

Abstract
A cytochrome P450 was engineered to selectively incorporate Ir(Me)-deuteroporphyrin IX (Ir(Me)-DPIX), in lieu of heme, in bacterial cells. Cofactor selectivity was altered by introducing mutations within the heme-binding pocket that discriminate the deuteroporphyrin macrocycle, in combination with mutations to the P450 axial cysteine to accommodate a pendant methyl group on the Ir(Me)-center. This artificial metalloenzyme was investigated for activity in non-native metallocarbenoid-mediated olefin cyclopropanation reactions and showed enhanced activity for aliphatic and electron-deficient olefins when compared to the native heme enzyme. This work provides a general strategy to augment the chemical functionality of heme enzymes in cells with application towards abiotic catalysis.
Keywords: artificial metalloenzyme, biocatalysis, cyclopropanation, Ir(Me)-porphyrin, orthogonal enzyme/cofactor
TOC image
Engineering an (Ir)regular Cytochrome P450: Mutations within the heme-binding pocket of a Cytochrome P450 enabled the selective incorporation of an artificial Ir-porphyrin cofactor into the protein, in cells. This orthogonal metalloprotein shows enhanced behavior in non-natural carbene-mediated cyclopropanation of aliphatic and electron deficient olefins.
Artificial metalloenzymes have emerged as promising tools to expand the reaction space available for biocatalysis.[1–3] A common strategy for the construction of artificial metalloenzymes makes use of existing molecular recognition motifs in protein active sites to immobilize new metallocofactors.[4–9] These metal-substituted enzymes often possess activity for abiotic reactions that can be optimized through mutation of the protein host.[10–13] Methods for introducing artificial cofactors into protein scaffolds, however, are often labor-intensive, requiring in vitro manipulation and reconstitution, which may limit engineering efforts and prevent the application of these systems in whole-cells.[1,10,14]
We recently introduced a strategy for the selective introduction of a synthetic heme analogue, Fe-deuteroporphyrin IX (Fe-DPIX, Figure 1), into proteins through the evolution of an orthogonal enzyme/cofactor pair.[15] To establish the utility of this approach, the heme-binding domain (BM3h) of a cytochrome P450 (P450) from Bacillus megaterium was evolved to selectively bind Fe-DPIX, even in the presence of native heme.[15] This altered selectivity was achieved via mutations that discriminate subtle structural differences between the two porphyrin macrocycles (i.e. the removal of two vinyl groups, Figure 1). Since Fe-DPIX and heme both possess an iron center, however, the orthogonal enzyme displayed similar reactivity compared to the native enzyme.[15] Here, we extend the application of orthogonal/enzyme cofactor pairs to the in cell production of an artificial Ir-Me-substituted P450 and demonstrate its activity towards non-native metallocarbenoid-mediated cyclopropanation of olefins that are challenging substrates for the native iron enzyme.
Figure 1. Generation of an orthogonal metal-substituted enzyme.
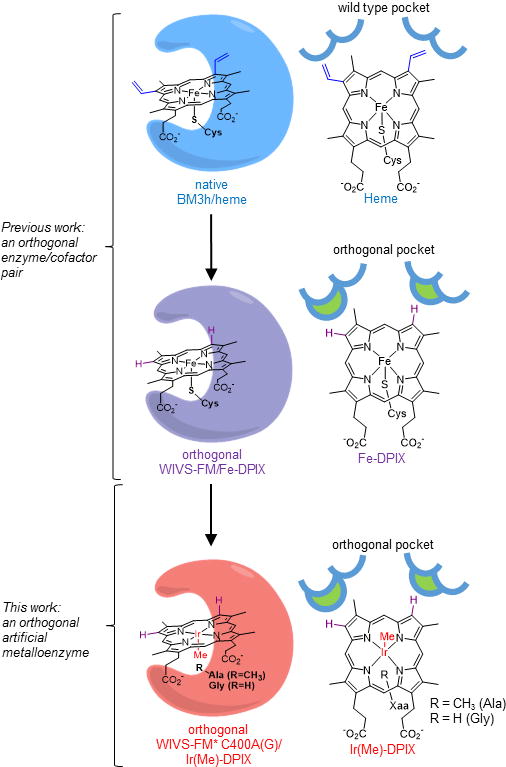
Heme proteins have evolved active site pockets and axial amino acid ligands for the selective binding and chemical tuning of heme (top). In a previous report, the active site of cytochrome BM3h was reshaped by mutation to create an orthogonal scaffold (WIVS-FM) selective for a non-native iron-porphyrin (Fe-DPIX, middle). Herein, further refinement of WIVS-FM through mutation of the axial Cys ligand (C400), permits expression of an artificial Ir(Me)-porphyrin (Ir(Me-DPIX) in cells under native growth conditions (bottom).
Heme proteins have gained recent, intensive attention due to the promiscuous ability of the heme cofactor to carryout abiological metallocarbenoid and –nitrenoid insertions into olefins and X-H bonds (X = C, N, S, Si).[16–28] Through mutagenesis of the protein scaffold, highly active and stereoselective catalysts have been obtained for these transformations. However, in many of these studies the substrate scope is limited to activated olefins or X-H bonds.[29,30] Metal substitution can significantly expand the reaction scope of heme proteins.[5–8] For example, the groups of Hartwig and Fasan have recently shown that Ir(Me)-containing myoglobin and P450 variants can access carbenoid and –nitrenoid insertions that are difficult to achieve using the native heme cofactor.[6,11,13,25,31] Inspired by these results, we sought to create an orthogonal platform for efficient, in cell assembly of iridium enzymes. We reasoned that DPIX-selective recognition motifs identified in our previously evolved orthogonal enzyme (denoted WIVS-FM) would enable substitution of Fe-DPIX for Ir(Me)-DPIX, while maintaining orthogonal recognition of the cofactor (Figure 1, bottom).
To begin this study, Ir(Me)-DPIX was synthesized in two steps and 38% overall yield from deuteroporphyrin IX dimethyl ester by adapting literature procedures for the preparation of Ir(Me)-heme analogs (see Supplementary Methods, Scheme S1). [6,31] With the non-natural cofactor in hand, we tested for cellular uptake of Ir(Me)-DPIX and its incorporation into engineered P450-BM3h variants, in vivo. To permit transport of Ir(Me)-DPIX into the cell, we made use of a natural Escherichia coli heme transport protein, ChuA, that has demonstrated promiscuous activity for heme derivatives bearing modifications to the metal center and/or porphyrin scaffold.[15,31–33]
We first compared the incorporation of Ir(Me)-DPIX in DPIX-selective WIVS-FM as well as the native (non-orthogonal) BM3h scaffold. (For all proteins examined in this report, we also included a T268A mutation that has been shown to be generally activating in P450-mediated carbenoid and nitrenoid transfer reactions[16,17]). Due to decreased protein stability in the presence of mutations that contribute to DPIX-orthogonality, only low levels of Ir-bound-WIVS-FM-T268A were isolated (< 2 mg/mL). Accordingly, for WIVS-FM variants, we also introduced two additional mutations (WIVS-FM-L52I/I366V, denoted here as WIVS-FM*) that have been shown to stabilize a number of BM3h-variants.[15,34] ChuA was coexpressed in E. coli with either BM3h-T268A or WIVS-FM*-T268A using iron-deficient minimal media (to limit heme biosynthesis) supplemented with 10 μM Ir(Me)-DPIX. Analytical HPLC of the purified protein showed the presence of Ir(Me)-DPIX with little heme content (< 5 %) under these stringent expression conditions (Table 1, Figure S2 and 2C) and protein yields for Ir-bound BM3h-T268A and WIVS-FM*-T268A were comparable (6.8 and 9.4 mg/mL, respectively, Table 1).The UV-Vis absorbance spectra for both Ir(Me)-DPIX-bound variants were comprised of two prominent Soret bands at ~ 406 nm and 438 nm (Figure 2A and S1 and Table S2). These absorbance bands are distinct from those observed for the free Ir(Me)-DPIX cofactor, heme-containing BM3h, and the Fe-DPIX-bound enzyme, which display single peaks at 386, 421, and 413 nm, respectively (Figure 2, Figure S1 and Table S2). Taken together, these results provide evidence for the promiscuous ChuA-mediated transport of Ir(Me)-DPIX and its incorporation into BM3h variants inside of cells.
Table 1.
Selectivity of BM3h variants for Ir(Me)-DPIX with respect to heme in minimal and rich mediaa
| Variant | M9 media Ir(Me)-DPIX : hemeb |
TB media Ir(Me)-DPIX : hemeb |
M9 yield Ir(Me)-DPIX (mg/L)c |
|---|---|---|---|
| BM3h-T268A | 95.0:5.0 ± 3.2 | 37.8:62.2 ± 3.6 | 6.8 |
| BM3h-T268A/C400A | 98.4:1.6 ± 2.2 | 80.2:19.8 ± 2.8 | 3.5 |
| BM3h-T268A/C400G | 99.5:0.5 ± 0.5 | 81.9:18.1 ± 6.2 | 4.1 |
| WIVS-FM*-T268A | 96.7:3.3 ± 2.1 | 51.8:48.2 ± 9.6 | 9.4 |
| WIVS-FM*-T268A/C400A | 99.7:0.3 ± 0.5 | 97.2:2.8 ± 1.1 | 11.5 |
| WIVS-FM*-T268A/C400G | >99.9:n.d. ± n/a | 98.3:1.7 ± 1.5 | 14.3 |
All values reported are the average and standard deviations from three independent experiments.
Determined by HPLC analysis of purified proteins from expressions in the denoted media, reported as molar ratios.
Yields were determined via HPLC analysis or from absorbance spectra using ε402 nm = 107.7 mM−1cm−1
Figure 2. Incorporation of Ir(Me)-DPIX into proteins in cells.
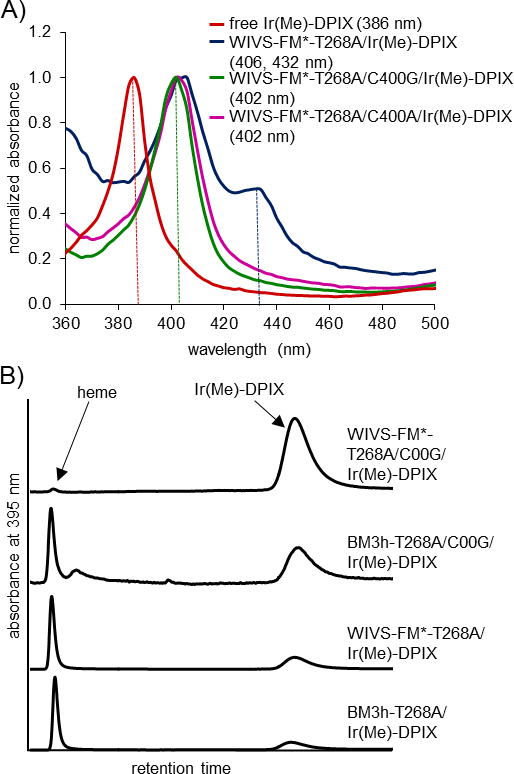
(A) Absorbance spectra of free Ir(Me)-DPIX (red) and Ir(Me)DPIX-bound BM3h variants: WIVS-FM*-T268A (blue), WIVS-FM*-T268A/C400G (green) and WIVS-FM*-T268A/C400A (violet). Absorbance maxima are indicated by dotted lines and denoted in the legend. (B) Representative HPLC traces for BM3h and WIVS-FM* variants expressed in rich (TB) media supplemented with 10 μM Ir(Me)-DPIX.
Unlike heme and Fe-DPIX, one of the apical coordination sites of Ir(Me)-DPIX is occupied by a pendant methyl group whereas the opposing coordination site remains unoccupied (Figure 1). Due to this asymmetry, we hypothesized that the two Soret peaks observed for Ir(Me)-DPIX-bound WIVS-FM*-T268A and BM3h-T268A might result from different coordination environments of the Ir(Me)-cofactor: one in which the metal is ligated by the conserved iron-coordinating axial cysteine (C400 in BM3h, Figure 1) and one in which the cofactor is not coordinated by C400, potentially due to a ring flipped conformation in which the methyl group faces the proximal binding pocket. To support this hypothesis, mutation of C400 to non-coordinating Gly or Ala residues resulted in Ir(Me)-DPIX-bound protein with a single Soret band at ~ 402 nm (Figure 1A and S1 and Table S2). These results suggest that the peak at 406 nm represents protein-bound Ir(Me)-DPIX that is not ligated by an amino acid side chain, whereas the additional peak at 432 nm is indicative of C400 ligated to the Ir(Me)-DPIX cofactor.
With the development of an effective means of introducing Ir(Me)-DPIX into proteins in cells, we next sought to determine whether the orthogonal enzyme maintained selective recognition of the metal-substituted DPIX cofactor in the presence of endogenous heme. Selectivity was analyzed by HPLC (Figure 2B) for proteins expressed in rich media (TB), to permit normal levels of heme biosynthesis. When expressed under these conditions, native BM3h-T268A showed a preference for heme over Ir(Me)-DPIX (Figure 2, Table 1 and Figure S2). Unexpectedly, orthogonal WIVS-FM*-T268A also contained a considerable amount of contaminating heme (heme:Ir(Me)-DPIX = 48.2:51.8; Figure 2, Table 1 and Figure S2). In contrast to our previous findings,[15] these results show that Ir(Me)-DPIX competes less effectively against heme, even in the presence of mutations that provide orthogonality for the DPIX macrocycle.
Introduction of axial C400A or C400G mutations led to improvements in Ir(Me)-DPIX selectivity in both BM3h and WIVS-FM* scaffolds. The C400A and C400G variants of BM3h-T268A displayed moderate enhancements towards Ir(Me)-DPIX selectivity in rich media (80.2 mol% and 81.9 mol% respectively; Figure 2B, Table 1 and Figure S2). In contrast, addition of C400A or C400G mutations to DPIX-selective WIVS-FM* afforded highly selective P450 variants for Ir(Me)-DPIX, showing >97 mol % enrichment in Ir(Me)-DPIX compared to endogenous heme (Figure 2, Table 1 and Figure S2). These data show that the identity of the metal center and axial ligand influence the efficiency of non-natural cofactor incorporation in heme proteins. In this case, orthogonality was achieved through mutations that provide selective recognition of the deuteroporphyrin macrocycle in addition to mutations that accommodate of the axial methyl group of the cofactor. Due to the high yields and selectivity obtained with WIVS-FM*-T268A/C400G/Ir(Me)-DPIX, this variant was chosen for subsequent catalysis experiments.
With an orthogonal Ir-P450 in hand, we next compared its activity with respect to Fe-containing BM3h variants for abiotic olefin cyclopropanation using diazoacetate reagents. Heme-mediated cyclopropanation has garnered significant attention due to the high yields and stereoselectivities that can be achieved by native and engineered heme proteins; however, the accessible substrate scope has been restricted primarily to electron rich aryl olefins.[16,25–27,29,35–37]
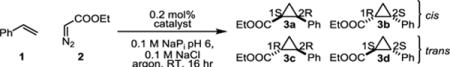
The activity of the orthogonal Ir-enzyme was first investigated for the model cyclopropanation reaction of styrene (1) with ethyl diazo acetate (2, EDA) (Table 2), a benchmark reaction for metallocarbenoid biocatalysis.[16,18] While the isolated Ir(Me)-DPIX cofactor (200 TTN) was significantly more active than free heme (80 total turnover, TTN), the Ir-bound enzyme showed marginally lower activity when compared to BM3h-T268A (240 and 339 TTN, respectively, Table 2, Figure S4). This result is consistent with work by Fasan and coworkers who found native myoglobin outperformed Ir-bound variants in this reaction.[31] Similar activity and stereoselectivities were observed for free Ir(Me)-DPIX and WIVS-FM*-T268A/C400G/Ir(Me)-DPIX. This result could be due to protein unfolding and liberation of the Ir-cofactor. However, absorbance measurements of the enzymatic reaction after 16 hours, which showed the characteristic Soret band of the protein-bound cofactor, suggested that the cofactor remained associated with protein throughout the reaction (Figure S5).
Table 2.
Activities and stereoselectivities of biocatalysts for the reaction of styrene with ethyl diazoacetate.a
| |||||
|---|---|---|---|---|---|
| catalyst | yield [%] | TTNb | dr (cis:trans) | ee cis [%]c | ee trans [%]d |
| heme | 16 | 80 | 13:87 | 0 | −3 |
| Ir(Me)-DPIX | 40 | 200 | 29:71 | −1 | −4 |
| BM3h-T268A/ heme | 68 | 339 | 1:99 | −11 | −97 |
| WIVS-FM*-T268A/C400G/Ir(Me)-DPIX | 48 | 240 | 29:71 | 0 | 0 |
reaction conditions: 7.5 mM olefin, 10 mM EDA, 15 μM catalyst, in 0.1 M NaPI pH 6.0 100 mM NaCl buffer, 5-% DMF as cosolvent (reactions with heme and heme enzymes also contain 10 mM dithionite). TTN and stereoselectivities determined by chiral GC analysis.
TTN = total turnover number.
(1S, 2R) – (1R, 2S).
(1R, 2R) – (1S, 2S).
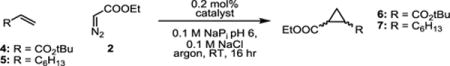
Encouraged by this initial activity, we sought to investigate the potential of the metal-substituted enzyme on more challenging olefins. In particular, the activity of WIVS-FM*-T268A/C400G/Ir(Me)-DPIX was investigated for the cyclopropanation of an electron deficient olefin, tert-butyl acrylate (4), and an aliphatic oliefin, 1-octene (5, Table 3), by EDA. Cyclopropanation of 1-octene has previously been shown to be catalyzed, in low yield, by Ir(Me)-substituted myoglobin and CYP119,[6,25] while tert-butyl acrylate is an untested substrate in biocatalytic cyclopropanation reactions.
Table 3.
Activities and diastereoselectivities of biocatalysts for the reaction of electron-deficient and aliphatic olefins with ethyl diazoacetate.a
| ||||
|---|---|---|---|---|
| catalyst | substrate | yield [%] | TTNb | drc,d (cis:trans) |
| heme | 4 | 6 | 29 | 31:69 |
| BM3h-T268A/heme | 4 | 7 | 35 | 43:57 |
| Ir(Me)-DPIX | 4 | 8 | 40 | 14:86 |
| WIVS-FM*-T268A/C400G/Ir(Me)-DPIX | 4 | 29 | 147 | 13:87 |
| heme | 5 | n.d.e | n.d.e | n.d.e |
| BM3h-T268A/heme | 5 | n.d.e | n.d.e | n.d.e |
| Ir(Me)-DPIX | 5 | 5 | 23 | 30:70 |
| WIVS-FM*-T268A/C400G/Ir(Me)-DPIX | 5 | 11 | 55 | 26:74 |
| WIVS-FM*-F87W/T268A/C400G/Ir(Me)-DPIX | 5 | 19 | 95 | 26:74 |
reaction conditions: 100 mM olefin, 10 mM EDA, 20 μM catalyst, in 0.1 M NaPI pH 6.0 100 mM NaCl buffer, 5% DMF as cosolvent (reactions with heme and heme enzymes also contain 10 mM dithionite). TTN and diastereoselectivities determined by chiral GC analysis. Enantiomers could not be resolved by chiral GC.
TTN = total turnover number.
Diastereomer identity determined by comparison to authentic standards.
Enantioselectivities are reported in table S4 and S5.
n.d. = not detected.
In the reaction between 4 and EDA, free Ir(Me)-DPIX (40 TTN) was observed to have slightly higher activity than free heme (29 TTN) and heme-bound BM3h-T268A (35 TTN). In contrast, WIVS-FM*-T268A/C400G/Ir(Me)-DPIX showed > 4 fold enhancement in activity over the heme-bound enzyme. (29 % yield, 147 TTN). Furthermore, the orthogonal enzyme maintained good selectivity for the trans diastereomer, whereas diastereoselectivity was attenuated in BM3h-T268A/heme (Table 3, Figure S6 and S7). In the case of the reaction between octene (5) and EDA, no product was observed with either free heme or the heme-bound enzyme. While low conversion were observed with free Ir(Me)-DPIX cofactor (23 TTN), orthogonal WIVS-FM*-T268A/C400G/Ir(Me)-DPIX provided a > 2 fold improvement in activity (55 TTN).
In an initial effort to improve the activity of WIVS-FM*-T268A/C400G/Ir(Me)-DPIX against 4 and 5, we screened a small panel of mutations focused around the active site and tailored to include bulky amino acids (W, F, etc.) or mutations known to increase cyclopropanation activity against styrene (Table S4, Figure S6 and S7). While no enhancements were observed for 4, a single amino acid substitution, F87W, was identified that afforded a ~ 2 fold improvement in yield for the cyclopropanation of octene (19 % yield, 95 TTN, Table 3). These results provide further evidence that reactions with the protein-bound iridium catalyst are sequestered in the active site of the protein. No improvements in diastereoselectivity were observed upon mutation, and no pronounced enantioinduction has been observed with the IR-bound BM3h variants (Table S4 and S5, Figure S8 and S9). Efforts to further improve activity and stereoselectivities on these and other substrates via directed evolution are currently underway.
In summary, we report the design of an orthogonal P450 scaffold for the selective production of an Ir(Me)-DPIX metalloenzyme in E. Coli. Selectivity over endogenous heme was achieved by introducing mutations within the heme-binding domain that contribute to the recognition of the altered deuteroporphyrin macrocycle, in combination with mutations to the P450 axial cysteine to accommodate the Ir(Me)-center. The iridium-substituted enzyme displayed abiotic carbenoid-transfer activity for the cyclopropanation of olefins by ethyl diazoacetate, and demonstrated improved activity for substrates that are challenging or inaccessible to the native iron cofactor. We anticipate that this engineered artificial metalloenzyme will provide an advantageous starting point for further protein engineering to improve activity and selectivity for a wealth of carbenoid and nitrenoid insertion reactions. Importantly, this work demonstrates that metal substitution in orthogonal cofactor/enzyme pairs is a promising strategy to expand the chemical functionality of proteins inside the cell. In the future, we envision combining strategies aimed at altering the cofactor metal center with modifications to the porphyrin backbone to create a suite of tunable, artificial metalloenzymes for diverse applications.
Supplementary Material
Acknowledgments
This work was supported by a DARPA Young Faculty Award (D13AP00024) and NSF Career Award (CHE-1552718) to EMB. EWR was supported by an NSF Graduate Research Fellowship (DGE1144081) and the Carol and Edward Smithwick Dissertation Completion Fellowship (UNC-Chapel Hill). We thank Dr. Brandie Ehrmann and the University of North Carolina Department of Chemistry Mass Spectrometry Core Laboratory for assistance with GC/MS analysis (supported by the NIH-NIGMS under award number R35GM118055).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Heinisch T, Ward TR. Curr Opin Chem Biol. 2010;14:184–99. doi: 10.1016/j.cbpa.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 2.Lewis JC. ACS Catal. 2013;3:2954–2975. [Google Scholar]
- 3.Pàmies O, Diéguez M, Bäckvall JE. Adv Synth Catal. 2015;357:1567–1586. [Google Scholar]
- 4.Jing Q, Kazlauskas RJ. ChemCatChem. 2010;2:953–957. [Google Scholar]
- 5.Oohora K, Kihira Y, Mizohata E, Inoue T, Hayashi T. J Am Chem Soc. 2013;135:17282–5. doi: 10.1021/ja409404k. [DOI] [PubMed] [Google Scholar]
- 6.Key HM, Dydio P, Clark DS, Hartwig JF. Nature. 2016;534:534–537. doi: 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Angew Chemie - Int Ed. 2003;42:1005–1008. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 8.Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. J Am Chem Soc. 2004;126:10812–3. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 9.Kawakami N, Shoji O, Watanabe Y. ChemBioChem. 2012;13:2045–2047. doi: 10.1002/cbic.201200446. [DOI] [PubMed] [Google Scholar]
- 10.Hyster TK, Ward TR. Angew Chem Int Ed. 2016;55:2–16. doi: 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- 11.Dydio P, Key HM, Nazarenko A, Rha JYE, Seyedkazemi V, Clark DS, Hartwig JF. Science (80-) 2016;354:102–106. doi: 10.1126/science.aah4427. [DOI] [PubMed] [Google Scholar]
- 12.Jeschek M, Reuter R, Heinisch T, Trindler C, Klehr J, Panke S, Ward TR. Nature. 2016;537:661–665. doi: 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- 13.Dydio P, Key HM, Hayashi H, Clark DS, Hartwig JF. J Am Chem Soc. 2017;139:1750–1753. doi: 10.1021/jacs.6b11410. [DOI] [PubMed] [Google Scholar]
- 14.Brustad EM, Arnold FH. Curr Opin Chem Biol. 2011;15:201–10. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds EW, McHenry MW, Cannac F, Gober JG, Snow CD, Brustad EM. J Am Chem Soc. 2016;138:12451–12458. doi: 10.1021/jacs.6b05847. [DOI] [PubMed] [Google Scholar]
- 16.Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–10. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- 17.Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, Brustad EM. ChemBioChem. 2015;17:394–397. doi: 10.1002/cbic.201500624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bordeaux M, Tyagi V, Fasan R. Angew Chemie Int Ed. 2014;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, Fasan R. Angew Chemie Int Ed. 2016;55:13562–13566. doi: 10.1002/anie.201607278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh R, Kolev JN, Sutera PA, Fasan R. ACS Catal. 2015;5:1685–1691. doi: 10.1021/cs5018612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sreenilayam G, Fasan R. Chem Commun. 2015;51:1532–4. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prier CK, Hyster TK, Farwell CC, Huang A, Arnold FH. Angew Chemie Int Ed. 2016;55:4711–4715. doi: 10.1002/anie.201601056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kan SBJ, Lewis RD, Chen K, Arnold FH. Science (80-) 2016;354:1048–1051. doi: 10.1126/science.aah6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chemie. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Key HM, Dydio P, Liu Z, Rha JYE, Nazarenko A, Seyedkazemi V, Clark DS, Hartwig JF. ACS Cent Sci. 2017 doi: 10.1021/acscentsci.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandenberg OF, Fasan R, Arnold FH. Curr Opin Biotechnol. 2017;47:102–111. doi: 10.1016/j.copbio.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gober JG, Ghodge SV, Bogart JW, Wever WJ, Watkins RR, Brustad EM, Bowers AA. ACS Chem Biol. 2017;12:1726–1731. doi: 10.1021/acschembio.7b00358. [DOI] [PubMed] [Google Scholar]
- 28.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH. Nat Chem. 2017;9:629–634. doi: 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gober JG, Brustad EM. Curr Opin Chem Biol. 2016;35:124–132. doi: 10.1016/j.cbpa.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Prier CK, Arnold FH. J Am Chem Soc. 2015;137:13992–14006. doi: 10.1021/jacs.5b09348. [DOI] [PubMed] [Google Scholar]
- 31.Fasan R, Sreenilayam G, Moore E, Steck V. Adv Synth Catal. 2017;359:2076–2089. doi: 10.1002/adsc.201700202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lelyveld VS, Brustad E, Arnold FH, Jasanoff A. J Am Chem Soc. 2011;133:649–651. doi: 10.1021/ja107936d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bordeaux M, Singh R, Fasan R. Bioorg Med Chem. 2014;22:5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Proc Natl Acad Sci U S A. 2006;103:5869–74. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajaj P, Sreenilayam G, Tyagi V, Fasan R. Angew Chemie Int Ed. 2016;55:16110–16114. doi: 10.1002/anie.201608680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renata H, Wang ZJ, Kitto RZ, Arnold FH. Catal Sci Technol. 2014;4:3640–3643. doi: 10.1039/C4CY00633J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernandez KE, Renata H, Lewis RD, Kan SBJ, Zhang C, Forte J, Rozzell D, Mcintosh JA, Arnold FH. ACS Catal. 2016;6:7810–7813. doi: 10.1021/acscatal.6b02550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.