

Abstract
There is an urgent need to develop protective vaccines for high priority viral pathogens. One approach known to enhance immune responses to viral proteins is to display them on a nanoparticle (NP) scaffold. However, little is known about the effect of protein density on the B cell response to antigens displayed on NPs. To address this question HIV-1 Envelope (Env) and influenza hemagglutinin (HA) were displayed on a polystyrene-based NP scaffold at various densities - corresponding to mean antigen distances that span the range encountered on naturally occurring virions. Our studies revealed that NPs displaying lower densities of Env or HA more efficiently stimulated antigen-specific B cells in vitro, as measured by calcium flux, than did NPs displaying higher antigen densities. Similarly, NPs displaying a low density of Env or HA also elicited higher titers of antigen-specific serum IgG in immunized BALB/c mice (including elevated titers of hemagglutination-inhibiting antibodies), as well as an increased frequency of antigen-specific antibody secreting cells in the lymph node, spleen and bone marrow. Importantly, our studies showed that the enhanced B cell response elicited by the lower density NPs is likely secondary to more efficient development of follicular helper CD4 T cells and germinal center B cells. These findings demonstrate that the density of antigen on a NP scaffold is a critical determinant of the humoral immune response elicited, and that high density display does not always result in an optimal response
Keywords: HIV, Envelope, influenza, hemagglutinin, nanoparticle, humoral immunity, antigen display, antigen density
Introduction
The development of effective vaccines for viral pathogens such as HIV-1 and pandemic influenza represents a major unmet public health need [1-3]. Unfortunately, recent vaccination approaches, such as adjuvanted protein and viral delivery vectors, have met with limited success, particularly in the case of HIV-1 [4, 5]. One approach which has been shown to enhance the immune response against pathogens is to display their antigens on the surface of a nanoparticle (NP) scaffold [6-10]. However, little is known about the effect of antigen density on the magnitude and quality of the immune response to antigens displayed on NPs. This is an important question because the density of antigen display on human viruses – which represent naturally occurring biological NPs - varies considerably. For example, the number of Envelope glycoprotein (Env) spikes on the surface of the HIV-1 virion is very low (only ∼14 copies/virion) [11], whereas hemagglutinin (HA) is very densely displayed on the influenza A virus particle (∼500 copies/virion) [12]. As a result, the mean distance between individual antigens on the surface of different viruses varies greatly. In the case of HIV-1, this exceeds the separation between the two antigen-binding sites present on IgG antibodies (10-15 nm), thereby ensuring monovalent binding of Env-specific IgGs, and potentially contributing to viral immune evasion [11, 13, 14].
To better understand how antigen density affects the developing immune response we generated NPs on which viral glycoproteins (Env and HA) were displayed at densities that span the range of antigen densities encountered on HIV-1 and influenza virus particles. We then tested the effect of antigen density on the generation of antigen-specific humoral and cellular immune responses using both in vitro and in vivo approaches. Antigen-decorated NPs were evaluated for their ability to stimulate the activation of antigen specific B cell lines in vitro (as assessed by calcium flux) [15, 16]. Additionally, BALB/c mice were immunized with antigen-decorated NPs at multiple densities and the resulting serologic and cellular response was evaluated using a number of approaches that evaluate B cell signaling, priming of follicular helper cell and germinal responses, production of serum antibody and development of antibody secreting cells. Collectively, these data suggest that the density of antigen display on NPs is an important factor for controlling the magnitude and quality of the immune response elicited, and that increased antigen density does not always result in a more robust response.
Materials and Methods
Production of Recombinant HIV-1 Env and Influenza HA
Trimeric recombinant HIV-1 Env (strain YU2/426c) [17-19] and Influenza HA (strain A/New Caledonia/20/1999) [20] containing a C-terminal hexahistidine tag and Avitag sequence were produced by transient transfection of human 293F cells, as described [21]. After protein purification using metal ion chromatography [21], Bradford reagent (BioRad) was used to quantify recovered protein (in conjunction with a bovine serum albumin standard curve). Protein was quality controlled by analysis using a 7.5% SDS-PAGE gel to observe trimeric structure and purity of the antigens.
Biotinylation of Recombinant Env and HA
Site-specific biotinylation of Env and HA was accomplished using the C-terminal Avitag motif and a BirA enzyme kit (Avidity). Proteins were then concentrated and depleted of biotin and BirA enzyme through use of a 50K molecular weight cutoff centrifugal filter (Millipore).
NP Decoration with Env and HA
Polystyrene nanoparticles of multiple sizes; 200, 400, 500nm, functionalized with streptavidin (Bangs Laboratories), were used for all decorations. Nanoparticles were washed twice in PBS and then combined with biotinylated antigens. This mixture was rotated for 1 hour at room temperature, spun down at 9000g and washed once. Supernatant and wash were collected and analyzed with the o-Phthaldialdehyde assay (Anaspec) to measure the amount of unbound protein, and thereby infer the protein decoration efficiency. Results were confirmed by using an antigen-specific ELISA to measure Env and HA levels in the supernatants of decorated NPs (see below).
Cell Lines and Single Cell Suspension Protocol
Parental or transduced DG75 (ATCC CRL-2625) cells were maintained in RPMI 1640 supplemented with L-glutamine and 10% FBS [17]. Single cell suspensions were generated from spleen via mechanical disruption through a 40μm filter, and bone marrow suspensions were generated as described [22]. Lymph nodes were processed into single cell suspensions using frosted glass tissue disruptors and then passed through a 40μm filter. Samples were then treated with red blood cell lysis buffer (Biolegend), washed and counted via trypan blue dye exclusion. All cells were plated in IMDM medium (Invitrogen) with L-glutamine/10% FBS/streptomycin and penicillin.
Transfection of Cells with Antigen-Specific B Cell Receptors
DG75 cells were transiently transfected with mammalian expression plasmids encoding human B cell receptors (BCR) specific for HIV-1 Env (germline NIH45-46 and NIH45-46) and influenza HA (FI6), as described [17]. To confirm expression of desired BCRs, aliquots of DG75 cells (corresponding to both transfected and untransfected cells) were stained with anti-human IgG antibody conjugated to APC (BD clone G18-145) at a 1:100 dilution in RPMI medium for 30 minutes on ice. Cells were then washed with 1 ml of RPMI, resuspended in 350 μl of fresh media and analyzed on a BD LSR 12-color flow cytometer.
Analysis of BCR-Mediated Intracellular Signaling (Calcium Flux)
Aliquots of DG75 cells (corresponding to both transfected and untransfected cells) were loaded with 1 μM of Fura-Red AM (ThermoFisher), according to the manufacturer's instructions. Samples were then analyzed for 30 seconds to measure baseline signaling, prior to ligand treatment (i.e., addition of undecorated NPs and HA/Env-decorated NPs) and subsequent analysis for an additional 270-330 seconds. In the final 30 seconds, cells were treated with ionomycin at 10μM to determine maximum calcium release. Samples were analyzed using kinetic analysis on FlowJo v9.8.5, where samples were adjusted to baseline signaling levels. The resulting stimulation curve was then used to determine area under the curve, which was normalized to cells exposed to undecorated NPs.
Mouse Immunization
In vivo studies were approved by the University of Rochester's Committee on Animal Research (UCAR), and conducted in compliance with local, state and federal regulations. Female BALB/c mice (Charles River) were housed in the UR vivarium prior to use, and were immunized at 6-8 weeks of age in the right calf muscle and then boosted 21 days later at the same site. All mice received an equal mass of antigen, delivered on NP bearing different densities of protein. Serial bleeds were collected via the submandibular vein at day 14 and 28, and animals were sacrificed at day 35; a terminal blood sample was collected via cardiac puncture and immune organs of interest were harvested for subsequent analysis.
ELISA
Assays for antigen (Env, HA) specific serum IgG antibodies were conducted as published [21].
B Cell ELISpot
Antigen-specific antibody secreting cells (IgG) from spleen, lymph node and bone marrow were enumerated via ELISpot assay as reported [23]. Briefly, wells were coated with 1μg/well of recombinant antigen (Env, HA) or 1μg/well of anti-mouse IgG antibody. Cells from immune organs were plated based on expected frequency of antigen specific cells (1×106 for spleen/bone marrow and 2×105 for lymph node) and total IgG producing cells (2×105 for spleen/bone marrow and 2×104 for lymph node) and incubated for 6 hours. Anti-mouse IgG alkaline phosphatase was used at a 1:1500 dilution to detect antibody foci (ThermoFisher). Spots were visualized using the Vector Blue Substrate Kit (Vectorlabs), which was used according to the manufacturer's instructions, and enumerated using Cellular Technologies LTD ELISpot counting software.
Flow cytometry
Single-cell suspensions were plated in a 96-well microtiter plate and stained with purified rat anti-mouse CD16/32 (clone 2.4G2) Fc Block (BD Biosciences) for 20 minutes at 4°C, followed by addition of antibody cocktails to detect Tfh and B cell subsets. Cells were incubated for an additional 30 minutes at 4°C and protected from light. Following incubation, cells were washed twice in stain buffer (Dulbecco's PBS [DPBS] plus 2% FBS and 0.01% NaN3) and either re-suspended in stain buffer for data acquisition or secondary antibody cocktail for an additional 15 minute incubation at 4°C, protected from light. Cells incubated in secondary stain were washed an additional two times and re-suspended in stain buffer for data acquisition. Immediately prior to data acquisition, single-cell suspensions were spiked with 7-aminoactinomycin D (7-AAD) reagent (BD Biosciences) to exclude non-viable cells. Samples were acquired on a BD LSR-II instrument with 405, 488 and 633-nm lasers using FACSDiva software. Data files were analyzed using FlowJo, version 10 software (Tree Star, Inc.). Antibodies and reagents, purchased from BD Biosciences unless otherwise noted, were as follows: CD19 (1D3), B220 (RA3-6B2), CD4 (RM4-5), CD44 (IM7), CD95 (Jo2), CXCR5-biotin (2G8), PD-1 (J43, eBiosciences), T and B cell activation antigen (GL-7), and streptavidin-phycoerythrin (PE). For gating schematic see supplemental figure 1.
Results
To investigate the effect of antigen density on the humoral immune response against viral antigens, we generated recombinant HIV-1 Env and influenza HA trimers bearing a C-terminal biotin tag, and then attached these proteins to polystyrene NPs functionalized with streptavidin. Holding the amount of antigen constant and titrating in increasing amounts of NPs allowed for the generation of NPs bearing HIV-1 Env and influenza HA at high (dense), intermediate (moderate), low (sparse), and extremely low (very sparse) density - corresponding to estimated mean antigen distances of 9, 12, 17 and 32nm on the NP surface (Figure 1A). The distance between antigens was estimated using a modified algorithm that calculates the “nearest neighbor” distance between all antigens present on a NP and has been found to correlate highly with measurements derived from electron microscopy of viral particles [11]. Using this analysis we determined that the antigen densities on our NP (very sparse, sparse, moderate, dense) spanned the range of mean antigen distances encountered on naturally occurring viruses - as exemplified by native HIV-1 (26nm) and influenza virus particles (5nm) (Figure 1B). To confirm that proteins were displayed as expected on our NP, control experiments were also conducted in which GFP was bound to NPs and then detected using flow cytometry. This analysis revealed the expected hierarchy of antigen display, as shown in Figure 1C.
Figure 1.
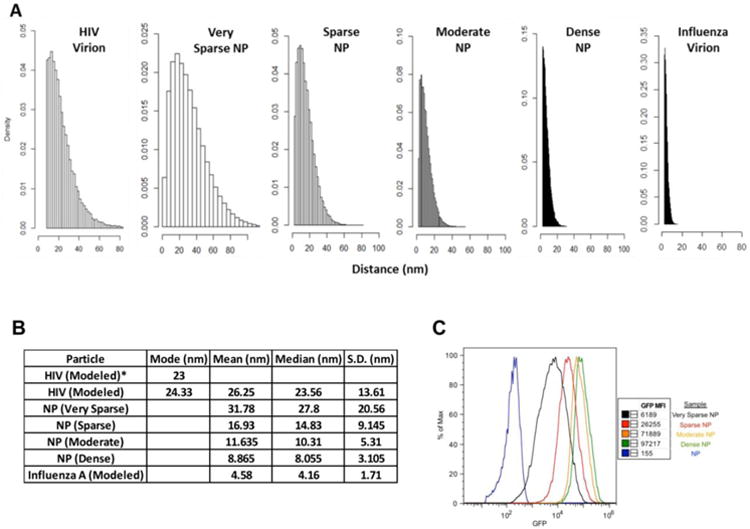
Analysis of antigen density on 500nm nanoparticles (NPs). Distances between antigens on the indicated particles (virions or nanoparticles) were estimated, using the nearest neighbor algorithm published by Zhu et al [11]. The amount of bound antigen was empirically measured for all decorated NP preparations (see Methods), and was estimated for the virus particles based on published studies [11, 12]. A) Panels show the results of 1500 simulations to determine the distribution of antigen distances on the surface of the NP preparations, versus naturally occurring virions. B) The table summarizes the estimated mean, mode and median distances between individual antigens on the surface of the indicated NPs and virions (along with the corresponding standard deviation). The left column shows the mode values for antigen distance on HIV-1 virions, as previously measured by cryoEM (23 nm) and as measured using our modified simulation (24.3 nm). C) This panel illustrates the relative amount and distribution of antigen on NP preparations. NPs were decorated with biotinylated GFP and analyzed by flow cytometry.
NPs displaying HIV-1 Env at high (dense) and low (sparse) density were then injected intramuscularly into female BALB/c mice, along with a matched dose of soluble Env, and the humoral response was tracked over time (note that all animals received the same total amount of Env). NPs bearing HIV-1 Env at low density elicited higher titers of antigen-specific serum IgG antibodies at every time point analyzed, when compared to NPs bearing a matched Env dose at high density or to soluble Env alone (Figure 2A, 2B). Consistent with this, the total number of Env-specific antibody secreting cells (ASC) in the bone marrow and spleen of immunized mice at day 35 (i.e., 14 days after the final boost) was greatest in mice immunized with “sparse” NPs (Figures 2C, 2D, left panels). Similarly, the frequency of Env-reactive ASCs (expressed as a fraction of total IgG+ ASCs) was also greatest in mice immunized with “sparse” NPs (Figures 2C, 2D, right panels). This indicated that sparse NPs appear to be more efficient at generating a long-lived antibody response, which can be measured by antibody secreting (plasma) cells in the bone marrow.
Figure 2.
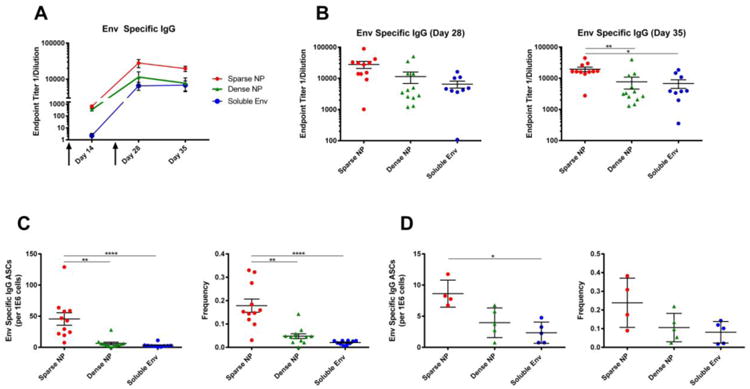
Humoral response to NPs (400 nm) displaying HIV-1 Env (Clade B, strain YU2). 6-8 week old female BALB/c mice were immunized intramuscularly with NPs displaying HIV-1 Env at high (dense) or low (sparse) density, or with soluble Env alone, at day 0 and then boosted at day 21 (arrows). Note that all animals received the same total mass of Env antigen. A) Endpoint titers of Env-specific serum IgG antibodies were measured at day 14, 28 and 35. B) Env-specific IgG titers for day 28 and 35 (sacrifice) are shown to highlight the distribution of responses within each group and facilitate comparisons between each of the formulations of antigen. C, D) Analysis of Env-reactive antibody secreting cells (ASC) in the bone marrow and spleen of immunized mice. The total number of Env-reactive IgG producing cells is shown (left) along with the frequency of these cells, relative to the total number of IgG producing cells in the bone marrow (right). Data for individual mice are shown (9-12/group), along with the mean and standard error of the mean. Significance was determined between groups using Kruskal Wallis (ANOVA) analysis. * p<0.05 **p<0.01 ****p<0.0001
To determine whether our findings were unique to the HIV-1 Env antigen, or more generalizable, we performed similar experiments using the influenza HA antigen. To do this, nanoparticles were decorated with recombinant influenza A virus (IAV) hemagglutinin (HA) trimers (A/New Caledonia/20/1999 (H1N1)) using the same method as described for HIV-1 Env. Once again, mice were immunized via the intramuscular route, with “sparse” and “dense” NPs and the resulting humoral response was assessed over time.
The results were very similar to those obtained using HIV-1 Env. NPs bearing IAV HA at low (sparse) density elicited higher titers of both HA-binding serum IgG antibodies (Figure 3A, 3B) and hemagglutination-inhibiting (HAI) serum antibodies (Figure 3C). HA-specific antibody secreting cells (ASC) were also quantified in the bone marrow (Figure 3D) and spleen (Figure 3E) of immunized mice at day 35, and the total number of HA-specific ASCs was greatest in mice immunized with “sparse” NPs.
Figure 3.
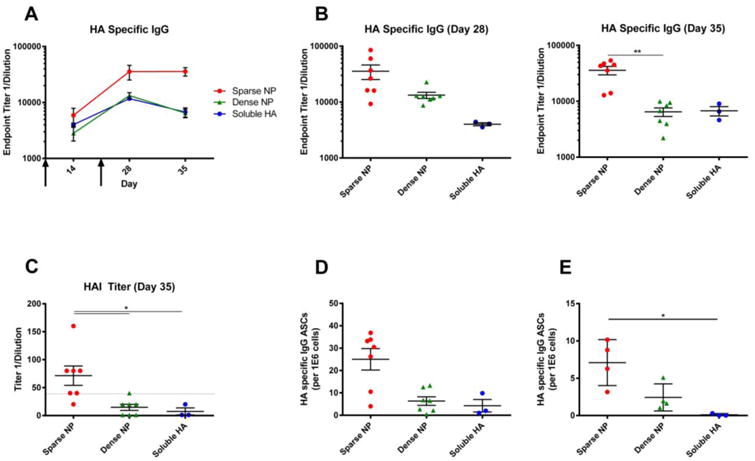
Humoral response to NPs (400 nm) displaying influenza HA (A/New Caledonia/20/1999). 6-8 week old female BALB/c mice were immunized intramuscularly with NPs displaying influenza HA at high (dense) or low (sparse) density, or with soluble HA alone, at day 0 and then boosted at day 21 (arrows); all animals received the same total mass of HA antigen. A) The endpoint titers of HA-specific serum IgG antibodies were measured at day 14, 28 and 35. B) HA specific IgG titers for day 28 and 35 (sacrifice) are shown to highlight the distribution of responses within each group and facilitate comparisons between each of the formulations of antigen. C) Hemagglutination-inhibiting (HAI) serum antibody titers were measured using influenza virus A/New Caledonia/20/1999 (H1N1). Analysis of HA-reactive ASC in the bone marrow (D) and spleen (E) of immunized mice. The total number of HA-specific IgG producing cells is shown. Data for individual mice are shown (3-7/group, as shown in panels B-E), along with the mean and the standard error of the mean. Significance was determined between groups using Kruskal Wallis (ANOVA) analysis. * p<0.05 ** p<0.01
Collectively, these observations suggest that not only is the magnitude of the response controlled by antigen density - but also the quality of the antibodies elicited (as reflected by the increase in titers of HAI serum antibodies, which typically correlate with the ability to neutralize virus [24].) In addition, the fact that our findings were similar for both Env and HA suggests that the superior humoral response elicited by NPs displaying “sparse” antigens may be a generalizable phenomenon - at least in the case of highly glycosylated proteins.
To determine if the enhanced in vivo immunogenicity of “sparse” NPs was due to more efficient stimulation of individual B cells, we performed in vitro experiments using a human B cell line (DG75) that was transfected to express B cell receptors (BCRs) specific for HIV-1 Env (or HA) [15, 17]. To do this, DG75 cells were transiently transfected with BCR expression plasmids corresponding to (i) wild-type NIH45-46, a high affinity (3.66nM) broadly neutralizing antibody specific for recombinant 426c HIV-1 Env used in our experiments [17], (ii) a germline reverted derivative of the NIH45-46 BCR, which has low (693nM) affinity for 426c Env [17], and (iii) FI6, a high affinity antibody specific for IAV HA. Briefly, cells were loaded with Fura Red, exposed to the NP preparations of interest, and intracellular calcium flux was quantitated by flow cytometry, as a measure of BCR-mediated cell activation.
This experiment showed that the efficiency of antigen-specific BCR activation was greatest when cells were treated with “sparse” NPs compared to “dense” NPs - both for HIV-1 Env (Figure 4A, left/center) and for flu HA (Figure 4A, right). Collectively, these data suggest that “sparse” antigen density on NPs (defined here as a mean antigen distance of 17 nm) elicits more efficient stimulation of antigen-specific B cells than high antigen density (defined here as a mean antigen distance of 9 nm).
Figure 4.
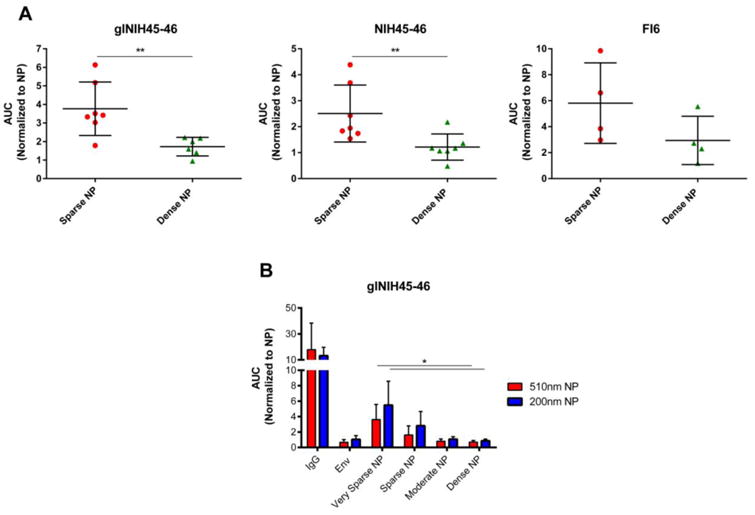
Stimulation of antigen-specific B cell lines by NPs displaying HIV-1 Env. DG-75 cells were transfected with plasmids expressing the following B cell receptors: A) Left: a germline reverted derivative of the broadly neutralizing antibody NIH45-46 (low affinity). Center: the broadly neutralizing HIV-1 Env specific antibody NIH45-46 (high affinity). Right: a high affinity influenza specific antibody (FI6). Cells were loaded with Fura-red, a calcium reactive fluorescent dye, and then exposed to NPs (500 nm) displaying HIV-1 Env at high (dense) or low (sparse) density, or to NPs displaying influenza HA at the same densities. B) DG75 cells stably transduced to express the glNIH45-46 receptor were treated with NPs of 200nm or 500nm diameter decorated with various densities of HIV-1 Env (dense, moderate, sparse, very sparse; as defined in Figure 1). Data represents resulting mean calcium flux measurements, reported as Area Under the Curve (AUC), and normalized to results from cells exposed to NPs alone. Each data point represents an individual experiment with cells exposed to each of the treatments. The data is represented as the mean and standard deviation of all experimental observations. Significance was calculated using the Mann-Whitney analysis **p<0.01 (A) or the Kruskal-Wallis (ANOVA) analysis *p<0.05 (B).
Our initial experiments (Figures 2 thru 4A) examined the effect of antigen density using large NPs (400nm in vivo and 500nm in vitro), which are bigger than most viruses. We therefore next tested whether similar results would be obtained using smaller NPs, with a diameter (200 nm) similar to that of naturally occurring viruses [11, 25, 26]. We also decided to interrogate the full range of available antigen densities (i.e., very sparse, sparse, moderate and dense; see Figure 1). As shown in Figure 4B, 200 nm and 500 nm NPs stimulated stably transduced antigen specific B cells (glNIH45-46) comparably at each of the multiple antigen densities tested, and sparse antigen densities consistently elicited stronger levels of cell activation. Thus, NP size did not affect the ability of sparse antigen densities to stimulate B cells more efficiently in vitro.
To determine whether NP size affected the in vivo immunogenicity of NPs decorated at various antigen densities, mice were immunized with 200nm NPs displaying dense, moderate, sparse and very sparse displays of HIV-1 Env. As was seen with larger NPs (Figures 2, 3) sparse displays of antigen on NPs significantly increased the levels of antigen specific antibody (Figure 5, left) in a density dependent manner. Sparse displays of antigen also significantly increased the level of antibody secreting (plasma) cells in the bone marrow compared to dense displays of antigen (Figure 5, right).
Figure 5.
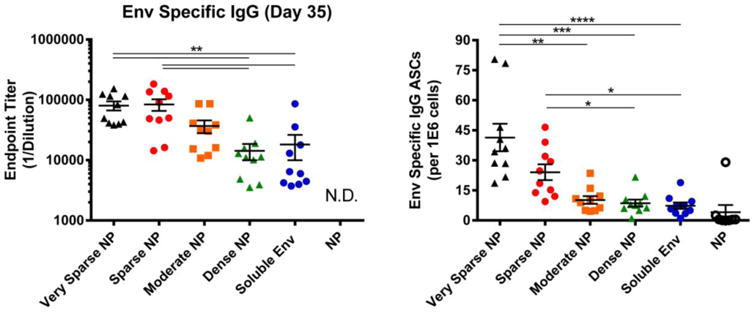
B cell response to NPs (200 nm) displaying HIV-1 Env. 6-8 week old female BALB/c mice were immunized intramuscularly with NPs displaying HIV-1 Env at high (dense), intermediate (moderate), low (sparse), extremely low (very sparse) density. Control mice were immunized with either undecorated NPs or soluble Env alone; all animals received the same total mass of Env antigen. All groups were immunized at day 0 and then boosted at day 21; at day 35, mice were sacrificed and the sera and bone marrow were collected. Left: Serum from individual animals was screened via an Env specific ELISA to determine relative levels (endpoint titer) of antigen specific IgG antibodies from all groups. N.D., not detected. Right: Bone marrow was isolated from the tibia and femur of individual animals. After depletion of red blood cells, single cell suspensions were screened for Env-specific ASCs. In both panels, data for individual mice are shown, along with the mean and the standard error of the mean for sera and bone marrow (n=10 animals). Significance was determined between groups using Kruskal Wallis (ANOVA) analysis. * p<0.05 **p<0.01
Finally, we addressed the possible mechanism behind these observations, by examining the germinal center response after immunization. A key consequence of the germinal center reaction is production of high-affinity, class-switched memory B cells and antibody secreting cells that can home to the bone marrow where they persist for long periods of time [27, 28]. Therefore, elevated levels of CD4+ T follicular helper (Tfh) and germinal center B cells (GC B cells) could explain the increased level of antibody and ASCs observed in our experiments.
To test this prediction, Tfh and GC B cells were measured by flow cytometry, following prime/boost immunization with Env-decorated NPs. Immunization with sparse NP elicited a higher frequency (Figure 6A, left, center) and number (data not shown) of Tfh and GC B cells than dense NPs; moreover, the level of these two cell populations was positively correlated (Figure 6A right). In addition, sparse NP immunization elicited significantly more ASCs in the draining lymph nodes (dLN) at both day 3 and 5 post-boost, as determined by flow cytometry (Figure 6B left) and by B cell ELISpot (Figure 6B right) - suggesting that antigen density can positively regulate the formation of germinal centers and ASCs.
Figure 6.
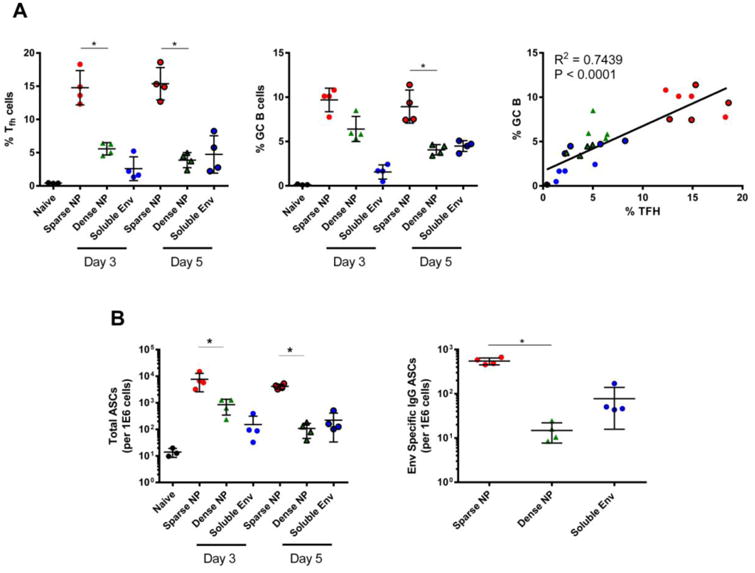
Frequency of CD4+ T follicular helper (Tfh) cells, germinal center B (GC B) cells and ASC elicited in the draining lymph node after NP (200 nm) immunization. Control animals were immunized with soluble Env or left naïve; all animals received the same total mass of Env antigen. All groups were immunized at day 0 and then boosted at day 21. Mice were sampled at day 3 and day 5 post-boost for Tfh, GC B cells and ASCs via flow cytometry and antigen specific B cell ELISpot assays. A) Left: frequency of Tfh cells (CD4+B220-CD44hiCXCR5+PD-1+), enumerated via flow cytometry. Center: frequency of GC B cells (CD19+CD4-B220+CD138+Fas/CD95+GL7+), enumerated via flow cytometry. Right: Correlation between Tfh and GC B cell responses. Pearson test was performed assuming Gaussian distribution, with P and R2 values shown. B) Left: the number of ASCs (CD19+CD4-B220loCD138+), measured by flow cytometry. Right: level of Env-specific IgG ASCs measured via antigen specific B cell ELISpot. In all panels, data for individual mice are shown, along with the mean and the standard deviation. Significance was determined between sparse NP and dense NP using the two tailed Mann-Whitney (t-test) analysis. * p<0.05
Discussion
Nanoparticle-based antigen display has been shown to result in an enhanced humoral immune response [6-8]. However, it is poorly understood how protein density influences the magnitude and quality of immune responses to NP displayed antigens [9]. Previous studies have used complex systems, including haptens, micron sized conjugates, or combinations of antigen/adjuvants to understand antigen density and its effects on host immunity [6, 29, 30]. To take a simpler and more direct approach to this question, we decorated NP with two model antigens (HIV-1 Env and influenza A virus HA) at a range of densities that spanned those encountered on naturally occurring viruses.
Immunization of BALB/c mice with NPs that were sparsely coated with antigen resulted in higher titers of antigen-specific serum IgG antibodies, and increased numbers of antigen-specific antibody-secreting cells (Figures 2 and 3), when compared to NPs that were densely coated with the same antigen. This was true for both Env and HA and was independent of NP size (400 nm NP being used in Figures 2 and 3, versus 200 nm NP in Figure 5), indicating that these finding are likely generalizable - both with respect to the antigen displayed, and the size of the nanoparticle on which that antigen is presented. In vitro studies using B cell line that expressed either Env- or HA- specific B cell receptors further supported our in vivo data, and revealed that NPs sparsely coated with antigen elicited higher levels of BCR-mediated B cell stimulation than NPs that were densely coated with the same antigen (Figure 4). Once again, this was true for both Env and HA decorated NP, and was unaffected by NP size.
To examine the underlying mechanism, we examined the germinal center (GC) response. The GC is a highly organized, T cell-dependent substructure that forms within secondary lymphoid tissues and is the main source of memory B cells and long-lived plasma cells, two cell types important in maintaining immune memory and protection upon reinfection [27, 28]. Plasma cells leaving the GC home to the bone marrow and spleen and are the source of long-lasting, affinity-matured antibodies. Our experiments showed that elevated levels of Tfh and GC B cells were elicited when mice were immunized with NP bearing sparsely displayed antigen (Figure 6); the level of ASCs was also increased (Figure 6). Taken together, these observations suggest that sparse antigen density on NPs allows improved GC reactions that, in turn, give rise to durable memory reservoirs and elevated, long-lived serum antibodies.
Overall, the data reported here are the first to our knowledge showing that antigen density is a critical determinant of the immune response to antigen-coated nanoparticles, and that high antigen density does not always result in improved antibody responses. It is noteworthy that recent findings have shown that influenza virions displaying reduced levels of HA (as achieved through codon deoptimization of the HA gene segment) elicited improved, long-lasting protection from a lethal virus challenge, at a reduced median protective dose [31]. Our data suggest that this may be in part a consequence of the reduced HA density on the virion surface. However, many questions still remain as to how antigen density influences protective immune responses. It is possible that surface virion protein density may influence antigen access and handling by innate immune cells localized at the site of immunization as well as within the draining lymph node. For example: soluble and membrane-associated C-type lectins can bind glycoprotein-containing NPs [32-35]. Therefore, a higher glycoprotein density could diminish the amount of NPs draining to the lymph node and modulate their ability to be efficiently captured and presented to B cells (e.g., subcapsular sinus macrophages, follicular dendritic cells) within the draining lymph node. It is also possible that by displaying antigen at a lower density, B cell epitopes become more accessible - and therefore potentially more stimulatory - through promoting activation of a larger percentage of the antigen-specific repertoire. Finally, through better signaling, B cell receptor engagement may augment the effector potential of helper T cells during the germinal center reaction, either at the level of T cell receptor signaling and/or co-stimulation [36]. Future experiments are planned to explore these multiple, non-exclusive mechanisms that underlie the advantages that low density NPs exhibit as immunogens.
Supplementary Material
Supplementary Figure 1. Gating scheme for identification of immune cell populations in the draining lymph node after immunization.
Acknowledgments
We would like to thank Dr. Leonidas Stamatatos at the University of Washington for his kind gift of expression plasmids encoding B cell receptors specific for HIV Env and Influenza HA. Additionally, we would like to thank Dr. Tim Bushnell and the University of Rochester (UR) Flow Cytometry Core facility for technical assistance, the UR Center for AIDS Research for assistance with protein production, as well as K.O. Knausgaard, J. Elliott and T. Walsh for helpful comments. This work was supported in part by the University of Rochester Center for AIDS Research grant P30 AI078498 (NIH/NIAID), the University of Rochester School of Medicine and Dentistry and NIH Predoctoral Training Grant T32 AI049815 (MGB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rerks-Ngarm S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361(23):2209–20. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 2.Gray GE, et al. Approaches to preventative and therapeutic HIV vaccines. Curr Opin Virol. 2016;17:104–109. doi: 10.1016/j.coviro.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14(3):167–82. doi: 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 4.Pitisuttithum P, et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194(12):1661–71. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- 5.Gray G, Buchbinder S, Duerr A. Overview of STEP and Phambili trial results: two phase IIb test-of-concept studies investigating the efficacy of MRK adenovirus type 5 gag/pol/nef subtype B HIV vaccine. Curr Opin HIV AIDS. 2010;5(5):357–61. doi: 10.1097/COH.0b013e32833d2d2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moon JJ, et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109(4):1080–5. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galloway AL, et al. Development of a nanoparticle-based influenza vaccine using the PRINT technology. Nanomedicine. 2013;9(4):523–31. doi: 10.1016/j.nano.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Sliepen K, et al. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology. 2015;12:82. doi: 10.1186/s12977-015-0210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, et al. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220(Pt A):141–8. doi: 10.1016/j.jconrel.2015.09.069. [DOI] [PubMed] [Google Scholar]
- 10.Benson RA, et al. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife. 2015;4 doi: 10.7554/eLife.06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu P, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441(7095):847–52. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 12.Compans RW, et al. Influenza virus proteins. I. Analysis of polypeptides of the virion and identification of spike glycoproteins. Virology. 1970;42(4):880–9. doi: 10.1016/0042-6822(70)90337-5. [DOI] [PubMed] [Google Scholar]
- 13.Klein JS, et al. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc Natl Acad Sci U S A. 2009;106(18):7385–90. doi: 10.1073/pnas.0811427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein JS, Bjorkman PJ. Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 2010;6(5):e1000908. doi: 10.1371/journal.ppat.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGuire AT, et al. Diverse recombinant HIV-1 Envs fail to activate B cells expressing the germline B cell receptors of the broadly neutralizing anti-HIV-1 antibodies PG9 and 447-52D. J Virol. 2014;88(5):2645–57. doi: 10.1128/JVI.03228-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ota T, et al. Anti-HIV B Cell lines as candidate vaccine biosensors. J Immunol. 2012;189(10):4816–24. doi: 10.4049/jimmunol.1202165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuire AT, et al. Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med. 2013;210(4):655–63. doi: 10.1084/jem.20122824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, et al. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. J Virol. 1992;66(11):6587–600. doi: 10.1128/jvi.66.11.6587-6600.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs JM, et al. Stable, uncleaved HIV-1 envelope glycoprotein gp140 forms a tightly folded trimer with a native-like structure. Proc Natl Acad Sci U S A. 2014;111(52):18542–7. doi: 10.1073/pnas.1422269112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yassine HM, et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med. 2015;21(9):1065–70. doi: 10.1038/nm.3927. [DOI] [PubMed] [Google Scholar]
- 21.Mattiacio J, et al. Dense display of HIV-1 envelope spikes on the lambda phage scaffold does not result in the generation of improved antibody responses to HIV-1 Env. Vaccine. 2011;29(14):2637–47. doi: 10.1016/j.vaccine.2011.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutz MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 23.He XS, et al. Plasmablast-derived polyclonal antibody response after influenza vaccination. J Immunol Methods. 2011;365(1-2):67–75. doi: 10.1016/j.jim.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulligan MJ, et al. Serological responses to an avian influenza A/H7N9 vaccine mixed at the point-of-use with MF59 adjuvant: a randomized clinical trial. JAMA. 2014;312(14):1409–19. doi: 10.1001/jama.2014.12854. [DOI] [PubMed] [Google Scholar]
- 25.Bhella D, Rixon FJ, Dargan DJ. Cryomicroscopy of human cytomegalovirus virions reveals more densely packed genomic DNA than in herpes simplex virus type 1. J Mol Biol. 2000;295(2):155–61. doi: 10.1006/jmbi.1999.3344. [DOI] [PubMed] [Google Scholar]
- 26.Bialas KM, et al. Specific nucleoprotein residues affect influenza virus morphology. J Virol. 2014;88(4):2227–34. doi: 10.1128/JVI.03354-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith KG, et al. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16(11):2996–3006. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi Y, et al. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187(6):885–95. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dintzis RZ, Middleton MH, Dintzis HM. Studies on the immunogenicity and tolerogenicity of T-independent antigens. J Immunol. 1983;131(5):2196–203. [PubMed] [Google Scholar]
- 30.Paus D, et al. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med. 2006;203(4):1081–91. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang C, et al. Deliberate reduction of hemagglutinin and neuraminidase expression of influenza virus leads to an ultraprotective live vaccine in mice. Proc Natl Acad Sci U S A. 2013;110(23):9481–6. doi: 10.1073/pnas.1307473110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez SF, et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol. 2010;11(5):427–34. doi: 10.1038/ni.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Londrigan SL, et al. Cell-surface receptors on macrophages and dendritic cells for attachment and entry of influenza virus. J Leukoc Biol. 2012;92(1):97–106. doi: 10.1189/jlb.1011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng WC, et al. Soluble host defense lectins in innate immunity to influenza virus. J Biomed Biotechnol. 2012;2012:732191. doi: 10.1155/2012/732191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tate MD, et al. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses. 2014;6(3):1294–316. doi: 10.3390/v6031294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Gating scheme for identification of immune cell populations in the draining lymph node after immunization.