ovariectomized mice but not ovariectomized/adrenalectomized mice (Figure 1F). Livers were examined histologically with H&E and oil red O staining to mark neutral lipid deposition, which confirmed the presence of steatosis only in ovariectomized mice (Figures 1G and 1H). Intriguingly, we observed increased protein expression of all five mitochondrial oxidative phosphorylation complexes specifically in ovariectomized mice but not sham and OVX+ADX mice (Figure S2A), suggesting that overt mitochondrial dysfunction is likely not the primary pathogenic mechanism promoting hypogonadism-induced steatosis. No overt metabolic phenotype was present in mice receiving ADX alone (Figure S1).
Because ADX removes an array of hormones, we wanted to gauge whether hepatic GR signaling was responsible for OVX-induced steatosis. To determine this, we utilized hepatocyte-specific GR knockout mice (H-GRKO) (He et al., 2015; Quinn and Cidlowski, 2016; Figure 1I). Ovariectomized GRfl/fl and H-GRKO mice had comparable increases in body weight between the two strains (Figure 1J), indicating that hepatocyte-specific GR signaling is not the underlying mechanism for systemic weight gain in hypogonadal female mice. However, OVX-induced steatosis was attenuated by deleting GR from hepatocytes (Figure 1K). These data indicate that OVX-induced steatosis is due to inherent GR signaling within the liver and not a secondary phenotype to obesity and altered adipose tissue lipolysis.
We found that we could also reverse OVX-induced metabolic syndrome by targeting the GR pathway 3 months after OVX (Figure 1L). Adrenalectomizing ovariectomized mice led to a significant decrease in body weight and percent weight gained in response to OVX (Figures 1M and 1N). This resulted in a reduced hepatic TG burden (Figure 1O). These findings implicate the hepatic GR pathway as a pathogenic driver of steatosis in hypogonadal female mice, which may be of therapeutic relevance in reversing hypogonadism-induced steatosis.
GR-Governed Hepatic Lipogenic Pathways Are Reprogrammed in Hypogonadal Female Mice
We next determined the hepatic GR transcriptome in normal and hypogonadal female mice via RNA sequencing (RNA-seq). A correlation heatmap of our RNA-seq datasets revealed a low correlation score for ovariectomized mice treated with the synthetic GC dexamethasone compared with dexamethasone-treated ovary-intact mice (Figure 2A). Furthermore, principal-component analysis shows clear separation between dexamethasone-treated normal and hypogonadal mice, further confirming an altered transcriptional response to GCs in the absence of ovarian hormones (Figure S3A). To determine the transcriptional activity of GR, we plotted the fold change elicited by GC treatment in normal and hypogonadal mice. This revealed a large cohort of genes that respond differentially to GR activation in the absence of ovarian hormones (Figure 2B). Notably, approximately 600 GC-upregulated genes were hyper-induced (Figure 2B).
Figure 2. The GR-Governed Hepatic Lipid Metabolism Network Is Reprogrammed in Hypogonadal Female Mice.

(A) Correlation heatmap of adrenalectomized ovary-intact and ADX+OVX mice treated with vehicle or dexamethasone for 6 hr.
(B) Scatterplot of dexamethasone-regulated genes commonly regulated between ovary-intact and ovariectomized mice. Each dot represents a gene. The x axis represents the dexamethasone (dex)-induced fold change in ADX alone, and the y axis represents the dexamthasone-induced fold change in ADX+OVX mice.
(C) GSEA of the GC-controlled lipid metabolism reactome in adrenalectomized and ADX+OVX mice treated with dexamethasone.
(D) Heatmap of fragments per kilobase per million (FPKM) values from adrenalectomized and ADX+OVX mice treated with vehicle or dexamethasone, showing the lipogenic (top) (GO_0008610) and lipolytic pathways (bottom) (GO_0016042). Hyper-induced GC targets are outlined in a yellow box.
(E–G) FPKM fold change elicited by GCs in ADX and ADX+OVX mice of genes involved in lipid synthesis (E), lipid transport (F), and β-oxidation (G).
Network enrichment analysis of the biological pathways significantly regulated by hormone treatment in ovary-intact and hypogonadal female mice revealed that GCs regulate similar physiological processes, such as cell cycle regulation, translation/transcription regulation, and axon guidance, regardless of the ovarian hormonal milieu (Figures S3B and S3C). Despite no appreciable differences in the pathways regulated by GCs between normal and hypogonadal mice, GR-governed lipid metabolism networks are highly sensitive to dysregulation in hypogonadal female mice (Figures S3B and S3C, red circles). Gene set enrichment analysis (GSEA) indicated a negative enrichment score for ovary-intact dexamethasone-treated mice (Figure 2C). In stark contrast, dexamethasone-treated ovariectomized mice had a positive enrichment score for the lipid metabolism reactome (Figure 2C). This demonstrates that OVX alters the function of GR in regulating hepatic lipid metabolism. In-depth analysis of the lipolytic pathway (GO_0016042) indicated negligible differences in GC regulation of this pathway between normal and hypogonadal mice (Figure 2D). However, the lipogenic pathway (GO_0008610) displayed altered GC regulation, characterized by transcriptional hypersensitivity (Figure 2D, yellow box). De novo motif analysis of hypersensitive lipogenic genes revealed a high number of putative GC response elements (GREs) within their loci, indicating that these genes may be subject to direct regulation by GR (Table S1). To gain a better understanding of the net physiological effect GCs exert on regulating hepatic lipid metabolism, we surveyed genes involved in lipid synthesis, transport, and fatty acid β-oxidation in ovary-intact and ovariectomized mice. We found a gene expression profile consistent with the accumulation of lipids in the liver. The steatotic gene program was characterized by higher induction of genes involved in lipid synthesis and transport following GC treatment in hypogonadal mice (Figures 2E and 2F). Inversely, GC upregulation of β-oxidation genes in ovary-intact mice is severely dampened in ovariectomized mice, whereas mitochondrially encoded genes were not subject to GC regulation (Figure 2G). PPARα is the master regulator of hepatic fatty acid β-oxidation and has previously been shown to crosstalk with GR, which alters their transcriptional activity (Bougarne et al., 2009; Lee et al., 2015). Global analysis of the PPARα pathway indicates altered GR/PPARα crosstalk in ovariectomized mice (Figure S4, green and red boxes). Moreover, 3 GR target genes identified by RNA-seq as being transcriptionally hypersensitive to GC treatment in ovariectomized mice (PLIN5, LCN2, and LPIN1) are all transcriptionally upregulated in the long-term OVX model of steatosis in a GC-dependent manner (Figure S5).
OVX Enhances GR Transcriptional Activity and Recruitment to Chromatin
We used PLIN5, LCN2, and LPIN1 as model genes to further examine the molecular mechanism underlying GC hypersensitivity in hypogonadal mice, with particular emphasis on PLIN5 given its role in hepatic lipid storage. Real-time PCR confirmed hyper-induction of PLIN5, LCN2, and LPIN1 following GC treatment in ovariectomized mice (Figure 3A). We tested whether GC hypersensitivity was due to enhanced GR recruitment to GRE-containing loci. A chromatin immunoprecipitation (ChIP) assay revealed enhanced occupancy of GR to the GREs within the PLIN5, LCN2, and LPIN1 loci following GC treatment in ovariectomized compared with normal mice (Figure 3B). These data suggest that increased GR recruitment to target loci underlies GC hypersensitivity in hypogonadal female mice.
Figure 3. Ovariectomy Promotes Enhanced Chromatin Recruitment of GR.

(A) qPCR of PLIN5, LCN2, and LPIN1 mRNA in adrenalectomized ovary-intact and ADX+OVX mice treated with either vehicle or dexamethasone for 6 hr. PLIN5, LCN2, and LPIN1 were normalized to PPIB mRNA; n = 3 per group. Data are expressed as relative mRNA to PPIB ± SEM.
(B) Chromatin immunoprecipitation of GR to putative GREs in the PLIN5, LCN2, and LPIN1 loci in response to hormone treatment for 1 hr in adrenalectomized ovary-intact and ADX+OVX mouse livers. n = 4 per group. Data are expressed as fold recruitment of GR over adrenalectomized ovary-intact vehicle-treated mice ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Estradiol Antagonizes Hepatic GR Signaling Independent of Hepatic ERα
Our findings indicate that removal of ovarian hormones promotes hepatic GC hypersensitivity. We hypothesized that OVX alters the chromatin architecture of target loci, allowing increased GR recruitment following dexamethasone treatment. We performed formaldehyde-assisted isolation of regulatory elements (FAIRE) to survey the chromatin architecture in ovary-intact and ovariectomized mice treated with dexamethasone to test this hypothesis. FAIRE analysis of the PLIN5 loci revealed increased accessibility of this GRE in both ovary-intact and hypogonadal female mice following GC treatment; however, no differences were seen between normal and ovariectomized mice (Figure S5A). This result indicates that enhanced GR recruitment to chromatin in hypogonadal female mice is not due to increased accessibility to the DNA. We also did not observe any differences in the epigenetic marks H3K27me3 or H3K27ac between groups (Figure S5B).
Because OVX does not increase chromatin accessibility at the PLIN5 loci or alter the H3K landscape, we postulated that ER competes with GR for DNA binding, limiting the recruitment of GR to chromatin in ovary-intact mice. To test this, we primed hypogonadal female mice with either vehicle or estradiol for 3 days to activate ER before treatment with dexamethasone. Estradiol re-administration blunted GR recruitment to the PLIN5 loci compared with vehicle-primed hypogonadal mice (Figure 4A). We next assayed PLIN5 mRNA to see whether the reduced chromatin recruitment resulted in diminished transcriptional output to dexamethasone. Indeed, estradiol priming significantly decreased GC induction of PLIN5 (Figure 4B). These data indicate that activation of ER could potentially be a mechanism to limit GR recruitment to chromatin and transcriptional activity. We focused our attention on ERα given the lack of a metabolic phenotype in the global ERβ KO (Bryzgalova et al., 2006). Moreover, ERβ expression was undetectable in livers of mice in our RNA-seq dataset. To test whether ERα competes with GR for chromatin binding, we generated hepatocyte-specific ERα KO mice (H-ERKO) by crossing ERfl/fl mice (Hewitt et al., 2010) with Alb-cre mice. Repeating our estradiol replacement experiment in ERfl/fl and H-ERKO mice revealed that estradiol still retained its ability to antagonize hepatic GR signaling even in the absence of hepatic ERα (Figure 4C). Furthermore, we hypothesized that if ERα is playing a protective role against GC hypersensitivity, then a greater steatotic pathology would be observed in ovariectomized H-ERKO mice. Oil red O staining of liver sections from ovariectomized ERfl/fl and H-ERKO mice revealed a comparable hepatic TG burden between the genotypes (Figure 4D). These data are consistent with lack of a basal phenotype previously reported in H-ERKO mice (Hart-Unger et al., 2017). Our data demonstrate that hepatic ERα is dispensable for mediating the favorable metabolic effects of estradiol and indicate that the antagonistic effects of estradiol on hepatic GR signaling are mediated via an extrahepatic ERα mechanism.
Figure 4. Systemic but Not Hepatic Estrogen Signaling Deficiency Promotes GC Hypersensitivity.

(A) Chromatin immunoprecipitation of GR to the GRE in the PLIN5 loci in hypogonadal female mice treated with dexamethasone for 1 hr, primed with and without estradiol for 72 hr. n = 3–4 animals per group. Data are expressed as fold recruitment of GR over vehicle-treated ovariectomized mice ± SEM.
(B) PLIN5 mRNA in ovariectomized mice with and without estradiol priming for 72 hr following 6 hr dexamethasone treatment. Data are expressed as relative PLIN5 mRNA normalized to PPIB mRNA ± SEM. n = 4 animals per group.
(C) PLIN5 mRNA in vehicle- and dexamethasone-treated ER-floxed and H-ERKO mice with and without estradiol priming. Data are expressed as relative PLIN5 mRNA normalized to PPIB mRNA ± SEM. n = 3 independent animals per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(D) Representative oil red O staining of livers from ovariectomized ERflox/flox and H-ERKO mice 2 months after OVX. Scale bars represent 100 μm. n = 3–4 mice per group.
Estradiol Deficiency Promotes Hyperphosphorylation of Serine 211 in GR
Collectively, our data indicate that estrogen deficiency results in GR transcriptional hypersensitivity associated with increased GR recruitment to chromatin. Because the chromatin architecture at the PLIN5 loci did not appear to be altered following OVX, and ERα does not limit GR binding by competing with GR for chromatin binding, we speculated that estradiol deficiency promotes these effects via an alternative mechanism, perhaps in a multi-organ fashion.
One primary mechanism regulating the transcriptional activity of GR is through post-translational modifications such as phosphorylation (Galliher-Beckley and Cidlowski, 2009). Most notably, phosphorylation of GR at serine 211 in response to ligand has been shown to increase transcriptional output following hormone treatment (Chen et al., 2008). Furthermore, phosphorylation of GR on serine 211 has also been shown to promote recruitment to GRE-containing loci in response to GC treatment (Blind and Garabedian, 2008). We therefore hypothesized that GC hypersensitivity in hypogonadal female mice is a product of enhanced ligand-dependent phosphorylation of GR at serine 211. Indeed, serine 211 of GR was hyperphosphorylated following dexamethasone treatment in the livers of hypogonadal mice compared with ovary-intact mice (Figure 5A). We next wanted to determine whether phosphorylation of serine 211 was also subject to estradiol antagonism. Estradiol readministration to hypogonadal female mice led to a trend in decreased ligand-dependent phosphorylation of serine 211 of GR (p = 0.07) (Figure 5B). These data indicate that hyperphosphorylation of serine 211 of GR could be the potential mechanism underlying the increased recruitment of GR to chromatin and subsequent transcriptional hypersensitivity elicited by GCs in estrogen-deficient mice.
Figure 5. Systemic Estrogen Deficiency Promotes Ligand-Dependent Phosphorylation of GR at Serine211 in the Liver.

(A) Western blot analysis of phospho-serine 211 of GR in vehicle- and dexamethasone-treated (1 hr) adrenalectomized ovary-intact and ADX+OVX mice. Data are expressed as percent of vehicle-treated adrenalectomized ovary-intact mice of p211 normalized to total GR ± SEM. n = 4 individual animals per group.
(B) Immunoblot for phosphoserine 211 of GR in vehicle- and dexamethasone-treated (1 hr) ovari-ectomized mice with and without estradiol priming for 48/72 hr. Data are expressed as percent of vehicle-treated adrenalectomized ovary-intact mice of p211 normalized to total GR ± SEM. n = 5–6 individual animals per group.
**p < 0.01, ****p < 0.0001.
FSH Enhances GC-Mediated Signaling
Our results suggest that the antagonistic effects of estradiol on hepatic GR signaling occur via an extra-hepatic mechanism. One of the primary endocrine manifestations of losing ovarian function is aberrant production of FSH and luteinizing hormone (LH) in both humans and rodents (Parlow, 1964; Wise and Ratner, 1980; Yen and Tsai, 1971). We therefore speculated that enhanced production of these pituitary peptides in hypogonadal mice may be the underlying mechanism driving ligand-induced hyperphosphorylation of GR. We observed detectable levels of FSH receptor protein, but not LH receptor, in the livers of female mice (Figure 6A); therefore, we focused our efforts on determining whether FSH could promote GC hypersensitivity. Consistent with previous reports, we observed a significant increase in circulating FSH levels following OVX (Figure 6B). Moreover, circulating FSH levels decreased only at the time point when we observed the antagonistic effects of estradiol on GR phosphorylation (72 hr) (Figure 6B). To determine whether FSH has the capacity to enhance GR-mediated signaling in vivo, we pre-treated ovary-intact mice with a bolus of FSH 5 min prior to administering dexamethasone and measured the phosphorylation levels of serine 211 of GR. This revealed that FSH does indeed have the ability to boost the ligand-dependent phosphorylation of GR both in vivo (Figure 6C) as well as in vitro (Figure S6). We did not observe FSH-elicited phosphorylation of serine 211 of GR in the absence of dexamethasone (Figure S6). This likely points to a conformational change in GR elicited by ligand binding, exposing serine 211 to FSH-mediated enhanced phosphorylation. Importantly, enhanced phosphorylation of serine 211 of GR elicited by FSH pre-treatment in vivo resulted in a subsequent increase in GR-mediated induction of PLIN5, LCN2, and LPIN1 mRNA (Figure 6D). Our data reveal a regulatory loop between estradiol regulation of FSH production to maintain proper hepatic GR signaling.
Figure 6. FSH Enhances GR Phosphorylation and Transcriptional Induction of Target Genes.

(A) Immunoblot of FSHR and LHR in female mouse livers. β-Actin was used as a loading control, and mouse ovary was used as a positive control.
(B) Circulating FSH levels in sham-operated mice and ovariectomized mice treated with either vehicle or estradiol for 48 and 72 hr. Data are expressed as milli-international units per milliliter of FSH ± SEM. n = 5–9 individual animals per group.
(C) Immunoblot for p211 GR and total GR in livers of vehicle-, dexamethasone-, and FSH+dex-amethasone-treated adrenalectomized ovary-intact mice. FSH was administered 5 min prior to dexamethasone injection, and livers were harvested 1 hr after dexamethasone injection. Data are expressed as percent of vehicle-treated adrenalectomized ovary-intact mice of p211 normalized to total GR ± SEM. n = 3–4 animals per group.
(D) PLIN5, LCN2, and LPIN1 mRNA in vehicle-, dexamethasone-, and FSH+dexamethasone-treated adrenalectomized ovary-intact mice. FSH was administered 5 min prior to dexamethasone injection, and livers were harvested 1 hr after dexamethasone injection. Data are expressed as relative mRNA to PPIB mRNA ± SEM. n = 3–8 individual mice per group.
*p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
It is well established that aging promotes metabolic disturbances that manifest most evidently in females during the menopausal period. The molecular drivers of menopause-induced metabolic dysfunction are largely unknown; however, estrogen depletion is thought to be the underlying pathological mechanism. Here we report that GCs are the primary pathogenic driver of metabolic dysfunction and steatosis in estrogen-depleted mice. Targeting the GR pathway via ADX or genetic deletion of GR from the liver protects against OVX-induced steatosis. Furthermore, altered global transcriptional responses following GR activation was observed in estrogen-depleted mice. Transcriptional hypersensitivity of GC-governed lipogenic genes is a pathogenic mechanism underlying OVX-induced steatosis in mice. GC hypersensitivity in hypogonadal females is associated with enhanced GR:DNA binding and heightened ligand-dependent phosphorylation of GR on serine 211. The heightened ligand-dependent phosphorylation of GR in hypogonadal females is at least in part due to aberrant production of FSH and subsequent signaling in hepatocytes. Lowering FSH levels in vivo by estradiol re-administration reversed the heightened GR responsiveness in ovariectomized mice and diminished the transcriptional response elicited by GCs.
Here we show that hypogonadism dramatically reprograms the hepatic GR transcriptome in female mice. Analyzing the physiological pathways regulated by GCs in normal and hypogonadal mice revealed that the hepatic lipid metabolism network was subject to the most profound changes in transcriptional regulation. Interestingly, genes within the lipogenic pathway exhibited heightened dexamethasone-mediated induction, suggesting that hypersensitivity to GCs on this pathway is a pathophysiological mechanism driving steatosis in hypogonadal female mice.
Increasing evidence indicates that ER and GR crosstalk to alter the transcriptome of one another (Whirledge and Cidlowski, 2013; Whirledge et al., 2013; Gong et al., 2008; West et al., 2016). In the uterus, estradiol can antagonize the transcriptional effects of GR activation, a mechanism requiring ERα (Whirledge and Cidlowski, 2013; Whirledge et al., 2013). These studies provide support for the notion that GC hypersensitivity in hypogonadal females is due to loss of estradiol/ERα antagonism of the GR pathway. We tested this hypothesis through a multi-tiered approach, including estradiol co-administration with dexamethasone and H-ERKO mice. Our findings revealed that loss of hepatic estrogen signaling was not the underlying cause promoting enhanced GC sensitivity in ovariectomized mice, indicating that estradiol depletion alters the hepatic GR transcriptome through extrahepatic actions. Our results support previous findings in which the protective metabolic effects of estrogens are mediated via ERα in non-hepatic tissues (Hart-Unger et al., 2017; Matic et al., 2013). Estrogen is a potent repressor of FSH synthesis in the pituitary to limit its own synthesis in a negative feedback loop. Following estrogen depletion, FSH is increased systemically. We discovered that long-term estradiol readministration could lower FSH levels and, consequently, antagonize the transcriptional activity of GR. Furthermore, administration of FSH to ovary-intact mice could promote GC hypersensitivity, indicating that this pituitary peptide has the capacity to potentiate hepatic GR signaling.
FSH is now being appreciated for its extragonadal actions, most notably its effects in metabolic tissues. In bone, for example, FSH, acting through the FSH-R, mediates hypogonadism-induced bone loss (Sun et al., 2006). Recently, the metabolic effects of FSH have been extended beyond bone to adipose tissue, where administration of this peptide to young chickens leads to increased abdominal fat mass by increasing lipid biosynthesis (Cui et al., 2012). In the liver, FSH has been shown to have multiple functions. For example, cholangiocytes, through a paracrine loop, utilize FSH-R signaling to regulate proliferation (Mancinelli et al., 2009). In their immunohistochemical staining, Mancinelli et al. (2009) showed FSH-R-positive staining not only in cholangiocytes but also in hepatocytes, suggesting that FSH may also elicit biological effects in the liver outside of the biliary tract. A recent report confirmed the expression of FSH-R in hepatocytes and showed that this signaling cascade results in the downregulation of LDL-R in the liver, resulting in increased circulating total cholesterol (Song et al., 2016). OVX-induced hypercholesterinemia could be abated by the use of a GNRH receptor agonist to systemically lower FSH (Song et al., 2016). Furthermore, FSH-R-haploinsufficient female mice, also known as FORKO mice, have diminished estradiol production and mimic human menopause by developing a dys-regulated menstrual cycle and reproductive senescence (Danilovich et al., 2000). These mice develop metabolic dysfunction and obesity, although, not as severely as the global ERα KO mouse (Heine et al., 2000), which, in conjunction with ablated estrogen signaling, has increased FSH production (Emmen and Korach, 2003). The FORKO mouse suggests that a mere lack of estrogen is not sufficient to recapitulate the full metabolic phenotype observed in the global ERα KO mouse, and perhaps elevated FSH levels are required for the pathogenesis of metabolic syndrome in hypogonadal females. This can somewhat explain the lack of a basal phenotype observed in our H-ERKO mice and why the transcriptional response to GC treatment is similar to that of ERfl/fl, where FSH levels are presumed to be normal. Our data support a model in which hepatic ERα signaling is dispensable for proper GC regulation of gene expression, but, rather, hepatic FSH signaling can alter the course of the GC response. This has revealed a potential mechanism by which elevated FSH levels may dysregulate hepatic lipid metabolism through amplification of the transcriptional effects of GCs. These findings also support a role for FSH signaling beyond the reproductive tracts as a regulator of metabolism. Our discovery is highlighted by the recent finding that targeting FSH through an FSH-neutralizing antibody could correct metabolic dysfunction in ovariectomized mice and promote beiging of adipose tissue (Liu et al., 2017). Our current findings indicate that dysregulation of the GR pathway may be the underlying molecular mechanism promoting metabolic dysfunction in response to aberrant FSH production. In a physiological context, we hypothesize that this crosstalk between FSH and GCs in regulating hepatic lipid metabolism is to initiate a unique maternal metabolic program to support the implanting embryo and subsequent pregnancy and is an active area of ongoing research.
Serine 211 is a promiscuous residue on GR and can be phosphorylated by an array of different protein kinases. Furthermore, FSH has been shown to activate a number of similar protein kinases known to phosphorylate GR at serine 211. For example, FSH-R has been shown to activate both p38 mitogen-activated protein kinase (MAPK) and SGK1, which also has the ability phosphorylate serine 211 of GR. Other potential mechanisms, including FSH-mediated inhibition of protein phosphatases, could also contribute to the hyperphosphorylation of GR mediated by FSH. The mechanism linking the hepatic effects of FSH to GR signaling is of serious clinical significance and is a subject of our future studies.
In the clinical context, our data illuminate the GR pathway as a potential alternative therapeutic target for the treatment of post-menopausal steatosis as an alternative to traditional therapies such as HRT and selective estrogen receptor modulators. Furthermore, selective blockage of GCs could prevent weight gain and alterations in body composition, suggesting a pathogenic role for GCs in adipose tissue in hypogonadal females. We did not evaluate the role of GR in adipose tissue in the present study; however, there are many reports implicating stress hormones in the development of obesity (reviewed in Baudrand and Vaidya, 2015; Lee et al., 2014). These findings suggest that anti-GC therapy may have beneficial effects during menopause-induced metabolic syndrome beyond the liver. The second clinical implication our study highlights is that GR activation differs in females depending on the ovarian hormone milieu/HPG axis function. Given the GC hypersensitivity to genes involved in lipogenesis, we speculate that post-menopausal women on cortico-steroid therapy may potentially be more sensitive to developing the metabolic side effects associated with chronic steroid use more rapidly than a woman in her reproductive years per se. In fact, development of adverse side effects during long-term corti-costeroid therapy occurs in approximately 80% of autoimmune hepatitis patients after 2 years of therapy (Czaja, 2008; Summerskill et al., 1975; Uribe et al., 1984), which is the most common reason for premature drug withdrawal (Czaja, 2008; Czaja et al., 1984; Manns et al., 2010). Understanding if and how certain populations, such as postmenopausal women, may be more sensitive to developing adverse side effects while on corticosteroid therapy could prove beneficial in developing strategies to combat these side effects, such as initiating combination therapy.
In conclusion, we report that GCs drive metabolic abnormalities, specifically steatosis, in hypogonadal female mice. Moreover, we have uncovered a regulatory loop of extrahepatic estradiol inhibition of hepatic GR signaling involving estradiol antagonism of FSH production, which, in turn, limits the transcriptional activity of GCs by reducing GR:DNA binding via decreased ligand-dependent phosphorylation of GR. These findings challenge the current dogma that the metabolic syndrome accompanied by hypogonadism in females is merely due to a loss of ovarian hormones. We suggest that the GR pathway is responsible for the metabolic phenotype in hypogonadal females, and it may prove to be a more clinically efficacious target than classical HRT for the treatment of metabolic complications during menopause in humans.
EXPERIMENTAL PROCEDURES
Materials
FSH was purchased from Sigma (St. Louis, MO). Dexamethasone and 17β-estradiol were purchased from Steraloids (Newport, RI).
Animal Experiments
Female C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). H-GRKO mice were generated by crossing GRflox/flox mice (Oakley et al., 2013) with mice expressing CRE-recombinase under the liver-specific promoter albumin (Quinn and Cidlowski, 2016). H-ERKO mice were generated in a similar fashion as the H-GRKO mice by crossing ERflox/flox mice (a kind gift from Dr. Kenneth Korach, National Institute of Environmental Health Sciences (NIEHS)/NIH) to albumin-CRE mice. Female homozygous floxed CRE+ mice were used as controls, and homozygous floxed CRE+ animals were used as experimental groups in all experiments. All animals were subject to a 12:12 hr light/dark cycle and had ad libitum access to standard mouse chow and drinking water. Bilateral OVX was performed as a surgical model of menopause in both wild-type and H-GRKO mice. To reduce circulating GCs, bilateral ADX was performed in wild-type mice. Adrenalectomized mice were maintained on 0.9% saline to maintain salt levels. Surgically altered mice were used between the ages of 5 and 7 months. To determine GC-regulated genes, dexamethasone (1 mg/kg) was injected, and livers were harvested 6 hr after injection. Dexamethasone-treated mice were used at 3 months of age. For ChIP and FAIRE assays, mice were treated with dexa-methasone, and livers were harvested 1 hr after injection. Estradiol was injected at 100 μg/kg daily up to 3 days. FSH was injected at 60 μg/kg 5 min prior to dexamethasone injection. All experiments utilizing non-adrenalectomized mice were performed at zeitgeber time 3 (ZT3) during the nadir of circadian GC release. All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee at the NIEHS, NIH.
Corticosterone and FSH Measurement
Circulating corticosterone and FSH were measured via colorimetric ELISAs (corticosterone at ZT3, Arbor Assays, Ann Arbor, MI; FSH, Enzo Life Sciences, Farmingdale, NY).
Body Composition
Body composition was determined in sham, ovariectomized, and OVX+ADX mice via dual X-ray absorptiometry (DEXA) (Lunar Piximus 2, Fitchburg, WI) according to the manufacturer’s protocol.
Glucose Measurements
Glucose levels were measured in mice fasted overnight using the TrueTrack glucometer (CVS Pharmacy, Durham, NC).
Hepatic TG Measurement
TGs were determined with a TG colorimetric assay kit (Cayman Chemicals, Ann Arbor, MI). Oil red O staining (Sigma) was used to stain lipids in frozen liver sections as described previously (Mehlem et al., 2013).
qPCR
One hundred nanograms of total RNA was reverse-transcribed and amplified using the iScript One-Step RT-PCR kit for probes (Bio-Rad, Hercules, CA). Real-time qPCR was performed with the Bio-Rad CFX96 sequence detection system using predesigned primer/probe sets against PLIN5, LCN2, and LPIN1. ERα and PPIB were from Applied Biosystems (Foster City, CA). The relative fluorescence signal was normalized to PPIB using the ΔCT method.
Chromatin Immunoprecipitation Assay
Approximately 100 mg of liver from vehicle- and dexamethasone-treated mice was cross-linked, and isolated nuclei were subjected to sonication (15 cycles on high, 30 s on, 30 s off; Diagenode Bioruptor, Denville, NJ). Sonicated DNA was immunoprecipitated with rabbit anti-GR monoclonal antibody (Cell Signaling Technology, Danvers, MA), followed by isolation using the Magna-ChIP kit (Millipore, Billerica, MA). Isolated DNA was purified via the QIAquick PCR purification kit (QIAGEN, Valencia, CA) and eluted in 50 μL of elution buffer. The primers used are listed in Table 1.
Table 1.
ChIP Primers
| Loci | Forward | Reverse |
|---|---|---|
| PLIN5 | 5′- CCCACTGCAAGCTCTGT-3′ | 5′-CAGCTGCGAGAGGACATT-3′ |
| LCN2 | 5′-TAGACAGGGAAGAAGAGGACA-3′ | 5′-GGCTCAAGGTATTGGACACTT-3′ |
| LPIN1 | 5′-GTTTGTGACGAAAGCTGAGAAA-3′ | 5′-ACATGCTGCTCCAACACT-3′ |
Western Blotting and Immunohistochemistry
Protein lysates were prepared from livers of mice by homogenization in SDS sample buffer (Bio-Rad, Hercules, CA) containing β-mercaptoethanol (Sigma). Approximately 30 μg of total protein was resolved on a 4%–20% Tris-glycine gel (Bio-Rad) and transferred onto a 0.2 μM nitrocellulose membrane (Bio-Rad). Membranes were blocked with blocking buffer (LI-COR Biosciences, Lincoln, NE) and incubated overnight with either anti-GR 59 (Oakley et al., 2017), anti-phosphoserine 211 GR (Cell Signaling Technology), anti-FSHR (Abcam), anti-hCGR (LHR, Abcam), or anti-β-actin (Millipore). Total GR, phosphorylated GR, FSHR, and LHR were used at 1:1,000, and β-actin was used at 1:10,000. Protein was detected via fluorescent secondary antibody detection (1:10,000) (LI-COR Biosciences) and imaged on the LI-COR Odyssey (LI-COR Biosciences). Densitometry was performed with LI-COR Odyssey software, and β-actin was used to normalize loading.
RNA Sequencing
RNA was extracted from livers of mice with Qiazol, and purification of total RNA was performed with the QIAGEN RNeasy RNA isolation kit (Redwood City, CA) according to the manufacturer’s protocol. RNA-seq libraries were generated with 1 μg of RNA as input using the TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA) and poly(A)-enriched according to the TruSeq protocol. Indexed samples were sequenced using the 100-bp paired-end protocol via the NextSeq500 (Illumina) according to the manufacturer’s protocol. Reads (38–55 million reads per sample) were aligned to the University of California Santa Cruz (UCSC) mm9 reference genome using TopHat2. The quantification results from htseq-count were then analyzed with the Bioconductor package DESeq2, which fits a negative binomial distribution to estimate technical and biological variability. Comparisons were made between vehicle- and dexa-methasone-treated adrenalectomized ovary-intact and ADX+OVX mice. A gene was considered differentially expressed when the p value for differential expression was less than 0.01. The correlation heatmap was generated using the R software package (version 3.3.3) with the “ggplot2” package. GSEA was performed using GSEA v2.2.2 software. Genes were pre-ranked based on the fold change of gene expression. This application scores a sorted list of genes with respect to their enrichment of selected functional categories (Kyoto Encyclopedia of Genes and Genomes [KEGG], Biocarta, Reactome, and gene ontology [GO]). The significance of the enrichment score was assessed using 300 permutations. Benjamini and Hochberg’s false discovery rate (FDR) was calculated for multiple testing adjustments. q < 0.05 was considered significant. The resulting enriched pathways were visualized using the Cytoscape (v3.3.0) Enrichment map plugin.
Statistical Analysis
Statistical significance was detected between groups by GraphPad Prism 7 (La Jolla, CA) using a Student’s t test when comparing two groups or two-way ANOVA when comparing three or more groups, followed by a Sidak post hoc test to correct for multiple comparisons. A normality test was performed on the t test to determine whether data were normally distributed, and when failing the normality test, a Mann-Whitney post hoc test was performed. Statistical significance was defined as p < 0.05.
Supplementary Material
Highlights.
Hypogonadal female mice develop Cushing-like syndrome
The hepatic GR transcriptome is reprogrammed in the absence of estrogen
Ovariectomy promotes GC hypersensitivity
FSH potentiates hepatic GR signaling
Acknowledgments
We would like to acknowledge the NIEHS Epigenomics core for assistance with RNA sequencing experiments. We thank Dr. Robert Oakley, Dr. Xiaoling Li, and Dr. Michael Fessler for critical reading of the manuscript. We would also like to thank the Pathology Support Group at NIEHS for assistance with histological examinations. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (1ZIAES090057 to J.A.C.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE99309.
Supplemental Information includes Supplemental Experimental Procedures, six figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.041.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, M.A.Q. and J.A.C.; Methodology, M.A.Q. and M.R.; Software, Formal Analysis, M.A.Q. and X.X.; Investigation, M.A.Q. and X.X.; Data Curation, X.X.; Writing – Original Draft, M.A.Q., X.X., and J.A.C.; Visualization, M.A.Q. and X.X.; Supervision, J.A.C.; Funding Acquisition, J.A.C.
References
- Arnaldi G, Scandali VM, Trementino L, Cardinaletti M, Appolloni G, Boscaro M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl 1):86–90. doi: 10.1159/000314213. [DOI] [PubMed] [Google Scholar]
- Baudrand R, Vaidya A. Cortisol dysregulation in obesity-related metabolic disorders. Curr Opin Endocrinol Diabetes Obes. 2015;22:143–149. doi: 10.1097/MED.0000000000000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Gunenc Z, Yilmaz M, Biri A, Tiras B, Güner H. Effects of hormone replacement therapy on glucose and lipid profiles and on cardiovascular risk parameters in postmenopausal women. Arch Gynecol Obstet. 2010;281:857–864. doi: 10.1007/s00404-009-1169-y. [DOI] [PubMed] [Google Scholar]
- Blind RD, Garabedian MJ. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol. 2008;109:150–157. doi: 10.1016/j.jsbmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, Staels B, Haegeman G, De Bosscher K. PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB. Proc Natl Acad Sci USA. 2009;106:7397–7402. doi: 10.1073/pnas.0806742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CW. Liver disease in menopause. World J Gastroenterol. 2015;21:7613–7620. doi: 10.3748/wjg.v21.i25.7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia. 2006;49:588–597. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. doi: 10.1210/me.2007-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M, Lane D, Rodabough RJ, Gilligan MA, Cyr MG, Thomson CA, et al. WHI Investigators. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative Randomized Trial. JAMA. 2003;289:3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- Cui H, Zhao G, Liu R, Zheng M, Chen J, Wen J. FSH stimulates lipid biosynthesis in chicken adipose tissue by upregulating the expression of its receptor FSHR. J Lipid Res. 2012;53:909–917. doi: 10.1194/jlr.M025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja AJ. Safety issues in the management of autoimmune hepatitis. Expert Opin Drug Saf. 2008;7:319–333. doi: 10.1517/14740338.7.3.319. [DOI] [PubMed] [Google Scholar]
- Czaja AJ, Davis GL, Ludwig J, Taswell HF. Complete resolution of inflammatory activity following corticosteroid treatment of HBsAg-negative chronic active hepatitis. Hepatology. 1984;4:622–627. doi: 10.1002/hep.1840040409. [DOI] [PubMed] [Google Scholar]
- Danilovich N, Babu PS, Xing W, Gerdes M, Krishnamurthy H, Sairam MR. Estrogen deficiency, obesity, and skeletal abnormalities in follicle-stimulating hormone receptor knockout (FORKO) female mice. Endocrinology. 2000;141:4295–4308. doi: 10.1210/endo.141.11.7765. [DOI] [PubMed] [Google Scholar]
- Emmen JM, Korach KS. Estrogen receptor knockout mice: phenotypes in the female reproductive tract. Gynecol Endocrinol. 2003;17:169–176. [PubMed] [Google Scholar]
- Fenkci S, Fenkci V, Yilmazer M, Serteser M, Koken T. Effects of short-term transdermal hormone replacement therapy on glycaemic control, lipid metabolism, C-reactive protein and proteinuria in postmenopausal women with type 2 diabetes or hypertension. Hum Reprod. 2003;18:866–870. doi: 10.1093/humrep/deg146. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61:979–986. doi: 10.1002/iub.245. [DOI] [PubMed] [Google Scholar]
- Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song WC, DeFranco DB, Cheng SY, Xie W. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386–7393. doi: 10.1158/0008-5472.CAN-08-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart-Unger S, Arao Y, Hamilton KJ, Lierz SL, Malarkey DE, Hewitt SC, Freemark M, Korach KS. Hormone signaling and fatty liver in females: analysis of estrogen receptor α mutant mice. Int J Obes. 2017;41:945–954. doi: 10.1038/ijo.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human Glucocorticoid Receptor β Regulates Gluconeogenesis and Inflammation in Mouse Liver. Mol Cell Biol. 2015;36:714–730. doi: 10.1128/MCB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaunitz AM, Manson JE. Management of Menopausal Symptoms. Obstet Gynecol. 2015;126:859–876. doi: 10.1097/AOG.0000000000001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842:473–481. doi: 10.1016/j.bbadis.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, Lodish HF. PPAR-α and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. 2015;522:474–477. doi: 10.1038/nature14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke U, Krones-Herzig A, Berriel Diaz M, Narvekar P, Ziegler A, Vegio-poulos A, Cato AC, Bohl S, Klingmüller U, Screaton RA, et al. The glucocorticoid receptor controls hepatic dyslipidemia through Hes1. Cell Metab. 2008;8:212–223. doi: 10.1016/j.cmet.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Li CI, Malone KE, Porter PL, Weiss NS, Tang MT, Cushing-Haugen KL, Daling JR. Relationship between long durations and different regimens of hormone therapy and risk of breast cancer. JAMA. 2003;289:3254–3263. doi: 10.1001/jama.289.24.3254. [DOI] [PubMed] [Google Scholar]
- Liu P, Ji Y, Yuen T, Rendina-Ruedy E, DeMambro VE, Dhawan S, Abu-Amer W, Izadmehr S, Zhou B, Shin AC, et al. Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature. 2017;546:107–112. doi: 10.1038/nature22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo RA, Davis SR, De Villiers TJ, Gompel A, Henderson VW, Hodis HN, Lumsden MA, Mack WJ, Shapiro S, Baber RJ. Prevention of diseases after menopause. Climacteric. 2014;17:540–556. doi: 10.3109/13697137.2014.933411. [DOI] [PubMed] [Google Scholar]
- Macfarlane DP, Raubenheimer PJ, Preston T, Gray CD, Bastin ME, Marshall I, Iredale JP, Andrew R, Walker BR. Effects of acute glucocorticoid blockade on metabolic dysfunction in patients with Type 2 diabetes with and without fatty liver. Am J Physiol Gastrointest Liver Physiol. 2014;307:G760–G768. doi: 10.1152/ajpgi.00030.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancinelli R, Onori P, Gaudio E, DeMorrow S, Franchitto A, Francis H, Glaser S, Carpino G, Venter J, Alvaro D, et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest Liver Physiol. 2009;297:G11–G26. doi: 10.1152/ajpgi.00025.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, Vierling JM American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:2193–2213. doi: 10.1002/hep.23584. [DOI] [PubMed] [Google Scholar]
- Matic M, Bryzgalova G, Gao H, Antonson P, Humire P, Omoto Y, Portwood N, Pramfalk C, Efendic S, Berggren PO, et al. Estrogen signalling and the metabolic syndrome: targeting the hepatic estrogen receptor alpha action. PLoS ONE. 2013;8:e57458. doi: 10.1371/journal.pone.0057458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013;8:1149–1154. doi: 10.1038/nprot.2013.055. [DOI] [PubMed] [Google Scholar]
- Mittelstrass K, Ried JS, Yu Z, Krumsiek J, Gieger C, Prehn C, Roemisch-Margl W, Polonikov A, Peters A, Theis FJ, et al. Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet. 2011;7:e1002215. doi: 10.1371/journal.pgen.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Ren R, Cruz-Topete D, Bird GS, Myers PH, Boyle MC, Schneider MD, Willis MS, Cidlowski JA. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc Natl Acad Sci USA. 2013;110:17035–17040. doi: 10.1073/pnas.1302546110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Busillo JM, Cidlowski JA. Cross-talk between the glucocorticoid receptor and MyoD family inhibitor domain-containing protein provides a new mechanism for generating tissue-specific responses to glucocorticoids. J Biol Chem. 2017;292:5825–5844. doi: 10.1074/jbc.M116.758888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlow AF. Effect of Ovariectomy on Pituitary and Serum Gonadotrophins in the Mouse. Endocrinology. 1964;74:102–107. doi: 10.1210/endo-74-1-102. [DOI] [PubMed] [Google Scholar]
- Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, Magomedova L, Li T, Chan JF, Budd C, et al. LXRβ is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest. 2011;121:431–441. doi: 10.1172/JCI41681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn MA, Cidlowski JA. Endogenous hepatic glucocorticoid receptor signaling coordinates sex-biased inflammatory gene expression. FASEB J. 2016;30:971–982. doi: 10.1096/fj.15-278309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Gräf S, Huss M, Keefe D, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Wang ES, Xing LL, Shi S, Qu F, Zhang D, Li JY, Shu J, Meng Y, Sheng JZ, et al. Follicle-Stimulating Hormone Induces Postmenopausal Dyslipidemia Through Inhibiting Hepatic Cholesterol Metabolism. J Clin Endocrinol Metab. 2016;101:254–263. doi: 10.1210/jc.2015-2724. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Colditz GA. Estrogen replacement therapy and coronary heart disease: a quantitative assessment of the epidemiologic evidence. Prev Med. 1991;20:47–63. doi: 10.1016/0091-7435(91)90006-p. [DOI] [PubMed] [Google Scholar]
- Stefanska A, Bergmann K, Sypniewska G. Metabolic Syndrome and Menopause: Pathophysiology, Clinical and Diagnostic Significance. Adv Clin Chem. 2015;72:1–75. doi: 10.1016/bs.acc.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Summerskill WH, Korman MG, Ammon HV, Baggenstoss AH. Prednisone for chronic active liver disease: dose titration, standard dose, and combination with azathioprine compared. Gut. 1975;16:876–883. doi: 10.1136/gut.16.11.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, Zaidi S, Zhu LL, Yaroslavskiy BB, Zhou H, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- Sun X, Luo W, Tan X, Li Q, Zhao Y, Zhong W, Sun X, Brouwer C, Zhou Z. Increased plasma corticosterone contributes to the development of alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G849–G861. doi: 10.1152/ajpgi.00139.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R, Lozoya O, Wang Y, Cardinale V, Gaudio E, Alpini G, Mendel G, Wauthier E, Barbier C, Alvaro D, Reid LM. Human hepatic stem cell and maturational liver lineage biology. Hepatology. 2011;53:1035–1045. doi: 10.1002/hep.24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe M, Go VL, Kluge D. Prednisone for chronic active hepatitis: pharmacokinetics and serum binding in patients with chronic active hepatitis and steroid major side effects. J Clin Gastroenterol. 1984;6:331–335. [PubMed] [Google Scholar]
- Wang X, Magkos F, Mittendorfer B. Sex differences in lipid and lipoprotein metabolism: it’s not just about sex hormones. J Clin Endocrinol Metab. 2011;96:885–893. doi: 10.1210/jc.2010-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West DC, Pan D, Tonsing-Carter EY, Hernandez KM, Pierce CF, Styke SC, Bowie KR, Garcia TI, Kocherginsky M, Conzen SD. GR and ER coactivation alters the expression of differentiation genes and associates with improved ER+ breast cancer outcome. Mol Cancer Res. 2016;14:707–719. doi: 10.1158/1541-7786.MCR-15-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirledge S, Cidlowski JA. Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology. 2013;154:499–510. doi: 10.1210/en.2012-1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirledge S, Xu X, Cidlowski JA. Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol Reprod. 2013;89:66. doi: 10.1095/biolreprod.113.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise PM, Ratner A. Effect of ovariectomy on plasma LH, FSH, estradiol, and progesterone and medial basal hypothalamic LHRH concentrations old and young rats. Neuroendocrinology. 1980;30:15–19. doi: 10.1159/000122968. [DOI] [PubMed] [Google Scholar]
- Woods NF, Mitchell ES, Smith-Dijulio K. Cortisol levels during the menopausal transition and early postmenopause: observations from the Seattle Midlife Women’s Health Study. Menopause. 2009;16:708–718. doi: 10.1097/gme.0b013e318198d6b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen SS, Tsai CC. The effect of ovariectomy on gonadotropin release. J Clin Invest. 1971;50:1149–1153. doi: 10.1172/JCI106587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.