

Summary
Huntington's disease (HD) is a hereditary progressive neurodegenerative disorder caused by a CAG repeat expansion in the gene coding for the protein huntingtin, resulting in a pathogenic expansion of the polyglutamine tract in the N‐terminus of this protein. The HD pathology resulting from the mutation is most prominent in the striatal part of the basal ganglia, and progressive differential dysfunction and loss of striatal projection neurons and interneurons account for the progression of motor deficits seen in this disease. The present review summarizes current understanding regarding the progression in striatal neuron dysfunction and loss, based on studies both in human HD victims and in genetic mouse models of HD. We review evidence on early loss of inputs to striatum from cortex and thalamus, which may be the basis of the mild premanifest bradykinesia in HD, as well as on the subsequent loss of indirect pathway striatal projection neurons and their outputs to the external pallidal segment, which appears to be the basis of the chorea seen in early symptomatic HD. Later loss of direct pathway striatal projection neurons and their output to the internal pallidal segment account for the severe akinesia seen late in HD. Loss of parvalbuminergic striatal interneurons may contribute to the late dystonia and rigidity.
Keywords: corticostriatal, Huntington's disease, interneurons, pathology, premanifest, projection neurons, striatum, thalamostriatal
1. INTRODUCTION
The most prominent neuropathology in HD occurs in the striatal part of the basal ganglia, in which progressive loss of projection neurons leads to severe striatal atrophy.1 Although striatal projection neuron (SPN) loss is associated with the prominent motor symptoms as HD progresses, more subtle functional deficits are evident already in premanifest stages of HD. Premanifest HD individuals are impaired, for example, in the initiation and/or execution of motor tasks.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 As striatal neuron loss is minimal in premanifest HD,1, 14, 15 early symptoms are likely to be largely driven by striatal neuron circuit level connectivity loss and dysfunction. Consistent with this, the early motor symptoms in premanifest HD develop in parallel with gradual loss of cerebral and striatal white matter,16, 17, 18, 19, 20, 21, 22, 23 increasing striatal hypometabolism,17, 24 and reduced striatal activation during behavioral tasks,25, 26 all indicative of decreased cortical input to striatal neurons. Among the major sources of input to the striatum, shrinkage and eventually marked neuronal loss are seen in deep layers of the cerebral cortex that project to striatum and in cerebral white matter.27, 28, 29, 30 Thalamus as well is a source of major input to striatum, and it shows substantial neuronal loss by late in disease.27, 29, 30 Thus, in addition to the well‐documented loss of SPNs and their outputs, abnormalities in the inputs to SPNs are likely to also contribute to symptoms, particularly early in HD. Moreover, striatal interneurons, which regulate SPN behavior, are also affected in HD, and this may have a further impact on SPN function and thereby contribute to HD pathophysiology. The present manuscript provides an overview of neuropathology and dysfunction in SPNs, in the outputs of SPNs to globus pallidus and substantia nigra, and in the inputs of SPNs from cerebral cortex, thalamus, and substantia nigra pars compacta (SNc), as revealed in human studies of HD and as suggested by animal models of HD. We also summarize information on abnormalities in striatal interneurons in HD and animal models of HD, in the outputs of striatal interneurons to SPNs, and in the inputs to striatal interneurons from cortex, thalamus, and SNc.
2. STRIATAL NEURON OUTPUT ABNORMALITIES IN HD
2.1. Striatal projection neuron abnormalities in human HD
Striatal projection neurons are GABAergic, make up about 95% of all striatal neurons, and are distributed within 2 neurochemically distinct compartments of the striatum, called the patch (or striosome) and matrix.31, 32, 33 The matrix is by far the larger of these 2 territories and is involved in motor control, while the patch neurons regulate midbrain dopamine neurons. As discussed in more detail below, SPNs in both striatal territories are affected in HD. Two major SPN types mediate the role of the matrix compartment in motor control, those rich in substance P (SP) but poor in enkephalin (ENK), and those rich in ENK and poor in SP.31, 32, 33 The SP+ SPNs project to 2 different extrastriatal regions containing pallidal‐type neurons that project to motor thalamus, the internal segment of globus pallidus (GPi), and the substantia nigra pars reticulata (SNr).31, 34, 35, 36, 37, 38, 39 Because of this bisynaptic input to motor thalamus, the SP+ SPNs are commonly termed direct pathway SPNs, or dSPNs. Of note, single‐cell labeling studies in primates and rodents indicate that dSPNs projecting to GPi and SNr are largely separate populations, which nonetheless collateralize somewhat to each other's target.36, 38, 39 In rodents, for example, GPi‐preferring dSPNs project heavily to GPi and slightly to SNr, while SNr‐preferring dSPNs project exclusively or nearly so to SNr.36, 39
To further provide evidence for differences between these 2 dSPN types, we identified several GENSAT40 lines that distinguish between the 2 different dSPN types, based on the labeling patterns for terminals in GPi and SNr and axons of passage through GPi. For example, the Phactr1 and Rasgrp2 lines appear to have selective EGFP expression in striato‐SNr dSPNs, as axons of passage through GPi are labeled and many labeled terminals are seen in the neuropil of SNr but not in the neuropil of GPi (Figures 1 and 2). By contrast, the membrane‐associated ring finger 4 (C3HC4) line (also called BC056494) and the Avpr1b line appear to show selective EGFP expression in the striato‐GPi/SNr dSPN type, as neuropil labeling is seen in both GPi and SNr but not in the axons passing through GPi (Figures 1 and 2). These findings further support a dichotomy of dSPNs into types that we will call striato‐SNr and striato‐GPi, which is further supported by the differential effect of HD on them, as discussed below. By contrast, several other GENSAT lines such as the Drd1 and Chrm4 show labeling of both striato‐SNr and striato‐GPi dSPNs, because more matrix striatal neurons are EGFP+ than in any of the above 4 lines and labeling is present in the neuropil of both GPi and SNr and in the axons of passage traversing GPi on their way to SNr (Figures 1 and 2). Single‐cell filling data36, 39 and these 4 GENSAT lines indicate that striato‐SNr dSPNs outnumber striato‐GPi dSPNs in rodents, as would be expected from the relative sizes of GPe and GPi in rodents,41 although they are likely to be more equal in numbers in primates given the more equal sizes of GPe and GPi in primates.
Figure 1.
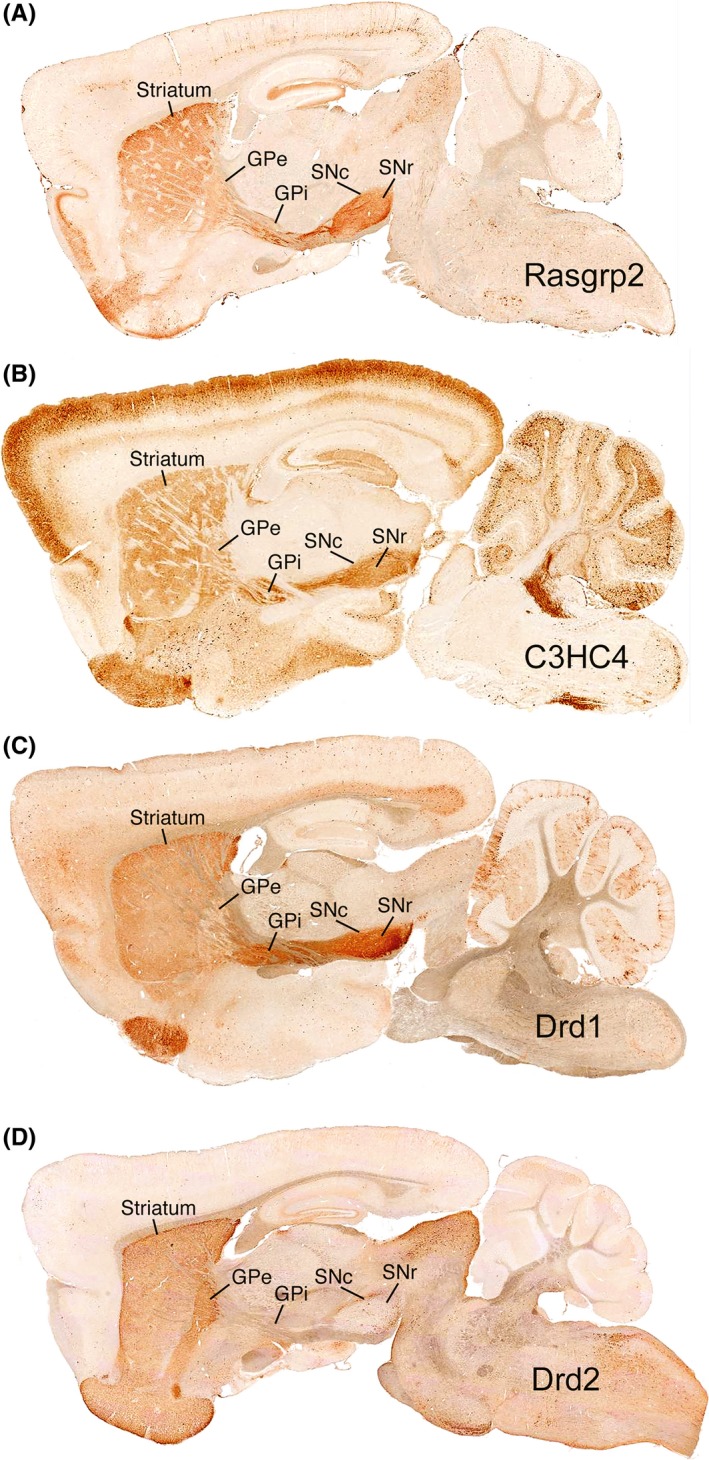
Images of matched sagittal levels for 4 different GENSAT lines (Rasgrp2, C3HC4, Drd1, and Drd2) that have GFP expression in different subsets of matrix SPNs. The images show striatum, GPe, GPi, SNc, and SNr in the single sagittal plane in all 4 cases, with GFP labeling of perikarya, axons, and terminals revealed by immunolabeling using diaminobenzidine (DAB) detection. Image A shows that labeling for the Rasgrp2 line is present minimally in terminals in GPe, but is present in axons passing through GPi and in terminals in the SNc and SNr neuropil. By contrast, image B for the C3HC4 mice shows some labeled terminals in the GPe neuropil and labeled terminals in the GPi, SNc, and SNr neuropil, but little labeling of axons passing through GPi. The Rasgrp2 mice thus appear to primarily have GFP labeling of striato‐SNr matrix dSPNs, while the C3HC4 mice have GFP labeling in matrix dSPNs projecting to both GPi and SNr. Image C for Drd1 mice shows GFP labeling of both sets of dSPNs, as they have labeling in both the GPi neuropil and in axons passing through to the nigra. Image D for Drd2 mice shows labeling of iSPNs with terminal labeling confined to GPe and some in SNc. Figure 2 shows higher magnification views of GPe, GPi, and SNr for these 4 GENSAT lines. Images were taken from the GENSAT website
Figure 2.
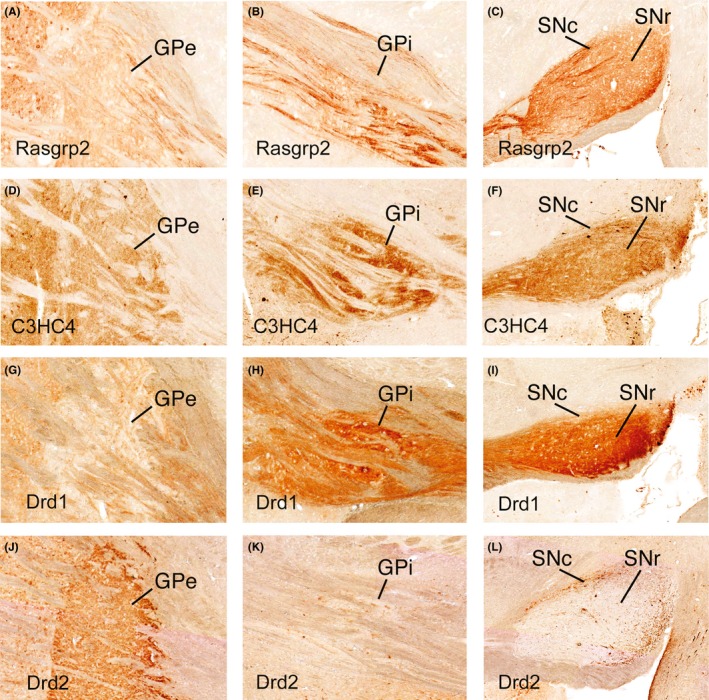
High magnification views of GPe, GPi, and SNr from the sagittal views shown in Figure 1. The images for the Rasgrp2 line (A‐C) show the labeled axons largely passing through GPi (B) on their way to the nigra, while C3HC4 mice (D‐F) have labeled terminals in the GPi neuropil, but little labeling of axons passing through GPi (E). The Drd1 mice (G‐I) show GFP labeling of both sets of dSPNs, as they show labeling in both the GPi neuropil and in axons passing through to the nigra (H). Drd2 mice (J‐L) show labeling of iSPNs with terminal labeling confined to GPe. Images were taken from the GENSAT website
The ENK+ SPNs of the matrix compartment project to the external segment of globus pallidus (GPe). Because GPe itself projects indirectly to motor thalamus via the subthalamic nucleus (STN), the ENK+ SPNs are commonly called indirect pathway SPNs, or iSPNs. A number of GENSAT mouse lines show selective and seemingly complete labeling of iSPNs and their projection to GPe, including pENK, Drd2, and GPR6 lines (Figures 1 and 2). The striatum also contains SPNs that project to SNc. These neurons also are rich in SP, but some are rich in ENK as well.31, 32 In rats, the striato‐SNc projection has been reported to arise exclusively from neurons of the striatal patch compartment,42 while in mice, a subset of neurons concentrated in the patch compartment but also present in the matrix compartment has been reported as the source of the striato‐SNc projection.43 The neurons of the striatal patch compartment and those SNc projecting neurons of the matrix appear to co‐contain SP and ENK more commonly than is the case for dSPNs and iSPNs.44
All SPNs share additional neurochemical features, but they differ in others that make it possible to distinguish dSPNs and iSPNs. Because SPNs are GABAergic, they all express the enzymes that convert glutamate to GABA, namely the 65‐kD and 67‐kD forms of glutamic acid decarboxylase (GAD).1, 31, 41 The perikarya of iSPNs and their terminals in GPe are also enriched in D2 dopamine and A2a adenosine receptors.45, 46, 47 By contrast, the perikarya of striato‐GPi dSPNs and their terminals in GPi are enriched in D1 dopamine receptors, as are the perikarya of striato‐SNr dSPNs and their terminals in SNr.45, 46, 47 Striato‐SNc neurons tend to express mu opiate receptors, resulting in the patch compartment being defined by its enrichment in such receptors,44 and coexpress D1 and D2 receptors.48 All SPN perikarya and terminals also possess CB1‐type cannabinoid receptors.49, 50, 51 Their various neurochemical traits make it possible to study the progressive effect of HD on the SPN populations, either by studying the loss of labeled terminals in SPN target areas or by loss of labeled perikarya. The iSPNs and 2 dSPN types play different roles in movement control, and it is thus valuable to characterize how HD affects them to better understand HD pathophysiology. As summarized below, the data indicate that while SPNs as a class are highly vulnerable in HD, and as a result, SPN markers are lost from striatum as disease progresses,52, 53, 54 iSPNs and striato‐SNr dSPNs are lost more rapidly than are striato‐GPi dSPNs in the human disease.
Immunohistochemical studies provided initial insight into the differential pattern of SPN loss in HD, indicating that ENK and GAD terminals in GPe, and SP and GAD terminals in the substantia nigra are lost sooner in the course of HD than are SP and GAD terminals in GPi (Figures 3, 4, 5). For example, depletion of ENK immunostaining from GPe was noted in premanifest HD.55, 56, 57 Consistent with this, striatal expression of the ENK precursor preproenkephalin (PPE) appears reduced in premanifest HD.14, 58, 59 By grade 1, ENK and GAD fibers in GPe are reduced to about 35% of control abundance and SP and GAD fibers in SNc and SNr are reduced to about 30% and 50%, respectively, of control abundance.60, 61, 62 By contrast, the loss of striatal terminals in GPi in grade 1 is considerably less, with SP and GAD fibers being 70%‐80% of control.61, 62 This pattern persists through grades 2 and 3,60, 61, 62, 63, 64 with loss of striatal terminals in GPe (~25% of normal), SNr (~25% of normal), and SNc (~30% of normal) much more substantial than that in GPi (~60% of normal). By grade 4, however, profound loss in all projection systems is apparent,63, 64 with striato‐GPe and striato‐GPi projections at about 5% of normal, and striato‐SNc and SNr projections at 10%,61 as consistent with neuropathological evidence of severe striatal neuron loss by grade 4,15 and severe loss of immunostaining for the pan‐SPN marker calcineurin in striatum and in terminals in SPN targets by grade 4.52, 65 The extent to which the projections to the striatal target areas dysfunction and reduce expression of terminal markers prior to their actual degeneration and elimination is uncertain, but clearly dysfunction and loss must be closely linked. Note that the pattern of differential loss we describe above is for adult‐onset HD, for which chorea is the presenting motor symptom. In the case of juvenile‐onset HD, rigidity is the presenting symptom, and the SPN neuropathology appears characterized by rapid and nondifferential loss of SPNs.63
Figure 3.
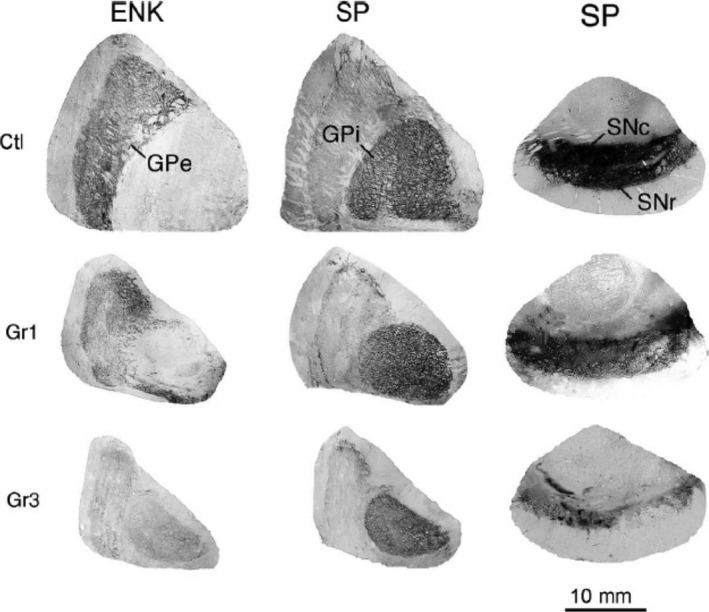
Images of immunohistochemically labeled sections showing GPi, GPe, and substantia nigra in control, grade 1 HD, and grade 3 HD cases, immunostained for SP in the case of GPi and the nigra and for ENK in the case of GPe. In the control, SP+ fibers abound in GPi, ENK+ fibers abound in GPe, and SP+ fibers abound in the nigra. In grade 1 HD, ENK+ fibers in GPe and SP+ fibers in the nigra are depleted, while SP+ fibers in GPi remain abundant. The contrast is even more evident in the grade 3 specimen, where ENK+ fibers are markedly depleted in the atrophied GPe and SP+ fibers in the nigra are sparse and patchy, but SP+ fibers in GPi are still quite prominent. This illustration is Figure 5 from Deng et al61
Figure 4.
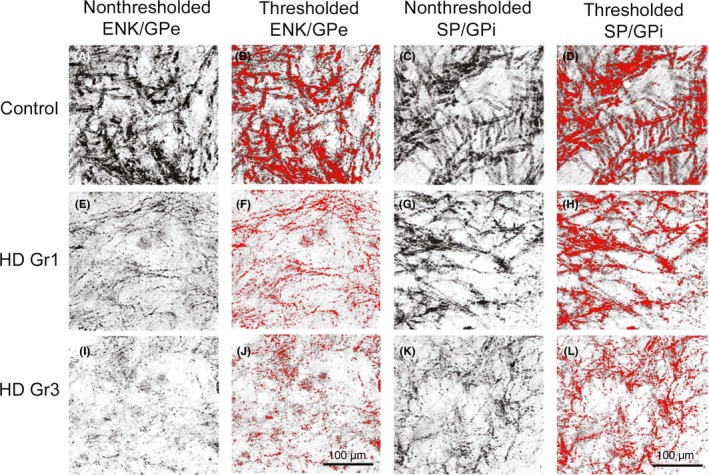
Images of ENK (A, B, E, F, I, J) and SP (C, D, G, H, K, L) immunolabeling of woolly fibers in GPe from control, grade 1 HD, and grade 3 HD brains. The images in the nonthresholded columns provide high magnification views of GPe and GPi immunolabeling, showing the great loss of immunolabeled fibers already evident by grade 1 HD in GPe but not in GPi. The thresholded columns show the same images as in nonthresholded columns, with the immunolabeled fibers thresholded in red using NIH Image. The thresholded images help to accentuate and thereby better appreciate the relative fiber loss in GPe, and similar images were used to quantify the percent of the field occupied by labeled fibers in control and HD cases. This illustration is a modified version of Figures 2 and 3 from Deng et al61
Figure 5.
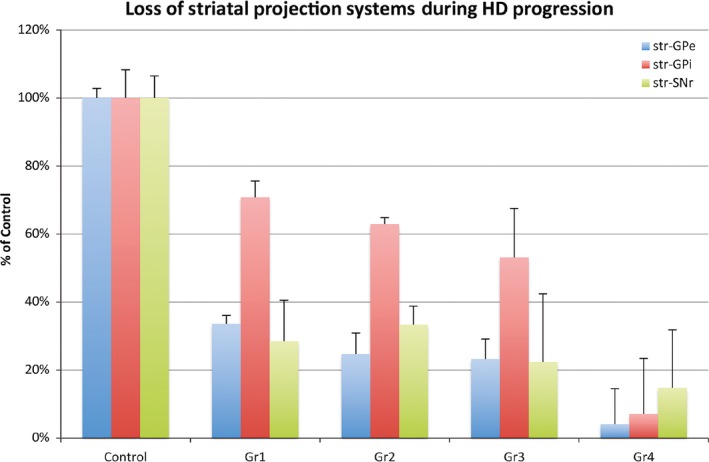
Graph showing the image analysis results for 17 Control, 4 Gr1 HD, 10 Gr2, 14 Gr3, and 7 Gr4 HD cases from Deng et al.61 Note the earlier grade‐wise loss of striato‐GPe and striato‐SNr projections than striato‐GPi projections
Further support for the differential pattern of SPN terminal loss from the striatal target areas comes from ligand binding studies for CB1 cannabinoid, D1 and D2 dopamine, and A2a adenosine receptors in striatal target areas,66 where they are found on striatal terminals. For example, CB1 cannabinoid, and D2 and A2a receptor binding is slightly reduced in GPe, but CB1 and D1 receptor binding is preserved in GPi at grade 0,66 indicating that dSPNs projecting to GPi are largely unaffected in premanifest HD, but iSPNs projecting to GPe are showing early signs of dysfunction or depletion. Consistent with this interpretation, grade 0 HD is characterized by a large increase in GABAA binding on pallidal neurons in GPe but not GPi.66 As upregulation of GABAA receptors is generally a postsynaptic response to reduced input, the preferential GABAA upregulation in grade 0 HD indicates specific reduction in iSPN input to GPe. Additionally, reduced D1 receptor binding has been observed in SNr at grade 0,66 suggesting that defects in striato‐SNr dSPNs are present already in premanifest HD. Grade 1 HD is characterized by near‐complete depletion of D2 and A2a adenosine receptors from GPe, continued upregulation of GABAA receptor binding in GPe, complete preservation of D1 receptors in GPi, greater preservation of cannabinoid receptors in GPi than GPe or SNr, and 20% loss of D1 receptors from SNr.49, 60, 67, 68 These findings are consistent with relative preservation of the striato‐GPi projection at grade 1 concomitant with considerable loss in the striato‐GPe projection, as also seen by immunolabeling. The loss in the striato‐SNr projection relative to the striato‐GPe projection is, however, relatively less than seen with immunolabeling. Grade 2 is characterized by near‐complete elimination of D2 and A2a adenosine receptors from GPe, 66% loss of D1 receptors from GPi, 69% loss of D1 receptors from SNr, and greater preservation of cannabinoid receptors in GPi than GPe.60, 66, 67 These findings are consistent with greater preservation of the striato‐GPi than the striato‐GPe projection at grade 2 seen by immunolabeling, although the finding by Glass et al60 of comparable preservation of D1 receptors in GPi and SNr at grade 2 is inconsistent with the greater vulnerability of the latter observed by immunolabeling. At grade 3, GPe is nearly devoid of cannabinoid, and D2 and A2a receptors, but about 30% of striatal D1 receptors remain in GPi, with cannabinoid receptor levels in GPi continuing to exceed those in GPe.60, 66, 67 Both GPi and SNr are, however, comparably depleted of D1 receptors by grade 3, and substantial upregulation of GABAA receptors is evident in GPi.60, 66 These findings are consistent with greater preservation of the striato‐GPi projection than the striato‐GPe at grade 3, but with evidence nonetheless of significant abnormality in the dSPN input to GPi. The data of Waeber and Palacios69 on 5HT‐1 receptors in grade 3 HD pallidum are also consistent with this conclusion, as they saw a 2‐fold preservation of presynaptic 5HT‐1 receptors in GPi compared to GPe. By grade 4, all of the various receptor markers are all greatly reduced in the striatal targets.66, 67
The immunohistochemical, in situ hybridization histochemistry (ISHH), and ligand binding findings for striato‐GPe and striato‐GPi projections in HD are consistent with biochemical assessments. For example, the radioimmunoassay (RIA) study of Seizinger et al70 reported that dynorphin32, which is colocalized with SP in striato‐GPi terminals (32), was undiminished in GPi in HD victims. By contrast, the PPE‐derived neuropeptide MERGL was only half its normal abundance in GPe in the HD brains they studied. Similarly, GABA and GAD are more greatly decreased in GPe than in GPi in symptomatic HD.71, 72, 73 Moreover, GABA is diminished in GPe in premanifest HD, while GABA in GPi remains normal.74 As striato‐GPi and striato‐GPe projection neurons are both GABAergic,31 these results too indicate a preferential loss of the striato‐GPe projection compared to the striato‐GPi projection during HD progression. Biochemical studies of SP, DYN, GABA, or GAD also indicate that striatal input to nigra is severely depleted in HD.70, 71, 72, 73, 75, 76, 77, 78, 79, 80, 81 Of note, Seizinger et al70 found that DYN in nigra and MERGL in GPe were halved in HD victims, but DYN in GPi was undiminished. The possibility that the striatal projection to SNc is differently affected in HD than that to SNr has been of interest because they arise from different striatal neuron types, and because Hedreen and Folstein57 reported that striosomal neurons, whose principal projection target is pars compacta,82 are already affected at grade 0. Judging whether the SP+ fiber loss is greater for SNc than for SNr is difficult, however, because dopaminergic neurons in primates, although concentrated in SNc, are also dispersed within the SNr as well, making it ambiguous to precisely define the boundaries of SNc.83, 84 Not surprisingly therefore, the available immunolabeling data do not unambiguously support the notion that presymptomatic HD is characterized by earlier loss in the striato‐SNc projection than the striato‐SNr projection.61 Similarly, by RIA, Beal et al75 observed extensive loss of SP from both SNr and SNc by grade 1, followed by further loss in subsequent grades, with no clear differences between them at any grade. Other biochemical studies have reported varied results, however, with some observing greater loss of SP or GABA from SNr than SNc,71, 76, 77, 79 and others the opposite.78 One study that distinguished HD cases as choreic (early to mid‐HD) vs rigid (late HD) reported greater loss of GAD from SNr than SNc in both.72 Given the inconsistent findings, it is useful to note that Tippett et al85 have reported that preferential striosomal loss (i.e, striato‐SNc neuron loss) is not invariably a trait of early HD, and appears associated with mood abnormality when it does occur. Thus, the inconsistencies regarding relative loss of the striato‐SNr vs striato‐SNc projection may stem from variation among HD victims in loss of striato‐SNc neurons, as well as difficulties in defining the boundary between SNc and SNr.
The loss of the iSPN ENK fibers from GPe, with the relative preservation of the dSPN SP fibers in GPi, predicts that SP neuron survival should be better than ENK neuron survival in HD striatum. Direct support for this comes from ISHH for SP and ENK mRNA in HD striatum14, 58, 59, 86, 87 and from ligand binding for D1 and D2 dopamine58, 59, 66 and A2a adenosine receptors66 in HD striatum. For example, neurons expressing mRNA for the SP precursor (i.e, preprotachykinin, or PPT) are more abundant in striatum during grades 1‐3 HD than are neurons expressing mRNA for PPE.86, 87 Similarly, grade 0 HD has been found to be characterized by loss of D2 and A2a receptor binding from striatum and relatively better preservation of D1 receptor binding in striatum, consistent with the idea that iSPNs but not dSPNs show dysfunction and possibly loss already in premanifest HD.66 The reductions in SP perikarya in striatum and D1 receptor binding in SNr already at grade 0,14, 49 however, suggest that some defects are present in premanifest HD in striato‐SNr dSPNs that are not yet evident in striato‐GPi dSPNs. Grades 1‐2 in HD are characterized by 80%‐95% loss of striatal D2 dopamine and A2a adenosine receptors (localized to iSPNs) but only 50% loss of striatal D1 receptors (localized to dSPNs).60, 66, 67, 68 At grade 3, striatum and GPe are nearly devoid of cannabinoid, D2 and A2a receptors, but about 30% of striatal D1 receptors remain.60, 66, 67 These findings are consistent with greater preservation of dSPNs than iSPNs over grades 1‐3. By grade 4, these various receptor markers are all greatly reduced in striatum and its targets,66, 67 consistent with the extensive loss of all SPN types by grade 4 suggested by studies of various striatal terminal markers.
2.2. Striatal projection neuron abnormalities in animal models of HD
The studies of human HD striatal neuropathology establish the greater vulnerability of iSPNs than dSPNs, beginning early in HD progression, with the dSPNs projecting to GPi being especially resistant compared to the striato‐SNr dSPNs. This feature of HD is then a hallmark neuropathology by which to judge the efficacy with which models of HD mimic the adult HD pathogenic process. Among the various rodent and large mammal models of HD, dSPN vs iSPN pathology has been most thoroughly assessed in mouse models, on which we will focus. In the case of the best‐studied mouse model of HD, the mutant exon‐1 transgene‐expressing R6/2 mouse, PPE mRNA is decreased in symptomatic 6‐ and 12‐week‐old R6/2 mice, while striatal PPT mRNA expression is better preserved (Figure 6).88, 89, 90, 91 Also consistent with the early preferential loss of iSPN markers from striatum in adult‐onset HD, Cha et al92 reported that both A2a and D2 receptors are decreased in 8‐ and 12‐week‐old R6/2 striatum. Our quantitative analysis showed that for 12‐week‐old R6/2 mice, the percentage of striatal neurons expressing PPE is halved, and the percentage expressing PPT is reduced by 25% by this age.91 We also found about a 20% decrease in ENK fiber staining in GPe at 12 weeks, but surprisingly about a 10%‐15% increase in SP fiber staining intensity in SN at 6 and 12 weeks. Note that this same greater iSPN neurochemical pathology is also evident in R6/2 mice with extreme expansions to greater than 325 CAG that slow progression and extend lifespan.93, 94 Thus, although the CAG repeat length, widespread aggregate formation, and early mortality of R6/2 mice simulate juvenile‐onset HD, their SPN pathology largely mimics adult‐onset HD. In contrast to R6/2 mice, R6/1 mice, which possess the same transgene as R6/2 mice but for unknown reasons are more slowly progressing, have been reported to show comparable levels of decline in striatum for iSPN markers (ENK, D2, and A2A) and dSPN markers (SP and D1).92, 95, 96, 97 In an HD mouse with a longer transgene fragment and 82 CAG repeats,98 4‐month symptomatic N171‐82Q mice showed greater iSPN than dSPN pathology compared to WT or N171‐18Q mice. For example, striatal D2 ligand binding was 61% of control, A2A ligand binding was 71% of control, D2 ISHH was 61% of control, and PPE ISHH was 59% of control. Abnormalities in dSPN striatal markers were not noted, suggesting that the reduction in DARPP‐32 ISHH to 62% of control mainly occurred in iSPNs.89
Figure 6.
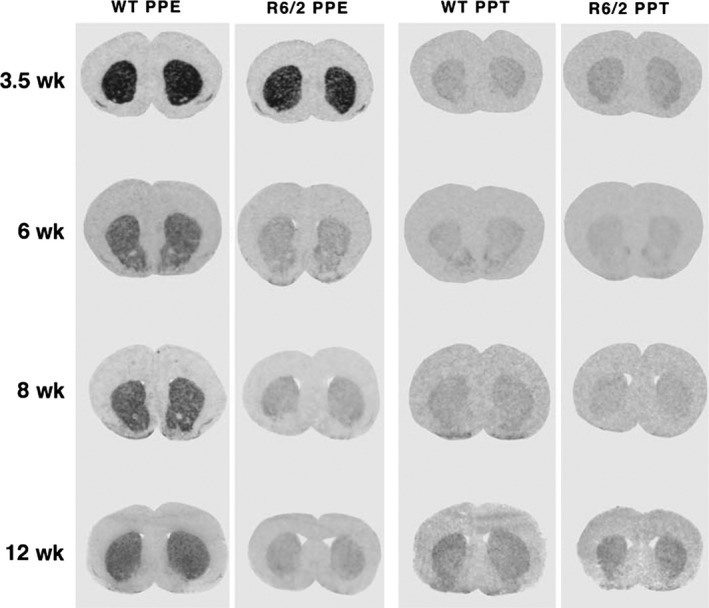
Images of coronal sections through rostral striatum labeled using in situ hybridization histochemistry, showing that striatal PPE mRNA levels but not PPT mRNA levels were reduced in R6/2 mice. Images of film autoradiograms for pairs of R6/2 and wild‐type littermate mice at 3.5, 6, 8, and 12, weeks of age are shown. A large PPE mRNA reduction is evident in R6/2 mice at 6‐12 weeks of age. No PPT mRNA changes in R6/2 mice are obvious at any age. This illustration is Figure 1 from Sun et al.91 Note that the autoradiographic analyses in this study were performed using radioactivity standards that allowed us to quantify the changes in PPE and PPT expression with age. This analysis showed PPE signal in WT declined from 3.5 to 6 weeks, but somewhat rebounded thereafter. In the case of PPT, signal was less at 6 and 8 weeks than at 3.5 weeks, but rebounded slightly at 12 weeks—see Figure 2 in Sun et al91
Several full‐length mouse models have been evaluated for differential effects on iSPNs and dSPNs as well, all of which show slower progression than is the case for the above‐truncated mutant huntingtin transgene models. In the case of YAC128 transgenic mice, Xie et al99 reported that message for D2 and PPE was down to 75% of WT in YAC128 striatum at 16 months of age, but D1 and PPT were relatively normal. Benn et al,100 however, reported no reductions in striatal D1 or D2 in 12‐month‐old YAC128 mice. Pouladi et al101 presented yet more discordant findings, reporting that striatal D1, D2, PPE, and DARPP‐32 were all reduced to around 50% of WT at 10 months of age in YAC128 mice. The basis of these inconsistencies is unclear, and it is thus uncertain whether YAC128 striatum shows differential effects on iSPNs and dSPNs, but the weight of evidence suggests not. In transgenic BACHD mice, however, it is clear that there is no evidence for differential SPN pathology, as Pouladi et al101 reported no D1, D2, PPE, or DARPP‐32 reduction at 10 months of age, and Miller et al102 saw no change in striatal D1 or D2 levels at 12 months. Thus, neither of these 2 full‐length transgenic models obviously replicates the hallmark differential SPN pathology of adult‐onset HD in humans. By contrast, full‐length knock‐in mouse models of HD do show such differential pathology. For example, Q140 mice form mutant huntingtin aggregates in striatum as early as 2 months of age,90 with mice clearly symptomatic by 6 months of age. For these mice, Stefanko et al103 showed by Western blot that at 4 and 6 months of age, striatal D1 receptors are normal in abundance, but D2 receptors are reduced by 15% and 20%, respectively. Similar results are seen in heterozygous Q175 knock‐in mice, with our studies showing significant reduction in ENK message in striatum by 6 months of age but no SP reduction.104, 105 The ENK reduction is progressive out to 18 months of age, but no SP message loss is observed even at 18 months. The ENK reduction is associated with a reduction in DARPP‐32 in iSPNs, but neuron counts show that these reductions reflect reduced expression rather than loss of iSPNs per se. Consistent with the idea that diminished ENK expression alone is sufficient for symptoms, Bissonnette et al106 showed that striatal overexpression of PPE reduces deficits in R6/2 mice. The overall results suggest that transgenic models expressing truncated mutant huntingtin and knock‐in models expressing full‐length mutant huntingtin from the endogenous mouse site produce differential SPN pathogenesis that mimics that in adult humans, but for uncertain reasons, the YAC128 and BACHD full‐length transgenic models do not. Nonetheless, YAC128 and BACHD mice do show SPN pathology and/or neuron loss,99, 101 albeit nondifferential, as characteristic of juvenile‐onset and end‐stage adult‐onset HD.
2.3. Functional implications of differential vulnerability of striatal projection neurons and their outputs in HD
The early loss of iSPNs and their output to the GPe are thought to account for the chorea seen as the presenting symptom in adult‐onset HD, according to the now standard direct‐indirect pathway model of basal ganglia function (Figure 7).61, 107, 108 The early loss of iSPN terminals in GPe was, in fact, part of the key early evidence in support of the model.107 Given that each type of SPN is organized into microzones that interweave with other types within striatum,109, 110 the preferential loss of some types of striatal projection neurons in early HD may be why diverse striatal projection neuron markers show nonuniform loss from striatum.58, 59, 65, 66, 67, 86, 87, 111 Loss of striato‐SNr neurons by grade 1 may cause the abnormalities in saccades seen in early HD, because SNr plays a role in saccadic eye movements.112 By grade 3, considerable loss of striato‐GPi dSPNs occurs, and this loss may contribute to the bradykinesia that develops late in HD, while the near‐complete loss of this projection system by grade 4 is likely to explain the akinesia in terminal adult‐onset grade 4 HD (Figure 7).63, 107 The apparently largely equal rapid pace of dSPN and iSPN loss in juvenile‐onset HD appears to explain why rigidity rather than chorea is the presenting symptom for this form of HD. The functional implications of striato‐SNc neuron loss are uncertain, but Tippett et al85 report that loss of these neurons is associated with mood abnormalities in HD patients. Moreover, loss of their inhibitory drive to SNc dopaminergic neurons may yield their hyperactivity, which would promote chorea by exacerbating hypoactivity of iSPNs and enhancing activity of dSPNs. Although differential loss is evident for the 4 main striatal projection systems, imaging studies assessing brain volume, glucose metabolism, or receptors on striatal projection neurons or their terminals emphasize that neither the striatum itself nor any one SPN type is completely normal even in premanifest HD.24, 58, 59, 66, 113, 114, 115, 116, 117
Figure 7.
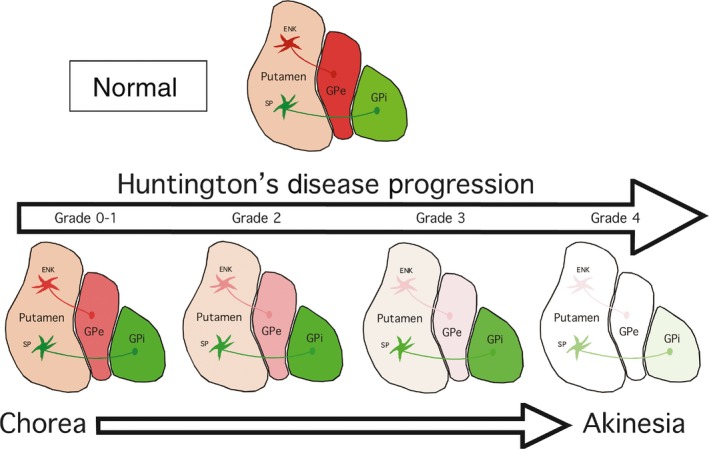
Schematic illustrating the relative grade‐wise loss of striato‐GPe iSPNs and striato‐GPi dSPNs in HD, and the relationship of this differential progression pattern to HD motor signs
The mutant rodent models provide an opportunity to examine the abnormal SPN physiology that may disrupt the output influence of striatum on its target areas prior to or during ongoing SPN loss and thereby contribute to pathophysiology. Many studies have reported increased excitability of SPNs in HD mouse models.118 For example, Klapstein et al119 reported that SPNs in R6/2 mice have depolarized resting membrane potentials, reduced inwardly rectifying potassium currents, and a lower threshold for action potentials by 12.5 weeks of age. Similarly, Rebec and co‐workers have reported increased firing of SPNs in intact and awake 6‐ to 9‐week‐old R6/2 mice, but reduced burst firing and reduced synchronous firing.120, 121 In vitro analysis has shown that the resting membrane potential of SPNs in Q175 mice also is more depolarized and more excitable as mice age.122 These studies, however, have not clarified whether the increased excitability is true of both types of SPN.119, 121 Sebastianutto et al123 recently examined the electrophysiological behavior of iSPNs in 2 of the mouse models showing greater iSPN than dSPN neurochemical pathology, namely 325CAG R6/2 mice and Q175 mice, with the HD mice crossed with a reporter line expressing EGFP in iSPNs (BAC‐adora2a‐eGFP) to allow identification of iSPNs during slice recordings in each model “before” and “after” the onset of motor deficits. In both models, iSPNs showed an early increase in excitability, driven by reduced inwardly rectifying potassium channel currents and a loss of primary dendrites. Following the onset of motor deficits, iSPNs in HD mice additionally showed loss of dendritic spines. Increased excitability of iSPNs predicts increased firing of these neurons in response to excitatory inputs in vivo. Note, however, as they did not examine dSPNs in this study, it is uncertain whether they do or do not show the enhanced excitability found in iSPNs.123 Thus, while increased excitability and disrupted burst firing would interfere with striatal control of its target areas, it is uncertain whether the disruption differs between dSPNs and iSPNs and is a cause of any differential functional deficits in the direct and indirect striatal output pathways.
2.4. Basis of differential striatal projection neuron vulnerability in HD
The attributes that make striato‐GPi dSPNs more resistant than striato‐GPe iSPNs, striato‐SNr dSPNs, and striato‐SNc neurons in adult‐onset HD and mouse models mimicking adult‐onset HD are not known, although considerable attention has focused on the role of glutamate receptor subunit configuration, free radical defenses, calcium sequestering, antiapoptotic mechanisms, BDNF dependence, and differences in their cortical input.57, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141 The mutant protein in its free or aggregated form might selectively reduce PPE by causing preferential injury to iSPNs, or initially by causing preferential dysregulation of PPE transcription.1, 89, 92, 142 With regard to whether cell autonomous pathogenesis differentially affects dSPNs and iSPNs, the work of Ruiz‐Calvo et al137 is of interest. They showed that viral vector‐driven expression of mutant huntingtin in dorsal striatum alone for 2 weeks produced parallel loss of dSPNs and iSPNs. They went on to show that the differential effect of HD on iSPNs and dSPNs appears driven from an action of the mutation on corticostriatal neurons. These findings will be discussed in more detail in the section below on evidence implicating disturbances in the cortical input to striatum in the genesis of differential SPN abnormalities.119, 135, 136, 137, 140, 143, 144, 145 Of further interest regarding SPN pathogenesis is that striatal pathology is more prominent caudally than rostrally in early disease, and striatal degeneration proceeds, for yet unknown reasons, in a dorsomedial to ventrolateral direction.146, 147 Consistent with this, shrinkage of the medially situated caudate is significant already 10 years from estimated disease onset, while shrinkage of the more laterally placed putamen is not significant until 3 years before estimated disease onset.115 The basis of this regional difference in SPN vulnerability is uncertain, but it could be that the tendency of striato‐SNr dSPNs to be more medially located in primates and striato‐GPi dSPNs more laterally may account for the greater vulnerability of the former compared to the latter.37, 148, 149
2.5. Striatal interneuron output abnormalities in human HD and animal models of HD
Striatal interneurons are said to make up about 5% of all striatal neurons and include 4 major well‐characterized types: (i) very large aspiny cholinergic neurons150, 151; (ii) large aspiny neurons that contain GABA and parvalbumin (PARV)152, 153; (iii) medium‐sized aspiny neurons that contain GABA, somatostatin (SS), neuropeptide Y (NPY), and nitric oxide synthase (NOS)151, 153; and (iv) medium‐sized aspiny neurons that contain GABA and calretinin (CALR).151, 154, 155, 156, 157 All striatal interneuron types are known to modulate striatal projection neurons, and thus, loss or dysfunction of their output to SPNs during HD would be expected to contribute to HD pathophysiology.151, 152, 158, 159 As discussed in more detail below, only PARV interneurons appear to be lost during the course of HD. Although cholinergic and SS/NPY striatal interneurons remain preserved throughout the course of HD, their connectivity and functioning are altered, nonetheless. The situation for calretinergic interneurons is more complex, as addressed below. The striatum also contains a small population of GABAergic interneurons that express tyrosine hydroxylase (TH). Little is known about this interneuron type,159 and they are further discussed in the section on the GABAergic interneurons cocontaining SS, NPY, and NOS.
Cholinergic interneurons represent 1%‐2% of all striatal neurons in humans and nonhuman mammals and correspond to the tonically active striatal neurons detected in electrophysiological studies.160, 161, 162 Cholinergic interneurons innervate both dSPNs and iSPNs and modulate SPN firing and reward‐related (i.e, dopamine release related) plasticity at corticostriatal synapses.161, 162, 163 Cholinergic interneurons exert differential effects on iSPNs and dSPNs in normal striatum, with cholinergic neuron activation increasing the responses of iSPNs relative to dSPNs to cortical drive.162 Cholinergic striatal interneurons survive in intact numbers late into human HD, as well as in diverse animal models of HD.63, 124, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174 The preservation of cholinergic interneurons in HD striatum is, nonetheless, accompanied by diminished expression of such cholinergic neuron markers as choline acetyltransferase (ChAT)173, 175 and the vesicular acetylcholine transporter (VAChT).176 Similarly, Smith et al176 reported that despite a normal number of cholinergic interneurons in R6/1 striatum, the levels of both ChAT and VAChT are markedly decreased early in the life span. Studies of other mouse HD models, as well, have reported preserved neuron numbers but depressed ChAT activity and VAChT binding.177, 178 Holley et al177 reported enhanced inhibitory responses in striatal cholinergic interneurons in R6/2 mice, which could exacerbate the presumed reduced acetylcholine release and influence on SPNs caused by the ChAT and VAChT reductions. Moreover, cholinergic interneurons contain VGLUT3 and have a glutamatergic influence on parvalbuminergic interneurons,179, 180 which could be diminished if cholinergic interneurons are less active and/or make less VGLUT3 in HD. We examined striatal cholinergic interneurons in heterozygous Q140 males at 1 and 4 months of age, using ChAT immunolabeling to identify cholinergic interneurons.166 Blinded neuron counts confirmed that ChAT+ perikarya were normal in abundance, but size measurements showed they were significantly smaller. Sholl analysis revealed that the dendrites of Q140 ChAT+ interneurons were significantly fewer and shorter (Figure 8), and their paucity was confirmed by ultrastructural analysis. As the diminished dendritic branching was associated with decreased net excitatory input per interneuron,166 as detailed in the section on thalamic inputs to interneurons, the thereby reduced excitatory drive is a likely contributor to reduced acetylcholine release from these neurons. Loss of excitatory drive would be predicted to more greatly lessen the responses of iSPNs than dSPNs to cortical drive,162 which models of basal ganglia function predict should cause hyperkinesia.107 As these defects in cholinergic neurons seem to be early occurring, they may contribute to the early hyperkinesia of adult‐onset HD. Moreover, since striatal cholinergic interneurons express high levels of huntingtin,143 these defects may be intrinsically driven by the mutation. Note, however, that any cell autonomous effect of the huntingtin mutation on cholinergic interneurons is not lethal for the neurons, despite the high production of mutant protein.
Figure 8.
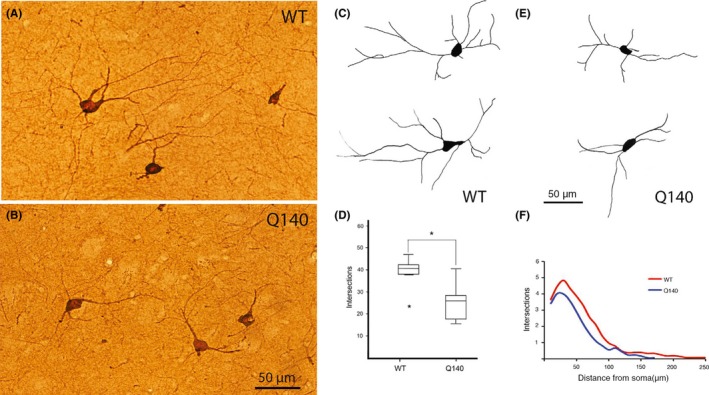
Images of cholinergic interneurons immunostained for ChAT in the striatum of WT (A) and Q140 (B) mice, showing the slightly smaller and lesser dendritic branching in the Q140 mice. Images C‐F show representative camera lucida drawings of cholinergic interneurons in the striatum of WT (C) and Q140 (E) mice, a box‐and‐whisker plot showing total dendrite intersections (D), and a graph showing dendrite intersections as a function of distance from the soma (F) in WT and mutant mice based on Sholl analysis. The results show that dendritic arborizations of cholinergic interneurons were significantly decreased in Q140 heterozygous mice. One outlier (asterisk) is evident for the WT mice in D. These illustrations are Figures 2 and 3 from Deng and Reiner166
The striatal interneurons cocontaining SS, NPY, and NOS, also called low‐threshold spiking (LTS) interneurons, are GABAergic and make up about 0.5%‐1% of all striatal neurons.159, 181 They receive a weak cortical input and have a modulatory somatostatinergic and weak GABAergic influence on SPNs.182 By means of NO release acting on SPN cGMP signaling, LTS interneurons also facilitate SPN responses to cortical activation.183, 184 No information is available on whether any of these LTS interneuron influences are differential for the 2 primary SPN types. As detected by somatostatin or NPY immunostaining or protein assays, LTS interneurons are resistant in HD, and survive seemingly undiminished late into disease (Figure 9).63, 124, 164, 165, 167, 168, 169, 170, 171, 172 This also appears true in animal models of HD.185, 186 Although somatostatinergic neuron abundance does not decline in HD striatum, NOS and somatostatin expression is progressively diminished.174 Moreover, LTS interneurons in at least some mouse HD models (R6/2 and BACHD) have been shown to be more spontaneously active than normal (R6/2 and BACHD).187 Nonetheless, SPN responses to activation of LTS interneurons were normal in these same mice.187 More recent studies using genetically engineered mice have found that about 20% of NPY‐expressing interneurons have unique neurochemistry (i.e, are devoid of SS and nNOS) and distinct electrophysiological properties, and are termed neurogliaform cells (NGF) due to their morphology.185, 188 NPY‐NGF interneurons are connected to SPNs with high probability and give rise to long‐lasting IPSCs (inhibitory postsynaptic currents) in SPNs with very slow decay. Although the fate of NPY‐NGF interneurons in HD in humans or animal models has not been determined, the observation that NPY‐NGF interneurons are activated by acetylcholine released from cholinergic interneurons indicates that HD‐related abnormalities in striatal cholinergic interneurons would lead to abnormalities in the influence of NPY‐NGF interneurons on SPNs. Note that Muñoz‐Manchado et al189 recently used the 5HT3aEGFP mouse to show that striatum also contains NPY‐negative neurogliaform cells and SS/NPY/nNOS‐negative LTS cells. The unique roles of each of these interneuron types in striatal function and the impact of the HD mutation on them remain to be determined. With regard to TH+ striatal interneurons, which may be a subtype of LTS interneuron,185 whose role is also uncertain, they appear to be reduced in HD striatum,190 although this may represent expression loss rather than neuron loss per se.
Figure 9.
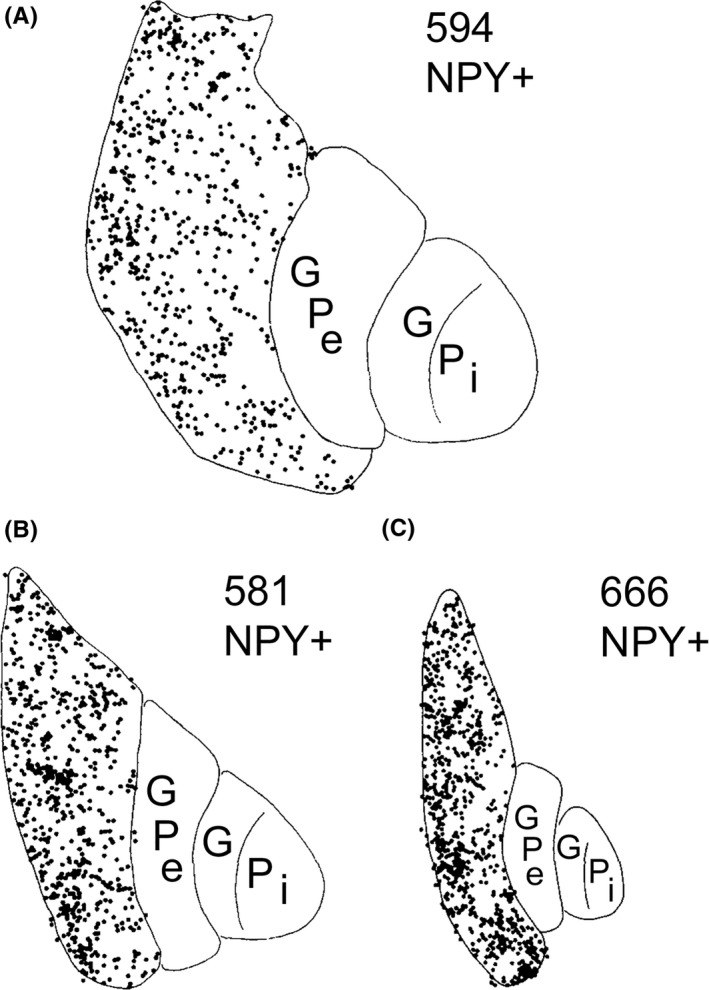
Camera lucida reconstructions of the distributions of neuropeptide Y‐immunoreactive (NPY+) neurons at comparable levels of the basal ganglia in a normal juvenile (A), a choreic juvenile‐onset HD grade 3 case (B), and a rigid juvenile‐onset HD grade 4 case (C). Although the number of NPY‐immunoreactive perikarya in putamen is similar for all 3 cases, shrinkage of the putamen greatly increases the packing density of these neurons in grade 3 and 4 HD. This illustration is Figure 2 from Albin et al63
PARV+ interneurons, also called fast‐spiking interneurons (FSIs), make up about 1% of striatal neurons, receive prominent cortical input, have heavy input to the proximal dendrites of nearby dSPNs and iSPNs, and exert strong inhibitory control over SPNs.181, 191, 192, 193, 194 Based on their connectivity and rapid cortical responses, FSIs are thought to inhibit striatal projection neurons in a feedforward manner as part of the process of switching from one movement to the next in a motor sequence.195, 196 Two types of FSIs have been identified based on their responses to depolarizing current injection (continuous vs stutter firing) in rats and mice, although the stuttering type is less common in mice.159 It is uncertain whether these 2 types have differential connectivity with dSPNs and iSPNs. Except for one study on R6/2 mice,197 PARV striatal interneurons have consistently been reported as preserved in mouse HD models.198, 199 We have made similar observations of PARV interneuron preservation in our own studies of R6/2 and Q175 mice,104, 105 although dendritic branch reduction has been reported.199 PARV interneurons do, however, show abnormal function in mouse models, as stimulation of PARV interneurons in R6/2 and BACHD mice induces significantly larger amplitude SPN inhibition than in control mice.187 Depending on whether this effect on SPNs is differential, it could contribute to early hyperkinesia if disproportionately affecting iSPNs early and late hypokinesia if subsequently disproportionately affecting dSPNs late. Until recently, published data on PARV striatal interneurons in human HD were limited but suggested that they may be lost from the striatum as HD progresses.200, 201 Given the important role such loss could play in the progression of HD symptoms, we used immunolabeling for PARV in fixed sections to assess this issue in more detail.202 In our study, we corrected for disease‐related striatal atrophy by expressing PARV+ interneuron counts in ratio to interneurons cocontaining somatostatin and neuropeptide Y (whose numbers are unaffected in HD). At all symptomatic HD grades, PARV+ interneurons were reduced to <26% of normal abundance in rostral caudate. In rostral putamen, loss of PARV+ interneurons was more gradual, not dropping off to <20% of control until grade 2. Loss of PARV+ interneurons was even more gradual in motor putamen at globus pallidus levels, with no loss at grade 1, and steady grade‐wise decline thereafter (Figure 10). Given the findings of some animal studies indicating that insufficient numbers of PARV+ striatal interneurons or their inactivation can cause dystonia,185, 192, 203, 204 and given the grade‐wise loss of PARV+ striatal interneurons in motor striatum in our human data in parallel with the grade‐wise appearance and worsening of dystonia, our results raise the possibility that loss of PARV+ striatal interneurons is a significant contributor to dystonia in HD. Note, however, other studies in mice have reported that inactivation of striatal PARV interneurons does not cause dystonia,205 and a 35% deficiency in PARV+ putamen interneurons was reported in Tourette syndrome patients without signs of dystonia.206, 207 Moreover, studies of mutant mouse Dyt1 dystonia models have suggested that it is loss of striatal cholinergic interneurons that causes dystonia in those models,208 although other researchers have not observed dystonia following selective ablation of striatal cholinergic interneurons.209, 210 Thus, the consequences of PARV interneuron loss for the pathophysiology of HD remain uncertain, as does the genesis of dystonia in later stages of HD.
Figure 10.
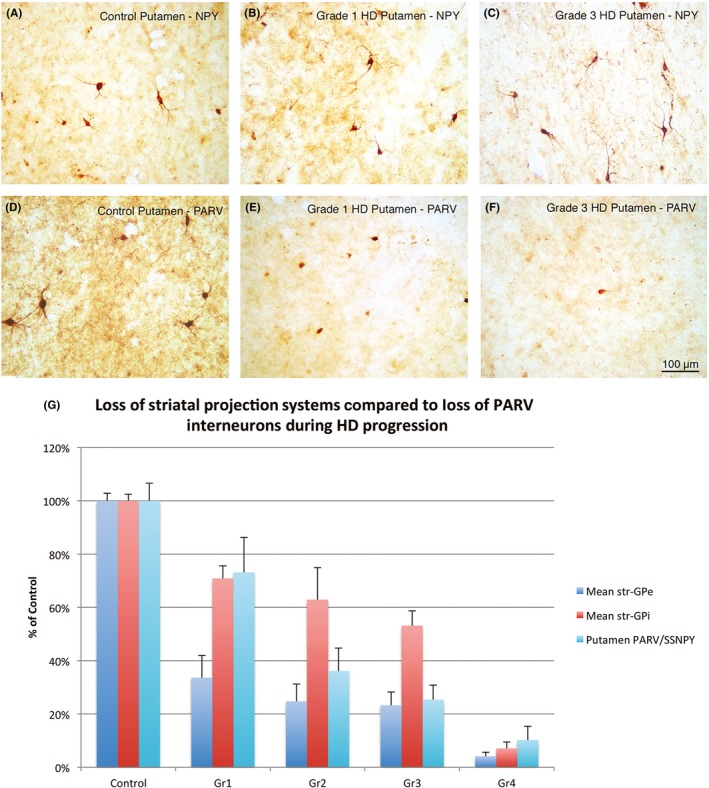
Series of images showing NPY striatal interneurons (A‐C) and PARV striatal interneurons (D‐F) in putamen of a control case (A, D), grade 1 HD case (B, E), and grade 3 HD case (C, F). Note that NPY neurons show no alteration in size, abundance, or labeling intensity with HD progression, while PARV interneurons show progressive decline in all 3 parameters. Magnification is the same in all images. Images A‐F represent Figure 1 in Reiner et al202 The graph (G) shows the relative grade‐wise loss of PARV striatal interneurons in putamen compared to grade‐wise loss of striato‐GPe and striato‐GPi projections, based on data in Reiner et al202 and Deng et al61
CALR+ interneurons are GABAergic, possess medium‐sized perikarya, and in rodents make up about 0.5% of all striatal neurons.151, 154, 156, 211 In rodent studies, the CALR+ interneurons have been shown to receive excitatory input and appear to form terminals on SPNs.154 In primate striatum, however, CALR+ neurons include both large and medium‐sized neurons as well and make up about 7% of all neurons.160, 212, 213, 214, 215 The vast majority of the large CALR+ neurons in normal human striatum coexpress ChAT and neurokinin‐1 receptor (~90%) and thus represent cholinergic interneurons,155, 213, 216, 217 with ~70% of striatal cholinergic neurons expressing CALR in humans173 and about 30% in rhesus monkeys.160 This may explain why the large CALR+ interneurons have been reported to survive well in HD, albeit with reduced ChAT expression, since as noted above cholinergic interneurons survive well in HD.155, 173, 216 The medium‐sized CALR+ interneurons of primate striatum appear to include both projection neurons and interneurons, with our studies in rhesus monkey indicating that CALR+ interneurons make up 1.5% of all striatal neurons and about 4% of SPNs being CALR+.160 The presence of CALR+ projection neurons in rhesus is consistent with evidence showing that some medium‐sized CALR+ neurons in humans possess spiny dendrites and thus appear to be SPNs.155, 214 Note that medium‐sized CALR neurons have also been reported to survive in HD.216, 217 It would be of interest to know whether both CALR+ interneurons and the CALR+ SPNs survive, because this could provide insight into pathogenesis, particularly if the CALR+ SPNs constitute an HD‐resistant subset of SPNs. Consistent with CALR+ interneuron survival in HD and a possible contribution to HD pathophysiology, striatal CALR expression is substantially increased in 3‐ to 6‐month‐old YAC128 mice.198
3. STRIATAL NEURON INPUT ABNORMALITIES IN HD
The striatum receives its major inputs from cerebral cortex, thalamus, and the SNc. Each of these is affected during HD progression, particularly the excitatory cortical and thalamic inputs, and this is likely to have an effect on striatal neurons and thus basal ganglia function. The input from cerebral cortex, which arises from all cortical regions to a greater or lesser extent, is the more substantial of the 2 excitatory inputs and is thought to provide striatum with an instructive signal for its role in motor control.82, 218 The thalamic input, which arises heavily from intralaminar, mediodorsal, and midline thalamic nuclei,219, 220 however, also provides drive to striatum that is essential to its role in movement selection and facilitation.162, 221, 222, 223 The dopaminergic input from SNc is critical for the activity of dSPNs and iSPNs and their opposing roles in motor control. Abnormalities in cortical, thalamic, and dopaminergic input to each major striatal neuron type are reviewed below.
3.1. Abnormalities in overall cortical input to striatum in human HD
Cerebral cortex undergoes neuron loss and shrinkage in HD, but more slowly and in the end less so than does striatum.15, 27, 224, 225, 226, 227 The loss occurs mainly among pyramidal projection neurons in layers 3, 5, and 6, is evident over grades 2‐4, and is prominent in grade 4.228, 229, 230 For example, Hedreen et al229 noted 57% neuron loss in layer 6, and 71% loss in layer 5 in grade 4 HD. Differences in regional neuron loss and thinning in HD cerebral cortex have been described by various authors. Primary motor and premotor cortices both consistently show 40%‐50% pyramidal neuron loss in late HD.230 On the other hand, Selemon et al227 reported that prefrontal cortex area 9 showed neuron loss but not prefrontal cortex area 46, but both showed shrinkage. Sotrel et al231 reported that surviving pyramidal neurons of layers 3 and 5 in prefrontal cortex showed dendritic augmentation, reflecting perhaps compensation for the loss of other neurons from those layers. MRI and CT imaging studies show results similar to these, revealing that the sensorimotor, insular, and opercular cortices show the most thinning, while frontal and temporal cortices show relatively less.231, 232, 233, 234 Of note, thinning in sensorimotor, insular, and opercular cortices was manifest even before overt HD motor symptoms and associated with decline in cognitive function as measured by the UHDRS.235 Heterogeneity in HD in motor vs mood symptoms appears in part attributable to regional variation in cortical neuron loss, as a significant association between motor dysfunction and neuronal loss in primary motor cortex is seen in HD.236 Moreover, mood disturbance is associated with neuronal loss in anterior cingulate cortex.236 Braak and Braak237 reported loss of entorhinal cortex neurons in advanced HD, suggesting a basis for memory deficits in late HD. MRI and fMRI studies show that the degree of cortical thinning is related to disease progress and to CAG repeat length232, 238 and seems to yield loss of input to striatum.239, 240
Alterations in neurotransmitter release also occur for neurons of cerebral cortex, as evidenced by biochemical analysis of cortical tissue. For example, a loss of various presynaptic proteins, such as the soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) protein, synaptosome‐associated protein 25 (SNAP25), and the vesicle docking and recycling protein rabphilin 3a, occurs in frontal cortex in HD grades 1‐4.241, 242 These losses are not due to a general loss of synapses in HD cortex.241 Similarly, Zucker et al243 showed that layer 5 motor cortex neurons in HD make less Lin7 homolog b (Lin7b), which is a scaffold protein implicated in synaptic plasticity and neurite outgrowth. In HD, uptake of glutamate was found to be reduced by 43% in prefrontal cortex, with the defect increasing in severity with CAG repeat expansion.244 These changes in synaptic proteins are likely to impair neuronal communication in cerebral cortex, and similar changes are likely for cortical terminals in striatum.
3.2. Abnormalities in cortical input to striatal projection neurons in humans and mutant mouse models of HD
Numerous studies have noted that premanifest HD individuals are slowed in the initiation and/or execution of a variety of motor tasks involving the eyes, hands, or lower limbs.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 This defect is mild in premanifest cases not yet near clinical onset, but more severe in those near onset.2, 8, 9, 10, 245 Diverse types of imaging studies (computed tomography, magnetic resonance imaging, positron emission tomography, and diffusion tensor imaging) show that these growing motor symptoms in premanifest HD occur in parallel with a slowly progressive loss of cerebral and striatal white matter,16, 17, 18, 19, 20, 21, 22, 23 striatal hypometabolism,17, 24 and reduced striatal activation during behavioral tasks.21, 26 Nonetheless, the available neuropathological studies of premanifest striatum have reported little or no neuronal loss, particularly in the motor striatum (i.e, putamen).1, 14, 15 These findings suggest that the very earliest motor defects in HD victims may be related to the loss of afferent connectivity of motor striatum with its major source of excitatory input—cerebral cortex. This input mainly ends as terminals that make asymmetric synaptic contact with dendritic spines of striatal projection neurons, which make up the vast majority of striatal neurons.31, 81, 107, 222, 246
Although it thus seems likely that SPNs lose cortical input beginning early in HD in humans, technical limitations make it difficult to perform the EM studies needed to ascertain this. In an effort to fill this gap, we carried out confocal light microscopic imaging (CLSM) studies and electron microscopic ultrastructural studies in male heterozygous Q140 HD knock‐in mice, to examine the possibility that loss of corticostriatal terminals prior to striatal neuron loss might underlie premanifest HD abnormalities.247 We used VGLUT1 immunolabeling to detect corticostriatal terminals in dorsolateral (motor) striatum over the first year of life in these mice, prior to striatal projection neuron pathology. We found that VGLUT1+ axospinous corticostriatal terminals represented about 55% of all excitatory synaptic terminals in striatum, with thalamic axospinous terminals accounting for the remainder of axospinous terminals (about 35% of all excitatory synaptic terminals in striatum). Many fewer cortical terminals ended on dendrites (about 5% of all cortical terminals in mice). In Q140 mice, VGLUT1+ axospinous and axodendritic terminals were reduced by 30% at 12 months (Figure 11), but showed no reduction at 1 or 4 months. No striatal shrinkage was observed even at 12 months, and no change was seen in immunolabeling of striatal projections systems,247 suggesting no SPN pathology. Although loss of corticostriatal input prior to significant striatal neuron loss has not been demonstrated neuropathologically in prior studies of HD mouse models, neurochemical and electrophysiological data from diverse such models are consistent with our findings in Q140 mice. For example, loss of presynaptic markers such as Lin7b and synaptophysin from cortex, loss of postsynaptic markers such as PSD‐95 from striatum, loss of dendritic spines from striatal projection neurons, and/or loss of excitatory synaptic terminals in striatum are observed in early symptomatic R6/2, Q175, and YAC128 mice.119, 248, 249, 250, 251, 252 Moreover, SPNs show electrophysiological signs of cortical disconnection in R6/2, Q175, or YAC128 mice at symptomatic ages at which SPNs do not yet show loss.100, 135, 136, 251, 253
Figure 11.
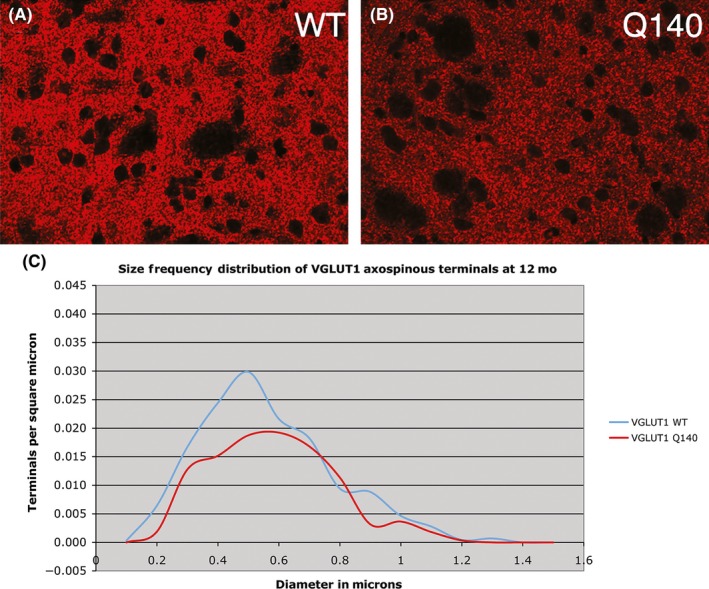
CLSM views of immunofluorescence labeling for VGLUT1 corticostriatal terminals in dorsolateral striatum of 12 month‐old male wild‐type (A) and 12 month‐old male heterozygous Q140 Huntington's disease knock‐in (B) mice. Both images are at the same magnification. Image C shows a graph of the size frequency distributions for VGLUT1+ corticostriatal axospinous synaptic terminals in striatum of 12 month‐old wild‐type and heterozygous Q140 Huntington knock‐in male mice, as determined from ultrastructural studies. Note that at 12 months a noteworthy shortfall in small corticostriatal terminals is evident in Q140 mice. These illustrations are Figures 1E,F, and 5C in Deng et al247
The corticostriatal input arises from 2 neuron types, an intratelencephalically projecting (IT) type residing predominantly in layer III and upper layer V, and a pyramidal tract (PT) type located primarily in lower layer V.254, 255, 256, 257, 258, 259, 260, 261, 262 We and other investigators have found in rats and monkeys that PT‐type corticostriatal neurons preferentially contact striatal neurons projecting to GPe with larger terminals, while IT‐type cortical neurons preferentially target striatal neurons projecting to GPi or SNr with smaller terminals (Figure 12).263, 264, 265, 266 In our study in 12‐month‐old Q140 mice, the loss of corticostriatal terminals occurred selectively for smaller terminals, suggesting they might be IT‐type terminals and preferentially lost from direct pathway type striatal neurons.247 If this was the case, this loss of drive to the “go” neurons of the direct pathway would be expected to cause behavioral hypoactivity, which is observed as a major symptom as Q140 mice age.90, 267, 268 To directly assess the loss of corticostriatal terminals from dSPNs vs iSPNs, we performed double immunolabeling to identify corticostriatal (VGLUT1+) axospinous terminals and D1 receptor immunolabeling to distinguish dSPN (D1+) and iSPN (D1−) synaptic targets and examined tissue at the electron microscopic (EM) level.269 We found that the loss of corticostriatal terminals at 12 months of age was preferential for D1+ spines and especially included smaller terminals (Figure 13). By contrast, presumptive iSPN D1− spines showed little loss of axospinous terminals at the same age. Regression analysis showed that the loss of VGLUT1+ terminals on D1+ spines was correlated with a slight decline in open field motor parameters at 12 months. Our overall results raise the possibility that preferential cortical input loss to dSPNs is an early event in human HD, which may contribute to the demonstrated premanifest motor slowing.
Figure 12.
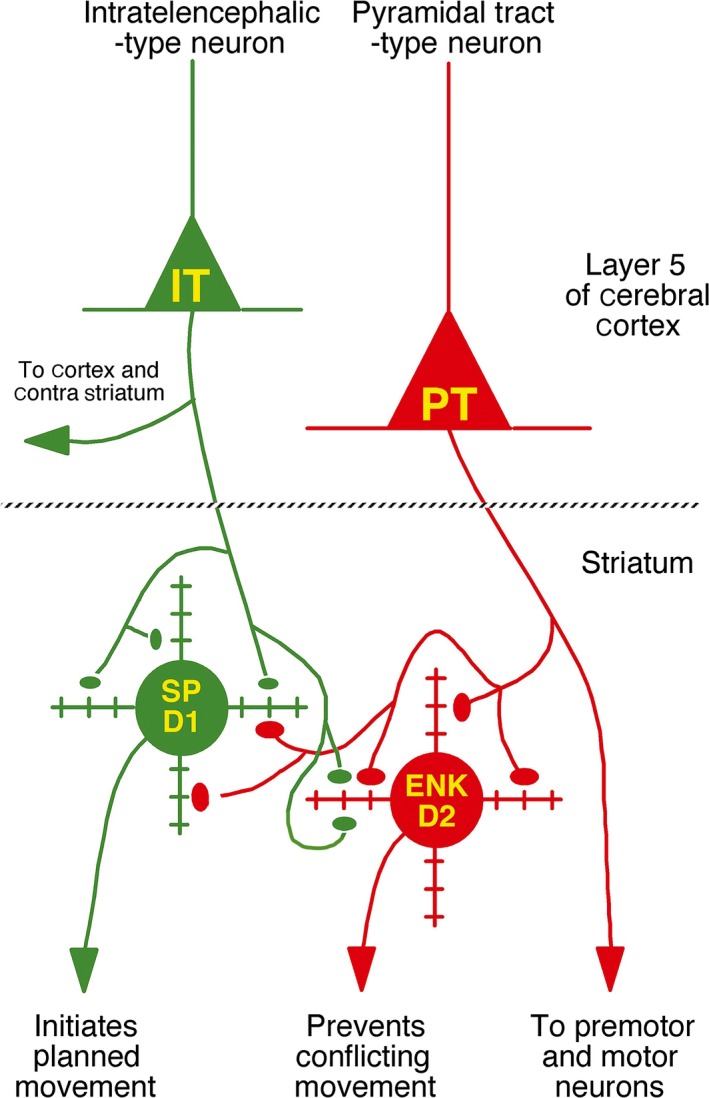
Schematic illustration of the relative abundances of axospinous IT‐type and PT‐type inputs to dSPNs and iSPNs. Both dSPNs and iSPNs receive axospinous IT‐type and PT‐type inputs, with dSPNs receiving about twice as much IT‐type and iSPNs receiving twice as much PT‐type axospinous input. Note that, as depicted, PT‐type axospinous terminals are uniformly larger than IT‐type axospinous terminals, and IT‐type axospinous terminals on iSPNs are slightly larger than IT‐type axospinous terminals on dSPNs. This illustration is Figure 4 from Deng et al264
Figure 13.
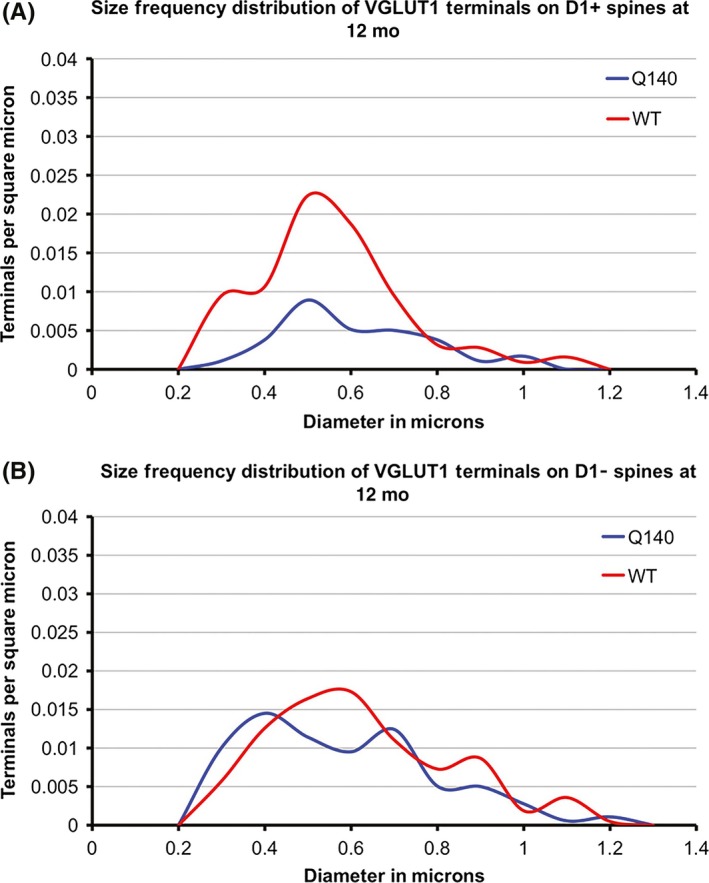
Graphs showing the size frequency distributions for VGLUT1+ axospinous synaptic terminals on D1+ (A) and D1− (B) SPNs in striatum of 12 month‐old WT and heterozygous Q140 male mice. D1+ spines belong largely to dSPNs and D1− spines to iSPNs. Note that the large shortfall in small terminals on D1+ spines (i.e, dSPNs) in Q140 mice. This illustration is Figure 5 from Deng et al269
The basis of the selective reduction in cortical terminals on dSPNs as HD symptoms progress is uncertain. In R6/2, Q175, and YAC128 HD mice, the loss in corticostriatal drive to the striatum is preceded by earlier corticostriatal hyperactivity.121, 248, 251, 270, 271 The dSPNs in particular show early enhanced and later reduced corticostriatal excitation.253, 270, 272 The reduced corticostriatal excitation appears to reflect the loss of corticostriatal input rather than reduced striatal excitability, because dSPNs remain more depolarized at rest and have elevated membrane input resistances.248, 252, 273 The preferential loss of cortical input to dSPNs we observed is of interest in light of the possibility that the loss is neuroprotective. The dSPNs projecting to GPi are the most resistant striatal projection neuron type in HD,61 and a downregulation in excitatory cortical input to them could help explain not only why striato‐GPi dSPNs resist death better in human HD than do other striatal SPN types, but also may explain the emergence of resistance to corticostriatal excitotoxicity as HD mice age.250, 274, 275 The corticostriatal synaptic pruning may, thus, involve activity‐dependent mechanisms, rather than an HD‐driven cortical pathology selective for the cortical input to dSPNs.276, 277 Stevens and co‐workers have suggested, on the other hand, that the corticostriatal pruning represents a complement‐mediated process involving microglial removal of defective corticostriatal terminals, with the mutation acting both pre‐ and postsynaptically to cause corticostriatal terminal defects and a process engendering their removal.278, 279 They also report that rescuing the terminals from microglial pruning by interfering with complement function reduces HD functional deficits. Foster et al,280 however, reported findings that appear to suggest that elimination of defective corticostriatal terminals improves motor performance in Q175 mice. Further studies are thus needed to determine the mechanisms underlying the pruning of corticostriatal terminals in HD and the therapeutic value of preventing their pruning without additionally taking measures to mitigate the potentially excitotoxic consequences of SPN hyperexcitability.
Prior to corticostriatal terminal loss, dysfunction in the corticostriatal input may contribute to disturbed SPN function, as well as to pathogenesis. Studies in mouse models have shown earlier and more prominent electrophysiological abnormalities in the cortical input to iSPNs than to dSPNs. For example, evoked glutamate currents in BACHD and YAC128 mice were found to be larger in iSPNs than dSPNs.270, 272 Moreover, iSPNs but not dSPNs show early deficits in corticostriatal synaptic plasticity in BACHD mice, and an earlier increase in extrasynaptic GluN2B NMDA receptor subunits.281 The defects in iSPN corticostriatal plasticity appear to stem from defective BDNF‐TrkB signaling in iSPNs.281 These findings on TrkB involvement are of interest in light of the evidence that normal huntingtin promotes BDNF production by cortical neurons but polyglutamine‐expanded huntingtin promotes BDNF production less effectively, and accordingly cortical BDNF production is reduced in human HD and in transgenic HD mice.139, 140, 141, 282, 283 TrkB on striatal neurons also appears to be reduced by the HD mutation in humans and mice,139, 141, 284 and accordingly, TrkB signaling by striatal neurons is reduced.285 For these reasons, considerable attention has focused on the possibly important role of diminished corticostriatal BDNF signaling in the pathogenesis of striatal injury in HD. The iSPNs seem especially dependent on BDNF signaling, as embryonic deletion of the BDNF receptor (TrkB) from striatal neurons or hemizygous knockout of BDNF in R6/1 mice results in the preferential reduction in iSPNs.95, 286 Moreover, BDNF overexpression rescues iSPNs in YAC128 mice,99 and drugs that boost cortical BDNF production appear to rescue iSPNs in proportion to the boost in BDNF production (Figure 14).136 In this regard, it is also of interest to note that BDNF production by the type of neuron whose axospinous input to dSPNs that we have shown appears to be preferentially lost in Q140 mice (i.e, the IT‐type)269 appears to be less than for PT‐type corticostriatal neurons,287 which are the preferential source of axospinous input to iSPNs.264, 265, 266 A particular role of diminished corticostriatal BDNF signaling in iSPNs may explain why presynaptic CB1 receptors on corticostriatal terminals can protect dSPNs against the excitotoxic effects of the HD mutation but not iSPNs.137 The possible existence of differing pathogenic mechanisms for iSPNs and dSPNs (BDNF deprivation for iSPNs and excitotoxicity for dSPNs) could help explain why they are differentially affected in HD.
Figure 14.
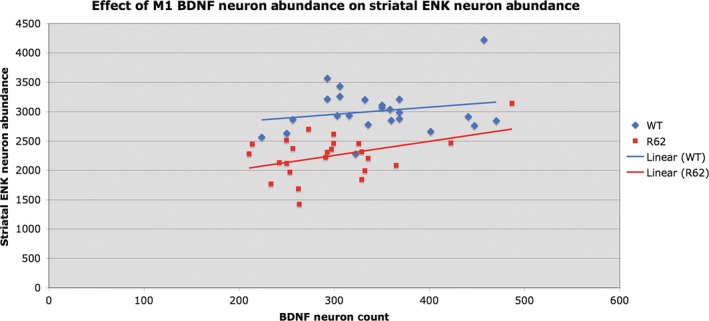
Graph showing the relationship between M1 motor cortex layer 5 BDNF neuron abundance and the abundance of striatal ENK neurons. For these graphs, all WT mice are pooled (i.e, vehicle‐treated and LY379268‐treated together) as a group, and all R6/2 are pooled (i.e, vehicle‐treated and LY379268‐treated together) as a group, because trends were similar irrespective of drug treatment. Note that ENK neuron abundance in R6/2 mice is significantly and positively correlated with cortical layer 5 BDNF neuron abundance (r = 0.4299). This illustration is Figure 5 from Reiner et al,136 in which we showed that daily LY379268 treatment increased cortical BDNF and mitigated deficits and pathology in R6/2 mice
Corticostriatal neurons themselves show reduced inhibition and increased activation in symptomatic R6/2, Q140, and YAC128 mice.288, 289 To better understand corticostriatal abnormalities in HD, it would also be useful to know whether HD differentially affects the 2 types of corticostriatal neurons, the IT‐type and the PT‐type,264, 265, 266 and their synapse formation with their striatal target neurons. For example, it could be the case that the HD mutation more greatly affects the behavior of IT‐type neurons, rendering them more active than PT‐type neurons, which ultimately then leads to the preferential loss of IT‐type terminals from dSPN spines.
3.3. Abnormalities in cortical input to striatal interneurons in humans and mutant mouse models of HD
Far less is known about abnormalities in the cortical input to striatal interneurons, and most of what is known comes from studies of animal models. Cholinergic striatal interneurons survive in intact numbers late into human HD as well as in diverse animal models of HD,124, 164, 166, 168, 170, 172, 173 but they show diminished expression of cholinergic neuron markers such as ChAT and VAChT.72, 170, 173, 175, 176, 177, 178, 290 Studies of symptomatic R6/2 mice indicate that diminished activity of cholinergic neurons, due to increased inhibitory input177 and decreased excitatory input,291 may underlie this reduction in cholinergic markers and be associated with a diminished influence of cholinergic interneurons in HD striatum on their target striatal neurons. Anatomical studies suggest that the excitatory input to cholinergic interneurons arises more from thalamus than cortex,222, 292, 293 and as discussed in the next section, thalamic input to cholinergic interneurons is reduced early in the lifespan of heterozygous Q140 mice,166 and thalamic responses are reduced in cholinergic interneurons in young heterozygous Q175 mice.294
The striatal LTS interneurons receive a weak cortical input and have a modulatory somatostatinergic influence and a weak GABAergic inhibitory effect on SPNs.182 Although LTS interneurons are resistant in HD and survive undiminished late into disease,164, 167, 169, 172 expression of NOS and somatostatin in these neurons is progressively diminished.174 Nonetheless, LTS+ neurons in at least some mouse HD models (R6/2 and BACHD) have been shown to be more spontaneously active than normal, but SPN responses to activation of LTS interneurons in these models was, nonetheless, normal.187 Although LTS neurons receive some limited cortical input,295 they appear to modulate SPNs in a cortical input‐dependent manner.183, 184 How cortical inputs to LTS interneurons or the other related striatal interneurons (i.e, NPY‐NGF interneurons, NPY‐negative neurogliaform cells, and TH interneurons) might be affected in HD, and how this might affect the function of their projection targets is unknown. Similarly, although it is known that PARV interneurons receive prominent cortical input,193 how HD might affect that input is uncertain. Moreover, it is unclear whether PARV interneurons receive input differentially from IT‐type and PT‐type corticostriatal neurons. The observation of dendritic branch reduction in parvalbuminergic interneurons in R6/2 mice199 suggests a likely loss of cortical input for this neuron type as disease progresses, which together with their loss would contribute to impairment in their feedforward inhibition to SPNs. Note that stimulation of PARV interneurons in R6/2 mice induces significantly larger amplitude SPN inhibition than in control mice,187 suggesting that at some stages of disease, PARV interneurons may compensate for the loss of excitatory drive by increasing their efficacy in inhibiting SPNs. Finally, although the calretinergic neurons are known to receive cortical input,154 the effect of HD on this or any other input to this interneuron type in HD has not been investigated.
3.4. Abnormalities in overall thalamic input to striatum in humans and mutant mouse models of HD
The striatum also receives a substantial excitatory input from the thalamus, which ends in large part on the spines and dendrites of SPNs, with cholinergic interneurons being a prominent target as well.222, 246, 292, 293 The thalamic projection is topographically organized and arises heavily from the intralaminar nuclei,219, 220 but also from specific sensory nuclei of the thalamus. The intralaminar thalamic nuclei projecting to striatum receive polysensory cortical and brainstem input and a feedback projection from GPi. By the topographically ordered polysensory input, the intralaminar thalamus detects diverse behaviorally relevant events and serves to signal the neurons in the appropriate part of striatum of this behaviorally relevant event. Accordingly, the thalamic input is thought to play a role in attentional mechanisms involved in motor planning and preparedness.162, 221, 222, 223 The intralaminar nuclei show differing evolutionary elaborations in primates and rodents, with the main intralaminar nuclei in primates being the more laterally situated centromedian and the more medially situated parafascicular nucleus. In rodents, the medial part of the parafascicular nucleus corresponds to the primate parafascicular nucleus and the lateral part of the rodent parafascicular nucleus corresponds to the primate centromedian.222, 296 Additional nuclei, however, are also part of the intralaminar complex that targets striatum, and they are divergent among mammals in name and number. Importantly, there appear to be variations among the intralaminar nuclei in their targeting of striatal neurons as well as their spiking characteristics, and thus, perhaps functional differences may exist among them. For example, parafascicular/centromedian terminals in rats and monkeys have been reported to preferentially contact dendrites, while terminals arising from the other intralaminar nuclei have been reported to favor spines.297, 298, 299, 300 Moreover, parafascicular neurons in rodents have been observed to differ in their firing properties from other intralaminar nuclei.297
The thalamus, including the intralaminar nuclei, undergoes shrinkage and cell loss in human HD.15, 301, 302, 303 The centromedian, for example, shows evident neuronal loss by grade 3,147 and together the centromedian/parafascicular nuclear complex shows about a 25% volume loss and 50% neuron loss in advanced HD,304 while only 15% neuron and volume loss are seen in the mediodorsal nucleus of the intralaminar thalamus.305 Various morphological and neurochemical abnormalities have also been observed in intralaminar thalamus in mouse models of HD, including increased GFAP expression, loss of the adhesion molecule tenascin‐C, and loss of the synaptic protein complexin II.306, 307 Given the role of thalamic input to the striatum in attentional mechanisms concerning motor planning and preparedness,222 the HD‐related neuronal loss, shrinkage, and pathology in intralaminar thalamus are likely to be associated with loss of thalamic input to striatum. This loss and the possible dysfunction preceding it are likely to be significant contributors to HD symptoms, particularly if there is a neuron‐type specific pattern to the loss. As in the case of the corticostriatal input, however, data from human HD on the neuron‐type specificity of the thalamic input loss and dysfunction is limited, and insight is mainly provided by mouse models.
3.5. Abnormalities in thalamic input to striatal projection neurons in humans and mutant mouse models of HD
To address the likely loss of thalamic input to SPNs in human HD, especially as it might occur prior to striatal neuron loss or clinically evident motor symptoms, we carried out CLSM imaging studies and electron microscopic ultrastructural studies in male heterozygous Q140 HD knock‐in mice.247 We used VGLUT2 immunolabeling to detect thalamostriatal terminals in dorsolateral (motor) striatum over the first year of life, prior to striatal projection neuron pathology. We found that the VGLUT2+ axospinous thalamostriatal terminals represented about 35% of all asymmetric synaptic terminals in normal striatum, with VGLUT2+ axodendritic terminals present as well (making up about 5% of all asymmetric synaptic terminals in normal striatum). In Q140 mice, a significant 40% shortfall in VGLUT2+ axodendritic thalamostriatal terminals and a 20% shortfall in axospinous thalamostriatal terminals were already observed at 1 month of age, at which time VGLUT1+ corticostriatal terminals were normal in abundance. The 20% deficiency in VGLUT2+ thalamostriatal axospinous terminals persisted at 4 and 12 months in Q140 mice (Figure 15), but loss of VGLUT1+ corticostriatal terminals was not observed until 12 months of age, as noted above. At no age was there evidence of striatal neuron loss. Consistent with our finding of early abnormalities in thalamostriatal input to SPNs in Q140 mice, Kolodziejczyk and Raymond308 reported increased probability of glutamate release from thalamostriatal terminals in 2‐month YAC128 mice. They suggested that this could be compensatory for an early occurring deficiency in thalamostriatal terminals. Kolodziejczyk and Raymond308 also observed that SPNs showed increased responses to thalamic activation at extrasynaptic NMDA receptors, which they suggested might drive excitotoxic SPN injury. Similar results for glutamate release and extrasynaptic NMDA receptors were observed in 6‐week‐old R6/2 mice.309 With regard to a pathogenic effect of thalamostriatal abnormalities in HD, Samadi et al310 reported that R6/2 mice show progressive reduction in BDNF expression by the parafascicular nucleus, with 6‐week‐old R6/2 mice already showing a 25% reduction. As noted previously for corticostriatal BDNF, this BDNF reduction (compounded by thalamostriatal terminal loss) could contribute to neurodegeneration of iSPNs in particular, due to their apparent particular dependence on BDNF.
Figure 15.
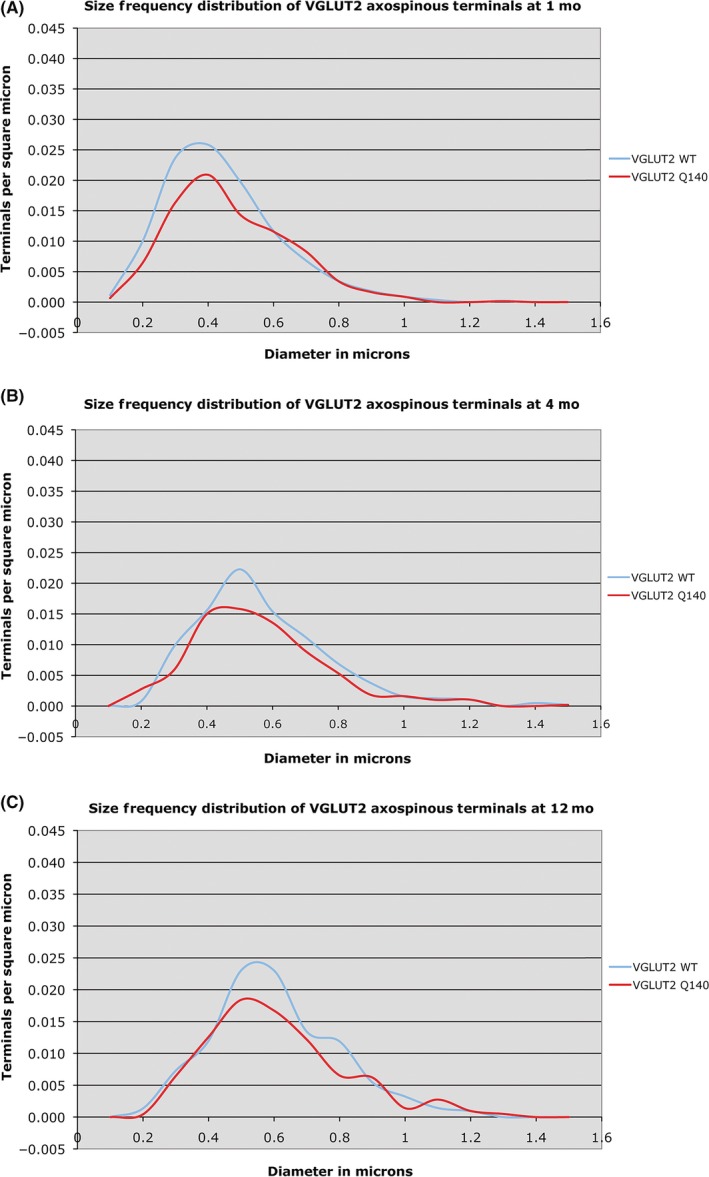
Graphs showing the size frequency distributions for VGLUT2+ thalamostriatal axospinous synaptic terminals in striatum of 1‐month‐ (A), 4‐month‐ (B), and 12 month‐old (C) wild‐type and heterozygous Q140 Huntington knock‐in male mice. Note that a shortfall in small terminals was found in Q140 mice at 1, 4 months, and 12 months. This illustration is Figure 9 from Deng et al247
Although the thalamic input to striatum targets both dSPNs and iSPNs, in primates and rats the input seems preferential for dSPNs,296, 299, 311, 312 but not in mouse.292 Additionally, the response of iSPNs to thalamic terminal activation differs from that for dSPNs, with iSPN responses more rapid and less sustained.295 To determine whether loss of thalamic input might differ between dSPNs and iSPNs and thus differentially affect HD pathophysiology, we carried out ultrastructural double‐label studies in male heterozygous Q140 mice, using immunolabeling to identify thalamostriatal (VGLUT2+) axospinous terminals, and D1 receptor immunolabeling to distinguish dSPN (D1+) and iSPN (D1−) synaptic targets (Deng et al269). We found that the thalamostriatal terminal loss was comparable for D1+ and D1− spines at both 4 and 12 months. Thus, although dSPNs have been reported to receive a greater thalamic input than iSPNs in rats and monkeys,296, 299, 312 no differential loss of thalamic input to SPNs was seen in our study of Q140 mice. Similar to Q140 mice, nondifferential thalamic terminal loss at D1/D2 spines was also reported in R6/2 mice by Parievsky et al.309
The finding that the deficiency in thalamostriatal axospinous terminals in Q140 mice is present already at 1 month of age suggests it may represent a development defect. Consistent with this, striatal expression of proteins critical to thalamostriatal synapse formation, such as the semaphorin‐3E receptor Plexin‐D1 signaling complex, is significantly reduced early in the lifespan of several transgenic or knock‐in HD mice,313, 314 and in human HD as well.315 In any case, the impoverishment of thalamic excitatory drive to striatum may help explain early motor defects in Q140 mice and in premanifest HD. Because of the low membrane excitability of SPNs, only temporally correlated excitatory input from a sufficiently large number of convergent excitatory inputs can depolarize SPNs to firing threshold.218, 246, 316, 317, 318, 319 Part of the needed activation may derive from the cortical inputs, but the attention‐related thalamic input may serve to ensure that the striatal neurons activated are those that drive the response appropriate to that environmental circumstance. This may be particularly true for dSPNs, which play a role in movement facilitation.107, 320 For any given striatal territory, the intermingled dSPNs and iSPNs play opposite roles in movement, with the direct facilitating desired and the indirect opposing unwanted movement. Thus, as for the input from any given part of cortex to any given part of striatum, the inputs to these 2 SPN types may arise preferentially from different thalamic neuron types. In this regard, it would be of interest to know whether there are differing types of thalamostriatal projection neurons and whether they are differently affected by the HD mutation. In our own work in Q140 mice,269 we found that dSPN spines in WT mice receive synaptic contact from thalamic terminals with a size peak at 0.4 μm, and iSPN spines in WT mice receive synaptic input from thalamic terminals with size peaks at 0.3‐0.4 μm and 0.5‐0.6 μm. This suggests that there may be thalamostriatal neuron subtypes that differ in their relative targeting of the 2 SPN types. Similarly, the monkey homolog of rat PFN (the centromedian/parafascicular complex) also consists of neuronal subtypes, as axonal reconstructions show that some of its neurons innervate cortex only, some striatum only, and some both.321 Moreover, neurons of the centromedian/parafascicular complex in primates have been divided into subtypes based on their responses to sensory stimuli, with some displaying short‐latency activation and others displaying long‐latency activation.322 These 2 populations are segregated in the centromedian/parafascicular complex of primates, with the short‐latency neurons predominantly found in the parafascicular nucleus and the long‐latency neurons in the centromedian nucleus.322 As noted above, the various intralaminar nuclei in rodents differ in the extent to which they target spines vs dendrites of striatal neurons, and they also differ in their perikaryal morphology and striatal arborization patterns.323 It would be of value to know if any of the apparent subtypes of intralaminar thalamic neurons differ in their targeting of dSPNs and iSPNs, above and beyond the preference of centromedian/lateral parafascicular complex for dSPNs in rats and primates. Note that the 2 SPN types both show depressed synaptic responsiveness to repetitive stimulation of thalamic input and thus do not differ in at least this one physiological regard.324
3.6. Abnormalities in thalamic input to striatal interneurons in humans and mutant mouse models of HD
No specific data are available on abnormalities in thalamic input to striatal interneurons in human HD. Farrar et al291 noted that activation of striatal cholinergic interneurons is dysfunctional in R6/2 HD mice and suggested that this may reflect abnormalities in the excitatory innervation of cholinergic interneurons. Accordingly, as the thalamus is the major source of excitatory input to cholinergic interneurons in striatum in rodents and primates222, 292, 293, 325 (and drives striatal acetylcholine release),326 we assessed whether there are changes in the VGLUT2 immunolabeled thalamic input to striatal cholinergic interneurons in heterozygous Q140 males at 1 and 4 months of age, using ChAT immunolabeling to identify cholinergic interneurons.166 Although blinded neuron counts showed that ChAT perikarya were normal in abundance in Q140 mice, the dendrites of Q140 ChAT interneurons were significantly fewer and shorter as early as 1 month of age, and individual ChAT interneurons as a consequence were likely to be contacted by fewer VGLUT2 thalamostriatal axodendritic terminals.247 These results show that the abundance of thalamic input to individual striatal cholinergic interneurons is reduced early in the lifespan of Q140 mice, raising the possibility that this may occur in human HD as well. Consistent with our findings, Tanimura et al294 reported reduced thalamic responsiveness in striatal cholinergic interneurons in R6/2 mice, and Holley et al177 reported enhanced inhibitory input to them, both of which would reduce their activity. As cholinergic interneurons differentially affect dSPNs and iSPNs,162 their reduced activation, especially by the thalamic input mediating attentional processes, may contribute to early abnormalities in movement onset and switching in HD in response to a changing stimulus context.326 No published information from studies of mouse models is available on how HD may affect thalamic inputs to the other striatal interneuron types, of which the parvalbuminergic interneurons and LTS interneurons but not calretininergic interneurons have been shown to receive and respond to thalamic input in rodents and/or primates.295, 325, 327
3.7. Abnormalities in overall nigral input to striatum in humans and mutant mouse models of HD
The dopaminergic input to the striatum from the SNc heavily targets SPNs and cholinergic interneurons and thereby plays a major role in motor plasticity and performance. The importance of this role is especially evidenced by the motor impairments associated with its extensive loss in Parkinson's disease. The loss of dopaminergic input to striatum and the contributions of any such loss to HD pathophysiology have not been as extensively investigated as other aspects of basal ganglia circuit pathology in HD. Neuropathological studies, nonetheless, show that the substantia nigra undergoes cell loss and shrinkage in HD, but much less so than the striatum.15, 27, 224, 328 Shrinkage of substantia nigra as detected in imaging studies also has been reported, which could stem from both dSPN input loss and nigral neuron death.301 Loss of both SNr GABAergic neurons and SNc dopaminergic neurons is evident in HD,147 and Oyanagi et al329 have reported 40% loss in both populations. Yohrling et al330 reported that in grade 4 HD tyrosine hydroxylase expression by dopaminergic neurons, as detected by in situ hybridization, is decreased by 46% per surviving dopaminergic neuron. Moreover, the dopaminergic neurons were 33% smaller than normal. Neuron counts were, however, not performed to assess dopaminergic neuron loss in their study. Consistent with the reduced tyrosine hydroxylase expression, Yohrling et al330 found that tyrosine hydroxylase protein in the nigra was reduced by 32%. As would be predicted from loss of nigral dopaminergic neurons and reduced tyrosine hydroxylase expression, terminals containing tyrosine hydroxylase appear to be reduced in abundance in advanced HD striatum.331, 332 Dopamine, homovanillic acid (HVA), the dopamine transporter (DAT), and the vesicular monoamine transporter‐2 (VMAT2) also have been reported to be reduced in HD striatum in later disease stages.290, 333, 334, 335 Nonetheless, increases in dopamine have actually been observed early in disease.336 Late loss of dopamine input could contribute to akinesia in HD, as it does in Parkinson's disease, while early increases could synergize with iSPN loss and dysfunction to exacerbate hyperkinesia. In the later regard, it is interesting to note that L‐DOPA provokes hyperkinesia in otherwise asymptomatic HD carriers.337 Dopaminergic neurons are also affected in transgenic mouse models in the late stages of the disease, consistent with what is seen in late human HD. In R6/2 and R6/1 mice, progressive reduction in striatal DA release is seen beginning at 6 weeks of age,338, 339, 340, 341 as well as reduction in striatal homovanillic acid in later symptomatic stages.342
3.8. Abnormalities in overall nigral input to striatal projection neurons in humans and mutant mouse models of HD
As the dopaminergic input to striatum heavily targets SPNs,41 the apparent biphasic change in dopaminergic input to them may contribute to HD pathophysiology in humans.343 Although no specific data are available for human HD on the extent to which the early increase in DA release and later loss of dopamine input may be preferential for either SPN type, even equal early increase and late loss would have differential effects on motor function due to the opposing actions of dopamine on iSPNs and dSPNs.82 For example, iSPN loss and dysfunction early in HD are associated with the early dominance of chorea as a symptom. Increased dopamine release would reduce the activity of surviving iSPNs and enhance the activity of dSPNs, with both effects exacerbating the hyperkinesia associated with iSPN loss and dysfunction alone. The late reduction in nigrostriatal dopamine would work in the opposite direction—the resulting reduced activity in the surviving dSPNs and increased activity in the surviving iSPNs would contribute to the bradykinesia of later HD. As >85% dopaminergic neuron loss is needed for the bradykinesia in Parkinson's disease, it seems unlikely that the dopaminergic input loss to SPNs in late HD by itself is great enough to have a symptomatic impact, but in combination with SPN loss could exert effects.
3.9. Abnormalities in overall nigral input to striatal interneurons in humans and mutant mouse models of HD
As the dopaminergic input to striatum also heavily targets cholinergic interneurons,344 it is likely that the dopaminergic input to them is affected in human HD and animal models of HD. No specific data are, however, available to confirm loss of dopaminergic input to cholinergic interneurons. Given that >85% dopaminergic neuron loss is needed for the motor dysfunction in Parkinson's disease, it seems unlikely that the dopaminergic input loss to cholinergic interneurons in HD alone is great enough to have a symptomatic impact. In combination with the reduced excitatory drive and reduce acetylcholine production and release, early increased dopamine release might exacerbate D2‐receptor‐mediated cholinergic interneuron hypofunction early in HD. The later loss might compromise their dopamine‐dependent role in reward‐mediated motor learning.
4. SUMMARY AND CONCLUSIONS
The pathophysiology of motor impairment in HD involves dysfunction and loss of striatal projection neurons (Figure 16). Dysfunction occurs early and stems from a loss of cortical and thalamic inputs to SPNs, abnormalities in their inputs from striatal interneurons, and dysregulation of intrinsic SPN neuronal excitability. Actions of the mutant protein on the neurons projecting to SPNs and on SPNs themselves are likely to contribute to the SPN dysfunction, which accounts for the early mild symptoms of HD such as motor slowing. By contrast, the later loss of SPNs and their outputs to GPe, GPi, and SNr is associated with the clinically prominent motor abnormalities of HD. More rapid loss of iSPNs projecting to GPe than dSPNs projecting to GPi accounts for early chorea that is gradually overlain with bradykinesia as striato‐GPi dSPN loss approximates iSPN loss. A greater vulnerability of iSPNs than dSPNs to BDNF deprivation stemming from reduced cortical production and delivery appears to account for the earlier and greater vulnerability of iSPNs. The basis of the emergence of dystonia late in HD is uncertain, but may relate to the late loss of striatal PARV interneurons, as well as to the then extensive dSPN loss.345 The extent to which slowing or preventing early input loss and/or correcting early dysregulation of SPN intrinsic excitability would mitigate early deficits and slow progression to SPN neuron and output loss remains to be determined, but insights into this could provide clues for pathogenesis and treatment approaches.
Figure 16.
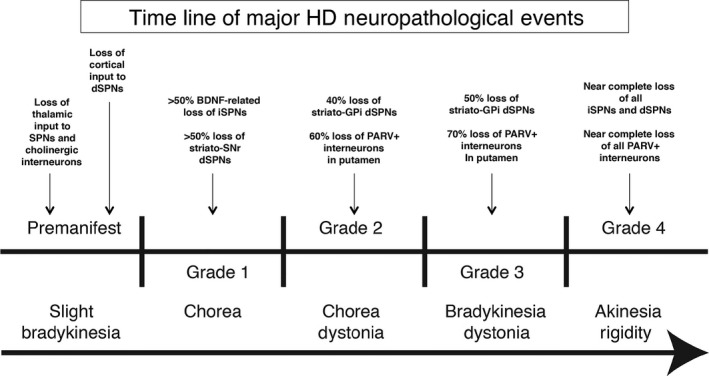
Time line of major changes in striatal projection neurons and interneurons and their inputs during HD progression, as they relate to the appearance of the major motor symptoms in adult‐onset HD. Note that in the case of juvenile HD, progression is very rapid, SPN loss appears to be nondifferential, and dystonia and rigidity are the presenting symptoms
CONFLICT OF INTEREST
The authors have no financial interest in the research reported here.
ACKNOWLEDGMENTS
We thank Marion Joni, Kathy Troughton, and Yunming Hu for technical assistance during the course of our studies. This article was supported by the Cure Huntington's Disease Initiative Foundation (AR), NIH NS28721 (AR), NIH NS57722 (AR), and The Methodist Hospitals Endowed Professorship in Neuroscience (AR).
Reiner A, Deng Y‐P. Disrupted striatal neuron inputs and outputs in Huntington's disease. CNS Neurosci Ther. 2018;24:250–280. 10.1111/cns.12844
REFERENCES
- 1. Vonsattel JP, DiFiglia M. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1998;57:369‐384. [DOI] [PubMed] [Google Scholar]
- 2. Bechtel N, Scahill RI, Rosas HD, et al. Tapping linked to function and structure in premanifest and symptomatic Huntington disease. Neurology. 2010;75:2150‐2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biglan KM, Ross CA, Langbehn DR, et al. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT‐HD study. Mov Disord. 2009;24:1763‐1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blekher TM, Yee RD, Kirkwood SC, et al. Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington's disease. Vision Res. 2004;44:2729‐2736. [DOI] [PubMed] [Google Scholar]
- 5. de Boo GM, Tibben A, Lanser JB, et al. Early cognitive and motor symptoms in identified carriers of the gene for Huntington disease. Arch Neurol. 1997;54:1353‐1357. [DOI] [PubMed] [Google Scholar]
- 6. Delval A, Bleuse S, Simonin C, et al. Are gait initiation parameters early markers of Huntington's disease in pre‐manifest mutation carriers? Gait Posture. 2011;34:202‐207. [DOI] [PubMed] [Google Scholar]
- 7. Kirkwood SC, Siemers E, Stout JC, et al. Longitudinal cognitive and motor changes among presymptomatic Huntington disease gene carriers. Arch Neurol. 1999;56:563‐568. [DOI] [PubMed] [Google Scholar]
- 8. Kirkwood SC, Siemers E, Bond C, Conneally PM, Christian JC, Foroud T. Confirmation of subtle motor changes among presymptomatic carriers of the Huntington disease gene. Arch Neurol. 2000;57:1040‐1044. [DOI] [PubMed] [Google Scholar]
- 9. Rao AK, Muratori L, Louis ED, Moskowitz CB, Marder KS. Spectrum of gait impairments in presymptomatic and symptomatic Huntington's disease. Mov Disord. 2008;23:1100‐1107. [DOI] [PubMed] [Google Scholar]
- 10. Rao AK, Gordon AM, Marder KS. Coordination of fingertip forces during precision grip in premanifest Huntington's disease. Mov Disord. 2011;26:862‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siemers E, Foroud T, Bill DJ, et al. Motor changes in presymptomatic Huntington disease gene carriers. Arch Neurol. 1996;53:487‐492. [DOI] [PubMed] [Google Scholar]
- 12. Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol. 2011;10:31‐42. [DOI] [PubMed] [Google Scholar]
- 13. Turner TH, Goldstein J, Hamilton JM, et al. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT‐HD study. J Mot Behav. 2011;43:295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Albin RL, Qin Y, Young AB, Penney JB, Chesselet MF. Preproenkephalin messenger RNA‐containing neurons in striatum of patients with symptomatic and presymptomatic Huntington's disease: an in situ hybridization study. Ann Neurol. 1991;30:542‐549. [DOI] [PubMed] [Google Scholar]
- 15. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559‐577. [DOI] [PubMed] [Google Scholar]
- 16. Aylward EH, Nopoulos PC, Ross CA, et al. Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry. 2011;82:405‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ciarmiello A, Cannella M, Lastoria S, et al. Brain white‐matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med. 2006;47:215‐222. [PubMed] [Google Scholar]
- 18. Dumas EM, van den Bogaard SJ, Ruber ME, et al. Early changes in white matter pathways of the sensorimotor cortex in premanifest Huntington's disease. Hum Brain Mapp. 2012;33:203‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hobbs NZ, Barnes J, Frost C, et al. Onset and progression of pathologic atrophy in Huntington disease: a longitudinal MR imaging study. AJNR Am J Neuroradiol. 2010;31:1036‐1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kipps CM, Duggins AJ, Mahant N, Gomes L, Ashburner J, McCusker EA. Progression of structural neuropathology in preclinical Huntington's disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry. 2005;76:650‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paulsen JS, Magnotta VA, Mikos AE, et al. Brain structure in preclinical Huntington's disease. Biol Psychiatry. 2006;59:57‐63. [DOI] [PubMed] [Google Scholar]
- 22. Reading SA, Yassa MA, Bakker A, et al. Regional white matter change in pre‐symptomatic Huntington's disease: a diffusion tensor imaging study. Psychiatr Res. 2005;140:55‐62. [DOI] [PubMed] [Google Scholar]
- 23. Rosas HD, Tuch DS, Hevelone ND, et al. Diffusion tensor imaging in presymptomatic and early Huntington's disease: selective white matter pathology and its relationship to clinical measures. Mov Disord. 2006;21:1317‐1325. [DOI] [PubMed] [Google Scholar]
- 24. Grafton ST, Mazziotta JC, Pahl JJ, et al. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol. 1992;49:1161‐1167. [DOI] [PubMed] [Google Scholar]
- 25. Paulsen JS, Zimbelman JL, Hinton SC, et al. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington's Disease. AJNR Am J Neuroradiol. 2004;25:1715‐1721. [PMC free article] [PubMed] [Google Scholar]
- 26. Wolf RC, Grön G, Sambataro F, et al. Brain activation and functional connectivity in premanifest Huntington's disease during states of intrinsic and phasic alertness. Hum Brain Mapp. 2012;33:2161‐2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De La Monte SM, Vonsattel JP, Richardson EP Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol. 1988;47:516‐525. [DOI] [PubMed] [Google Scholar]
- 28. Heinsen H, Strik M, Bauer M, et al. Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol. 1994;88:320‐333. [DOI] [PubMed] [Google Scholar]
- 29. Lange H, Thörner G, Hopf A, Schröder KF. Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci. 1976;28:401‐425. [DOI] [PubMed] [Google Scholar]
- 30. Lange H. Quantitative changes of telencephalon, diencephalon and mesencephalon in Huntington's chorea, postencephalitic, and idiopathic Parkinsonism. Verh Anat Ges. 1981;75:923‐925. [Google Scholar]
- 31. Reiner A, Anderson KD. The patterns of neurotransmitter and neuropeptide co‐occurrence among striatal projection neurons: conclusions based on recent findings. Brain Res Rev. 1990;15:251‐265. [DOI] [PubMed] [Google Scholar]
- 32. Reiner A, Medina L, Haber SN. The distribution of dynorphinergic terminals in striatal target regions in comparison to the distribution of substance P‐containing and enkephalinergic terminals in monkeys and humans. Neuroscience. 1999;88:775‐793. [DOI] [PubMed] [Google Scholar]
- 33. Wang HB, Laverghetta AV, Foehring R, et al. Single‐cell RT‐PCR, in situ hybridization histochemical, and immunohistochemical studies of substance P and enkephalin co‐occurrence in striatal projection neurons in rats. J Chem Neuroanat. 2006;31:178‐199. [DOI] [PubMed] [Google Scholar]
- 34. Beckstead RM, Cruz CJ. Striatal axons to the globus pallidus, entopeduncular nucleus and substantia nigra come mainly from separate cell populations in cat. Neuroscience. 1986;19:147‐158. [DOI] [PubMed] [Google Scholar]
- 35. Feger J, Crossman AR. Identification of different subpopulations of neostriatal neurons projecting to globus pallidus or substantia nigra in the monkey: a retrograde fluorescence double‐labeling study. Neurosci Lett. 1984;49:7‐12. [DOI] [PubMed] [Google Scholar]
- 36. Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 1990;10:3421‐3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parent A, Smith Y, Filion M, Dumas J. Distinct afferents to the internal and external pallidal segments in the squirrel monkey. Neurosci Lett. 1989;96:140‐144. [DOI] [PubMed] [Google Scholar]
- 38. Parent A, Charara A, Pinault D. Single striatofugal axons arborizing in both pallidal segments and in the substantia nigra in primates. Brain Res. 1995;698:280‐284. [DOI] [PubMed] [Google Scholar]
- 39. Wu Y, Richard S, Parent A. The organization of the striatal output system: a single‐cell juxtacellular labeling study in the rat. Neurosci Res. 2000;38:49‐62. [DOI] [PubMed] [Google Scholar]
- 40. Gong S, Zheng C, Doughty ML, Losos K, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917‐925. [DOI] [PubMed] [Google Scholar]
- 41. Reiner A, Medina L, Veenman CL. Structural and functional evolution of the basal ganglia in vertebrates. Brain Res Rev. 1998;28:235‐285. [DOI] [PubMed] [Google Scholar]
- 42. Fujiyama F, Sohn J, Nakano T, et al. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron‐tracing study using a viral vector. Eur J Neurosci. 2011;33:668‐677. [DOI] [PubMed] [Google Scholar]
- 43. Smith JB, Klug JR, Ross DL, et al. Genetic‐based dissection unveils the inputs and outputs of striatal patch and matrix compartments. Neuron. 2016;91:1069‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang HB, Deng YP, Reiner A. In situ hybridization histochemical and immunohistochemical evidence that striatal projection neurons co‐containing substance P and enkephalin are overrepresented in the striosomal compartment of striatum in rats. Neurosci Lett. 2007;425:195‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fink JS, Weaver DR, Rivkees SA, et al. Molecular cloning of the rat A2 adenosine receptor: selective co‐expression with D2 dopamine receptors in rat striatum. Mol Brain Res. 1992;14:186‐195. [DOI] [PubMed] [Google Scholar]
- 46. Le Moine C, Bloch B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol. 1995;355:418‐426. [DOI] [PubMed] [Google Scholar]
- 47. Schiffmann SN, Jacobs O, Vanderhaeghen JJ. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem. 1991;57:1062‐1067. [DOI] [PubMed] [Google Scholar]
- 48. Biezonski DK, Trifilieff P, Meszaros J, Javitch JA, Kellendonk C. Evidence for limited D1 and D2 receptor coexpression and colocalization within the dorsal striatum of the neonatal mouse. J Comp Neurol. 2015;523:1175‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glass M, Dragunow M, Faull RLM. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299‐318. [DOI] [PubMed] [Google Scholar]
- 50. Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267‐274. [DOI] [PubMed] [Google Scholar]
- 51. Mailleux P, Vanderhaeghen JJ. Localization of cannabinoid receptor in the human developing and adult basal ganglia. Higher levels in the striatonigral neurons. Neurosci Lett. 1992;148:173‐176. [DOI] [PubMed] [Google Scholar]
- 52. Goto S, Hirano A, Rojas‐Corona RR. An immunohistochemical investigation of the human neostriatum in Huntington's disease. Ann Neurol. 1989;25:298‐304. [DOI] [PubMed] [Google Scholar]
- 53. Seto‐Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calcium binding protein containing neurons in Huntington's disease. Lancet. 1988;1:1252‐1255. [DOI] [PubMed] [Google Scholar]
- 54. Reiner A, Dragatsis I, Dietrich P. Genetics and neuropathology of Huntington's disease In: Jenner P, Brotchie J, Bezard E, eds. Pathophysiology, Pharmacology And Biochemistry of Dyskinesia. Oxford: Elsevier Academic Press; 2011; 98: 325‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albin RL, Young AB, Penney JB, et al. Abnormalities of striatal projection neurons and N‐methyl‐D‐aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;332:1923‐1928. [DOI] [PubMed] [Google Scholar]
- 56. Albin RL, Reiner A, Anderson KD, et al. Preferential loss of striato‐external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425‐430. [DOI] [PubMed] [Google Scholar]
- 57. Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54:105‐120. [DOI] [PubMed] [Google Scholar]
- 58. Augood SJ, Faull RLM, Love DR, Emson PC. Reduction in enkephalin and substance P mRNA in the striatum of early grade Huntington's disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023‐1036. [DOI] [PubMed] [Google Scholar]
- 59. Augood SJ, Faull RL, Emson PC. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann Neurol. 1997;42:215‐221. [DOI] [PubMed] [Google Scholar]
- 60. Allen KL, Waldvogel HJ, Glass M, Faull RL. Cannabinoid (CB1), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington's disease. J Chem Neuroanat. 2009;37:266‐281. [DOI] [PubMed] [Google Scholar]
- 61. Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A. Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27:143‐164. [DOI] [PubMed] [Google Scholar]
- 62. Sapp E, Ge P, Aizawa H, et al. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grad Huntington's disease using high resolution image analysis. Neuroscience. 1995;64:397‐404. [DOI] [PubMed] [Google Scholar]
- 63. Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity‐akinesia. Ann Neurol. 1990;27:357‐365. [DOI] [PubMed] [Google Scholar]
- 64. Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85:5733‐5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goto S, Hirano A, Rojas‐Corona RR. Immunohistochemical visualization of afferent nerve terminals in human globus pallidus and its alteration in neostriatal neurodegenerative disorders. Acta Neuropathol. 1989;78:543‐550. [DOI] [PubMed] [Google Scholar]
- 66. Glass M, Dragunow M, Faull RLM. The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience. 2000;97:505‐519. [DOI] [PubMed] [Google Scholar]
- 67. Richfield EK, Herkenham M. Selective vulnerability in Huntington's disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann Neurol. 1994;36:577‐584. [DOI] [PubMed] [Google Scholar]
- 68. Walker FO, Young AB, Penney JB, Dovorini‐Zis K, Shoulson I. Benzodiazepine and GABA receptors in early Huntington's disease. Neurology. 1984;34:1237‐1240. [DOI] [PubMed] [Google Scholar]
- 69. Waeber C, Palacios JM. Serotonin‐1 receptor binding sites in the human basal ganglia are decreased in Huntington's chorea but not in Parkinson's disease: a quantitative in vitro autoradiography study. Neuroscience. 1989;32:337‐347. [DOI] [PubMed] [Google Scholar]
- 70. Seizinger BR, Liebisch DC, Kish SJ, Arendt RM, Hornykiewicz O, Herz A. Opioid peptides in Huntington's disease: alterations in prodynorphin and proenkephalin system. Brain Res. 1986;378:405‐408. [DOI] [PubMed] [Google Scholar]
- 71. Ellison DW, Beal MF, Mazurek MF, Malloy JR, Bird ED, Martin JB. Amino acid neurotransmitter abnormalities in Huntington's disease and the quinolinic acid animal model of Huntington's disease. Brain. 1987;110:1657‐1673. [DOI] [PubMed] [Google Scholar]
- 72. Spokes EGS. Neurochemical alterations in Huntington's chorea: a study of post‐mortem brain tissue. Brain. 1980;103:179‐210. [DOI] [PubMed] [Google Scholar]
- 73. Storey E, Beal MF. Neurochemical substrates of rigidity and chorea in Huntington's disease. Brain. 1993;116:1201‐1222. [DOI] [PubMed] [Google Scholar]
- 74. Reynolds GP, Pearson SJ. Brain GABA levels in asymptomatic Huntington's disease. N Engl J Med. 1990;323:682. [DOI] [PubMed] [Google Scholar]
- 75. Beal MF, Ellison DW, Mazurek MF, et al. A detailed examination of substance P in pathologically graded cases of Huntington's disease. J Neurol Sci. 1988;84:51‐61. [DOI] [PubMed] [Google Scholar]
- 76. Buck SH, Burks TF, Brown MR, Yamamura HI. Reduction in basal ganglia and substantia nigra substance P level in Huntington's disease. Brain Res. 1981;209:464‐469. [DOI] [PubMed] [Google Scholar]
- 77. Emson PC, Arregui A, Clement‐Jones V, Sandberg BEB, Rossor M. Regional distribution of methionine‐enkephalin and substance P‐like immunoreactivity in normal human brain and in Huntington's disease. Brain Res. 1980;199:147‐160. [DOI] [PubMed] [Google Scholar]
- 78. Gale GS, Bird ED, Spokes EG, Iversen LL, Jessell T. Human brain substance P: distribution in controls and Huntington's chorea. J Neurochem. 1977;30:633‐634. [DOI] [PubMed] [Google Scholar]
- 79. Kanazawa I, Bird E, O'Connell R, Powell D. Evidence for a decrease in substance P content of substantia nigra in Huntington's chorea. Brain Res. 1977;120:387‐392. [DOI] [PubMed] [Google Scholar]
- 80. Kanazawa I, Bird ED, Gale JS, et al. Substance P: decrease in substantia nigra and globus pallidus in Huntington's disease. Adv Neurol. 1979;23:495‐504. [Google Scholar]
- 81. Spokes EGS, Garrett NJ, Rossor MN, Iversen LL. Distribution of GABA in post‐mortem brain tissue from control, psychotic and Huntington's chorea subjects. J Neurol Sci. 1980;48:303‐313. [DOI] [PubMed] [Google Scholar]
- 82. Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285‐320. [DOI] [PubMed] [Google Scholar]
- 83. Arsenault MY, Parent A, Seguela P, Descarries L. Distribution and morphological characteristics of dopamine‐immunoreactive neurons in the midbrain of the Squirrel monkey (Saimiri sciureus). J Comp Neurol. 1988;267:489‐506. [DOI] [PubMed] [Google Scholar]
- 84. Hökfelt T, Martensson R, Björklund A, Kleinau S, Goldstein M. Distributional maps of tyrosine hydroxylase‐immunoreactive neurons in the rat brain In: Björklund A, Hökfelt T, eds. Handbook of Chemical Neuroanatomy, vol. 2 Classical Transmitter in the CNS. Part I Amsterdam, The Netherlands: Elsevier; 1984:277‐379. [Google Scholar]
- 85. Tippett LJ, Waldvogel HJ, Thomas SJ, et al. Striosomes and mood dysfunction in Huntington's disease. Brain. 2007;130:206‐221. [DOI] [PubMed] [Google Scholar]
- 86. Richfield EK, Maguire‐Zeiss KA, Cox C, Gilmore J, Voorn P. Reduced expression of preproenkephalin in striatal neurons from Huntington's disease patients. Ann Neurol. 1995;37:335‐343. [DOI] [PubMed] [Google Scholar]
- 87. Richfield EK, Maguire‐Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol. 1995;38:852‐861. [DOI] [PubMed] [Google Scholar]
- 88. Bibb JA, Yan Z, Svenningsson P, et al. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci USA. 2000;97:6809‐6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Luthi‐Carter R, Strand A, Peters NL, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259‐1271. [DOI] [PubMed] [Google Scholar]
- 90. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock‐in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11‐26. [DOI] [PubMed] [Google Scholar]
- 91. Sun Z, Del Mar N, Meade C, Goldowitz D, Reiner A. Differential changes in striatal projection neurons in R6/2 transgenic mice for Huntington's disease. Neurobiol Dis. 2002;11:369‐385. [DOI] [PubMed] [Google Scholar]
- 92. Cha JHJ, Kosinski CM, Kerner JA, et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. J Neurosci. 1998;19:1189‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dragatsis I, Goldowitz D, Del Mar N, et al. CAG repeat lengths ≥335 attenuate the phenotype in the R6/2 Huntington's disease transgenic mouse. Neurobiol Dis. 2009;33:315‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Morton AJ, Glynn D, Leavens W, et al. Paradoxical delay in the onset of disease caused by super‐long CAG repeat expansions in R6/2 mice. Neurobiol Dis. 2009;33:331‐341. [DOI] [PubMed] [Google Scholar]
- 95. Canals JM, Pineda JR, Torres‐Peraza JF, et al. Brain‐derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci. 2004;24:7727‐7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cha JH, Frey AS, Alsdorf SA, et al. Altered neurotransmitter receptor expression in transgenic mouse models of Huntington's disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:981‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pineda JR, Canals JM, Bosch M, et al. Brain‐derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington's disease. J Neurochem. 2005;93:1057‐1068. [DOI] [PubMed] [Google Scholar]
- 98. Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N‐terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397‐407. [DOI] [PubMed] [Google Scholar]
- 99. Xie Y, Hayden MR, Xu B. BDNF Overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J Neurosci. 2010;30:14708‐14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Benn CL, Slow EJ, Farrell LA, et al. Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington's disease. Neuroscience. 2007;147:354‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pouladi MA, Stanek LM, Xie Y, et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full‐length BACHD and YAC128 mice. Hum Mol Genet. 2012;21:2219‐2232. [DOI] [PubMed] [Google Scholar]
- 102. Miller S, Hill Della Puppa G, Reidling J, Marcora E, Thompson LM, Treanor J. Comparison of phosphodiesterase 10A, dopamine receptors D1 and D2 and dopamine transporter ligand binding in the striatum of the R6/2 and BACHD mouse models of Huntington's disease. J Huntingtons Dis. 2014;3:333‐341. [DOI] [PubMed] [Google Scholar]
- 103. Stefanko DP, Shah VD, Yamasaki WK, Petzinger GM, Jakowec MW. Treadmill exercise delays the onset of non‐motor behaviors and striatal pathology in the CAG140 knock‐in mouse model of Huntington's disease. Neurobiol Dis. 2017;105:15‐32. [DOI] [PubMed] [Google Scholar]
- 104. Deng Y, Wang H, Joni M, Reiner A. Basal ganglia pathology in heterozygous 18 month‐old Q175 knock‐in Huntington's disease mice. Soc Neurosci Abst 2016; # 226.09. [DOI] [PMC free article] [PubMed]
- 105. Deng Y, Wang HB, Joni M, Reiner A. Striatal projection neuron pathology in heterozygous Q175 knock‐in Huntington's disease mice. Soc Neurosci Abst 2017; # 575.08.
- 106. Bissonnette S, Vaillancourt M, Hébert SS, Drolet G, Samadi P. Striatal pre‐enkephalin overexpression improves Huntington's disease symptoms in the R6/2 mouse model of Huntington's disease. PLoS One. 2013;8:e75099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366‐375. [DOI] [PubMed] [Google Scholar]
- 108. Crossman AR. Primate models of dyskinesia: the experimental approach to the study of basal ganglia‐related involuntary movement disorders. Neuroscience. 1987;21:1‐40. [DOI] [PubMed] [Google Scholar]
- 109. Flaherty AW, Graybiel AM. Output architecture of the primate putamen. J Neurosci. 1993;13:3222‐3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gimenez‐Amaya JM, Graybiel AM. Modular organization of projection neurons in the matrix compartment of the primate striatum. J Neurosci. 1991;11:779‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Richfield EK, O'Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington's disease. Neurosci Lett. 1991;132:121‐126. [DOI] [PubMed] [Google Scholar]
- 112. Hikosaka O. Role of basal ganglia in saccades. Rev Neurol (Paris). 1989;145:580‐586. [PubMed] [Google Scholar]
- 113. Antonini A, Leenders KL, Spiegel R, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996;11:2085‐2095. [DOI] [PubMed] [Google Scholar]
- 114. Aylward EH, Brandt J, Codori AM, Mangus RS, Barta PE, Harris GJ. Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at‐risk persons. Neurology. 1994;44:823‐828. [DOI] [PubMed] [Google Scholar]
- 115. Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch Neurol. 1996;53:1293‐1296. [DOI] [PubMed] [Google Scholar]
- 116. Kuwert T, Lange HW, Boecker H, et al. Striatal glucose consumption in chorea‐free subjects at risk of Huntington's disease. J Neurol. 1993;241:31‐36. [DOI] [PubMed] [Google Scholar]
- 117. Weeks RA, Piccini P, Harding AE, Brooks DJ. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington's disease. Ann Neurol. 1996;40:49‐54. [DOI] [PubMed] [Google Scholar]
- 118. Bunner KD, Rebec GV. Corticostriatal dysfunction in Huntington's disease: the basics. Front Hum Neurosci. 2016;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Klapstein GJ, Fisher RS, Zanjani H, et al. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667‐2677. [DOI] [PubMed] [Google Scholar]
- 120. Miller BR, Walker AG, Shah AS, Barton SJ, Rebec GV. Dysregulated information processing by medium spiny neurons in striatum of freely behaving mouse models of Huntington's disease. J Neurophysiol. 2008;100:2205‐2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 2006;137:327‐336. [DOI] [PubMed] [Google Scholar]
- 122. Heikkinen T, Lehtimäki K, Vartiainen N, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock‐in mouse model of Huntington's disease. PLoS One. 2012;7:e50717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sebastianutto I, Cenci MA, Fieblinger T. Alterations of striatal indirect pathway neurons precede motor deficits in two mouse models of Huntington's disease. Neurobiol Dis. 2017;105:117‐131. [DOI] [PubMed] [Google Scholar]
- 124. Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci. 1991;11:1649‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Calabresi P, Centonze D, Pisani A, et al. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann Neurol. 1998;43:586‐597. [DOI] [PubMed] [Google Scholar]
- 126. Chen Q, Veenman CL, Reiner A. Cellular expression of ionotropic glutamate receptor subunits on specific striatal neuron types and its implication for striatal vulnerability in glutamate receptor‐mediated excitotoxicity. Neuroscience. 1996;73:715‐731. [DOI] [PubMed] [Google Scholar]
- 127. Chen Q, Veenman L, Knopp K, et al. Evidence for the preferential localization of glutamate receptor‐1 subunits of AMPA receptors to the dendritic spines of medium spiny neurons in rat striatum. Neuroscience. 1998;83:749‐761. [DOI] [PubMed] [Google Scholar]
- 128. DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci. 1990;13:286‐289. [DOI] [PubMed] [Google Scholar]
- 129. Gervais FG, Singaraja R, Xanthoudakis S, et al. Recruitment and activation of caspase‐8 by the Huntingtin‐interacting protein Hip‐1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95‐105. [DOI] [PubMed] [Google Scholar]
- 130. Figueredo‐Cardenas G, Harris C, Anderson KD, Reiner A. Relative resistance of striatal neurons containing calbindin or parvalbumin to quinolinic acid‐mediated excitotoxicity compared to other striatal neuron types. Exp Neurol. 1998;149:356‐372. [DOI] [PubMed] [Google Scholar]
- 131. Hackham AS, Yassa AS, Singaraja R, et al. Huntingtin interacting protein 1 induces apoptosis via novel caspase‐dependent death effector domain. J Biol Chem. 2000;275:41299‐41308. [DOI] [PubMed] [Google Scholar]
- 132. Huang Q, Zhou D, Sapp E, et al. Quinolinic acid induced increases in calbindin D28k immunoreactivity in rat striatal neurons in vivo and in vitro mimic the pattern seen in Huntington's disease. Neuroscience. 1995;65:397‐407. [DOI] [PubMed] [Google Scholar]
- 133. Medina L, Figueredo‐Cardenas G, Reiner A. Differential abundance of superoxide dismutase in interneurons versus projection neurons in patch versus matrix neurons in monkey striatum. Brain Res. 1996;708:59‐70. [DOI] [PubMed] [Google Scholar]
- 134. Reiner A, Dragatsis I, Zeitlin SO, Goldowitz D. Wild‐type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28:259‐275. [DOI] [PubMed] [Google Scholar]
- 135. Reiner A, Lafferty DC, Wang HB, Del Mar N, Deng YP. The group 2 metabotropic glutamate receptor agonist LY379268 rescues neuronal, neurochemical and motor abnormalities in R6/2 Huntington's disease mice. Neurobiol Dis. 2012;47:75‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Reiner A, Wang HB, Del Mar N, Sakata K, Yoo W, Deng YP. BDNF may play a differential role in the protective effect of the mGluR2/3 agonist LY379268 on striatal projection neurons in R6/2 Huntington's disease mice. Brain Res. 2012;1473:161‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ruiz‐Calvo A, Maroto IB, Bajo‐Grañeras R, et al. Pathway‐specific control of striatal neuron vulnerability by corticostriatal cannabinoid CB1 receptors. Cereb Cortex. 2018;28:307‐322. [DOI] [PubMed] [Google Scholar]
- 138. Zeron MM, Hansson O, Chen N, et al. Increased sensitivity to N‐methyl‐d‐aspartate receptor‐mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849‐860. [DOI] [PubMed] [Google Scholar]
- 139. Zuccato C, Cattaneo E. Role of brain‐derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294‐330. [DOI] [PubMed] [Google Scholar]
- 140. Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin‐mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493‐498. [DOI] [PubMed] [Google Scholar]
- 141. Zuccato C, Marullo M, Conforti P, McDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008;18:225‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55‐66. [DOI] [PubMed] [Google Scholar]
- 143. Fusco FR, Chen Q, Lamoreaux WJ, et al. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M. Axonal transport of N‐terminal huntingtin suggests early pathology of corticostriatal projections in Huntington's disease. J Neuropathol Exp Neurol. 1999;58:165‐173. [DOI] [PubMed] [Google Scholar]
- 145. Nic‐Niocaill B, Haraldsson B, Hansson O, O'Connor WT, Brundin P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur J Neurosci. 2001;13:206‐210. [DOI] [PubMed] [Google Scholar]
- 146. Roos RA, Pruyt JF, de Vries J, Bots GT. Neuronal distribution in the putamen in Huntington's disease. J Neurol Neurosurg Psychiatry. 1985;48:422‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Parent A, Bouchard C, Smith Y. The striatopallidal and striatonigral projections: two distinct fiber systems in primate. Brain Res. 1984;303:385‐390. [DOI] [PubMed] [Google Scholar]
- 149. Smith Y, Parent A. Differential connections of caudate nucleus and putamen in the squirrel monkey (Saimiri sciureus). Neuroscience. 1986;18:347‐371. [DOI] [PubMed] [Google Scholar]
- 150. Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493‐8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurons: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527‐535. [DOI] [PubMed] [Google Scholar]
- 152. Kita H, Kosaka T, Heizmann CW. Parvalbumin‐immunoreactive neurons in the rat neostriatum: a light and electron microscopic study. Brain Res. 1990;536:1‐15. [DOI] [PubMed] [Google Scholar]
- 153. Figueredo‐Cardenas G, Morello M, Sancesario G, Bernardi G, Reiner A. Colocalization of somatostatin, neuropeptide Y, neuronal nitric oxide synthase and NADPH‐diaphorase in striatal interneurons in rats. Brain Res. 1996;735:317‐324. [DOI] [PubMed] [Google Scholar]
- 154. Bennett BD, Bolam JP. Characterization of calretinin‐immunoreactive structures in the striatum of the rat. Brain Res. 1993;609:137‐148. [DOI] [PubMed] [Google Scholar]
- 155. Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Rev. 2000;34:80‐101. [DOI] [PubMed] [Google Scholar]
- 156. Figueredo‐Cardenas G, Medina L, Reiner A. Calretinin is localized to a unique population of striatal interneurons in rats. Brain Res. 1996;709:145‐150. [DOI] [PubMed] [Google Scholar]
- 157. Kubota Y, Mikawa S, Kawaguchi Y. Neostriatal GABAergic interneurons contain NOS, calretinin or parvalbumin. NeuroReport. 1993;5:205‐208. [DOI] [PubMed] [Google Scholar]
- 158. Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908‐4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tepper JM, Tecuapetla F, Koós T, Ibáñez‐Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Deng YP, Shelby E, Reiner A. Immunohistochemical localization of AMPA‐type glutamate receptor subunits in the striatum of rhesus monkey. Brain Res. 2010;1344:104‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Pisani A, Bernardi G, Ding J, Surmeier DJ. Re‐emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545‐553. [DOI] [PubMed] [Google Scholar]
- 162. Smith Y, Surmeier DJ, Redgrave P, Kimura M. Thalamic contributions to basal ganglia‐related behavioral switching and reinforcement. J Neurosci. 2011;31:16102‐16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168‐171. [DOI] [PubMed] [Google Scholar]
- 165. Dawbarn D, DeQuidt ME, Emson PC. Survival of basal ganglia neuropeptide Y‐somatostatin neurons in Huntington's disease. Brain Res. 1985;340:251‐260. [DOI] [PubMed] [Google Scholar]
- 166. Deng Y, Reiner A. Cholinergic interneurons in the Q140 knock‐in mouse model of Huntington's disease: reductions in dendritic branching and thalamostriatal input. J Comp Neurol. 2016;524:3518‐3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ferrante RJ, Kowall NW, Beal MF, Richardson EP Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;230:561‐563. [DOI] [PubMed] [Google Scholar]
- 168. Ferrante RJ, Kowall NW, Richardson EP Jr, Bird ED, Martin JB. Topography of enkephalin, substance P, and acetylcholinesterase staining in Huntington's disease striatum. Neurosci Lett. 1986;71:283‐288. [DOI] [PubMed] [Google Scholar]
- 169. Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP Jr. Morphological and histochemical characteristics of a spared subset of striatal neurons in Huntington's disease. J Neuropathol Exp Neurol. 1987;46:12‐27. [DOI] [PubMed] [Google Scholar]
- 170. Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Martin JB Jr. Sparing of acetylcholinesterase‐containing striatal neurons in Huntington's disease. Brain Res. 1987;411:162‐166. [DOI] [PubMed] [Google Scholar]
- 171. Hawker K, Lang AE. Hypoxic‐ischemic damage of the basal ganglia: case reports and a review of the literature. Mov Disord. 1990;5:219‐224. [DOI] [PubMed] [Google Scholar]
- 172. Kowall NW, Ferrante RJ, Martin JB. Pattern of cell loss in Huntington's disease. Trends Neurosci. 1987;10:24‐29. [Google Scholar]
- 173. Massouh M, Wallman MJ, Pourcher E, Parent A. The fate of the large striatal interneurons expressing calretinin in Huntington's disease. Neurosci Res. 2008;62:216‐224. [DOI] [PubMed] [Google Scholar]
- 174. Norris PJ, Waldvogel HJ, Faull RL, Love DR, Emson PC. Decreased neuronal nitric oxide synthase messenger RNA and somatostatin messenger RNA in the striatum of Huntington's disease. Neuroscience. 1996;72:1037‐1047. [DOI] [PubMed] [Google Scholar]
- 175. Aquilonius AM, Eckernas SA, Sundwall A. Regional distribution of choline acetyltransferase in the human brain: changes in Huntington's chorea. J Neurol Neurosurg Psychiatry. 1975;38:669‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Smith R, Chung H, Rundquist S, et al. Cholinergic neuronal defect without cell loss in Huntington's disease. Hum Mol Genet. 2006;15:3119‐3131. [DOI] [PubMed] [Google Scholar]
- 177. Holley SM, Joshi PR, Parievsky A, et al. Enhanced GABAergic inputs contribute to functional alterations of cholinergic interneurons in the R6/2 mouse model of Huntington's disease. eNeuro. 2015;2:e0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Vetter JM, Jehle T, Heinemeyer J, et al. Mice transgenic for exon 1 of Huntington's disease: properties of cholinergic and dopaminergic pre‐synaptic function in the striatum. J Neurochem. 2003;85:1054‐1063. [DOI] [PubMed] [Google Scholar]
- 179. Higley MJ, Gittis AH, Oldenburg IA, et al. Cholinergic interneurons mediate fast VGluT3‐dependent glutamatergic transmission in the striatum. PLoS One. 2011;6:e19155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Nelson AB, Bussert TG, Kreitzer AC, Seal RP. Striatal cholinergic neurotransmission requires VGLUT3. J Neurosci. 2014;4:8772‐8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467‐472. [DOI] [PubMed] [Google Scholar]
- 182. Galarraga E, Vilchis C, Tkatch T, et al. Somatostatinergic modulation of firing pattern and calcium‐activated potassium currents in medium spiny neostriatal neurons. Neuroscience. 2007;146:537‐554. [DOI] [PubMed] [Google Scholar]
- 183. Sammut S, Park DJ, West AR. Frontal cortical afferents facilitate striatal nitric oxide transmission in vivo via a NMDA receptor and neuronal NOS‐dependent mechanism. J Neurochem. 2007;103:1145‐1156. [DOI] [PubMed] [Google Scholar]
- 184. West AR, Grace AA. The nitric oxide‐guanylyl cyclase signaling pathway modulates membrane activity states and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J Neurosci. 2004;24:1924‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35:557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Menalled L, Zanjani H, MacKenzie L, et al. Decrease in striatal enkephalin mRNA in mouse models of Huntington's disease. Exp Neurol. 2000;162:328‐342. [DOI] [PubMed] [Google Scholar]
- 187. Cepeda C, Galvan L, Holley SM, et al. Multiple sources of striatal inhibition are differentially affected in Huntington's disease mouse models. J Neurosci. 2013;33:7393‐7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Ibáñez‐Sandoval O, Tecuapetla F, Unal B, Shah F, Koós T, Tepper JM. A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J Neurosci. 2011;31:16757‐16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Muñoz‐Manchado AB, Foldi C, Szydlowski S, et al. Novel striatal GABAergic interneuron populations labeled in the 5HT3a(EGFP) mouse. Cereb Cortex. 2016;26:96‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Huot P, Levesque M, Parent A. The fate of striatal dopaminergic neurons in Parkinson's disease and Huntington's chorea. Brain. 2007;130:222‐232. [DOI] [PubMed] [Google Scholar]
- 191. Bennett BD, Bolam JP. Synaptic input and output of parvalbumin‐immunoreactive neurons in the neostriatum of the rat. Neuroscience. 1994;62:707‐719. [DOI] [PubMed] [Google Scholar]
- 192. Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Lapper SR, Smith Y, Sadikot AF, Parent A, Bolam JP. Cortical input to parvalbumin‐immunoreactive neurons in the putamen of squirrel monkey. Brain Res. 1992;580:215‐224. [DOI] [PubMed] [Google Scholar]
- 194. Planert H, Szydlowski SN, Hjorth JJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast‐spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499‐3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Berke JD. Uncoordinated firing rate changes of striatal fast‐spiking interneurons during behavioral task performance. J Neurosci. 2008;40:10075‐10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Berke JD. Functional properties of striatal fast‐spiking interneurons. Front Syst Neurosci. 2011;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Giampà C, Middei S, Patassini S, et al. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902‐910. [DOI] [PubMed] [Google Scholar]
- 198. Czeredys M, Gruszczynska‐Biegala J, Schacht T, Methner A, Kuznicki J. Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington's disease. Front Mol Neurosci. 2013;6:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Simmons DA, Belichenko NP, Yang T, et al. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington's disease. J Neurosci. 2013;33:18712‐18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ferrer I, Kulisevsky J, Gonzalez G, Escartin A, Chivite A, Casas R. Parvalbumin‐immunoreactive neurons in the cerebral cortex and striatum in Huntington's disease. Neurodegeneration. 1994;3:169‐173. [Google Scholar]
- 201. Harrington KM, Kowall NW. Parvalbumin immunoreactive neurons resist degeneration in Huntington's disease striatum. J Neuropathol Exp Neurol. 1991;50:309. [DOI] [PubMed] [Google Scholar]
- 202. Reiner A, Shelby E, Wang H, et al. Striatal parvalbuminergic neurons are lost in Huntington's Disease: implications for dystonia. Mov Disord. 2013;28:1691‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Gernert M, Hamann M, Bennay M, Löscher W, Richter A. Deficit of striatal parvalbumin‐reactive GABAergic interneurons and decreased basal ganglia output in a genetic rodent model of idiopathic paroxysmal dystonia. J Neurosci. 2000;20:7052‐7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC. Selective inhibition of striatal fast‐spiking interneurons causes dyskinesias. J Neurosci. 2011;31:15727‐15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Xu M, Li L, Pittenger C. Ablation of fast‐spiking interneurons in the dorsal striatum, recapitulating abnormalities seen post‐mortem in Tourette syndrome, produces anxiety and elevated grooming. Neuroscience. 2016;324:321‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kalanithi PSA, Zheng W, Kataoka Y, et al. Altered parvalbumin‐positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci USA. 2005;102:13307‐13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Kataoka Y, Kalanithi PS, Grantz H, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette Syndrome. J Comp Neurol. 2010;518:277‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Pappas SS, Darr K, Holley SM, et al. Forebrain deletion of the dystonia protein torsinA causes dystonic‐like movements and loss of striatal cholinergic neurons. eLife. 2015;4:e08352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Martos YV, Braz BY, Beccaria JP, Murer MG, Belforte JE. Compulsive social behavior emerges after selective ablation of striatal cholinergic interneurons. J Neurosci. 2017;37:2849‐2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Rapanelli M, Frick LR, Xu M, et al. Targeted interneuron depletion in the dorsal striatum produces autism‐like behavioral abnormalities in male but not female mice. Biol Psychiat. 2017;82:194‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Deng YP, Xie JP, Wang HB, Lei WL, Chen Q, Reiner A. Differential localization of the GluR1 and GluR2 subunits of the AMPA‐type glutamate receptor among striatal neuron types in rats. J Chem Neuroanat. 2007;33:167‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Fortin M, Parent A. Patches in the striatum of squirrel monkeys are enriched with calretinin fibers but devoid of calretinin cell bodies. Neurosci Lett. 1994;182:51‐54. [DOI] [PubMed] [Google Scholar]
- 213. Petryszyn S, Beaulieu JM, Parent A, Parent M. Distribution and morphological characteristics of striatal interneurons expressing calretinin in mice: a comparison with human and nonhuman primates. J Chem Neuroanat. 2014;59–60:51‐61. [DOI] [PubMed] [Google Scholar]
- 214. Prensa L, Giménez‐Amaya JM, Parent A. Morphological features of neurons containing calcium‐binding proteins in the human striatum. J Comp Neurol. 1998;390:552‐563. [PubMed] [Google Scholar]
- 215. Wu Y, Parent A. Striatal interneurons expressing calretinin, parvalbumin or NADPH‐diaphorase: a comparative study in the rat, monkey and human. Brain Res. 2000;863:182‐191. [DOI] [PubMed] [Google Scholar]
- 216. Cicchetti F, Parent A. Striatal interneurons in Huntington's disease: selective increase in the density of calretinin‐immunoreactive medium‐sized neurons. Mov Disord. 1996;11:619‐626. [DOI] [PubMed] [Google Scholar]
- 217. Cicchetti F, Beach TG, Parent A. Chemical phenotype of calretinin interneurons ion the human striatum. Synapse. 1998;30:284‐297. [DOI] [PubMed] [Google Scholar]
- 218. Wilson CJ. Dendritic morphology, inward rectification and the functional properties of neostriatal neurons In: McKenna T, Davis J, Zornetzer SF, eds. Single Neuron Computation. San Diego, CA: Academic Press; 1992:141‐171. [Google Scholar]
- 219. Berendse HW, Groenewegen HJ. Organization of the thalamostriatal projections in the rat, with special emphasis on the ventral striatum. J Comp Neurol. 1990;299:187‐228. [DOI] [PubMed] [Google Scholar]
- 220. Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci. 1994;17:52‐57. [DOI] [PubMed] [Google Scholar]
- 221. Kato S, Kuramochi M, Kobayashi K, et al. Selective neural pathway targeting reveals key roles of thalamostriatal projection in the control of visual discrimination. J Neurosci. 2011;31:17169‐17179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Smith Y, Raju DV, Pare JP, Sidibe M. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004;27:520‐527. [DOI] [PubMed] [Google Scholar]
- 223. Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and Parkinsonian states. Brain Res Bull. 2009;78:60‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Byers RK, Gilles FH, Fung C. Huntington's disease in children. Neuropathological study of four cases. Neurology. 1983;23:561‐569. [DOI] [PubMed] [Google Scholar]
- 225. Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200‐204. [DOI] [PubMed] [Google Scholar]
- 226. Passani LA, Vonsattel JP, Carter RE, Coyle JT. N‐acetylaspartylglutamate, N‐acetylaspartate, and N‐acetylated alpha‐linked acidic dipeptidase in human brain and their alterations in Huntington and Alzheimer's diseases. Mol Chem Neuropathol. 1997;31:97‐118. [DOI] [PubMed] [Google Scholar]
- 227. Selemon LD, Rajkowska G, Goldman‐Rakic PS. Evidence for progression in frontal cortical pathology in late‐stage Huntington's disease. J Comp Neurol. 2004;468:190‐204. [DOI] [PubMed] [Google Scholar]
- 228. Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH. Morphometric analysis of the prefrontal cortex in Huntington's disease. Neurology. 1991;41:1117‐1123. [DOI] [PubMed] [Google Scholar]
- 229. Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett. 1991;133:257‐261. [DOI] [PubMed] [Google Scholar]
- 230. Macdonald V, Halliday G. Pyramidal cell loss in motor cortices in Huntington's disease. Neurobiol Dis. 2002;10:378‐386. [DOI] [PubMed] [Google Scholar]
- 231. Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington's disease: a quantitative Golgi study. Neurology. 1993;43:2088‐2096. [DOI] [PubMed] [Google Scholar]
- 232. Kassubek J, Juengling FD, Kioschies T, et al. Topography of cerebral atrophy in early Huntington's disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry. 2004b;75:213‐220. [PMC free article] [PubMed] [Google Scholar]
- 233. Mühlau M, Weindl A, Wohlschläger AM, et al. Voxel‐based morphometry indicates relative preservation of the limbic prefrontal cortex in early Huntington disease. J Neural Transm. 2007;114:367‐372. [DOI] [PubMed] [Google Scholar]
- 234. Rosas HD, Koroshetz WJ, Chen YI, et al. Evidence for more widespread cerebral pathology in early HD: an MRI‐based morphometric analysis. Neurology. 2003;60:1615‐1620. [DOI] [PubMed] [Google Scholar]
- 235. Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745‐747. [DOI] [PubMed] [Google Scholar]
- 236. Thu DC, Oorschot DE, Tippett LJ, et al. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington's disease. Brain. 2010;133:1094‐1110. [DOI] [PubMed] [Google Scholar]
- 237. Braak H, Braak E. Allocortical involvement in Huntington's disease. Neuropathol Appl Neurobiol. 1992;18:539‐547. [DOI] [PubMed] [Google Scholar]
- 238. Jech R, Klempír J, Vymazal J, et al. Variation of selective gray and white matter atrophy in Huntington's disease. Mov Disord. 2007;22:1783‐1789. [DOI] [PubMed] [Google Scholar]
- 239. Klöppel S, Stonnington CM, Barnes J, et al. Accuracy of dementia diagnosis: a direct comparison between radiologists and a computerized method. Brain. 2008;131:2969‐2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Wolf RC, Sambataro F, Vasic N, Schönfeldt‐Lecuona C, Ecker D, Landwehrmeyer B. Aberrant connectivity of lateral prefrontal networks in presymptomatic Huntington's disease. Exp Neurol. 2008;213:137‐144. [DOI] [PubMed] [Google Scholar]
- 241. Morton AJ, Faull RL, Edwardson JM. Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease. Brain Res Bull. 2001;56:111‐117. [DOI] [PubMed] [Google Scholar]
- 242. Smith R, Klein P, Koc‐Schmitz Y, et al. Loss of SNAP‐25 and rabphilin 3a in sensory‐motor cortex in Huntington's disease. J Neurochem. 2007;103:115‐123. [DOI] [PubMed] [Google Scholar]
- 243. Zucker B, Kama JA, Kuhn A, et al. Decreased Lin7b expression in layer 5 pyramidal neurons may contribute to impaired corticostriatal connectivity in Huntington disease. J Neuropathol Exp Neurol. 2010;69:880‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem Res. 2008;33:232‐237. [DOI] [PubMed] [Google Scholar]
- 245. Rupp J, Blekher T, Jackson J, et al. Progression in prediagnostic Huntington disease. J Neurol Neurosurg Psychiatry. 2010;81:379‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Wilson CJ, Chang HT, Kitai ST. Origins of postsynaptic potentials evoked in identified neostriatal neurons by stimulation in substantia nigra. Exp Brain Res. 1982;45:157‐167. [DOI] [PubMed] [Google Scholar]
- 247. Deng YP, Wong T, Bricker‐Anthony C, Deng B, Reiner A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington's disease mice. Neurobiol Dis. 2013;60:89‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Cepeda C, Hurst RS, Calvert CR, et al. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Cummings DM, Cepeda C, Levine MS. Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington's disease. ASN Neuro. 2010;2:e00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Graham RK, Pouladi MA, Joshi P, et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J Neurosci. 2009;29:2193‐2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Indersmitten T, Tran CH, Cepeda C, Levine MS. Altered excitatory and inhibitory inputs to striatal medium‐sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington's disease. J Neurophysiol. 2015;113:2953‐2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Singaraja RR, Huang K, Sanders SS, et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20:3899‐3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Galvan L, André VM, Wang EA, Cepeda C, Levine MS. Functional differences between direct and indirect striatal output pathways in Huntington's disease. J Huntingtons Dis. 2012;1:17‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Cowan RL, Wilson CJ. Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J Neurophysiol. 1994;71:17‐32. [DOI] [PubMed] [Google Scholar]
- 255. Levesque M, Charara A, Gagnon S, Parent A, Deschenes M. Corticostriatal projections from layer V cells in rat are collaterals of long‐range corticofugal axons. Brain Res. 1996;709:311‐315. [DOI] [PubMed] [Google Scholar]
- 256. Lévesque M, Gagnon S, Parent A, Deschênes M. Axonal arborizations of corticostriatal and corticothalamic fibers arising from the second somatosensory area in the rat. Cereb Cortex. 1996;6:759‐770. [DOI] [PubMed] [Google Scholar]
- 257. Lévesque M, Parent A. Axonal arborization of corticostriatal and corticothalamic fibers arising from prelimbic cortex in the rat. Cereb Cortex. 1998;8:602‐813. [DOI] [PubMed] [Google Scholar]
- 258. Parent M, Parent A. Single‐axon tracing study of corticostriatal projections arising from primary motor cortex in primates. J Comp Neurol. 2006;496:202‐213. [DOI] [PubMed] [Google Scholar]
- 259. Reiner A, Jiao Y, Del Mar N, Laverghetta AV, Lei WL. Differential morphology of pyramidal tract‐type and intratelencephalically projecting‐type corticostriatal neurons and their intrastriatal terminals in rats. J Comp Neurol. 2003;457:420‐440. [DOI] [PubMed] [Google Scholar]
- 260. Wilson CJ. Morphology and synaptic connections of crossed corticostriatal neurons in the rat. J Comp Neurol. 1987;263:567‐580. [DOI] [PubMed] [Google Scholar]
- 261. Wright AK, Norrie L, Ingham CA, Hutton EA, Arbuthnott GW. Double anterograde tracing of outputs from adjacent “barrel columns” of rat somatosensory cortex. Neostriatal projection patterns and terminal ultrastructure. Neuroscience. 1999;88:119‐133. [DOI] [PubMed] [Google Scholar]
- 262. Wright AK, Ramanathan S, Arbuthnott GW. Identification of the source of the bilateral projection system from cortex to somatosensory neostriatum and an exploration of its physiological actions. Neuroscience. 2001;103:87‐96. [DOI] [PubMed] [Google Scholar]
- 263. Cepeda C, André VM, Yamazaki I, Wu N, Kleiman‐Weiner M, Levine MS. Differential electrophysiological properties of dopamine D1 and D2 receptor‐containing striatal medium‐sized spiny neurons. Eur J Neurosci. 2008;27:671‐682. [DOI] [PubMed] [Google Scholar]
- 264. Deng Y, Lanciego J, Kerkerian‐Le Goff L, et al. Differential organization of cortical inputs to striatal projection neurons of the matrix compartment in rodents. Front Syst Neurosci. 2015;9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Lei W, Jiao Y, Del Mar N, Reiner A. Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J Neurosci. 2004;24:8289‐8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Reiner A, Hart NM, Lei W, Deng Y. Corticostriatal projection neurons ‐ dichotomous types and dichotomous functions. Front Neuroanat. 2010;4:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Hickey MA, Kosmalska A, Enayati J, et al. Extensive early motor and non‐motor behavioral deficits are followed by striatal neuronal loss in knock‐in Huntington's disease mice. Neuroscience. 2008;157:280‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Rising AC, Xu J, Carlson A, Napoli VV, Denovan‐Wright EM, Mandel RJ. Longitudinal behavioral, cross‐sectional transcriptional and histopathological characterization of a knock‐in mouse model of Huntington's disease with 140 CAG repeats. Exp Neurol. 2011;228:173‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Deng YP, Wong T, Wan JY, Reiner A. Differential early loss of thalamostriatal and corticostriatal input to striatal projection neuron types in the Q140 Huntington's disease knock‐in mouse model. Front Syst Neurosci. 2014;8:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. André VM, Cepeda C, Fisher YE, et al. Differential electrophysiological changes in striatal output neurons in Huntington's disease. J Neurosci. 2011;31:1170‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Joshi PR, Wu NP, André VM, et al. Age‐dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29:2414‐2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. André VM, Fisher YE, Levine MS. Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington's disease. Front Syst Neurosci. 2011b;5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Estrada‐Sanchez A, Rebec VG. Role of cerebral cortex in the neuropathology of Huntington's disease. Front Neural Circuits. 2013;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Hansson O, Petersén A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid‐induced striatal excitotoxicity. Proc Natl Acad Sci USA. 1999;96:8727‐8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Hansson O, Guatteo E, Mercuri NB, et al. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur J Neurosci. 2001;14:1492‐1504. [DOI] [PubMed] [Google Scholar]
- 276. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron. 2012;74:691‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ. MEF‐2 regulates activity‐dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci. 2010;44:94‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Gu X, Wilton D, Daggett A, Lee C, Stevens BA, Yang X. Complement c3 deficiency modifies BACHD disease phenotypes. Soc Neurosci Abst 2017; # 212.01.
- 279. Wilton D, Heller M, Daggett A, et al. Microglia mediate early loss of specific synaptic connections in Huntington's disease. Soc Neurosci Abst 2017; # 564.18.
- 280. Foster N, Becerra M, Bowman I, et al. Pathological changes in descending cortical projections in the zQ175 mouse model of Huntington's disease. Soc Neurosci Abst 2017; #575.12.
- 281. Plotkin JL, Day M, Peterson JD, et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington's disease. Neuron. 2014;83:178‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24:182‐188. [DOI] [PubMed] [Google Scholar]
- 283. Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919‐930. [DOI] [PubMed] [Google Scholar]
- 284. Ginés S, Bosch M, Marco S, et al. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci. 2006;23:649‐658. [DOI] [PubMed] [Google Scholar]
- 285. Nguyen KQ, Rymar VV, Sadikot AF. Impaired TrkB signaling underlies reduced BDNF‐mediated trophic support of striatal neurons in the R6/2 mouse model of Huntington's disease. Front Cell Neurosci. 2016;10:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Baydyuk M, Russell T, Liao GY, et al. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci USA. 2011;108:1669‐1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Doyle JP, Dougherty JD, Heiman M, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Cepeda C, Cummings DM, André VM, Holley SM, Levine MS. Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms. ASN Neuro. 2010;2:e00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Cummings DM, André VM, Uzgil BO, et al. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci. 2009;29:10371‐10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Suzuki M, Desmond TJ, Albin RL, Frey KA. Vesicular neurotransmitter transporters in Huntington's disease: initial observations and comparison with traditional synaptic markers. Synapse. 2001;41:329‐336. [DOI] [PubMed] [Google Scholar]
- 291. Farrar A, Callahan JW, Abercrombie ED. Reduced striatal acetylcholine efflux in the R6/2 mouse model of Huntington's disease: an examination of the role of altered inhibitory and excitatory mechanisms. Exp Neurol. 2011;232:119‐125. [DOI] [PubMed] [Google Scholar]
- 292. Doig NM, Moss J, Bolam JP. Cortical and thalamic innervation of direct and indirect pathway medium‐sized spiny neurons in mouse striatum. J Neurosci. 2010;30:14610‐14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. 1992;51:533‐545. [DOI] [PubMed] [Google Scholar]
- 294. Tanimura A, Lim SA, Aceves Buendia JJ, Goldberg JA, Surmeier DJ. Cholinergic interneurons amplify corticostriatal synaptic responses in the Q175 model of Huntington's disease. Front Syst Neurosci. 2016;10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Arias‐García MA, Tapia D, Laville JA, et al. Functional comparison of corticostriatal and thalamostriatal postsynaptic responses in striatal neurons of the mouse. Brain Struct Funct. 2017; 10.1007/s00429-017-1536-6 [DOI] [PubMed] [Google Scholar]
- 296. Lei W, Deng YP, Liu BB, et al. A confocal laser scanning microscopy and ultrastructural study of VGLUT2 thalamic input to striatal projection neurons in rats. J Comp Neurol. 2013;521:1354‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci. 2007;27:4374‐4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Sadikot AF, Parent A, Smith Y, Bolam JP. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a light and electron microscopic study of the thalamostriatal projection in relation to striatal heterogeneity. J Comp Neurol. 1992;320:228‐242. [DOI] [PubMed] [Google Scholar]
- 299. Sidibé M, Smith Y. Differential synaptic innervation of striatofugal neurones projecting to the internal or external segments of the globus pallidus by thalamic afferents in the squirrel monkey. J Comp Neurol. 1996;365:445‐465. [DOI] [PubMed] [Google Scholar]
- 300. Xu ZC, Wilson CJ, Emson PC. Restoration of thalamostriatal projections in rat neostriatal grafts: electron microscopic analysis. J Comp Neurol. 1991;303:22‐34. [DOI] [PubMed] [Google Scholar]
- 301. Douaud G, Gaura V, Ribeiro MJ, et al. Distribution of grey matter atrophy in Huntington's disease patients: a combined ROI‐based and voxel‐based morphometric study. NeuroImage. 2006;32:1562‐1575. [DOI] [PubMed] [Google Scholar]
- 302. Byers RK, Gilles FH, Fung C. Huntington's disease in children. Neuropathologic study of four cases. Neurology. 1973;23:561‐569. [DOI] [PubMed] [Google Scholar]
- 303. Mann DM, Oliver R, Snowden JS. The topographic distribution of brain atrophy in Huntington's disease and progressive supranuclear palsy. Acta Neuropathol. 1993;85:553‐559. [DOI] [PubMed] [Google Scholar]
- 304. Heinsen H, Rüb U, Gangnus D, et al. Nerve cell loss in the thalamic centromedian‐parafascicular complex in patients with Huntington's disease. Acta Neuropathol. 1996;91:161‐168. [DOI] [PubMed] [Google Scholar]
- 305. Heinsen H, Rüb U, Bauer M, et al. Nerve cell loss in the thalamic mediodorsal nucleus in Huntington's disease. Acta Neuropathol. 1999;97:613‐622. [DOI] [PubMed] [Google Scholar]
- 306. Freeman W, Morton AJ. Regional and progressive changes in brain expression of complexin II in a mouse transgenic for the Huntington's disease mutation. Brain Res Bull. 2004;63:45‐55. [DOI] [PubMed] [Google Scholar]
- 307. Kusakabe M, Mangiarini L, Laywell ED, et al. Loss of cortical and thalamic neuronal tenascin‐C expression in a transgenic mouse expressing exon 1 of the human Huntington disease gene. J Comp Neurol. 2001;430:485‐500. [DOI] [PubMed] [Google Scholar]
- 308. Kolodziejczyk K, Raymond LA. Differential changes in thalamic and cortical excitatory synapses onto striatal spiny projection neurons in a Huntington disease mouse model. Neurobiol Dis. 2016;86:62‐74. [DOI] [PubMed] [Google Scholar]
- 309. Parievsky A, Moore C, Kamdjou T, Cepeda C, Meshul CK, Levine MS. Differential electrophysiological and morphological alterations of thalamostriatal and corticostriatal projections in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2017;108:29‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Samadi P, Boutet A, Rymar VV, et al. Relationship between BDNF expression in major striatal afferents, striatum morphology and motor behavior in the R6/2 mouse model of Huntington's disease. Genes Brain Behav. 2013;12:108‐124. [DOI] [PubMed] [Google Scholar]
- 311. Giorgi S, Rimoldi M, Consolo S. Parafascicular thalamic nucleus deafferentation reduces c‐fos expression induced by dopamine D‐1 receptor stimulation in rat striatum. Neuroscience. 2001;103:653‐661. [DOI] [PubMed] [Google Scholar]
- 312. Lanciego JL, Gonzalo N, Castle M, Sanchez‐Escobar C, Aymerich MS, Obeso JA. Thalamic innervation of striatal and subthalamic neurons projecting to the rat entopeduncular nucleus. Eur J Neurosci. 2004;19:1267‐1277. [DOI] [PubMed] [Google Scholar]
- 313. Ding JB, Oh WJ, Sabatini BL, Gu C. Semaphorin 3E‐Plexin‐D1 signaling controls pathway‐specific synapse formation in the striatum. Nat Neurosci. 2011;15:215‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Kuhn A, Goldstein DR, Hodges A, et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild‐type huntingtin dosage. Hum Mol Genet. 2007;16:1845‐1861. [DOI] [PubMed] [Google Scholar]
- 315. Strand AD, Baquet ZC, Aragaki AK, et al. Jones KR Expression profiling of Huntington's disease models suggests that brain‐derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758‐11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Kawaguchi Y, Wilson CJ, Emson PC. Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs. J Neurophysiol. 1989;62:1052‐1068. [DOI] [PubMed] [Google Scholar]
- 317. Mahon S, Deniau JM, Charpier S. Relationship between EEG potentials and intracellular activity of striatal and cortico‐striatal neurons: an in vivo study under different anesthetics. Cereb Cortex. 2001;11:360‐373. [DOI] [PubMed] [Google Scholar]
- 318. Nisenbaum ES, Wilson CJ. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci. 1995;15:4449‐4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Stern EA, Kincaid AE, Wilson CJ. Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol. 1997;77:1697‐1715. [DOI] [PubMed] [Google Scholar]
- 320. DeLong M. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281‐285. [DOI] [PubMed] [Google Scholar]
- 321. Parent M, Parent A. Single‐axon tracing and three‐dimensional reconstruction of centre median‐parafascicular thalamic neurons in primates. J Comp Neurol. 2005;481:127‐144. [DOI] [PubMed] [Google Scholar]
- 322. Matsumoto N, Minamimoto T, Graybiel AM, Kimura M. Neurons in the thalamic CM–Pf complex supply striatal neurons with information about behaviorally significant sensory events. J Neurophysiol. 2001;85:960‐976. [DOI] [PubMed] [Google Scholar]
- 323. Deschênes M, Bourassa J, Parent A. Two different types of thalamic fibers innervate the rat striatum. Brain Res. 1995;701:288‐292. [DOI] [PubMed] [Google Scholar]
- 324. Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28:6483‐6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Sidibé M, Smith Y. Thalamic inputs to striatal interneurons in monkeys: synaptic organization and co‐localization of calcium binding proteins. Neuroscience. 1999;89:1189‐1208. [DOI] [PubMed] [Google Scholar]
- 326. Brown HD, Baker PM, Ragozzino ME. The parafascicular thalamic nucleus concomitantly influences behavioral flexibility and dorsomedial striatal acetylcholine output in rats. J Neurosci. 2010;30:14390‐14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Rudkin TM, Sadikot AF. Thalamic input to parvalbumin‐immunoreactive GABAergic interneurons: organization in normal striatum and effect of neonatal decortication. Neuroscience. 1999;88:1165‐1175. [DOI] [PubMed] [Google Scholar]
- 328. Sharp AH, Ross CA. Neurobiology of Huntington's disease. Neurobiol Dis. 1996;3:3‐15. [DOI] [PubMed] [Google Scholar]
- 329. Oyanagi K, Takeda S, Takahashi H, Ohama E, Ikuta F. A quantitative investigation of the substantia nigra in Huntington's disease. Ann Neurol. 1989;26:13‐19. [DOI] [PubMed] [Google Scholar]
- 330. Yohrling GJ 4th, Jiang GC, DeJohn MM, et al. Analysis of cellular, transgenic and human models of Huntington's disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Mol Brain Res. 2003;119:28‐36. [DOI] [PubMed] [Google Scholar]
- 331. Bedard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson's disease and Huntington's chorea. Parkinsonism Relat Disord. 2011;17:593‐598. [DOI] [PubMed] [Google Scholar]
- 332. Ferrante RJ, Kowall NW. Tyrosine hydroxylase‐like immunoreactivity is distributed in the matrix compartment of normal human and Huntington's disease striatum. Brain Res. 1987;416:141‐146. [DOI] [PubMed] [Google Scholar]
- 333. Bohnen NI, Koeppe RA, Meyer P, et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54:1753‐1759. [DOI] [PubMed] [Google Scholar]
- 334. Ginovart N, Lundin A, Farde L, et al. PET study of the pre‐ and post‐synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain. 1997;120:503‐514. [DOI] [PubMed] [Google Scholar]
- 335. Kish SJ, Shannak K, Hornykiewicz O. Elevated serotonin and reduced dopamine in subregionally divided Huntington's disease striatum. Ann Neurol. 1987;22:386‐389. [DOI] [PubMed] [Google Scholar]
- 336. Garrett MC, Soares‐da‐Silva P. Increased cerebrospinal fluid dopamine and 3,4‐dihydroxyphenylacetic acid levels in Huntington's disease: evidence for an overactive dopaminergic brain transmission. J Neurochem. 1992;58:101‐106. [DOI] [PubMed] [Google Scholar]
- 337. Klawans HC, Paulson GW, Barbeau A. Predictive test for Huntington's chorea. Lancet. 1970;2:1185‐1186. [DOI] [PubMed] [Google Scholar]
- 338. Callahan JW, Abercrombie ED. In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full‐Length (YAC128) transgenic mouse models of Huntington's disease. Front Syst Neurosci. 2011;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem. 2002;81:46‐59. [DOI] [PubMed] [Google Scholar]
- 340. Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem. 2006;97:737‐746. [DOI] [PubMed] [Google Scholar]
- 341. Petersen A, Puschban Z, Lotharius J, et al. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington's disease mice. Neurobiol Dis. 2002;11:134‐146. [DOI] [PubMed] [Google Scholar]
- 342. Mochel F, Durant B, Durr A, Schiffmann R. Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS One. 2011;6:e18336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Cepeda C, Murphy KP, Parent M, Levine MS. The role of dopamine in Huntington's disease. Prog Brain Res. 2014;211:235‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Girasole AE, Nelson AB. Probing striatal microcircuitry to understand the functional role of cholinergic interneurons. Mov Disord. 2015;30:1306‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Gantois I, Fang K, Jiang L, et al. Ablation of D1 dopamine receptor‐expressing cells generates mice with seizures, dystonia, hyperactivity, and impaired oral behavior. Proc Natl Acad Sci USA. 2007;104:4182‐4187. [DOI] [PMC free article] [PubMed] [Google Scholar]