

Abstract
Phytosterols, the plant analogues of cholesterol, widely occur in the human diet. In this study, we investigated and compared the effects of stigmasterol and β-sitosterol (both with purities ≥ 95%) on dextran sulfate sodium (DSS)-induced colitis in C57BL/6J male mice fed a high fat western-style diet. Mice treated with DSS developed severe mucosal colitis, with a marked distortion and crypt loss of colonic surface epithelium. Both β-sitosterol and stigmasterol significantly inhibited colon shortening, lowered fecal hemoglobin contents, and reduced the severity of colitis in the middle and distal colon (p < 0.05). They also significantly suppressed the activation of inflammatory master regulator nuclear factor-kappa B. Stigmasterol significantly lowered colonic inflammation score and the expression of cyclooxygenase-2 and colony stimulating factor-1, while β-sitosterol was less or not effective. These results suggest that dietary intake of stigmasterol and β-sitosterol ameliorate colitis. Such activities of stigmasterol and β-sitosterol in humans remain to be investigated.
Keywords: phytosterols, NF-κB, colitis, high fat western-style diet, inflammatory bowel disease
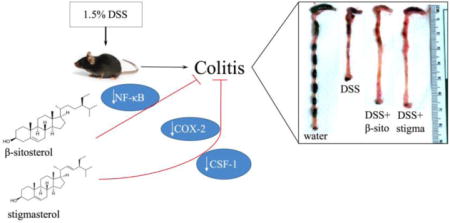
Graphical abstract
Stigmasterol and β-sitosterol ameliorate DSS-induced colitis in mice by suppressing the activation of NF-κB; stigmasterol also downregulates COX-2 and CSF-1.
1. Introduction
Phytosterols, the analogues of cholesterol in plants, are composed of a tetracyclic cyclopenta[α]phenanthrene ring and a long flexible side chain at the C-17 carbon atom1. The molecular structures of cholesterol, stigmasterol and β-sitosterol are shown in Fig. 1. Phytosterols are quite prevalent in the human diet. It was reported that the total sterol contents of rye, wheat, barley and oats in Finland were 95.5, 69.0, 76.1 and 44.7 mg/100 g, respectively2. The phytosterol concentrations in cereal foods in Sweden and Netherlands ranged from 4.1 to 344 mg/100 g. The daily dietary intake of plant sterols was estimated to be 140 to 360 mg in Finland, 163 mg in the United Kingdom, 359 mg in Netherlands and 375 mg in Japan3.
Fig. 1.
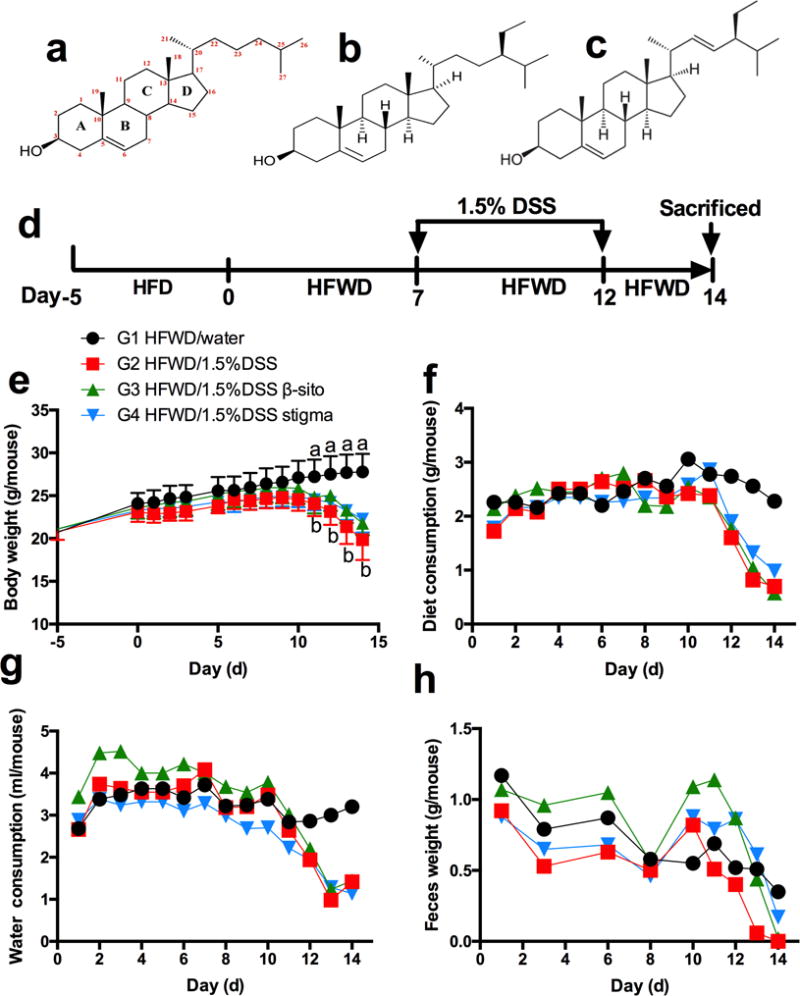
chemical structures, scheme of experimental design and general observations. chemical structures of cholesterol (a), β-sitosterol (b), and stigmasterol (c), scheme of experimental design (d). also shown are body weight (e), (mean ± sd, n=5), as well as dietary consumption (f), water consumption (g), and fecal weight (h), (one measurement per day of each cage contained 5 mice). “a, b” indicating significant difference (p<0.05) between g1 and g2 by anova.
In recent decades, phytosterols have received much attention due to their capability to inhibit cholesterol intestinal absorption, resulting in lower serum total cholesterol and low-density lipoprotein cholesterol levels4. Phytosterols have also been reported to have anti-inflammatory effects in commonly used animal models for colitis5–9. It was reported that β-sitosterol attenuates high-fat diet-induced colitis in C57BL/6J mice5. The anti-colitic benefit of β-sitosterol was also observed in the 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis model6. Other phytosterols, such as guggulsterone and phytosteryl ferulates (γ-oryzanol), were also reported to ameliorate acute colitis7–9. However, whether different types of phytosterols have different effects on colitis and the molecular mechanisms involved are still unclear.
In recent decades, the incidence of inflammatory bowel disease (IBD) has increased throughout the world. The dextran sulfate sodium (DSS) and TNBS-induced mouse models of colitis are the two most widely used models to mimic IBD in humans9. Oral administration of DSS (0.7% to 3% in drinking water) for several days leads to an ulcerative colitis-like colonic inflammation, while administration of TNBS initiates a Crohn’s disease-like intestinal inflammation10–14. A high-fat diet has also been reported to induce intestinal inflammation in mice5. Combination of DSS and a high-fat diet to induce colitis could be a relevant model for studying IBD. A large number of pro-inflammatory cytokines, such as tumor necrosis factor (TNF-α) and interleukin (IL)-6 and the enzyme cyclooxygenase (COX)-2, have been reported to be upregulated in colitis7, 14–16. While several studies demonstrated that phytosterols ameliorate colitis by inhibiting the nuclear factor-kappa B (NF-κB) pathway5, 6, 8, their mechanisms of action remain to be investigated.
In order to study the colitis preventive activities of individual phytosterols, we selected β-sitosterol and stigmasterol as examples and compared their activities. These are the two most abundant phytosterols. For example, our previous study showed that β-sitosterol and stigmasterol were the two major phytosterols in sugarcane, with contents (μg/g dry weight) of 118 ± 20 to 801 ± 34 and 883 ± 24 to 1824 ± 25, respectively17. β-Sitosterol was reported to prevent TNBS-induced colitis. However, whether it has the same effects in a DSS-induced colitis model is not known. The aim of the present study was to compare the effects of pure β-sitosterol and stigmasterol on colitis induced by DSS in C57BL/6J mice fed the high fat western-style diet (HFWD).
2. Materials and Methods
2.1. Animals and diets
A total of twenty 6-week-old male C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). All animal experiments were carried out at Rutgers University under protocol 02-027, approved by the Institutional Animal Care and Use Committee of Rutgers University. Mice were maintained in our animal facility under standard conditions (temperature 24–25°C, humidity 70–75% and lighting regimen of 12-hour light-dark cycles), with free access to food and water. The basal diet was the HFWD. This HFWD was modified from the Western-style diet designed by Newmark et al18 (which mimics dietary risk factors of Western populations for colon cancer) by increasing the total fat content from 40% to 60% of total calorie. The diet contains a higher level of fat and lower amounts of calcium, vitamin D3, choline, folate, and fiber than the normal AIN76A diet. We previously used the HFWD to induce metabolic syndrome in mice, and the composition of the diet has been published19. β-sitosterol and stigmasterol were added to the basal diet at 0.4% to make the HFWD+β-sito and HFWD+stigma, respectively. The level of 0.4% in the diet was based on the conclusion of the FDA that a daily dietary intake of 2g of phytosterols is needed to lower low-density lipoprotein cholesterol and reduce cardiovascular risk20. Assuming a person consumes 500g of food in dry weight (2000 to 2500 kcal), then 2g/500g = 0.4%. These diets were prepared by Research Diets, Inc. (New Brunswick, NJ), and the compositions are shown in Supplementary Table 1.
2.2. Chemicals
DSS (MW=36,000-50,000) was purchased from MP Biomedicals, LLC (Solon, OH). Stigmasterol and β-sitosterol (both with purities≥95%), hematoxylin, 3,3′,5,5′-tetramethylbenzidine (TMB), hematoxylin and eosin (H&E) were purchased from Sigma Chemical Co. (St. Louis, MO). Other chemicals used in the study were all of analytical grade.
2.3. Experimental procedure with animals
After 1 week of acclimation on the chew diet, the mice were pre-treated with a high-fat diet (60% energy as fat)21 for 5 days. Then, the mice were allocated into 4 groups (each group with 5 mice), and maintained on the HFWD (G1, negative control), HFWD (G2, DSS control), HFWD+β-sito (G3) and HFWD+stigma (G4) throughout the experiment. After receiving the experimental diet for 1 week, G2, G3 and G4 were treated with 1.5% DSS in drinking water for 5 days to induce colitis. Body weight, fecal weight, diet consumption and water consumption were monitored every day. Two days after the termination of the DSS treatment, the mice were euthanized by CO2 asphyxiation. Blood samples were taken by cardiac puncture. Serum was prepared and stored frozen at −80°C. Colon was resected, washed with ice-cold saline, and divided longitudinally into two halves. The proximal colon (~2 cm segment), which was much less affected by DSS compared to middle or distal colon22, was cut off and not analyzed. Half of the colon was flattened on filter paper and fixed in 10% buffered formalin for 24 hours for histopathological analyses. The other half of the colon was stored in RNAlater solution (Qiagen, Valencia, CA) and stored at −80°C. The feces were collected every morning. After freeze drying with a vacuum freeze dryer (Vir Tis benchtop K, Gardner, NY), the fecal samples were weighted and stored frozen at −80°C.
2.4. Fecal hemoglobin content determination
A method based on the heme-catalyzed oxidation of TMB by H2O2 was used to determine fecal hemoglobin content. In brief, 0.3 g frozen dry fecal samples from each group were mixed with 6 ml H2O, and the mixtures were put in boiling water for 10 minutes to inactivate plant peroxidases. Two minutes later, the mixtures were put on ice, and 3 ml of 30% acetic acid was added. Then, 4.5 ml ethyl acetate was added to extract heme. The aqueous and organic layers were separated by centrifugation. The organic layer (0.5 ml) was mixed with 0.5 ml of TMB solution (0.6 mmol/L TMB in acetic acid/ddH2O/ethanol=20/30/50 (volume/volume/volume)) and 0.5 ml of 3% H2O2. The absorbance was measured with a spectrophotometer (Beckman coulter DU800, Fullerton, CA) at 660 nm. The fecal hemoglobin levels were calculated by comparing with a standard curve.
2.5. Histopathological characterization
The formalin-fixed colon was swiss-rolled, paraffin-embedded, and serially sectioned at 4-μm thickness. The histopathological alterations in the colon were examined in two H&E stained sections per colon. A modified histological score system based on a previous publication21 was used to analyze the severity of colitis. The scoring was determined separately for the middle colon and distal colon in a blind manner. The inflammation score was the sum of four individual inflammatory parameters: inflammation severity (0, 1, 2 or 3), extent of inflammation (0,1 or 2), crypt damage (0, 1, 2, 3 or 4), and inflammation area involved (0, 1, 2, 3 or 4) as detailed in Table 1. Hyperplasia and dysplasia were not observed in this evaluation, because the mice were sacrificed after only 7 days of DSS treatment.
Table 1.
Histological score system of colitis
| Score | inflammation severity | inflammation extent | crypt damage | percent area involvement |
|---|---|---|---|---|
| 0 | normal colonic mucosa; | normal colonic mucosa and submucosa; | normal colonic mucosa; | 0% |
| 1 | mild inflammation: either focal or wildly separated multifocal inflammation; | inflammation extent to mucos; | shortening and loss of the basal one-third of the actual crypts with mild inflammation and edema; | 1–25% |
| 2 | moderate inflammation: either multifocal or locally extensive inflammation; | inflammation extent to mucosa and submucosa; | loss of the basal two-thirds of the crypts with moderate inflammation; | 26–50% |
| 3 | severe inflammation: mucosal ulcers with monocytes and polymorphonuclear leukocytes; | loss of the entire crypts with severe inflammation in the mucosa, but with retention of the surface epithelium; | 51–75% | |
| 4 | loss of the entire crypt and surface epithelium; | 76–100% |
2.6. Immunohistochemical analysis for NF-κB p65 positive cells
One section of each colon was used for immunohistochemistry (IHC) with a specific antibody for NF-κB p65 as described previously21. In brief, the sections were deparaffinated in xylene and rehydrated, unmasked in antigen unmasking solution (DAKO, Copenhagen, Denmark). Endogenous peroxidase activity was quenched by 3% H2O2 in distilled water. Sections were then treated for 1 hour at room temperature in PBS containing 10% normal serum and incubated with primary antibodies: NF-κB p65 (1:2000, Abcam, Cambridge, MA) overnight at 4°C and incubated with biotinylated secondary antibody (1:200 in 10% normal serum). Then, the sections were treated with avidin-biotinylated enzyme complex (Vector Laboratories, Burlingame, CA) and 3,3′-diaminobenzidine (Vector Laboratories). Finally, the sections were counterstained with hematoxylin and mounted with Permount (Sigma-Aldrich, St. Louis, MO). Negative controls were processed in the absence of the primary antibody. The total number of cells and number of positively stained cells were quantified using the Aperio ScanScope GL system (Aperio, Vista, CA). The results were presented as the percentage of the cells with positive stained cells.
2.7. Total RNA extraction, reverse transcription and quantitative PCR analysis
Total RNA was extracted from colon samples stored in RNAlater solution using RNeasy mini kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s protocol. Reverse transcription reaction was performed using a Superscript kit (Qiagen Inc.) according to the manufacturer’s protocol. Quantitative PCR was performed using a Power SYBR® Green PCR Master Mix kit (ABI Co., Ltd., Nagareyama, Japan) with an ABI ViiATM 7 system. The sequences of primers were as follows: COX-2: forward, 5′-TTCAACACACTCTATCACTGGC-3′; reverse, 5′-AGAAGCGTTTGCGGTACTCAT-3′; TNF-α: forward, 5′-CCCTCACACTCAGATCATCTTCT-3′; reverse, 5′-GCTACGACGTGGGC -TACAG-3′; colony stimulating factor (CSF)-1: forward, 5′-GGCTTGG CTTGGGATGATTCT-3′; reverse, 5′-GAGGGTCTGGCAGGTACTC-3′; interleukin (IL)-6: forward, 5′-TAGTCCTTCCTACCCCAATTTCC-3′ reverse; 5′-TTGGTCCTTAGCCACTCCTTC-3′. The PCR reaction was initiated by a 5-minute activation of hot-start DNA polymerase at 95°C followed by 49 cycles of target cDNA amplification. The template was initially denatured at 95°C for 15 seconds, followed by 49-cycle program with 45 seconds annealing at 65°C and 10 seconds elongation at 72°C and acquisition of SYBR Green fluorescence. The results were normalized with the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
2.8 Tissue lysis and ELISA
Frozen colon tissue samples were weighted and added into ice-cold Cell Extraction Solution (the lysis buffer in mouse COX-2 ELISA kit; Abcam) at 0.1 g/ml. The tissue samples were then lysed using Omni Bead Raptor 24 (Omni International, Kennesaw, GA) at ice temperature. The supernatant was collected after the tissue lysis was centrifuged at 12,000×g for 10 min at 4°C and stored at −20°C. Protein concentrations of these samples were determined by BCA protein assay kit (Pierce/Thermo Scientific, Rockford, IL). The levels of COX-2 and CSF-1 in these samples were determined using Mouse COX-2 and Mouse CSF-1 ELISA kits (Abcam) according to the protocols provided by the manufacturer. The values of COX-2 and CSF-1 were normalized by the protein concentration.
2.9. Statistical analysis
Data were presented as mean ± standard deviation (SD) and analyzed using one-way ANOVA followed by Tukey’s post hoc test among multiple groups. Student’s t-test was used to determine the differences between two groups. A significance level of p < 0.05 was set for all the tests.
3. Results
3.1. General observations
Mean body weights, diet consumption, water consumption and fecal weight were measured daily (Fig. 1). When mice were treated with 1.5% DSS in drinking water, all these parameters decreased markedly. The mice in HFWD (G2, DSS control) group had bloody feces on day 10 (3 days after DSS treatment). Apparent diarrhea was also observed on day 11, and there were almost no solid feces observed by day 13 in G2. In HFWD+β-sito (G3) group, there were no solid feces observed on day 14 (2 days after secession of DSS treatment). Liquid consumption was not different among the three DSS-treated groups (Fig. 1c), indicating that the presence of phytosterols in the diet did not affect liquid consumption. The DSS consumption (in μg/day/mouse) was calculated to be 43.32±9.17 in G2, 48.66±9.79 in G3 and 37.56±6.25 in G4 during the treatment period. There was no significant difference among different groups.
3.2. Stigmasterol and β-sitosterol prevent colitis-induced colon length shortening and decrease fecal hemoglobin
Colon length, a commonly used indicator of colitis severity, was measured (Fig. 2a). DSS treatment caused a significant reduction in colon length (5.48 ± 0.49 cm in G2 vs 8.56 ± 0.31 cm in G1), and both stigmasterol and β-sitosterol significantly prevented colon shortening (p < 0.05), showing colon lengths of 6.89 ± 0.38 cm in G3 and 7.02 ± 0.25 cm in G4.
Fig. 2.
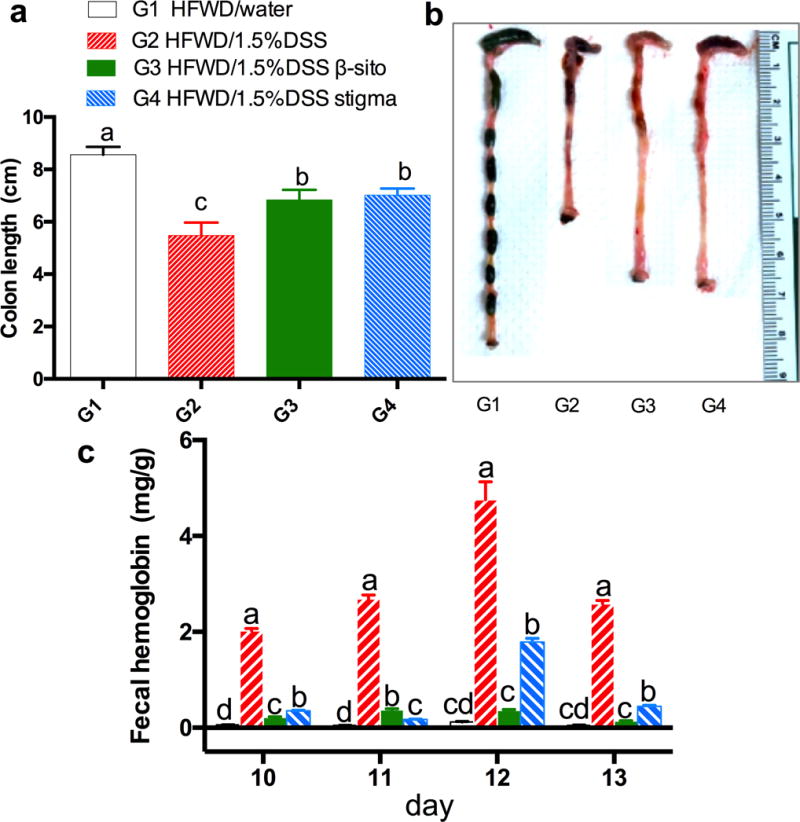
effects of phytosterol treatments on colon length and fecal hemoglobin contents; (a) colon length, (b) picture of colon from 4 groups and (c) fecal hemoglobin contents on day 10 to 13 of each group (one sample per cage each day; the error bars are sd of 3 determinations); a, b, c and d on top of bars indicating significant difference (p<0.05) by anova.
Fecal hemoglobin content in G2 was found to be much higher than that in G1, G3 and G4 (Fig. 2b). In G2, the fecal hemoglobin increased from 2.01 ± 0.05 (day 10) to 4.76 ± 0.38 mg/g (day 12), suggesting an increase in the severity of colitis. After secession of DSS treatment, the fecal hemoglobin decreased to 2.58 ± 0.07 mg/g (day 13). β-Sitosterol and stigmasterol were effective in reducing fecal hemoglobin levels on days 10 to 13. Compared to G2, the average values of fecal hemoglobin of the four time points were significantly lower in G3 and G4 (p<0.05, ANOVA). This finding indicated that both phytosterols ameliorate colon ulceration in colitis mice.
3.3. Stigmasterol and β-sitosterol decrease colon inflammation score
The effects of β-sitosterol and stigmasterol on DSS-induced colitis were determined after H&E staining of colon tissue slides, and the histopathology of the middle colon is shown in Fig. 3a. The colon from the negative control mice (G1) was histologically normal. DSS treatment (G2) induced a moderately severe colitis with severe inflammation in mucosa and submucosa, and loss of the entire crypts and surface epithelium in almost all areas of the middle and distal colon. However, no transmural phenomenon was observed. Moderate inflammation in mucosa and submucosa was observed in the colons from mice treated with 0.4% β-sitosterol (G3). The colons from mice treated with 0.4% stigmasterol (G4) have a mild inflammation in mucosa. As compared to G2, the inflammation scores of middle colon, as well as the distal colon, were significantly lower in G4 (p < 0.05), while β-sitosterol appeared to be less effective (Fig. 3b and c). As shown in Table 2, stigmasterol significantly decreased inflammation severity, inflammation extent, crypt damage and percent area involvement compared to G2 in middle colon. However, β-sitosterol treatment only significantly decreased crypt damage in middle and distal colon, but have no effect in other individual parameters in Table 2.
Fig. 3.
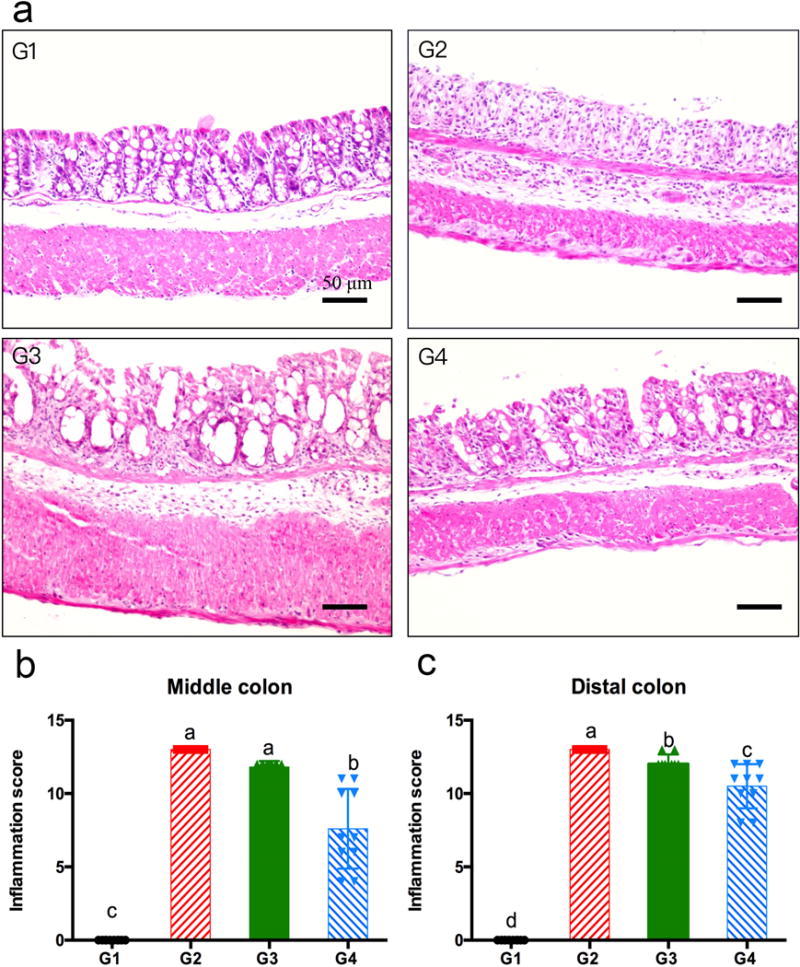
h&e staining (panel a, 200×) of middle colon tissues of dss-treated mice. panel a: g1, a middle colon tissue slide from g1 showing normal colonic mucosa; g2, a middle colon tissue slide from g2 showing severe inflammation in mucosa and submucosa; g3, a middle colon tissue slide from g3 showing moderate inflammation in mucosa and submucosa; g4, a middle colon tissue slide from g4 showing mild inflammation in mucosa. panel b and c: inflammation scores of middle and distal colon, respectively (mean ± sd, n=5); a, b, c on top of bars indicating significant difference (p<0.05) by anova, ** indicating difference (p<0.01) by t-test, while not significantly different by anova.
Table 2.
Histological scores of middle and distal colon tissue
| group | inflammation severity | inflammation extent | crypt damage | percent area involvement | Sum | |
|---|---|---|---|---|---|---|
| Middle colon | G1 | 0.0±0.0c | 0.0±0.0c | 0.0±0.0d | 0.0±0.0c | 0.0±0.0c |
| G2 | 3.0±0.0a | 2.0±0.0a | 4.0±0.0a | 4.0±0.0a | 13.0±0.0a | |
| G3 | 2.8±0.4a | 2.0±0.0a | 3.0±0.0b | 4.0±0.0a | 11.8±0.4a | |
| G4 | 1.6±0.8b | 1.0±0.0b | 2.0±0.9c | 3.0±1.2b | 7.6±2.7b | |
| Distal colon | G1 | 0.0±0.0c | 0.0±0.0c | 0.0±0.0d | 0.0±0.0b | 0.0±0.0d |
| G2 | 3.0±0.0a | 2.0±0.0a | 4.0±0.0a | 4.0±0.0a | 13.0±0.0a | |
| G3 | 2.8±0.4ab | 2.0±0.0a | 3.2±0.4b | 4.0±0.0a | 12.0±0.6b | |
| G4 | 2.5±0.5b | 1.4±0.5b | 2.8±0.4c | 3.8±0.4a | 10.5±1.5c | |
indicating significant difference (p<0.05) by ANOVA analysis.
3.4. Stigmasterol and β-sitosterol decrease nuclear levels of NF-κB p65
To determine the mechanisms involved in the anti-inflammatory activities of phytosterols, we investigated the inhibitory effects of β-sitosterol and stigmasterol on the expression of NF-κB p65 by IHC. Positive NF-κB p65 staining was observed in the cytoplasm of colon epithelium in G1 (Fig. 4a G1). High levels of positive NF-κB p65 staining was observed in the nucleus in G2 (Fig. 4a G2), whereas lower levels of nuclear NF-κB p65 staining were observed in G3 and G4 (Fig. 4a G3, G4). Fig. 4b shows that both β-sitosterol and stigmasterol significantly decreased the percentage of NF-κB positive cells in the colons of DSS-treated mice.
Fig. 4.
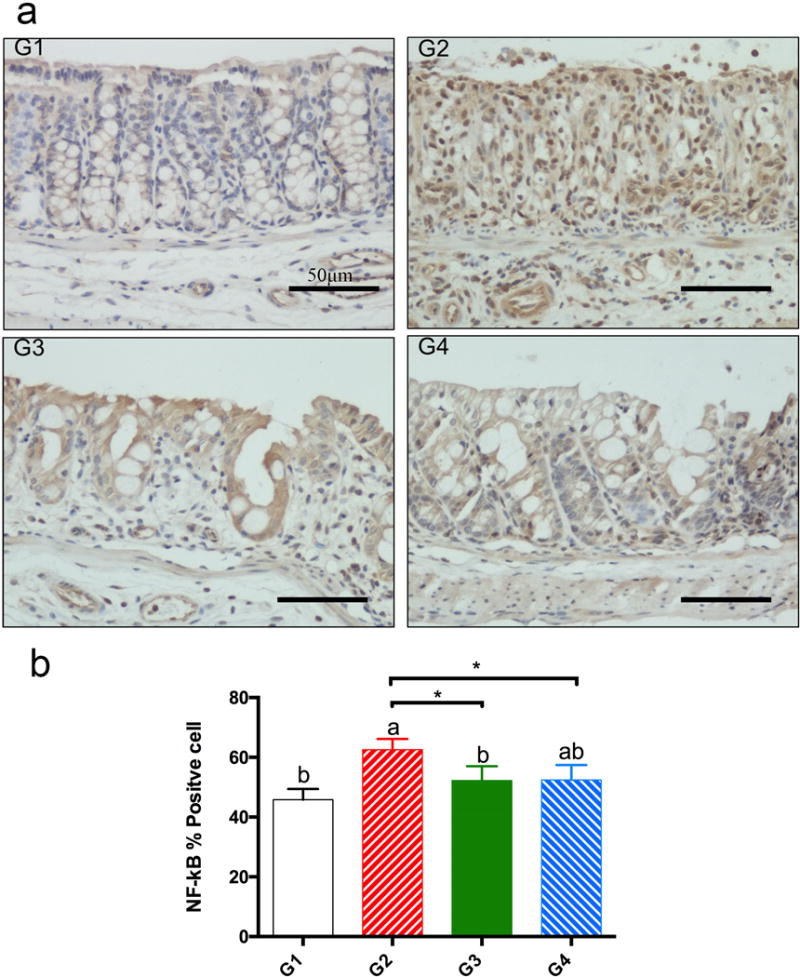
nf-κb p65 immunohistochemical staining (panel a, 400×). panel a: ihc staining of nf-κb p65: g1, normal colonic mucosa from g1 with positive staining in the cytoplasm and low positive staining in the nucleus; g2, colitic colon mucosa from g2 with high positive staining in the nucleus; g3, colitic colon mucosa from g3 with lower positive staining in the nucleus; g4, colitic colon mucosa from g4 with lower positive nf-kb p65 staining in the nucleus. panel b: percentage of nf-κb p65 positively stained cells (mean ± sd, n=5). a, b on top of bars indicating significant difference (p<0.05) by anova, * indicating difference (p<0.05) by t-test.
3.5. Suppression of mRNA levels of COX-2 and CSF-1
Quantitative real-time PCR assays were performed for the inflammatory enzyme – COX-2 as well as pro-inflammatory cytokines – TNF-α and CSF-1 (Fig. 5). Administration of 1.5% DSS, which induced inflammation in colon tissue, increased expression of COX-2, CSF-1 and TNF-α by 23-fold, 3-fold and 41-fold, respectively. As compared to G2, stigmasterol inhibited the expression of COX-2 by 34% (p<0.01, by t-test), CSF-1 by 75% (p<0.05 by ANOVA) and TNF-α by 58% (p > 0.05, by ANOVA); and β-sitosterol inhibited the expression of CSF-1 by 65% (p < 0.05, by ANOVA) and non-significantly inhibited TNF-α by 45% (p > 0.05, by ANOVA). Administration of DSS also increased the expression of IL-6; however, both β-sitosterol and stigmasterol have no significant inhibitory effects on the expression of this cytokine (Fig. 5d).
Fig. 5.
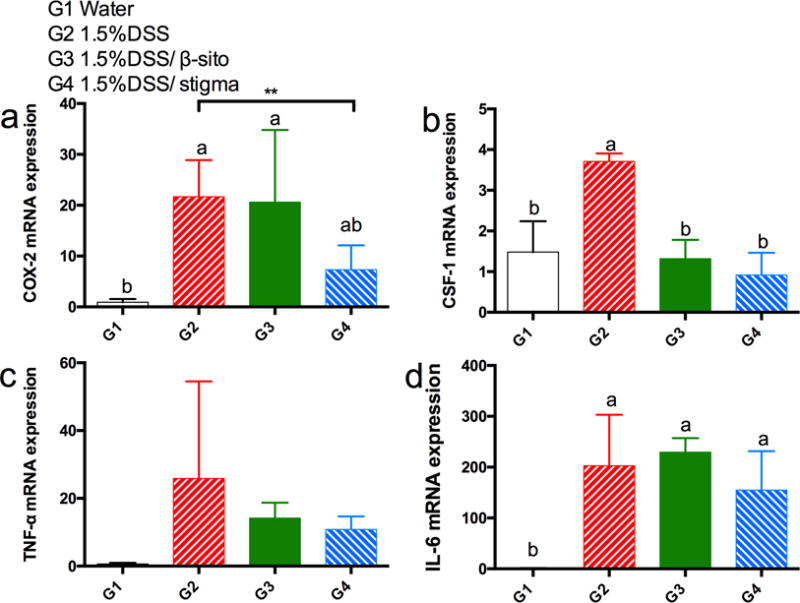
effects of β-sitosterol and stigmasterol on the expression of cox-2 (a), tnf-α (b), csf-1 (c), and il-6 (d) in dss-treated mice (mean ± sd, n=5). a, b on top of bars indicating significant difference (p<0.05) by anova, ** indicating difference (p<0.01) by t-test, while not significantly different by anova.
3.6 Suppression of protein levels of COX-2 and CSF-1
Protein levels of COX-2 and CSF-1 in colon tissues are reported in Figure 6. Administration of 1.5% DSS significantly increased the protein levels of COX-2 and CSF-1. Stigmasterol effectively decreased COX-2 and CSF-1 protein expression (p<0.05 by ANOVA), consistent with the effects on their mRNA expression; however, the inhibitory effect of β-sitosterol was not statistically significant.
Fig. 6.
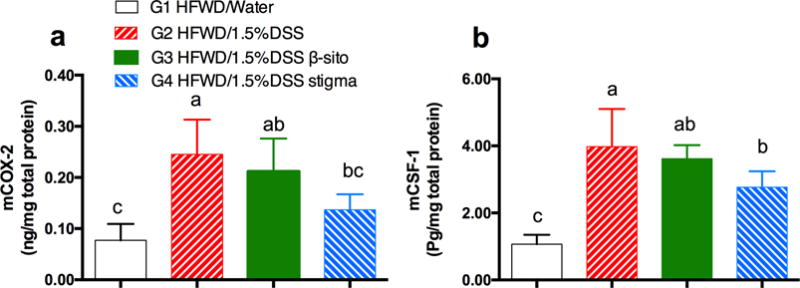
effects of β-sitosterol and stigmasterol on the expression of cox-2 (a) and csf-1 (b) in dss-treated mice as determined by elisa (mean ± sd, n=5). the letter “m” is used to signify mouse proteins. a, b, and c on top of bars indicating significant difference (p<0.05) by anova.
4. Discussion
In the present study, both stigmasterol and β-sitosterol treatments significantly attenuated DSS-induced colitis in mice. Infiltration of leukocytes and mucosal damage were significantly ameliorated (Fig. 3a,b). The inhibitory mechanisms are likely through suppression of NF-κB activation, COX-2 expression and CSF-1 expression. β-Sitosterol appeared to be less effective in decreasing inflammation score and COX-2 expression.
In this study, HFWD was used as the basal diet. In theory, low concentrations of calcium/vitamin D3/folate/choline in the HFWD should contribute to colon inflammation18. In this experiment, it took 5 days of 1.5% DSS treatment to produce colitis. In other studies in our laboratory with C57BL/6J mice on the AIN93M diet (10% calories from fat), it took 10 days to produce less severe colitis by 1.5% DSS (Yang, C.S. et al., unpublished results). However, this effect needs to be substantiated in additional experiments. In colitis, colon length shortening is usually associated with body weight loss23, and this was clearly shown in G2. Both β-sitosterol and stigmasterol appeared to prevent body weight loss. However, the preventive effects were not statistically significant, possibly due to the smaller changes in this short time experiment and the large standard deviations involved. It also reflects that the phytosterols did not completely prevent colitis development.
NF-κB complexes are held in the cytoplasm in an inactive state by members of the NF-κB inhibitor (I-κB) family. In a conventional activation pathway, I-κB is phosphorylated by I-κ-B kinases (IKKs) and degraded in response to different activators. The liberated active NF-κB complexes then translocate to the nucleus24. Our results showed that DSS treatment caused higher levels of NF-κB p65 in the nucleus of colonic cells. However, the elevated nuclear NF-κB p65 levels were only moderately decreased by stigmasterol or β-sitosterol (p<0.05 by t-test), suggesting other factors may be involved in the anti-inflammatory effects. Our results are consistent with other studies showing that β-sitosterol attenuated colitis by inhibiting NF-κB signaling5, 6. The suppression of NF-κB signaling pathway by stigmasterol has not been reported in a colitis model; however, it has been reported in an osteoarthritis model25.
The enzyme COX-2 transforms arachidonic acid to pro-inflammatory prostaglandins and plays an important role in inflammatory processes. Excessive production of COX-2 is usually observed in uncontrolled inflammation26, 27. In our study, COX-2 was found to be upregulated in colon tissues by DSS. Stigmasterol significantly inhibited the expression of COX-2 both at the mRNA and protein levels, while β-sitosterol did not. A possible mechanism for stigmasterol to inhibit colonic expression of COX-2 is via suppression of the activation of NF-κB, similar to the proposed action of guggulsterone28. β-Sitosterol did not significantly affect COX-2 expression. The possibility that the inhibitor action of β-sitosterol was counter balanced by its stimulation of ROS formation, which has been reported in cell lines29–31, remains to be explored.
The cytokine CSF-1 is important in the regulation of macrophage differentiation, proliferation, activation and migration. CSF-1 has been shown to be involved in mucosal inflammation, and an anti-CSF-1 antibody significantly suppressed DSS-induced colitis in mice32–34. The DSS-induced expression of CSF-1 was suppressed by stigmasterol at both the mRNA and protein levels, while suppressed by β-sitosterol only at the mRNA level. This may contribute to the less effectiveness of β-sitosterol in decreasing the histological score of colitis. The inhibition of macrophage function by phytosterols through the suppression of CSF-1 and TNF-α could be important mechanisms for the protection against DSS-induced colitis.
Another hypothesis for the anti-inflammatory action of phytosterols is the lowering of plasma cholesterol concentrations. This may reduce the extent of LDL cholesterol oxidation and the recruitment of immune cells to the arterial intima; leading to lower levels of production of cytokines, ROS and adhesion molecules35. In our study, with a duration of only 14 days, plasma lipids (total cholesterol, triglyceride, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol) did not significantly change (Supplementary Fig. 1). Therefore, in this short-term study, the anti-inflammatory effect of phytosterols appears to be not related to the hypocholesterolemic effect.
Many phytosterols such as β-sitosterol6, guggulsterone36 and phytosteryl ferulates7 were reported to attenuate colitis. Both β-sitosterol and stigmasterol are structurally similar to cholesterol, except the side chains contain an ethyl group37. Compared to β-sitosterol, stigmasterol has a double-bond in the side chain. This added double bond of stigmasterol likely contributes to the higher effects in decreasing inflammatory score and COX-2 expression. In phytosteryl ferulates, a ferulic acid moiety bonds to the hydroxyl group of C-3 in the sterol moiety. The anti-inflammatory effects were believed to be at least partially due to the antioxidant effect of the ferulic acid moiety in the structure of phytosteryl ferulates7. β-Sitosterol and stigmasterol are not known to be strong antioxidants and their treatments did not change the serum levels of 8-isoprostane, a marker of oxidative stress (G3 and G4 versus G2, data not shown).
5. Conclusion
Dietary phytosterols, β-sitosterol and stigmasterol, significantly attenuated DSS-induced colitis in mice, possibly through the suppression of NF-κB activation. Stigmasterol also significantly decreased the expression of COX-2 and CSF-1, while β-sitosterol was less or not effective. The anti-inflammatory activities of these phytosterols in humans remain to be investigated.
Supplementary Material
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (CA120915 and shared facilities funded by CA72720 and ES05022), the John L. Colaizzi Chair Endowment fund. The authors thank the personnel of Laboratory Animal Service in the Laboratory for Cancer Research for taking care of our research mice, and thank Ms. Vi P. Dan for assistance in the preparation of this manuscript.
Abbreviations
- COX-2
cyclooxygenase-2
- CSF-1
colony stimulating factor-1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H&E
hematoxylin & eosin
- HFWD
high-fat western-style diet
- IBD
inflammatory bowel disease
- IHC
immunohistochemistry
- I-κB
NF-κB inhibitor
- IKKs
I-kappa-B kinases
- IL-6
interleukin-6
- NF-κB
nuclear factor-kappa B
- TMB
3,3′,5,5′-tetramethylbenzidine
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- TNF-α
tumor necrosis factor
Footnotes
Conflict of interest
The authors have declared no conflict of interest.
References
- 1.Georges P, Sylvestre M, Ruegger H, Bourgeois P. Steroids. 2006;71:647–652. doi: 10.1016/j.steroids.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Piironen V, Toivo J, Lampi AM. Cereal Chem. 2002;79:148–154. [Google Scholar]
- 3.Lagarda MJ, Garcia-Llatas G, Farre R. J Pharmaceut Biomed. 2006;41:1486–1496. doi: 10.1016/j.jpba.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 4.Vanstone CA, Raeini-Sarjaz M, Jones PJH. J Nutr Biochem. 2001;12:565–574. doi: 10.1016/s0955-2863(01)00175-9. [DOI] [PubMed] [Google Scholar]
- 5.Kim KA, Lee IA, Gu W, Hyam SR, Kim DH. Mol Nutr Food Res. 2014;58:963–972. doi: 10.1002/mnfr.201300433. [DOI] [PubMed] [Google Scholar]
- 6.Lee IA, Kim EJ, Kim DH. Planta Med. 2012;78:896–898. doi: 10.1055/s-0031-1298486. [DOI] [PubMed] [Google Scholar]
- 7.Islam MS, Murata T, Fujisawa M, Nagasaka R, Ushio H, Bari AM, Hori M, Ozaki H. Brit J Pharmacol. 2008;154:812–824. doi: 10.1038/bjp.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheon JH, Kim JS, Kim JM, Kim N, Jung HC, Song IS. Inflamm Bowel Dis. 2006;12:1152–1161. doi: 10.1097/01.mib.0000235830.94057.c6. [DOI] [PubMed] [Google Scholar]
- 9.Aldini R, Micucci M, Cevenini M, Fato R, Bergamini C, Nanni C, Cont M, Camborata C, Spinozzi S, Montagnani M, Roda G, D’Errico-Grigioni A, Rosini F, Roda A, Mazzella G, Chiarini A, Budries R. Plos One. 2014;9 doi: 10.1371/journal.pone.0108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boismenu R, Chen Y. J Leukoc Biol. 2000;67:267–278. doi: 10.1002/jlb.67.3.267. [DOI] [PubMed] [Google Scholar]
- 11.Seril DN, Liao J, Ho KLK, Yang CS, Yang GY. Carcinogenesis. 2002;23:993–1001. doi: 10.1093/carcin/23.6.993. [DOI] [PubMed] [Google Scholar]
- 12.Seril DN, Liao J, Ho KLK, Warsi A, Yang CS, Yang GY. Digest Dis Sci. 2002;47:1266–1278. doi: 10.1023/a:1015362228659. [DOI] [PubMed] [Google Scholar]
- 13.Wen XD, Wang CZ, Yu CH, Zhao L, Zhang ZY, Matin A, Wang YW, Li P, Xiao SY, Du W, He TC, Yuan CS. Phytother Res. 2014;28:892–898. doi: 10.1002/ptr.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez-Fidalgo S, Cardeno A, Sanchez-Hidalgo M, Aparicio-Soto M, Villegas I, Rosillo MA, de la Lastra CA. Eur J Pharm Sci. 2013;48:572–581. doi: 10.1016/j.ejps.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Komaki Y, Komaki F, Sakuraba A, Cohen R. Curr Treat Options Gastroenterol. 2016;14:83–90. doi: 10.1007/s11938-016-0079-x. [DOI] [PubMed] [Google Scholar]
- 16.Mudter J, Neurath MF. Inflamm Bowel Dis. 2007;13:1016–1023. doi: 10.1002/ibd.20148. [DOI] [PubMed] [Google Scholar]
- 17.Feng S, Liu S, Luo Z, Tang K. Food Chem. 2015;181:9–14. doi: 10.1016/j.foodchem.2015.02.073. [DOI] [PubMed] [Google Scholar]
- 18.Newmark HL, Yang K, Lipkin M, Kopelovich L, Liu Y, Fan K, Shinozaki H. Carcinogenesis. 2001;22:1871–1875. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 19.Chen YK, Cheung C, Reuhl KR, Liu AB, Lee MJ, Lu YP, Yang CS. Journal of agricultural and food chemistry. 2011;59:11862–11871. doi: 10.1021/jf2029016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao S, Liu MJ, Lee B, Besecker B, Lai JP, Guttridge DC, Knoell DL. Am J Physiol Lung Cell Mol Physiol. 2010;298:L744–754. doi: 10.1152/ajplung.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li GX, Lee MJ, Liu AB, Yang ZH, Lin Y, Shih WJ, Yang CS. Free Radical Bio Med. 2012;52:1151–1158. doi: 10.1016/j.freeradbiomed.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Kitajima S, Takuma S, Morimoto M. J Vet Med Sci. 1999;61:67–70. doi: 10.1292/jvms.61.67. [DOI] [PubMed] [Google Scholar]
- 23.Fitzpatrick LR, Wang J, Le T. Digest Dis Sci. 2000;45:2327–2336. doi: 10.1023/a:1005630723111. [DOI] [PubMed] [Google Scholar]
- 24.Hayden MS, West AP, Ghosh S. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 25.Gabay O, Sanchez C, Salvat C, Chevy F, Breton M, Nourissat G, Wolf C, Jacques C, Berenbaum F. Osteoarthritis Cartilage. 2010;18:106–116. doi: 10.1016/j.joca.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Cao LJ, Wang H, Cheng XF, Wang L, Zhu L, Yan TT, Xie Y, Wu YZ, Zhao M, Ma SJ, Wu MQ, Wang GJ, Hao HP. Drug Metab Dispos. 2015;43:1181–1189. doi: 10.1124/dmd.115.063800. [DOI] [PubMed] [Google Scholar]
- 27.Chassin C, Vimont S, Cluzeaud F, Bens M, Goujon JM, Fernandez B, Hertig A, Rondeau E, Arlet G, Hornef MW, Vandewalle A. J Am Soc Nephrol. 2008;19:2364–2374. doi: 10.1681/ASN.2007121273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DG, Bae GS, Jo IJ, Choi SB, Kim MJ, Jeong JH, Kang DG, Lee HS, Song HJ, Park SJ. Inflammation. 2016;39:87–95. doi: 10.1007/s10753-015-0226-x. [DOI] [PubMed] [Google Scholar]
- 29.Awad AB, Burr AT, Fink CS. Prostag Leukotr Ess. 2005;72:219–226. doi: 10.1016/j.plefa.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Awad AB, Von Holtz RL, Cone JP, Fink CS, Chen YC. Anticancer Res. 1998;18:471–473. [PubMed] [Google Scholar]
- 31.Iwai K, Kondo T, Watanabe M, Yabu T, Kitano T, Taguchi Y, Umehara H, Takahashi A, Uchiyama T, Okazaki T. J Biol Chem. 2003;278:9813–9822. doi: 10.1074/jbc.M201867200. [DOI] [PubMed] [Google Scholar]
- 32.Marshall D, Cameron J, Lightwood D, Lawson ADG. Inflamm Bowel Dis. 2007;13:219–224. doi: 10.1002/ibd.20055. [DOI] [PubMed] [Google Scholar]
- 33.Chitu V, Stanley ER. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Wiktor-Jedrzejczak W, Ansari AA, Szperl M, Urbanowska E. Eur J Immunol. 1992;22:1951–1954. doi: 10.1002/eji.1830220743. [DOI] [PubMed] [Google Scholar]
- 35.Nashed B, Yeganeh B, HayGlass KT, Moghadasian MH. J Nutr. 2005;135:2438–2444. doi: 10.1093/jn/135.10.2438. [DOI] [PubMed] [Google Scholar]
- 36.Kim JM, Kang HW, Cha MY, Yoo D, Kim N, Kim IK, Ku J, Kim S, Ma SH, Jung HC, Song IS, Kim JS. Lab Invest. 2010;90:1004–1015. doi: 10.1038/labinvest.2010.54. [DOI] [PubMed] [Google Scholar]
- 37.Brufau G, Canela MA, Rafecas M. Nutr Res. 2008;28:217–225. doi: 10.1016/j.nutres.2008.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.