

Abstract
Monoclonal antibodies that target CD20 expressing B cells represent an important new treatment option for patients with multiple sclerosis (MS). B cell depleting therapy is highly effective against relapsing forms of the disease, and is also the first treatment approach proven to protect against disability worsening in primary progressive MS. Moreover, evolving clinical experience with B-cell therapy, combined with a more sophisticated understanding of humoral immunity in preclinical models and in patients with MS, has led to major progress in deciphering the immune pathogenesis of MS. Here, we review the nuanced roles of B cells in MS autoimmunity, the clinical data supporting use of ocrelizumab and other anti-CD20 therapies in the treatment of MS, as well as safety and practical considerations for prescribing. Lastly, we summarize remaining unanswered questions regarding the proper role of anti-CD20 therapy in MS, its limitations, and the future landscape of B cell based approaches to treatment.
INTRODUCTION
Paradigm shifts in the understanding of disease usually occur at the intersection of the laboratory and the bedside, but the key conceptual advances – eureka moments – are more often made when real-life clinical trial data are reported. The clinical trials of B cell therapy in multiple sclerosis (MS) are an example of this principle,1–3 unifying decades of observation, associations, and speculation that B-lymphocytes, the central actors of humoral immunity, are critical to the pathogenesis of MS.4 Indeed, B cells have now emerged as the important target for our most highly effective therapeutics. The recently reported trials of the humanized anti-CD20 monoclonal antibody (mAb) ocrelizumab revealed dramatic effects on all key clinical and magnetic resonance imaging (MRI) outcomes in relapsing MS (RMS), and also demonstrated clear benefits for the previously untreatable form of the disease, primary progressive MS (PPMS). Ocrelizumab was recently approved by the US Food and Drug Administration (FDA), and decisions by other regulatory agencies are expected to be forthcoming. In this review we will summarize emerging concepts of B cell biology relevant to MS, outline likely mechanisms of action of CD20 therapies, and speculate on the proper role of ocrelizumab in the therapeutic arsenal.
THE NEUROIMMUNOLOGY OF B CELLS
As is true of many cells and molecules of the immune system, B cells can function in either pro- or anti-inflammatory roles, depending on their subtype and context.5 The pro-inflammatory functions of B cells, including presentation of critical antigens to Th17 and Th1 cells, secretion of cytokines and other molecules, as well as antibody production (Figure 1), have generally received the most attention as mediators of tissue damage in many neurologic disorders. There is also increasing recognition of the clinical importance of countervailing regulatory B cells (B-regs) that can dampen excessive inflammatory responses. Additional roles for B cells in the processes of growth, remodeling and repair have also been identified. The multifaceted biology of B cells underlies their varied roles as a primary or secondary player in human disease.
Figure 1. Landscape of B-cell therapies and possible mechanisms of action.
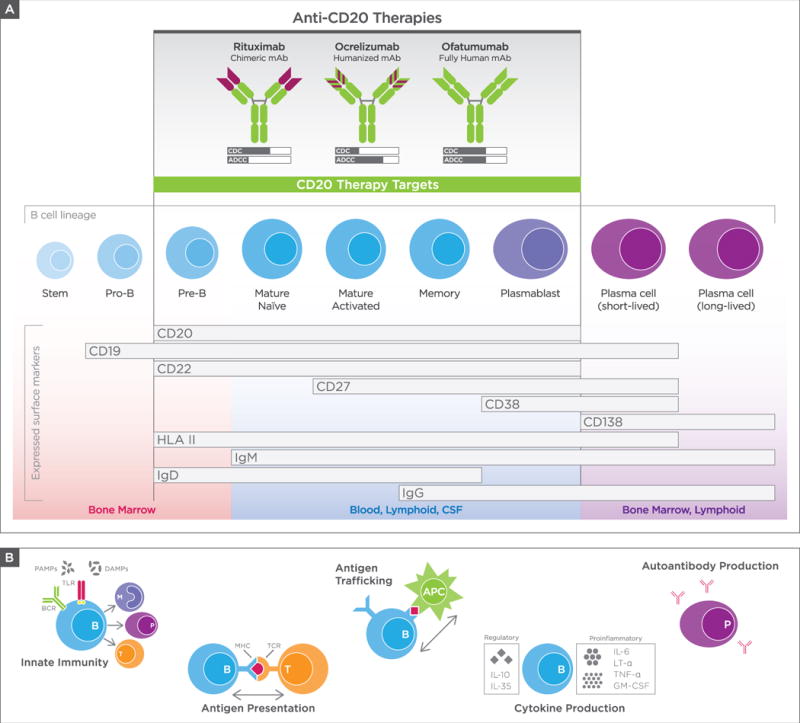
(A) Anti-CD20 mAbs in clinical use and summary of expression of CD20 and other B-cell surface antigens. Top: Structure of anti-CD20 mAbs in clinical use, with mechanisms of action summarized as relative degrees of complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC). Middle: B-cell maturation stages, defined by cell-surface antigens, highlighting B-cell subsets most depleted by anti-CD20 therapies (shaded region in center). Bottom: Common tissue locations for B-cell subsets. Of note: heterogeneity in surface-marker expression across previously defined B-cell subsets is recognized, as are exceptions to defined tissue locations of B-cell subsets. (B) Diverse functional roles of B cells in immunity and autoimmunity. The many roles of B cells, including participation in innate immunity, antigen presentation, antigen trafficking, cytokine production, and autoantibody production. The mechanism(s) responsible for the rapid onset and nearly complete protection against development of new focal lesions in MS is/are unknown, but may be attributed to effects of anti-CD20 therapy on antigen presentation and cytokine production. APC = antigen-presenting cell; BCR = B-cell receptor; CSF = cerebrospinal fluid; DAMPs = damage-associated molecular pattern molecules; GM-CSF = granulocyte-macrophage colony-stimulating factor; HLA = human leukocyte antigen; IgG = immunoglobulin G; IL = interleukin; LT-α = lymphotoxin-alpha; MHC = major histocompatibility complex; PAMPs = pathogen-associated molecular pattern molecules; TCR = T-cell receptor; TLR = Toll-like receptor; TNFα, tumor necrosis factor alpha.
Why Target B Cells in MS?
Traditional murine models of MS, known as experimental autoimmune encephalitis (EAE), were mediated largely or exclusively by pathogenic T cells, with little or no participation by B cells or antibodies. T cells removed from paralyzed animals were sufficient to “adoptively transfer” EAE to unimmunized littermates; similarly, transgenic mice engineered to express myelin-recognizing T cells spontaneously developed EAE. However, EAE is not a unitary syndrome, but rather encompasses a spectrum of pathologies that vary by the antigen, adjuvant, and strain of animal employed.6–10 Some of the most useful laboratory models result from autoimmunity to myelin oligodendrocyte glycoprotein (MOG), a quantitatively minor CNS-restricted protein expressed on the outermost lamellae of the myelin sheath. MOG is highly immunogenic in most species studied, and produces an inflammatory white matter pathology superficially resembling MS.
A modern reassessment of the potential role of humoral immunity in EAE began with a search to replicate a more distinctive pattern of demyelination present in MS lesions and not found in T cell mediated EAE models, specifically the presence of large, sharply circumscribed, macrophage-rich demyelinating lesions with vesicular disruption of myelin membranes.11–14 A close recapitulation of this characteristic MS-like lesion was produced in marmosets15 and also in rats,16 although an earlier description may date back more than 50 years in a guinea pig optic neuritis model.17 Surprisingly, in these MS-like models, antibodies and B cells, acting in concert with T cells, were found to be required for full disease expression.18 These observations provided a new theoretical framework for the use of B cell based therapeutics in MS.14
One of the hallmarks of MS, used as a diagnostic tool for nearly 75 years, is the presence of oligoclonal bands (OCB), representing clonally-restricted antibodies found in the cerebrospinal fluid (CSF). These secreted immunoglobulins produced by B cells and plasma cells are largely stable in an individual over the course of disease,19 acting as a private ‘fingerprint’ for each MS patient.20 Despite many decades of work, however, the antigen targets of OCB have never been convincingly identified21 and their role in MS pathophysiology is uncertain. Although some OCB have been reported to recognize known myelin proteins, these appear to represent the exception rather than the rule.22, 23 In one recent report, some CSF derived antibodies were found to recognize ubiquitous cellular debris,23 suggesting that they represent a secondary phenomenon in response to tissue injury rather than having a primary pathogenic role. Because antibodies and complement are commonly found deposited along myelin sheaths and on the surface of macrophages in active,24 and even newly forming,25 MS lesions, however, the search for autoantigens recognized by CNS antibodies in MS continues. New technologies such as phage-display technology now make possible screening with levels of resolution not previously achievable.26,27
Increased numbers of activated B cell derived plasmablasts, capable of antibody secretion, can remain in the CSF of MS patients for many years,28 in contrast to their transient elevations in self-limited neurologic inflammations such as neuroborreliosis or viral encephalitis. Plasmablasts appear to be an important source of secreted immunoglobulin (Ig) in the CSF,29, 30 as well as a correlate of disease severity28 and gadolinium-enhancing lesions.28, 31 Sequencing studies of B cell receptor (BCR) genes from CSF plasmablasts and B cells have revealed that: (1) CSF plasmablasts are derived from a small number of clonally-related B cells within the CNS compartment that have undergone antigen-driven activation and somatic mutation;31 (2) similar B cell clones are present in the CSF and brain parenchyma;30 (3) the same unique clonal families can also be found in peripheral blood (PBL) and draining cervical lymph nodes in MS, with antigen-driven stimulation likely occurring in both compartments, revealing an active immune axis with dynamic recirculation of B cells between the periphery and the CNS;32, 33 (4) identical clonally-related memory B cells and plasmablasts persist in the CSF over time;34 (5) oligoclonal plasmablasts produce OCBs,29 and in an individual MS patient distinct OCBs have highly similar amino acid sequences.35 Remarkably, OCBs appear to be largely resistant to all interventions for MS, including cyclophosphamide, brain irradiation, autologous bone marrow transplantation, the anti-CD20 mAb rituximab, as well as all FDA-approved therapies.20, 36–45 These observations emphasize the persistent and refractory nature of these highly selective compartmentalized engines of humoral immunity in the CNS of individuals with MS.
In addition to differentiation to antibody secreting plasmablasts and plasma cells, functions of B cells that may be most relevant to MS pathogenesis include roles in antigen presentation and/or cytokine production.
B cells represent a unique population of antigen presenting cells (APCs). Most APCs recognize a range of exogenous and endogenous antigens, internalizing and presenting these to T cells in the context of class II major histocompatibility complex (MHC) and co-stimulatory molecules. In contrast, B cells efficiently recognize only those antigens bound to their specialized, unique surface BCR. Thus, while most APCs are promiscuous antigen presenters, B cells are highly selective ones. Upon binding a specific antigen to the B cell’s surface Ig, the antigen is internalized, processed in the endosome, its constituent peptides are then complexed with MHC class II molecules, and the antigen-MHC complex is transported to the cell surface where it can activate T cells via engagement of the T cell receptor and costimulatory molecules.46, 47 In a B cell dependent EAE model, selective elimination of MHC class II expression on B cells – but not on other APCs – conferred resistance to disease, indicating that the APC function of B cells was critical.48 Furthermore, EAE studies indicate that only some autoantigens, including the highly immunogenic CNS protein MOG, require antigen presentation by B cells to activate autoreactive T cells. It is reasonable, although speculative, to consider the possibility that the antigen or antigens that trigger human MS are similarly B cell dependent.
Stimulated in vitro, B cells from MS patients upregulate MHC class II and co-stimulatory molecules to a greater extent than B cells from healthy individuals or individuals with other neuroinflammatory diseases.49 Genes that are active in B cells also represent a major component of all variants, now numbering more than 200,50 known to increase MS risk. Notably, the gene encoding the MHC class II DR beta chain, critical for APC function, is the strongest MS susceptibility signal genome-wide. It is likely that the net effect of this MS genetic burden is to bias B cell biology towards a pro-inflammatory phenotype, perhaps promoting presentation of self-antigens to effector T cells or augmenting autoimmune responses via production of cytokines and other mediators.
B cells also regulate a wide range of effector immune functions mediated by both B and T cells through secretion of pro-inflammatory and regulatory cytokines. Tumor necrosis factor alpha (TNFα), lymphotoxin, and granulocyte macrophage colony-stimulating factor (GM-CSF) are produced by pro-inflammatory B cells, while B-regs produce interleukin (IL)-10 and IL-35.51–53 Not surprisingly, MS B cells cultured in vitro have been found to secrete higher levels of pro-inflammatory, and lower levels of regulatory cytokines.54 A role for B cell derived pro-inflammatory cytokines in MS is supported indirectly by the clinical finding that B cell depletion rapidly eliminates gadolinium enhancement on MRI,3 suggesting that B cell mediators could be responsible for disruptions of endothelial tight junctions in active MS.
IL-10 secreting B-regs normally act in the setting of inflammation to inhibit pro-inflammatory T cell responses mediated in part through interferon gamma and IL17.55 However, in the face of innate immune system signaling through toll like receptor 7, B-regs of MS patients do not respond and differentiate to the extent they do in healthy controls.55 Interestingly, in MS patients, abnormalities in B cell tolerance appear to predominate in MS patients only in the periphery, in contrast to other autoimmune diseases that involve defects in central B cell tolerance.56
B cells Are Targeted by Other Disease Modifying Therapies for MS
Although most disease-modifying therapies for MS have traditionally been conceptualized as functioning via T cell based mechanisms, a growing body of data indicates that all have demonstrable effects on B cells as well.57 Common themes include promoting naïve rather than memory or plasmablast B cells (alemtuzumab); shifting B-cell cytokines towards an anti-inflammatory tone (beta interferon, glatiramer acetate, fingolimod); increasing B-regs (beta interferon, glatiramer acetate, fingolimod and dimethyl fumarate); decreasing class II MHC expression and costimulatory molecules on B cells required for antigen presentation (beta interferon and dimethyl fumarate); sequestering B cells in lymphoid organs (fingolimod); blocking VLA-4 mediated B cell trafficking to the CNS (natalizumab); or direct cytolysis of B cells (alemtuzumab, teriflunomide, mitoxantrone). A detailed discussion of B-cell effects of MS disease modifying therapies can be found elsewhere.57, 58
Not All B Cell Based Approaches Will Be Beneficial in MS
In EAE, B cells can serve as APCs for large protein autoantigens including human MOG but not for myelin peptides, such as the amino acid 35–55 MOG fragment.59, 60 Peptide immunization models are B cell independent because the BCR, which binds conformational determinants of large protein antigens with highest affinity, is not involved in capture and presentation of these short, linear peptide epitopes.59 As noted above, B cells also regulate autoimmunity by provision of IL-10.60 Remarkably, when EAE was induced with whole MOG, B cell depletion was protective, but in MOG peptide-induced EAE, B cell depletion worsened disease severity,61, 62 probably by depleting IL-10 producing B-regs. In humans, a clinical trial of atacicept, a decoy receptor for the B cell growth factors B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), worsened RMS for uncertain reasons but possibly by increasing memory B cells or reducing B-reg tone.63
Other Therapeutic Approaches Targeting B cells
Inebilizumab (MEDI-551) is an anti-CD19 mAb that is designed to lead to more complete B cell depletion than CD20 approaches as it also targets pro-B cells, plasmablasts and some plasma cells (Figure 1). In a recently published small phase I study in relapsing MS, inebilizumab produced prolonged B cell depletion and had suggestive benefits on MRI activity.64 It is currently in late phase clinical trials for neuromyelitis optica.
Daratumumab is an anti-CD38 mAb that targets a surface antigen present on plasmablasts and some plasma cells (Figure 1). It is approved for the treatment of multiple myeloma, a plasma cell malignancy, and is a reasonable candidate for consideration as a treatment for progressive forms of MS. Other early stage trials targeting the BAFF pathway, including anti-BAFF and anti-BAFF-R (VAY736) are in progress. Small molecules targeting B cell signaling, including ibrutinib (Bruton’s tyrosine kinase inhibitor) and idealisib (PI3 kinase inhibitor) are additional options for targeting B cells involved in MS, and idealisib may possibly have the additional advantage, not present with monoclonal antibodies, of effective penetration across the blood brain barrier.
ANTI-CD20 THERAPY
Overview
Often referred to as B-cell depletion therapy, anti-CD20 mAbs rapidly and profoundly deplete circulating CD20+ B cells; following intravenous administration, these typically remain depleted in PBLs for 6 to 9 months.65 Spared from anti-CD20 lysis are stem cells (pro-B cells), many plasmablasts, and terminally differentiated antibody-producing plasma cells – B cells that do not express CD20. Plasma cells may be indirectly affected, as plasma cells in the circulation (a tiny fraction of total PBL lymphocytes) have been reported to decline65 during treatment. In non-blood patient tissues, the extent and duration of depletion is not fully known but is likely to be partial, to depend on the specific anti-CD20 mAb and dose, and to be modulated by individual factors such as genetic background. In general, preclinical studies indicate that depletion in various lymphoid tissues and bone marrow is less complete than in PBL. In primates, ocrelizumab administered at concentrations similar to those administered to patients (10mg/kilogram), is capable of at least transiently and partially depleting B cells from lymphoid tissues. At 5- and 10-fold higher concentrations than human dosing, more severe depletion was observed, but full reconstitution still occurred by 43 weeks.66–68 In a study of rituximab in humans, lymph nodes sampled four weeks post-treatment revealed a relative decrease in naïve B cells and an increase in the percentage of switched memory B cells; transitional (CD24hiCD38hi) cells became nearly absent.69 These lymph node B cells post-rituximab were less able to induce pro-inflammatory IL17 expression from T cells.69 It is not fully known whether and to what extent B cells are effectively depleted from the CNS. There are reports in MS patients that CD20+ B cells can continue to be detected in the brain parenchyma and perivascular cuffs in some MS patients after treatment with rituximab,70 therefore it is likely that CD20-expressing B cells in CNS tissue sites are at least somewhat protected from treatment effects.
There are important differences in various anti-CD20 mAb molecules based on their structure, predominant mechanism of action (MOA) of B cell killing, and on/off rate of binding to cell-surface CD20. Three mAbs, rituximab, ocrelizumab, and ofatumumab, are currently in clinical use for MS. Rituximab, a chimeric mouse-human monoclonal antibody, was approved in 1997 for B cell lymphoma and indeed was one of the first mAbs ever developed for clinical use.71 Rituximab was approved for rheumatoid arthritis (RA) in 2006, and has now been used in more than 184,000 patients with RA,72 as well as off-label use in IgG4 disease,73 pemphigus,74 ANCA vasculitis,75 neuromyelitis optica,76 and myasthenia gravis,77 in addition to widespread use in MS. Rituximab works primarily through complement-dependent cytotoxicity (CDC) of B cells, but also has significant antibody-dependent cellular cytotoxicity (ADCC) activity.
Ocrelizumab, now approved for relapsing and primary progressive forms of MS, differs from rituximab in that it has a humanized antibody backbone. Ocrelizumab exhibits greater ADCC compared to CDC than rituximab,78 and also depletes B cells through multiple mechanisms, including apoptosis and antibody-dependent cellular phagocytosis.
Ofatumumab, a fully human monoclonal antibody approved for refractory chronic lymphocytic leukemia,79 has greater CDC than ADCC activity80–82 and is the only anti-CD20 mAb now being tested using a subcutaneous, rather than intravenous, dosing regimen.
Other anti-CD20 mAbs include obinutuzumab, a humanized IgG1 targeting partly the same epitope of CD20 as rituximab, but designed to induce greater cell death due to its on/off binding rates,83 and ublituximab, an anti-CD20 antibody glycoengineered for higher affinity to all FcγRIIIa receptors, yielding greater ADCC than rituximab and ofatumumab,84 especially in cells with low CD20 expression.
Pharmacokinetics & Pharmacodynamics
In MS trials of ocrelizumab, B cell levels returned to either lower limit of normal or baseline at a median 72 (27–175) weeks, though the efficacy against disease lasted far longer than the duration of B cell depletion. In one small experience, approximately 10% of patients were not fully B cell repleted even 2.5 years after their last infusion. It should also be noted that no pharmacokinetic studies have been performed in patients >65 years old, an important consideration for an aging MS demographic. Similarly, while patients with mild renal and hepatic impairment in the trials did not show any change in pharmacokinetics, those with significant hepatic or renal impairment were not studied.
Efficacy of Anti-CD20 Therapies in MS (Table 1)
Table 1.
Pivotal MS Trials with Ocrelizumab
| Study | Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis1 | Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis2 |
|---|---|---|
| Design | Phase 3, randomized 2:1 OCR vs placebo | Tandem (2) phase 3 trials, randomized 1:1 OCR vs IFNβ-1a |
| Number of Patients | 732 (488 OCR; 244 placebo) | 821 (410 OCR; 411 IFNβ-1a) + 835 (417 OCR; 418 IFNβ-1a) |
| Key Inclusion Criteria | Age: 18–55; EDSS: 3.0–6.5; Motor FSS: 2+; Disease duration: <10yrs if EDSS 3–5, <15yrs if EDSS 5–6.5; Elevated IgG index or 1+ OCB; No prior B cell depletion or immune suppression | Age: 18–55; RMS; EDSS: 1–5.5 (unless EDSS <2 after 10yrs); No prior anti-CD20, ALEM; No recent IFN, GA, IVIg, PLEX; 2 or more relapses/2yrs or 1 relapse in past yr |
| Dose | All doses: OCR 600mg q24wks; each given as paired 300mg IV infusions 2wks apart; 100mg methylprednisolone IV before each infusion. | OCR 600mg q24wks (initial course given as paired 300mg IV infusions 2wks apart; 100mg methylprednisolone IV before each infusion) vs IFNβ-1a 44μg tiw |
| Duration | Event-driven; approx.120wks (2.3yrs) | 96wks (1.8yrs) |
| Primary Outcome | % pts with CDP-12 (24% reduction favoring OCR) | ARR (46% and 47% reductions favoring OCR) |
| Significant Secondary Outcomes | Clinical: CDP-24, T25W; MRI: T2 lesion volume, whole brain volume loss | Clinical: CDP-12, CDP-24, CDI-12, MSFC (pooled analyses); MRI: Gd+ lesions, new-enlarging lesions |
| Adverse Events | Infusion reactions; Minor URIs; Oral herpes infections (nonserious); Imbalance in breast cancer, possibly other neoplasms, of uncertain significance | Infusion reactions |
| Comments | First effective disease-modifying therapy for PPMS. FDA approval for dosing identical to RMS (single 600mg dose q24wks after initial paired 300mg infusions). | Highly effective against relapses and new MRI lesions; significant benefits against disability. |
Montalban, X, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med. 2017 Jan 19;376(3):209–220
Hauser, SL, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 2017 Jan 19;376(3):221–234 ALEM = alemtuzumab; ARR = annualized relapse rate; CDI-12 = 12-week confirmed disability improvement; CDP-12 = 12-week confirmed disability progression; CDP-24 = 24-week confirmed disability progression; EDSS = expanded disability status scale; FDA = Food & Drug Administration; FSS = functional systems score; GA = glatiramer acetate; Gd+ = gadolinium-enhancing; IFN = interferon; IFNβ-1a = interferon beta-1a; IgG = immunoglobulin G; IV = intravenous; IVIg = intravenous immunoglobulin; mg = milligram; MRI = magnetic resonance imaging; MSFC = multiple sclerosis functional composite; OCB = oligoclonal band; OCR = ocrelizumab; PLEX = plasma exchange; PPMS = primary progressive multiple sclerosis; pts = patients; RMS = relapsing multiple sclerosis; T25W = timed 25-foot walk; tiw = three times weekly; URI = upper respiratory tract infection; wks = weeks; yrs = years; μg = microgram
In relapsing MS, preliminary clinical trials of the anti-CD20 therapies rituximab, ocrelizumab, and ofatumumab have shown profound reductions in new brain MRI lesion formation, clinical relapse rates, prolonged duration of efficacy with no evidence of rebound activity after drug cessation, and a favorable safety profile. A PPMS trial of rituximab failed to reach its primary endpoint of 12 week confirmed disability progression (CDP-12), but there was a positive trend for this primary outcome measure, and pre-planned exploratory analyses suggested that younger patients and those with active (e.g. gadolinium enhancing) MRI scans at baseline might have responded favorably.85 Based on these studies, off-label use of rituximab for relapsing and primary progressive forms of MS has been used in some settings, and subsequent published observational data have, arguably, strengthened the evidence for its effectiveness and safety in RMS.104
Ocrelizumab: The Pivotal Trials
Relapsing MS
In two parallel Phase III studies3 of relapsing MS patients, ocrelizumab-treated patients receiving every 6-month infusions over a 2 year period had an annualized relapse rate (ARR), the primary endpoint, of 0.16 compared to 0.29 in the high dose interferon β‒1α (Rebif) comparator arms. Ocrelizumab produced stunning reductions in the MRI endpoint of gadolinium enhancement and new lesion formation approaching 99% compared with baseline levels, indicating nearly complete elimination of new focal lesion formation in brain white matter tissue. In addition, a range of secondary and exploratory outcomes favored ocrelizumab, including clinical disability (CDP-12 and 24 weeks [CDP-24]) revealing a risk reduction of 40% compared to interferon beta; confirmed disability improvement; and MRI endpoints of fewer new and/or enlarging lesions as well as reduced whole brain atrophy.
Primary Progressive MS
In a single pivotal study,2 ocrelizumab was compared to placebo infusions (2:1 randomization) over a 2.5 year period, an acceptable design as there are no approved therapies for PPMS. The primary endpoint, CDP-12, statistically favored ocrelizumab with a risk reduction of 24%, and multiple secondary clinical and MRI endpoints, including CDP-24, timed 25-foot walk, change in total T2 white matter lesion volume and reduction in whole brain atrophy, also showed benefits favoring treatment, as did the exploratory endpoint of new and enlarging T2 lesions. Furthermore, the clinical benefit of ocrelizumab was sustained and durable in the open label extension phase of the study.86
Although the ocrelizumab study was not powered to assess efficacy in subgroups of PPMS patients, in analyses of individuals with and without gadolinium-enhancing lesions at baseline,87 trends favoring ocrelizumab were present in both groups for multiple clinical and MRI outcomes, including CDP-12, CDP-24, 25 foot walk, T2 lesion volume and whole brain volume.2 In another subgroup analysis that looked at effects by gender, the primary endpoint of CDP-12 showed no difference in females receiving ocrelizumab compared with placebo (approximately 36% of patients in each group reached CDP-12), whereas a clear benefit favoring ocrelizumab was present in males (30% in the ocrelizumb group and 42% in the placebo arm reached CDP-12). This finding was unexpected and remains of uncertain significance; notably, trends favoring ocrelizumab in males and females were present for a number of other clinical and MRI outcomes, including annualized relapse rate, change in total T2 volume, and new and enlarging T2 lesions.
General Comments Regarding Efficacy, Safety, and Dosing
As noted above, the various anti-CD20 mAbs are structurally different molecules, each reacting with different epitopes of CD20 and each with distinct, if overlapping, MOAs. As such, they should not be considered “biosimilars”, and it is likely that as more experience is gained, nuances (and possibly more significant differences) in short- and long-term efficacy and safety will emerge.88 Given that rituximab is a relatively inexpensive chimeric molecule whose period of patent protection has expired, it is extremely unlikely that a pivotal registration trial of this therapy for MS will ever be undertaken to rigorously determine its efficacy in MS. Some differences observed in efficacy and safety that might emerge between the different mAbs could result from relative differences in dosing; for example ocrelizumab is probably 3–5 times more potent than rituximab on a milligram (mg) to mg basis, and a 600mg infusion of ocrelizumab is likely biologically a higher dose than a 1000mg infusion of rituximab. No other data exist to assess the minimal effective dose, optimal dose frequency, or duration of anti-CD20 therapy necessary to achieve sustained clinical outcomes.
These caveats aside, published studies of the anti-CD20 therapies reported to date – all administered by infusion – have revealed important principles. All appear to be astoundingly effective against focal brain inflammation measured by MRI – with risk reductions approaching 100% – and are also highly effective against new clinical relapses and relapse-associated disability accumulation. These effects are sustained over time, and unlike some other effective therapies, such as natalizumab and fingolimod, no rebound disease activity has been observed with anti-CD20 therapies following treatment withdrawal or B cell repletion.1, 78, 89 Furthermore, the benefits may be sustained well beyond the period of B cell repletion; in the phase 2 ocrelizumab study in RMS, gadolinium-enhancing MRI lesions still remained effectively undetectable 18 months following the 4th and final infusion cycle,90 similar to the 12 month data following a single infusion cycle in the phase 2 rituximab study.
The degree to which anti-CD20 therapy benefits progressive MS patients is less clear. The PPMS trial of ocrelizumab demonstrated clear evidence of benefit at the 120-week time point, but the risk reduction for CDP-12 was only 24%, very similar to the earlier trial of rituximab that failed to reach a significance level of p<0.05 prerequisite for approval by regulatory bodies.85 The survival curves from these two studies had similar trajectories (Figure 2), and ocrelizumab may have succeeded in PPMS while rituximab did not simply because the ocrelizumab study was larger (e.g. 732 vs 488 randomized patients) and had greater power to detect the primary endpoint. The MRI data from these PPMS trials, specifically the change in total T2 lesion load and whole brain volume, might favor ocrelizumab over rituximab; however, cross-trial comparisons of this type are extremely problematic and fraught with high potential for error and erroneous conclusions.
Figure 2. Cross-trial comparisons of placebo-controlled trials of rituximab and ocrelizumab for RMS1, 78 and PPMS.2, 85.
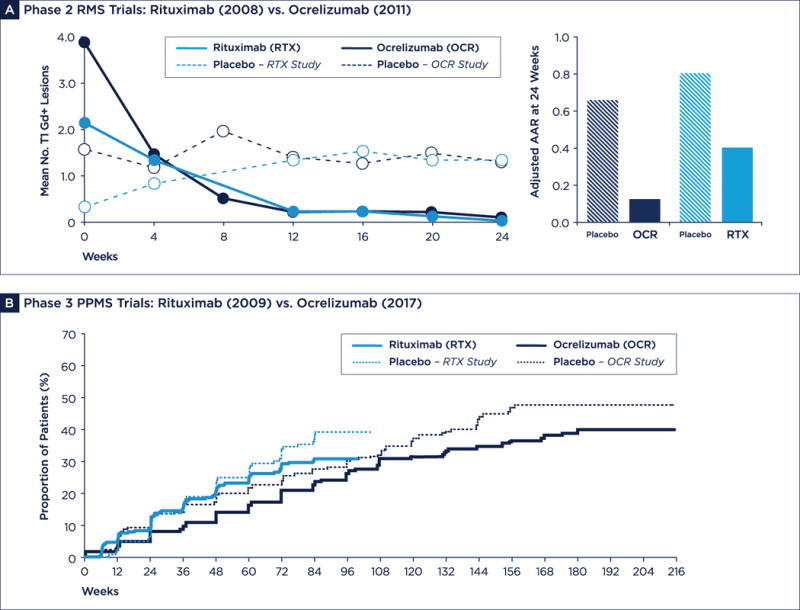
(A) Phase 2 RMS trials (600mg arm of OCR phase 2 trial). Gadolinium enhanced lesion counts at different time points are shown (left), and annualized relapse rates (ARR) (right). Data show similar levels of suppression of gadolinium disease activity, and also significant suppression of clinically observed relapse activity (arguably with a possible advantage favoring ocrelizumab)*. (B) Phase 3 PPMS trials. Disability progression confirmed at 12 weeks (CDP-12) show similar effect sizes on Kaplan-Meyer survival plots; however, the benefit of ocrelizumab was statistically significant (p = 0.03), whereas rituximab was not (p = 0.14), possibly attributed to differences in the size of the studies. *In the OCR trial, ARR was adjusted for exposure time. In the RTX trial, ARR was adjusted for exposure time, baseline EDSS, and previous exposure to glatiramer acetate or interferon. Gd+ = gadolinium positive; PPMS = primary progressive multiple sclerosis; RMS = relapsing multiple sclerosis.
What is clear from the PPMS studies is that the risk reduction against progression is relatively modest, and this could reflect suboptimal dosing of anti-CD20 mAb, an inadequacy of our measurement tools to detect progressive disease activity in a short-term study, or the possibility that treatment was begun at too advanced a stage of disease to have a more substantial effect. More likely, however, these data suggest that there is a ceiling effect for anti-CD20 therapy, and that the greater proportion of the underlying neurodegenerative process responsible for PPMS is not ameliorated by anti-CD20 therapy. This conclusion is also in line with the MRI whole brain atrophy data in the pivotal RMS and PPMS ocrelizumab trials revealing consistent but only partial reductions in atrophy progression. Similarly, the modest risk reductions in CDP and whole brain atrophy observed in the RMS trials of ocrelizumab also suggest that there is a significant underlying neurodegenerative component present, even in early RMS, that is resistant to anti-CD20 treatment. This concept was also highlighted in a case report of a patient who developed secondary progressive MS in the setting of longstanding rituximab treatment for his RMS.36
Safety Considerations
The generally favorable safety profile of anti-CD20 therapies may be due to several factors. First, although B cell depletion from the circulation is nearly complete, only 2% of the body’s total lymphocyte pool exists in peripheral blood,91 and depletion of the body’s major stores of B cells in lymphoid organs is only partial. Thus, a large reservoir of B lymphocytes likely remain, even after multiple doses. Early B cell precursors and late stage plasma cells are both CD20-negative92 and unaffected by treatment, meaning that immune reconstitution by stem cells and pre-existing humoral immunity mediated by long-lived plasma cells are both preserved.
Infusion Reactions
In the ocrelizumab trials, infusion reactions were mostly mild to moderate in severity and most often occurred with the first dose. The low incidence and severity of infusion reactions observed with ocrelizumab, compared to rituximab, may have been due to glucocorticoid pretreatment and to ocrelizumab’s mechanism of B cell lysis involving ADCC more than CDC. It is also noteworthy that fewer than 1% of ocrelizumab treated patients developed anti-drug antibodies during the 2 year treatment course, in contrast to 25% of rituximab treated MS patients treated with just a single infusion in the phase 2 trial. Premedication with glucocorticoids prior to ocrelizumab, but not rituximab, in these trials might have reduced the frequency of anti-drug antibodies, but the humanized and less immunogenic structure of ocrelizumab compared with chimeric rituximab was likely also responsible. With the anticipated need for chronic use, the advantages of a humanized (ocrelizumab) or fully human (ofatumamab) anti-CD20 molecule could become even more evident, although experience with rituximab as add-on therapy in RA indicates that only very rarely does formation of anti-drug antibodies degrade clinical efficacy.93
Serious and Opportunistic Infections Including Progressive Multifocal Leukoencephalopathy
To date, there has only been one reported case of progressive multifocal leukoencephalopathy (PML) observed in any MS patient on anti-CD20 therapy, and this single case was likely due to recent use of natalizumab therapy in a patient with positive antibody titers to John Cunningham (JC) virus. However, experience with ocrelizumab in MS (<14,000 patient years of use) is still quite limited, and with ofatumumab an even smaller experience exists. The annual incidence of PML in rituximab-treated RA patients is estimated at less than 1:25,000, and the portion of this risk that is due to rituximab is undoubtedly even lower: this is because rituximab is used in RA as add-on to a polypharmacy regimen including chronic glucocorticoids and antimetabolites, which are both known to increase PML risk, and because untreated RA is itself a PML-associated condition.94 Nonetheless, vigilance and careful reporting of all serious infections occurring in MS patients treated with anti-CD20 treatments will be important given the expected large-scale adoption of ocrelizumab in MS and the confounding effects of legacy immunosuppressives, such as natalizumab, in patients who are switching from other disease-modifying therapies.
No increase in other serious infections, including disseminated zoster and opportunistic infections, were observed in the ocrelizumab trials. Slight overall increases in upper respiratory infections and mild, cutaneous herpes infections were detected.2, 3 However, experience using anti-CD20 long term in a larger number of patients will be needed in order to identify low signal infections or those that develop over longer periods of exposure. It is noteworthy that a phase 2 trial of ocrelizumab as an add-on therapy in RA was stopped in 2011 because of a small cluster of opportunistic infections that appeared in older patients from Southeast Asia; however, these trials used doses of ocrelizumab that were higher than those approved for MS.95
Patients with chronic active hepatitis, including active hepatitis B and C, should not receive anti-CD20 therapy until infection is cleared. Similarly, live virus vaccines should be administered at least 6 weeks prior to initiating treatment.96–98 If inactivated or recombinant vaccines are given during B cell depletion, their efficacy may be reduced (influenza, Tdap, hepatitis).96–98 Ideally, vaccination status should be updated prior to beginning therapy. It is also important to screen for varicella zoster virus (VZV) IgG positivity to document immunity to VZV prior to starting anti-CD20 therapy and to consider vaccinating for VZV if patients are not immune; a new highly effective recombinant vaccine, Shingrix, is now available, eliminating the need for live virus vaccination.
Malignancy
There was a small but concerning imbalance in the number of malignancies, and breast cancer in particular, in MS patients who received ocrelizumab in the phase 3 trials. For a number of reasons, the observed imbalance may not signify a biologically meaningful difference: the total numbers of patients with breast or other cancers in the ocrelizumab-treated populations were not higher than epidemiological expectations, the incidence of cancer has fallen during the subsequent open-label extension studies,99 and diverse types breast cancers were observed that did not fall into a single pathologic category suggestive of a causative exposure. In addition, in the RA trials of ocrelizumab, no malignancy signal was present,95 and in limited experience in lymphoma there was no increase in the rate of secondary malignancy.100 Rituximab, after 20 years and > 4.8 million total infusions, has also not been associated with any increased risk of malignancy. There are no official recommendations for increased malignancy surveillance screening in patients receiving ocrelizumab; age-appropriate cancer screening guidelines should be followed, and patients should be cautioned that until further clarifying information is available, a possible increase in cancer risk should be considered before beginning treatment with ocrelizumab.
Pregnancy & Lactation
Women are advised to use contraception during and for 6 months after the last infusion of ocrelizumab.101 Currently available anti-CD20 therapies are all of the IgG1 isotype, which do not cross the placenta during the first trimester, but can cross after the 16th week of gestation and reduce B cell levels in the developing fetus. There is no evidence of maternal toxicity, infertility, teratogenicity or embryo toxicity in animal studies of ocrelizumab at doses similar to those used in humans, and a very limited experience in MS patients who conceived while taking rituximab102 and ocrelizumab was also favorable.103
Some MS patients have received anti-CD20 therapy prior to planning to conceive, either after several months post-infusion or at the time of B cell reconstitution. Here the rationale is that protection against MS relapses can last beyond B cell reconstitution, possibly throughout pregnancy and early postpartum period. In this way, the fetus is not exposed to circulating drug and the mother is protected against MS relapses. However, there is no systematic evidence to indicate that this is a safe or effective practice.
Mechanistic Insights
Ocrelizumab and rituximab have half-lives of ~400 hours (approximately the same as a typical endogenously synthesized IgG molecule), but pharamacodynamic effects of anti-CD20 therapy can last for many months to years. While long-term IgG levels generally do not change with initial courses of anti-CD20 therapy, a small decrease in IgM levels may occur.1–3, 78, 104 There has been speculation that the high apparent efficacy of anti-CD20 mAbs for IgG4-associated diseases could be explained by IgG4 secretion by short lived plasmablasts.105 When circulating B cells replete after anti-CD20 therapy, most represent a naïve or immature/transitional phenotype based on surface receptors and degree of somatic hypermutation, while potentially pathogenic memory B cells remain at reduced levels, even 2 years after treatment.65 There are fewer GM-CSF producing B cells, lower levels of IL-6, and higher levels of IL-10.53 Similarly, the T-cell compartment following anti-CD20 treatment is characterized by fewer proinflammatory Th1 and Th17 cells and larger numbers of CD251FOXP3 regulatory T cells.54,114 Myeloid cells cultured in the presence of B cell supernatants are also less likely to produce the pro-inflammatory cytokines IL-6 and IL-12, and more likely to produce IL-10 in the post-reconstitution phase.53 Together, these data suggest that a reprogramming of immunity from an activated to a resting state may occur following therapy and could account for the long-lived protection against MS attacks observed in the post-reconstitution period.
Surprisingly, in addition to B cells, approximately 6% of total circulating T cells express CD20; these cells, termed CD3+CD20dim because of their low density of CD20 expression, appear to be phenotypically and functionally heterogeneous and are slightly more often CD8 positive.106, 107 Transient drops in CD4 and CD8 counts occur for 3–6 months following anti-CD20 treatment,108 possibly relating to removal of CD20-positive T cells.
Intrathecal concentrations of systemically administered monoclonal antibodies, including anti-CD20 antibodies, are only approximately 0.1% of plasma levels.109 However, CSF B cells decrease dramatically with CD20 depletion therapy, and remarkably levels of CSF T cells also are reduced.37 The effectiveness of anti-CD20 therapy in reducing levels of cellular inflammation in the CSF is consistent with a disease model postulating that clonally restricted B cells migrating from the circulation into the CNS are the key triggers for RMS disease activity, and that they orchestrate an immune response mediated by both B and T cells.32, 33, 35
Possible Reasons for Limitations of Anti-CD20 Therapy
As summarized above, CNS B cells of MS patients are clonally restricted and appear to traffic from the cervical and other lymph nodes.33 Lymph node B cells are not fully depleted by anti-CD20 therapy, and this could provide an ongoing source of disease activity. CNS lymphatics could possibly provide a route for B cell trafficking that bypasses the peripheral circulation, allowing for continued B cell maturation in the lymph nodes.110 Perhaps more important, the failure of anti-CD20 therapy to deplete some B cells in the CNS parenchyma or meninges, or plasma cells anywhere in the body,70, 111 might be a source of underlying progressive disease activity even when relapses and focal plaque accumulation cease. These theoretical limitations of anti-CD20 therapy may also explain its relative safety, including the paucity of observed complications often associated with immunosuppressive states. Although therapies that target humoral immune system cells more broadly than anti-CD20 could possibly offer a higher level of efficacy, a less favorable side effect profile is expected with use of more potent immunosuppressive therapies directed against B cells and/or plasma cells for MS, including inhibitors of Bruton’s tyrosine kinase, inhibitors of proteasomes, and mAbs against plasma cell antigens.
Switching From Other Disease Modifying Therapies
There is short-term observational safety data on switching from natalizumab to rituximab. In one such study derived from registry data from three hospitals in Sweden, no significant safety signals were identified, and over a mean period of 21 months 1.8% of rituximabtreated patients experienced a relapse whereas 17.6% switched to fingolimod relapsed.115 In order to avoid combined immunosuppression, caution should be exercised when switching from medications such as fingolimod, dimethyl fumarate or other cytotoxic therapies that can produce lymphopenia, which can, particularly with dimethyl fumarate, be prolonged for many months. We generally recommend institution of anti-CD20 therapy when lymphopenia has resolved.
FUTURE QUESTIONS
The promise of ocrelizumab, the first FDA approved anti-CD20 therapy for RMS, is grounded in its marked and sustained high level of efficacy and favorable safety profile for RMS compared to other highly effective MS therapies. It is also the first FDA approved medication for PPMS. Anti-CD20 therapy nearly eliminates relapses and new MRI activity and has a modest albeit significant effect in PPMS. In a broader sense, although it is clear that B cell based therapies represent a significant conceptual advance in treating all forms of MS and in understanding the biology of this complex disease, many questions for future study remain.
If anti-CD20 therapy is initiated at disease onset, can secondary progression be prevented? The long-term course of MS has become less severe in the treatment era112 and it is possible that more effective therapies might have an even more pronounced beneficial effect. Many clinicians currently employ a “treat-to-target” therapeutic paradigm for patients with MS, with gradual escalation to more highly effective therapies prompted by on-treatment disease breakthroughs. Ocrelizumab’s high level of efficacy and reasonable safety record permits its use as a first-line agent, which should enable an understanding of how RMS behaves over the long-term when focal inflammation is shut down at the earliest possible time in the disease course.
What role do plasma cells and other CNS-resident B cells that escape anti-CD20 therapy have in MS, particularly with respect to clinical progression and measures of accelerated neurodegeneration across the disease spectrum? Do CSF OCBs correlate with treatment-resistant meningeal B cell and plasma cell aggregates, and if so can OCB be used as a surrogate marker for persistence of humoral autoimmunity in the CNS?
What is the optimal dosing frequency of anti-CD20 therapy, and can stop criteria for treatment be identified? Given the prolonged clinical effects observed, would a time-limited induction therapy – perhaps for 2 or 3 years – followed by active surveillance for clinical and MRI disease activity, and possibly also biomarkers such as returning memory B cells in PBL,106 permit the tailoring of anti-CD20 retreatment on an “as needed” basis? How would an episodic treatment regimen compare with continuous treatment in terms of preventing long-term disability?
Can more selective B cell therapies be developed against only the ‘bad actors’ as opposed to all CD20+ cells? For example, could new emigrants from the lymph nodes be depleted? Might it be possible to deplete subpopulations of B cells such as CD27+ plasmablasts or even particular B cell clones responsible for OCB production, while preserving protective B cells, such as B-regs, that are depleted by anti-CD20 therapy but may contribute to neural repair?113 Advances in knowledge of the specific B cell directed autoimmune mechanisms at play in MS, coupled with rapid progress in the biology of drug development and in CNS targeting, make these possibilities realistic ones for design of the next generation of B cell therapies.
Supplementary Material
Acknowledgments
A.L.G. is supported by the National Multiple Sclerosis Society through the Kathleen C. Moore Postdoctoral Fellowship. S.L.H. is supported by NIH grants R01NS092835, R01NS026799, and R01NS049477, and support from the Conrad N. Hilton Foundation. The authors also wish to thank Andrew Barnecut for outstanding editorial assistance and preparation of the figures used in this manuscript.
S.L.H serves on the scientific advisory boards for Symbiotix, Annexon, Bionure, and Molecular Stethoscope; board of trustees for Neurona; also reports receiving travel reimbursement and writing assistance from F. Hoffmann-La Roche for CD20-related meetings and presentations.
Footnotes
Author Contributions:
A.L.G. and S.L.H. both contributed to conception and design of the study; acquisition and analysis of data; drafting a significant portion of the manuscript.
Potential Conflicts of Interest:
No other disclosures were reported.
References
- 1.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. The New England journal of medicine. 2008 Feb 14;358(7):676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 2.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. The New England journal of medicine. 2017 Jan 19;376(3):209–20. doi: 10.1056/NEJMoa1606468. [DOI] [PubMed] [Google Scholar]
- 3.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. The New England journal of medicine. 2017 Jan 19;376(3):221–34. doi: 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- 4.Hauser SL. The Charcot Lecture | beating MS: a story of B cells, with twists and turns. Multiple sclerosis (Houndmills, Basingstoke, England) 2015 Jan;21(1):8–21. doi: 10.1177/1352458514561911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nature clinical practice Neurology. 2008 Oct;4(10):557–67. doi: 10.1038/ncpneuro0901. [DOI] [PubMed] [Google Scholar]
- 6.Korn T, Kallies A. T cell responses in the central nervous system. Nature reviews Immunology. 2017 Mar;17(3):179–94. doi: 10.1038/nri.2016.144. [DOI] [PubMed] [Google Scholar]
- 7.Hirota K, Duarte JH, Veldhoen M, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nature immunology. 2011 Mar;12(3):255–63. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivers TM, Schwentker FF. ENCEPHALOMYELITIS ACCOMPANIED BY MYELIN DESTRUCTION EXPERIMENTALLY PRODUCED IN MONKEYS. The Journal of experimental medicine. 1935 Apr 30;61(5):689–702. doi: 10.1084/jem.61.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Godec J, Ben-Aissa K, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity. 2014 Mar 20;40(3):355–66. doi: 10.1016/j.immuni.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivers TM, Sprunt DH, Berry GP. OBSERVATIONS ON ATTEMPTS TO PRODUCE ACUTE DISSEMINATED ENCEPHALOMYELITIS IN MONKEYS. The Journal of experimental medicine. 1933 Jun 30;58(1):39–53. doi: 10.1084/jem.58.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein LG, Prineas JW, Raine CS. Attachment of myelin to coated pits on macrophages in experimental allergic encephalomyelitis. Journal of the neurological sciences. 1983 Oct-Nov;61(3):341–8. doi: 10.1016/0022-510x(83)90167-3. [DOI] [PubMed] [Google Scholar]
- 12.Lucchinetti CF, Popescu BF, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. The New England journal of medicine. 2011 Dec 8;365(23):2188–97. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain : a journal of neurology. 2007 Apr;130(Pt 4):1089–104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 14.Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. Journal of neuroimmunology. 2011 Jan;230(1-2):1–9. doi: 10.1016/j.jneuroim.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.t Hart BA, van Meurs M, Brok HP, et al. A new primate model for multiple sclerosis in the common marmoset. Immunology today. 2000 Jun;21(6):290–7. doi: 10.1016/s0167-5699(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 16.Weissert R, Wallstrom E, Storch MK, et al. MHC haplotype-dependent regulation of MOG-induced EAE in rats. The Journal of clinical investigation. 1998 Sep 15;102(6):1265–73. doi: 10.1172/JCI3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao NA, Tso MO, Zimmerman EL. Experimental allergic optic neuritis in guinea pigs: preliminary report. Investigative ophthalmology & visual science. 1977 Apr;16(4):338–42. [PubMed] [Google Scholar]
- 18.Genain CP, Hauser SL. Allergic Encephalomyelitis in Common Marmosets: Pathogenesis of a Multiple Sclerosis-like Lesion. Methods (San Diego, Calif) 1996 Dec;10(3):420–34. doi: 10.1006/meth.1996.0120. [DOI] [PubMed] [Google Scholar]
- 19.Yu X, Burgoon M, Green M, et al. Intrathecally synthesized IgG in multiple sclerosis cerebrospinal fluid recognizes identical epitopes over time. Journal of neuroimmunology. 2011 Dec 15;240–241:129–36. doi: 10.1016/j.jneuroim.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh MJ, Tourtellotte WW. Temporal invariance and clonal uniformity of brain and cerebrospinal IgG, IgA, and IgM in multiple sclerosis. The Journal of experimental medicine. 1986 Jan 1;163(1):41–53. doi: 10.1084/jem.163.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owens GP, Bennett JL, Lassmann H, et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Annals of neurology. 2009 Jun;65(6):639–49. doi: 10.1002/ana.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Budingen HC, Harrer MD, Kuenzle S, Meier M, Goebels N. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. European journal of immunology. 2008 Jul;38(7):2014–23. doi: 10.1002/eji.200737784. [DOI] [PubMed] [Google Scholar]
- 23.Brandle SM, Obermeier B, Senel M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proceedings of the National Academy of Sciences of the United States of America. 2016 Jul 12;113(28):7864–9. doi: 10.1073/pnas.1522730113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popescu BF, Pirko I, Lucchinetti CF. Pathology of multiple sclerosis: where do we stand? Continuum (Minneapolis, Minn) 2013 Aug;19(4 Multiple Sclerosis):901–21. doi: 10.1212/01.CON.0000433291.23091.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Annals of neurology. 2004 Apr;55(4):458–68. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- 26.Larman HB, Zhao Z, Laserson U, et al. Autoantigen discovery with a synthetic human peptidome. Nature biotechnology. 2011 May 22;29(6):535–41. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larman HB, Laserson U, Querol L, et al. PhIP-Seq characterization of autoantibodies from patients with multiple sclerosis, type 1 diabetes and rheumatoid arthritis. Journal of autoimmunity. 2013 Jun;43:1–9. doi: 10.1016/j.jaut.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cepok S, Rosche B, Grummel V, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain : a journal of neurology. 2005 Mar 30;128:1667–76. doi: 10.1093/brain/awh486. [DOI] [PubMed] [Google Scholar]
- 29.von Budingen HC, Gulati M, Kuenzle S, Fischer K, Rupprecht TA, Goebels N. Clonally expanded plasma cells in the cerebrospinal fluid of patients with central nervous system autoimmune demyelination produce “oligoclonal bands”. Journal of neuroimmunology. 2010 Jan 25;218(1-2):134–9. doi: 10.1016/j.jneuroim.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Obermeier B, Lovato L, Mentele R, et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. Journal of neuroimmunology. 2011 Apr;233(1-2):245–8. doi: 10.1016/j.jneuroim.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eggers EL, Michel BA, Wu H, et al. Clonal relationships of CSF B cells in treatment-naive multiple sclerosis patients. J Clin Invest Insight. 2017;2(22) doi: 10.1172/jci.insight.92724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Science translational medicine. 2014 Aug 6;6(248):248ra106. doi: 10.1126/scitranslmed.3008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stern JN, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Science translational medicine. 2014 Aug 06;6(248):248ra107. doi: 10.1126/scitranslmed.3008879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenfield A, Eggers E, Wu H, et al. Clonal B cell persistence in multiple sclerosis: A longitudinal immune repertoire study (P1.383) Neurology. 2017 Apr 18;88(16 Supplement) 2017. [Google Scholar]
- 35.Bankoti J, Apeltsin L, Hauser SL, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Annals of neurology. 2014 Feb;75(2):266–76. doi: 10.1002/ana.24088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Budingen HC, Bischof A, Eggers EL, et al. Onset of secondary progressive MS after long-term rituximab therapy - a case report. Annals of clinical and translational neurology. 2017 Jan;4(1):46–52. doi: 10.1002/acn3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. Journal of neuroimmunology. 2006 Nov;180(1-2):63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trotter JL, Garvey WF. Prolonged effects of large-dose methylprednisolone infusion in multiple sclerosis. Neurology. 1980 Jul;30(7 Pt 1):702–8. doi: 10.1212/wnl.30.7.702. [DOI] [PubMed] [Google Scholar]
- 39.Confavreux C, Chapuis-Cellier C, Arnaud P, Robert O, Aimard G, Devic M. Oligoclonal “fingerprint” of CSF IgG in multiple sclerosis patients is not modified following intrathecal administration of natural beta-interferon. Journal of neurology, neurosurgery, and psychiatry. 1986 Nov;49(11):1308–12. doi: 10.1136/jnnp.49.11.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tourtellotte WW, Baumhefner RW, Potvin AR, et al. Multiple sclerosis de novo CNS IgG synthesis: effect of ACTH and corticosteroids. Neurology. 1980 Nov;30(11):1155–62. doi: 10.1212/wnl.30.11.1155. [DOI] [PubMed] [Google Scholar]
- 41.Rudick RA, Cookfair DL, Simonian NA, et al. Cerebrospinal fluid abnormalities in a phase III trial of Avonex (IFNbeta-1a) for relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group. Journal of neuroimmunology. 1999 Jan 01;93(1-2):8–14. doi: 10.1016/s0165-5728(98)00174-x. [DOI] [PubMed] [Google Scholar]
- 42.Petereit HF, Moeller-Hartmann W, Reske D, Rubbert A. Rituximab in a patient with multiple sclerosis–effect on B cells, plasma cells and intrathecal IgG synthesis. Acta neurologica Scandinavica. 2008 Jun;117(6):399–403. doi: 10.1111/j.1600-0404.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 43.Axelsson M, Mattsson N, Malmestrom C, Zetterberg H, Lycke J. The influence of disease duration, clinical course, and immunosuppressive therapy on the synthesis of intrathecal oligoclonal IgG bands in multiple sclerosis. Journal of neuroimmunology. 2013 Nov 15;264(1-2):100–5. doi: 10.1016/j.jneuroim.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Kowarik MC, Pellkofer HL, Cepok S, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology. 2011 Apr 05;76(14):1214–21. doi: 10.1212/WNL.0b013e3182143564. [DOI] [PubMed] [Google Scholar]
- 45.Piccio L, Naismith RT, Trinkaus K, et al. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Archives of neurology. 2010 Jun;67(6):707–14. doi: 10.1001/archneurol.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez-Pinto D. B cells as antigen presenting cells. Cellular immunology. 2005 Dec;238(2):67–75. doi: 10.1016/j.cellimm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nature reviews Immunology. 2009 Jan;9(1):15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- 48.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. The Journal of experimental medicine. 2013 Dec 16;210(13):2921–37. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathias A, Perriard G, Canales M, et al. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Multiple sclerosis (Houndmills, Basingstoke, England) 2017 May;23(6):802–9. doi: 10.1177/1352458516664210. [DOI] [PubMed] [Google Scholar]
- 50.IMSGC. Patsopoulos N. 200 loci complete the genetic puzzle of multiple sclerosis. American Society of Human Genetics 2016 Annual Meeting; Vancouver, BC, Canada. 2016. [Google Scholar]
- 51.Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Current opinion in immunology. 2008 Jun;20(3):332–8. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen P, Roch T, Lampropoulou V, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014 Mar 20;507(7492):366–70. doi: 10.1038/nature12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li R, Rezk A, Miyazaki Y, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Science translational medicine. 2015 Oct 21;7(310):310ra166. doi: 10.1126/scitranslmed.aab4176. [DOI] [PubMed] [Google Scholar]
- 54.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Annals of neurology. 2010 Apr;67(4):452–61. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 55.Giacomini E, Rizzo F, Etna MP, et al. Thymosin-alpha1 expands deficient IL-10-producing regulatory B cell subsets in relapsing-remitting multiple sclerosis patients. Multiple sclerosis (Houndmills, Basingstoke, England) 2017 Feb;01:1352458517695892. doi: 10.1177/1352458517695892. [DOI] [PubMed] [Google Scholar]
- 56.Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. The Journal of clinical investigation. 2013 Jun 3;123(6):2737–41. doi: 10.1172/JCI68775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabatino JJ, Zamvil SS, Hauser SL. B Cell Therapies in Multiple Sclerosis. Cold Spring Harbor Perspectives. 2017 doi: 10.1101/cshperspect.a032037. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longbrake EE, Cross AH. Effect of Multiple Sclerosis Disease-Modifying Therapies on B Cells and Humoral Immunity. JAMA neurology. 2016 Feb;73(2):219–25. doi: 10.1001/jamaneurol.2015.3977. [DOI] [PubMed] [Google Scholar]
- 59.Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. European journal of immunology. 1999 Nov;29(11):3432–9. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 60.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nature immunology. 2002 Oct;3(10):944–50. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 61.Weber MS, Prod’homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Annals of neurology. 2010 Sep;68(3):369–83. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. The Journal of clinical investigation. 2008 Oct;118(10):3420–30. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kappos L, Hartung HP, Freedman MS, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet neurology. 2014 Apr;13(4):353–63. doi: 10.1016/S1474-4422(14)70028-6. [DOI] [PubMed] [Google Scholar]
- 64.Agius MA, Klodowska-Duda G, Maciejowski M, et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Multiple sclerosis (Houndmills, Basingstoke, England) 2017 Nov;1:1352458517740641. doi: 10.1177/1352458517740641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis and rheumatism. 2006 Aug;54(8):2377–86. doi: 10.1002/art.22019. [DOI] [PubMed] [Google Scholar]
- 66.Vugmeyster Y, Howell K, Bakshi A, Flores C, Hwang O, McKeever K. B-cell subsets in blood and lymphoid organs in Macaca fascicularis. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2004 Sep;61(1):69–75. doi: 10.1002/cyto.a.20039. [DOI] [PubMed] [Google Scholar]
- 67.Gelzleichter T. Reduction and reconstitution of B-cells in peripheral blood and lymphoid tissues in cynomolgus monkeys following administration of ocrelizumab. ECTRIMS; 2014. July 27, 2017. [Google Scholar]
- 68.Brown PC. Ocrelizumab Pharmacology Review. FDA.gov. 2016 [July 27, 2017] Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761053Orig1s000PharmR.pdf.
- 69.Kamburova EG, Koenen HJ, Borgman KJ, ten Berge IJ, Joosten I, Hilbrands LB. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013 Jun;13(6):1503–11. doi: 10.1111/ajt.12220. [DOI] [PubMed] [Google Scholar]
- 70.esfandi S, Salimian S, Corboy J, Alvarez E. Persistent B lymphocytes in multiple sclerosis plaques after rituximab treatment (P5.341) Neurology. 2017 Apr 18;88(16 Supplement) 2017. [Google Scholar]
- 71.Coiffier B. Monoclonal antibody as therapy for malignant lymphomas. Comptes rendus biologies. 2006 Apr;329(4):241–54. doi: 10.1016/j.crvi.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 72.van Vollenhoven RF, Fleischmann RM, Furst DE, Lacey S, Lehane PB. Longterm Safety of Rituximab: Final Report of the Rheumatoid Arthritis Global Clinical Trial Program over 11 Years. The Journal of rheumatology. 2015 Oct;42(10):1761–6. doi: 10.3899/jrheum.150051. [DOI] [PubMed] [Google Scholar]
- 73.Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Annals of the rheumatic diseases. 2015 Jun;74(6):1171–7. doi: 10.1136/annrheumdis-2014-206605. [DOI] [PubMed] [Google Scholar]
- 74.Ran NA, Payne AS. Rituximab therapy in pemphigus and other autoantibody-mediated diseases. F1000Research. 2017;6:83. doi: 10.12688/f1000research.9476.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. The New England journal of medicine. 2014 Nov 06;371(19):1771–80. doi: 10.1056/NEJMoa1404231. [DOI] [PubMed] [Google Scholar]
- 76.Etemadifar M, Salari M, Mirmosayyeb O, et al. Efficacy and safety of rituximab in neuromyelitis optica: Review of evidence. Journal of research in medical sciences : the official journal of Isfahan University of Medical Sciences. 2017;22:18. doi: 10.4103/1735-1995.200275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tandan R, Hehir MK, 2nd, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: A systematic review. Muscle & nerve. 2017 Aug;56(2):185–96. doi: 10.1002/mus.25597. [DOI] [PubMed] [Google Scholar]
- 78.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011 Nov 19;378(9805):1779–87. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 79.ARZERRA (ofatumumab) Label. FDA.gov. 2016 [July 12, 2017]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125326s062lbl.pdf.
- 80.Li B, Shi S, Qian W, et al. Development of novel tetravalent anti-CD20 antibodies with potent antitumor activity. Cancer research. 2008 Apr 01;68(7):2400–8. doi: 10.1158/0008-5472.CAN-07-6663. [DOI] [PubMed] [Google Scholar]
- 81.Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004 Sep 15;104(6):1793–800. doi: 10.1182/blood-2004-01-0039. [DOI] [PubMed] [Google Scholar]
- 82.Teeling JL, Mackus WJ, Wiegman LJ, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. Journal of immunology (Baltimore, Md : 1950) 2006 Jul 01;177(1):362–71. doi: 10.4049/jimmunol.177.1.362. [DOI] [PubMed] [Google Scholar]
- 83.Valgardsdottir R, Cattaneo I, Klein C, Introna M, Figliuzzi M, Golay J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood. 2017 May 11;129(19):2636–44. doi: 10.1182/blood-2016-08-735605. [DOI] [PubMed] [Google Scholar]
- 84.Sharman JP, Farber CM, Mahadevan D, et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: results of a phase 2 trial. British journal of haematology. 2017 Feb;176(3):412–20. doi: 10.1111/bjh.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Annals of neurology. 2009 Oct;66(4):460–71. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 86.Wolinsky JS. Sustained and durable reduction in confirmed disability progression in patients with primary progressive multiple sclerosis receiving ocrelizumab: findings from the phase III ORATORIO study extended control period. ECTRIMS; Oct 27, 2017. 2017. [Google Scholar]
- 87.Wolinsky JS. Ocrelizumab Efficacy in PPMS Patients in the Presence/Absence of T1 Gadolinium-Enhancing Lesions at Baseline in a Phase III, Placebo-Controlled Trial. CMSC; Jun 3, 2016. 2016. [Google Scholar]
- 88.Smith P, Huck C, Schmid C, et al. Ofatumumab Differs from Rituximab by Effectively Targeting Lymph Node B cells and Achieving Faster Post-treatment Repletion (S24.003) Neurology. 2017 Apr 18;88(16 Supplement) 2017. [Google Scholar]
- 89.Sorensen PS, Lisby S, Grove R, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. 2014 Feb 18;82(7):573–81. doi: 10.1212/WNL.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 90.Kappos LLD, Calabresi P, O’Connor P, Bar-Or A, Barkhof F, Wells C, Leppert D, Masterman D, Tinbergen J, Hauser SL. Long-term safety and efficacy of ocrelizumab in patients with relapsing-remitting multiple sclerosis: week 144 results of a phase II, randomised, multicentre trial. Multiple sclerosis (Houndmills, Basingstoke, England) 2012;18(S4):140–1. 2012. [Google Scholar]
- 91.Dock J, Hultin L, Hultin P, et al. Human immune compartment comparisons: Optimization of proliferative assays for blood and gut T lymphocytes. Journal of immunological methods. 2017 Jun;445:77–87. doi: 10.1016/j.jim.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.St Clair EW. Good and bad memories following rituximab therapy. Arthritis and rheumatism. 2010 Jan;62(1):1–5. doi: 10.1002/art.25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haraoui B, Pelletier JP, Martel-Pelletier J. Immunogenicity of biologic agents: a new concern for the practicing rheumatologist? Current rheumatology reports. 2007 Aug;9(4):265–7. doi: 10.1007/s11926-007-0042-x. [DOI] [PubMed] [Google Scholar]
- 94.Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis and rheumatism. 2009 Dec;60(12):3761–5. doi: 10.1002/art.24966. [DOI] [PubMed] [Google Scholar]
- 95.Emery P, Rigby W, Tak PP, et al. Safety with ocrelizumab in rheumatoid arthritis: results from the ocrelizumab phase III program. PloS one. 2014;9(2):e87379. doi: 10.1371/journal.pone.0087379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nazi I, Kelton JG, Larche M, et al. The effect of rituximab on vaccine responses in patients with immune thrombocytopenia. Blood. 2013 Sep 12;122(11):1946–53. doi: 10.1182/blood-2013-04-494096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Friedman MA, Winthrop KL. Vaccines and Disease-Modifying Antirheumatic Drugs: Practical Implications for the Rheumatologist. Rheumatic diseases clinics of North America. 2017 Feb;43(1):1–13. doi: 10.1016/j.rdc.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 98.Tsigrelis C, Ljungman P. Vaccinations in patients with hematological malignancies. Blood reviews. 2016 Mar;30(2):139–47. doi: 10.1016/j.blre.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 99.Kappos L, Hauser S, Montalban X, et al. Safety of Ocrelizumab in Multiple Sclerosis: Updated Analysis in Patients with Relapsing and Primary Progressive Multiple Sclerosis (P5.407) Neurology. 2017 Apr;1888(16 Supplement) doi: 10.1212/WNL.0000000000012700. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Morschhauser F, Marlton P, Vitolo U, et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Annals of oncology : official journal of the European Society for Medical Oncology. 2010 Sep;21(9):1870–6. doi: 10.1093/annonc/mdq027. [DOI] [PubMed] [Google Scholar]
- 101.OCREVUS (ocrelizumab) Label. FDA.gov. 2017 [July 18, 2017]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761053lbl.pdf.
- 102.Ostensen M. Safety issues of biologics in pregnant patients with rheumatic diseases. Annals of the New York Academy of Sciences. 2014 May;1317:32–8. doi: 10.1111/nyas.12456. [DOI] [PubMed] [Google Scholar]
- 103.Wray SB-WS, Buffels R, Masterman D, Napieralski J, Hauser SL, Chhatwal I. Pregnancy Outcomes Following Ocrelizumab Treatment in Patients with Multiple Sclerosis and Other Autoimmune Diseases. 2017 Annual Meeting of the Consortium of Multiple Sclerosis Centers; May 25, 2017; New Orleans, Louisiana, USA. 2017. [Google Scholar]
- 104.Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. 2016 Nov 15;87(20):2074–81. doi: 10.1212/WNL.0000000000003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Annals of the rheumatic diseases. 2015 Jan;74(1):190–5. doi: 10.1136/annrheumdis-2014-205233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. Journal of immunology (Baltimore, Md : 1950) 2014 Jul 15;193(2):580–6. doi: 10.4049/jimmunol.1400118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Holley JE, Bremer E, Kendall AC, et al. CD20+inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Multiple sclerosis and related disorders. 2014 Sep;3(5):650–8. doi: 10.1016/j.msard.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 108.Graves J, Vinayagasundaram U, Mowry EM, et al. Effects of rituximab on lymphocytes in multiple sclerosis and neuromyelitis optica. Multiple sclerosis and related disorders. 2014 Mar;3(2):244–52. doi: 10.1016/j.msard.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 109.Batchelor TT, Grossman SA, Mikkelsen T, Ye X, Desideri S, Lesser GJ. Rituximab monotherapy for patients with recurrent primary CNS lymphoma. Neurology. 2011 Mar 08;76(10):929–30. doi: 10.1212/WNL.0b013e31820f2d94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015 Jul 16;523(7560):337–41. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liang A, Finney-Stable A, Lin J, Sadiq S. Cerebrospinal Fluid (CSF) Cellular Analysis of Patients with Multiple Sclerosis (MS) Treated with Anti B-cell Therapy – Correlated with Treatment Response (P5.331) Neurology. 2017 Apr 18;88(16 Supplement) 2017. [Google Scholar]
- 112.Cree BA, Gourraud PA, Oksenberg JR, et al. Long-term evolution of multiple sclerosis disability in the treatment era. Annals of neurology. 2016 Oct;80(4):499–510. doi: 10.1002/ana.24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang J, Jiang Z, Fitzgerald DC, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. The Journal of clinical investigation. 2009 Dec;119(12):3678–91. doi: 10.1172/JCI37914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 114.Vallerskog T, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clinical immunology (Orlando, Fla) 122:62–74. doi: 10.1016/j.clim.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 115.Alping P, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Annals of neurology. 79:950–958. doi: 10.1002/ana.24651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.