

Abstract
Objective
Autosomal recessive mutations in TBCK cause intellectual disability of variable severity. Although the physiologic function of TBCK remains unclear, loss-of-function mutations are associated with inhibition of mTORC1 signaling. As mTORC1 signaling is known to regulate autophagy, we hypothesized that TBCK-encephalopathy patients with a neurodegenerative course have defects in autophagic-lysosomal dysfunction.
Methods
Children (n=8) of Puerto Rican (Boricua) descent affected with homozygous TBCK p.R126X mutations underwent extensive neurological phenotyping and neurophysiological studies. We quantified autophagosome content in TBCK−/− patient-derived fibroblasts by immunostaining and assayed autophagic markers by Western assay. Free sialylated oligosaccharide profiles were assayed in patient’s urine and fibroblasts.
Results
The neurologic phenotype of children with TBCK p.R126X mutations, which we call TBCK-encephaloneuronopathy (TBCKE), include congenital hypotonia, progressive motor neuronopathy, leukoencephalopathy and epilepsy. Systemic features include coarse facies, dyslipidemia, and osteoporosis. TBCK−/− fibroblasts in vitro exhibit increased numbers of LC3+ autophagosomes, and increased autophagic flux by immunoblots. Free oligosaccharide profiles in fibroblasts and urine of TBCKE patients differ from control fibroblasts and are ameliorated by treatment with the mTORC1 activator leucine.
Interpretation
TBCKE is a clinically distinguishable syndrome with progressive central and peripheral nervous system dysfunction, consistently seen in patients with the p.R126X mutation. We provide evidence that inappropriate autophagy in the absence of cellular stressors may play a role in this disorder, and that mTORC1 activation may ameliorate the autophagic-lysosomal system dysfunction. Free oligosaccharide profiles could serve as a novel biomarker for this disorder as well as a tool to evaluate potential therapeutic interventions.
INTRODUCTION
We recently reported biallelic mutations in TBCK as a cause of intellectual disability (ID) and congenital hypotonia1. We now further define the neurodegenerative clinical phenotype in children of Puerto Rican (Boricua) descent sharing a homozygous founder mutation in TBCK (p.R126X) and define the neuromuscular features of this distinct phenotype, which we clinically termed TBCK-encephaloneuronopathy (TBCKE). The phenotypic spectrum reported with TBCK mutations is quite variable1–6. While some children have non-progressive, mild ID, autistic features and limited motor impairment2, others have profound ID, progressive leukoencephalopathy and brain atrophy along with extreme neuromuscular weakness, medication refractory epilepsy, and chronic respiratory failure.
Although little is known about the function of TBCK, previous studies link loss-of-function of TBCK to changes in mTOR (mechanistic target of rapamycin) signaling output7. The mTOR pathway regulates crucial cellular responses including growth, apoptosis, autophagy and energy metabolism8. mTOR pathway signaling activity is regulated by growth factors, insulin, and amino acids9. mTOR is a ser/thr kinase that interacts with select protein binding partners to form two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Augmented mTOR signaling is central to the pathogenesis of tuberous sclerosis complex and other neurodevelopmental disorders such as cortical dysplasia and megalencephaly that share symptoms of autism and epilepsy10, 11. To date, most neurologic disorders associated with aberrant mTORC1 signaling result from over-activation of the mTOR pathway10. On the other hand, TBCKE is distinctly characterized by inhibition of mTORC1 signaling1, 2. A few neurodevelopmental disorders have been linked to mTOR inhibition, including Rett syndrome12, 13, Phelan-McDermid syndrome14 and Galloway-Mowat15 syndrome. Clinically, all of these disorders are also associated with ID and epilepsy.
TBCK encodes its homonym protein, TBC1-domain containing Kinase which contains a TBC (Tre-2, Bub2, and Cdc16) domain flanked by an N-terminal kinase-like domain and a rhodanese homology domain at the C-terminus. Sequence homology suggests TBCK encodes a Rab GTPase-activating protein, although this remains unproven7. TBCK mRNA and protein appear to be expressed in most tissues of the human body and mouse brain transcriptome data suggests expression in astrocytes, neurons and oligodendrocytes.
Knockdown of TBCK expression using RNAi decreases phosphorylation of mTORC1 targets, such as 4eBP1 (Thr37/46) and p70S6K (Thr389) in HEK293 cells7. Consistently, a >70% decrease in pS6K phosphorylation was found in TBCK-encephalopathy patient’s lymphocytes2.
mTORC1 signaling is known to regulate autophagy, the process by which proteins, lipids and organelles are trafficked to lysosomes for degradation16, 17. Autophagy defects are emerging as a final common pathway in neurodegenerative disorders across the age spectrum, from lysosomal storage disorders to Parkinson’s disease17.
Since loss-of-function of TBCK leads to mTORC1 signaling inhibition and mTORC1 inhibitors are known to induce autophagy, we hypothesized that cells from patients with progressive neurodegeneration associated with TBCKE would exhibit enhanced autophagy activity. Here we report that patient-derived fibroblasts homozygous for TBCK p.R126X mutations have increased LC3b-positive autophagosomes and autophagic flux. In line with suspected autophagic-lysosomal dysfunction, we also found deficits in degradation of glycosylated proteins (as reflected on oligosaccharide profiles), thus constituting a novel disease biomarker for TBCK-encephalopathy.
SUBJECTS AND METHODS
Patients and Whole Exome Sequencing (WES)
All patients were diagnosed or referred to the pediatric neurogenetics program at the Children’s Hospital of Philadelphia. Patient 126-1 underwent research WES at the University of Washington, while patient 126-2 was diagnosed based on clinical WES. Both were part of the cohort leading to our earlier report that TBCK mutations cause congenital hypotonia and severe infantile encephalopathy1. Prior to our report, the TBCK p.R126X mutation was also identified in a family with two affected siblings (patients126-4 and 126-5) by research WES at the National Institutes of Health (NIH). All additional patients were diagnosed by clinical WES. The studies were approved by the corresponding IRB committees of the Children’s Hospital of Philadelphia, University of Washington and the NIH. Informed consent was obtained from all participants. All patients reported here are of Puerto Rican ancestry without a history of consanguinity, consistent with a founder mutation. We termed this the “Boricua” mutation, as this is the self-identification term often preferred by inhabitants of Puerto Rico and their descendants.
Characterization of Neurological Phenotype
All patients underwent diagnostic brain MRIs as part of neurological evaluations for severe encephalopathy and global developmental delays. Given clinical concerns for developmental regression, and in one patient metabolic stroke, some patients underwent muscle biopsies and evaluation for mitochondrial disorders. Due to progressive, severe neuromuscular weakness, which is not a consistent feature amongst TBCK-encephalopathy patients, we extensively characterized the neuromuscular phenotype by performing muscle ultrasound, nerve conduction studies (NCS) and electromyography (EMG) with standard techniques. Muscle ultrasound was performed during illness when possible due a history of clinically evident fasciculations in some patients which was more prominent with illness. Nerve and muscle biopsies were reviewed when available.
Skin Biopsy and Immunocytochemical Studies
Patients underwent skin biopsy (3mm punch biopsy) as part of their diagnostic workup. Primary fibroblasts were maintained in DMEM with 15% fetal bovine serum, non-essential amino acids, and glutamax. Control primary fibroblasts (n=3 lines, comparable ages) were commercially obtained (Coriell Institute) and genotyped (by TBCK targeted Sanger sequencing) to confirm normal sequence. Immunocytochemical staining was performed on fibroblasts plated onto #1.5 coverslips under basal conditions. Following 4% paraformaldehyde fixation, cells were blocked with 2% normal goat serum and .01% triton for 1 hour at room temperature, and then incubated with rabbit anti-Anti-LC3b (LC3II) (Abcam) overnight at 4°C. Secondary anti Rabbit Alexa555 was incubated for 2 hours in room temperature. Cells were imaged using a Zeiss LSM710 confocal system. Imaging analysis for quantification of LC3 puncta and co-localization analysis was performed in with the Zeiss Zen Blue analysis software. Statistical analysis was performed using Graph Pad Prism, with a p value <0.05 considered significant.
Western Assay
Fibroblasts were grown in routine culture conditions, seeded at 150,000 cells per 6-well plate and harvested 24 hours post plating. For serum-free experiments, cells were plated at the same density as above, maintained 24 hours in similar media except for removal of FBS, and subsequently treated with chloroquine 50μM (4 hours. Sigma) or L-Leucine 600 μg/mL (5 hours, Sigma). Proteins were extracted with ultra pure water and 1x Laemmli buffer (Bio-Rad #161-0747) and sonicated 25s at the power of 160 watts (in two rounds of ice).18 Membranes (PVDF) were probed with antibodies recognizing Beclin-1 (Cell Signaling technologies, 1:1000), LC3 (Cell Signaling #2775, 1:1000), GADPH-HRP (Cell Signaling Technologies #3683, 1:2000) and p62/SQSTM1 (Cell Signaling technologies #5114, 1:1000). Membranes were incubated with HRP conjugated anti-rabbit secondary antibodies and analyzed using a ChemiDoc™ MP Imaging System (Bio-Rad). Protein levels were normalized to GAPDH and densitometric analysis was performed using Image Studio (Li-Cor Biosciences).
Oligosaccharide analysis by MALDI-TOF Mass Spectrometry
TBCK−/− fibroblasts and controls were assayed to obtain oligosaccharide profiles by MALDI-TOF (matrix assisted laser desorption/ionization-time of flight) mass spectroscopy as previously described with minor modification. Free oligosaccharides from cell lysate of 400 μg total protein were separated from cellular proteins using a 10kDa size exclusion column, purified through C18 and carbograph solid phase extraction columns, and were permethylated before subject to MALDI-TOF analysis. Urine specimens were also analyzed. Fibroblasts were treated with rapamycin 100 nM or 600 μg/mL leucine for 16 hours before harvesting for oligosaccharide assays.
RESULTS
TBCK p.126X causes a distinct neurological phenotype with characteristic dysmorphic features
Clinical Features
The TBCK p.R126X genotype was identified in all 8 patients in our cohort (8 males, ages ranging 9-14 years; all of Puerto Rican descent). Although there are no females in our cohort, one female patient has been reported with the same TBCK genotype2, and one of the boys in our cohort had an older, similarly affected female sibling, who was deceased before prior to the availability of genetic testing. There is no known consanguinity, suggestive of a remote founder allele in this island population. Seven of eight boys presented with congenital hypotonia. Patient 5 was reported to have normal tone and neurological exam in the neonatal period but became profoundly hypotonic and less interactive around 6 weeks of age, coinciding with onset of infantile spasms. All patients have profound intellectual disability, with at most having few words of expressive language that were subsequently lost. All patients were found to be areflexic on neurological exam within the first year of life. All patients in our cohort had head circumferences within the normal range at birth and during childhood.
All children exhibited recognizable dysmorphic features, most remarkably showing progressive coarsening of facial features, bitemporal narrowing and macroglossia. Upper and lower limbs were grossly brachymelic and hands and feet demonstrated acromelia (not measured, by clinical impression, figure 1).
Figure 1. Clinical Features of TBCK-encephaloneuronopathy.
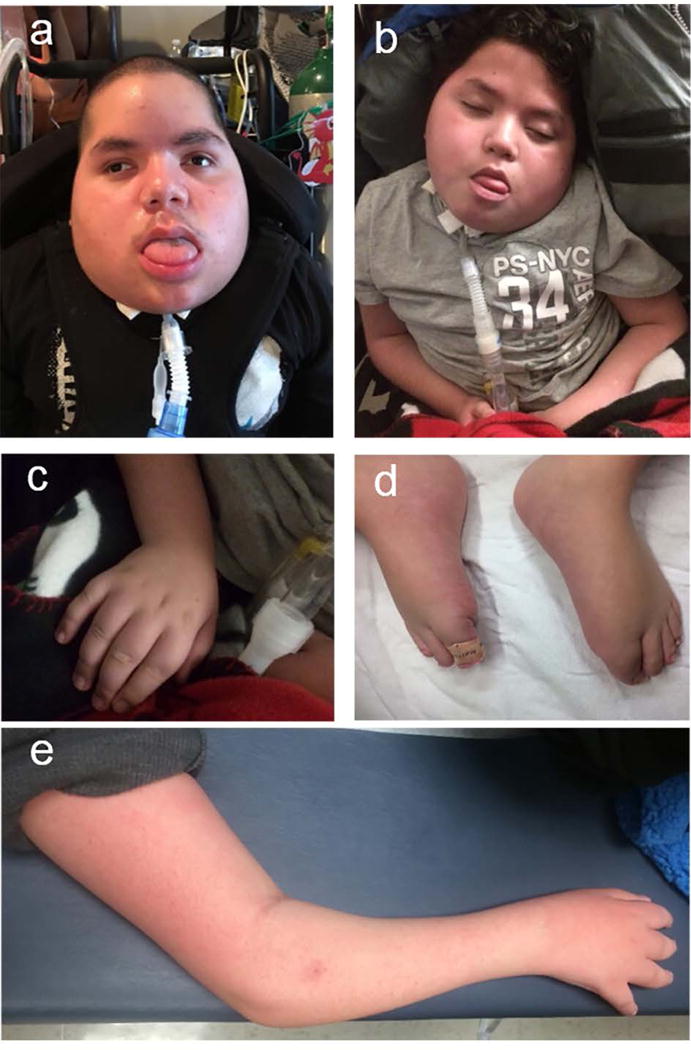
Coarse facial features in patients 126-3 (panel a, age 13 years old) and 126-4 (panel b, age 12 years old); tapered fingers (c) and distal muscle wasting (e). Upper and lower extremity limbs were grossly brachymelic and hands and feet demonstrated acromelia (c, d).
TBCK p. R126X causes progressive neuromuscular weakness and chronic respiratory failure
All patients had a clinical history of hypotonia and were areflexic since infancy, leading to initial genetic evaluations for spinal muscular atrophy (SMA) in some. Early motor milestones were significantly delayed in all, with only one of the children achieving the ability to sit unsupported. Beyond significant global delays since infancy, all children have exhibited some evidence of developmental regression by teenage years. Head control and voluntary reaching were achieved during first years of life and subsequently lost. All of them have distal > proximal muscle atrophy and profound muscle weakness. Six of the eight boys developed chronic respiratory failure, five are ventilator-dependent (with tracheostomy). The two patients without evidence of respiratory failure in our cohort are the youngest (3 and 5 years old), consistent with neuromuscular weakness progressing with age (Table 1).
Table 1.
Clinical Features of Children of Puerto Rican Descent with p.R126X homozygous “Boricua” mutation in TBCK
| Patient | 126-1 | 126-2 | 126-3 | 126-4 | 126-5 | 126-6 | 126-7 | 126-8 |
|---|---|---|---|---|---|---|---|---|
| Age, Sex | 15 yr, M | 15 yr, M | 13 yr, M | 13 yr, M | 11 yr, M | 18yr, M | 3yr, M | 5yr, M |
| MRI Findings | N/A | N/A | ||||||
| Abnormal White matter Corpus Callosum |
+ Partial ACC |
+ Thin |
+ Thin |
+ Thin |
+ Thin |
+ Thin |
||
| Global atrophy | + | + | + | + | mild | |||
| developmental regression | + | + | + | + | + | + | _ | _ |
| Seizures | ||||||||
| Epilepsy onset | 2.5 yr | 15 mo | 2yr | 2yr | 2mo | 4yr | 2yr | 1yr |
| EEG features | MFS | MFS | MFS, GPFA | MFS, GPFA | slowing | MFS | slowing | |
| Neuromuscular | ||||||||
| Congenital hypotonia | + | + | + | + | + | + | + | + |
| Progressive weakness/Muscle wasting | + | + | + | + | + | + | - | |
| Areflexia | + | + | + | + | + | + | + | + |
| Systemic/Other | ||||||||
| Chronic respiratory failure | Trach | BiPAP | Trach | Trach | Trach | Trach | no | no |
| Coarse Features | + | + | + | + | + | + | no | no |
| Hypothyroidism | - | + | + | - | - | + | N/A | - |
| Osteoporosis or Fractures | + | + | + | + | N/A | + | - | - |
| Hypothermia | + | + | + | + | + | + | - | - |
| Dyslipidemia | + | + | + | N/A | + | + | + | + |
| Short limbs | + | + | + | + | + | + | - | - |
yr= year, mo= month, M=male, ACC= agenesis of corpus callosum, MFS= multifocal sharps, GPFA=generalized paroxysmal fast activity, N/A= not available, trach=tracheostomy, BiPAP=bilelvel positive airway pressure
Patients underwent NCS, EMG and muscle ultrasound to further characterize the neuromuscular phenotype of the disease. Table 2 summarizes findings of the neurophysiological studies performed in 5 patients. Aside from patient 1 who had absent sural sensory response (likely from a technical reason) but normal median and ulnar sensory responses, all others 4 patients had preserved sensory responses. The peroneal and tibial compound muscle action potentials (CMAPs) were absent or markedly reduced in three patients, with normal distal motor latencies and conduction velocities. Patients 2 and 4 showed normal CMAPs but patient 2 had intermediate slowing in all motor nerves studied. Needle EMG showed evidence of various severity of chronic partial denervation/reinnervation in all patients and patients 3 and 5 had frequent fasciculation discharges. Thus, their electrophysiologic features are most consistent with an anterior horn cell disease or less likely a distal motor neuropathy. Available serial studies in the same patients are also consistent with progressive neuromuscular abnormalities, as electrophysiologic studies during infancy were essentially normal but later in childhood revealed a neurogenic process (Patients 1 and 2). Patients 3 and 5 showed mixed small and short and large and long polyphasic motor units with motor unit recruitment more than expected in some muscles giving the minimum contraction of muscles, suggesting possible co-existing myopathic process. Muscle ultrasound revealed severely atrophic and dense muscle with widespread evidence of denervation as evidenced by fasciculating intramuscular movements on real-time ultrasound (see supplemental video of muscle ultrasound). Fasciculations were also intermittently noted on exam in tongue and extremities, and were particularly prominent during times of illness. Muscle and nerve biopsies were obtained in infancy (when available) and were overall non-diagnostic.
Table 2.
Neurophysiological features of children TBCK p.R126X homozygous mutations
| Patient | 126-1 | 126-2 | 126-3 | 126-4 | 126-5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Age at study | 14 yr | 14 yr | 12 yr | 11 yr | 10 yr | |||||
| Amp/CV | mV | m/s | mV | m/s | mV | m/s | mV | m/s | mV | m/s |
| SENSORY | ||||||||||
| Sural | NR* | 31.7 | 38 | 9.5 | 60 | 12.5 | 43 | 7.1 | 52 | |
| Median | 43.5 | 64 | 38.1 | 66 | ||||||
| Ulnar | 23.2 | 64 | 16.7 | 66 | ||||||
| Radial | 42.1 | 49 | 30.0 | 58 | 70.8 | 88 | 49.3 | 56 | ||
| MOTOR | ||||||||||
| Peroneal | NR | 3.6 | 29 | 0.1 | 2.7 | 37 | NR | |||
| Tibial | NR | 11.5 | 29 | 0.5 | 11.0 | 37 | 0.8 | |||
| Median | 4.2 | 48 | 4.9 | 42 | 6.5 | 42 | 10.0 | 51 | 3.4 | 37 |
| Ulnar | 5.8 | 48 | 10.6 | 43 | 5.1 | 52 | 7.2 | 52 | 3.5 | 43 |
| EMG | ||||||||||
| Evidence of Denervation | Yes | Yes | Yes | Yes | Yes | |||||
| Fasc/myokymia | No | No | Yes | No | Yes | |||||
TBCK p.126X is associated with epilepsy with mixed focal and generalized features
All patients developed epilepsy with electroclinical features of focal and generalized seizures, although the age of onset and the severity of seizures were variable (see Table 1). Most children had a history of first seizure in the setting of fever, and then months to years later, onset of unprovoked seizures. One patient initially presented at 3 months with infantile spasms and hypsarrhythmia on EEG (electroencephalogram). EEG features include slow and disorganized background early on, with multifocal epileptiform discharges as well as generalized discharges in older patients. Of note, severe adverse reactions leading to prolonged hospitalizations were reported in the 2 patients that were treated with the ketogenic diet, despite normal acylcarnitine profiles.
TBCK 126X mutation leads to progressive leukoencephalopathy and global atrophy
Neuroimaging features include progressive volume atrophy, thinning of the corpus callosum and cerebellar atrophy. Interestingly, some patients who underwent brain MRI in the neonatal period were reported as unremarkable despite profound congenital hypotonia (no images available for review). All patients that underwent MRI after the first year of life had abnormal findings, including scattered abnormal T2 white matter hyperintensities consistent with a leukoencephalopathy. Patient 1 had an episode clinically characterized as a metabolic stroke, in the setting of RSV infection at age 2.5 years old, whose MRI findings we previously reported1.
Additional systemic features TBCKE patients
Hypothermia and Hypothyroidism
Some patients have a history of intermittent hypothermia, with documented temperatures as low as 33-34°C. Hypothermia occurred in the setting of illness, but not consistently, similar temperature fluctuations were also reported when patients were otherwise at baseline. Thyroid studies (TSH, thyroid stimulating hormone) were available in 7 patients. Three teenage patients were found to have subclinical hypothyroidism, with elevated TSH values ranging from 14.3 to 30.8 (normal 0.5-3.8 uIU/mL).
Dyslipidemia
We reviewed available lipid panel data given frequently reported history of hypercholesterolemia in our cohort. Table 1 lists a summary of findings, with “dyslipidemia” defined as elevated total cholesterol and/or triglycerides. Lipid profiles were available in 7/8 patients. Total cholesterol was elevated in 5/7 patients, with values ranging from 192 to >650 (mean 371, normal 107-225, mg/dL). Triglycerides were elevated in 5/7 available studies, with abnormal values exhibiting a broad range from mild elevations in young age to very significant in teenagers (>1500mg/dL) (normal 34-165 mg/dL).
Osteoporosis
Patients reported multiple fractures with minimal trauma, most commonly with femur fractures but also lumbar spine fractures in one patient after a prolonged hospitalization. Further evaluation was consistent with severely decreased bone mineralization. DEXA scans were available in two patients. Bone mineral density in non-ambulatory children with cerebral palsy have been reported, with mean lumbar Z-score of −1.719. TBCKE patients had remarkably lower scores (although only available in 2 patients), with mean lumbar Z-score of −5.7 (126- 2) and Z −5.8 (126-3). Consistent with our observations, a recent report also noted osteoporosis in affected patients with a different mutation in TBCK4.
Genitourinary
Four of the five teenage patients have been noted to have urinary retention and renal calculi. Those evaluated by urology were found to have a neurogenic bladder, requiring intermittent catheterization for management. They also experienced frequent recurrent urinary tract infections.
Fibroblasts homozygous for TBCK p.R126X have significant increase of autophagosomes
Since loss of TBCK function has been linked with mTORC1 signaling inhibition, and our previous studies showed that the p.R126X mutation results in premature protein truncation and undetectable levels of TBCK protein1, we expected to observe induction of autophagy in patient-derived fibroblasts. LC3 (microtubule-associated protein 1 light chain 3), the mammalian homologue of the autophagy-related Atg8 encoded product in yeast, is a marker for cellular autophagy. Following induction of autophagy, the soluble form LC3b-I (or LC3-I) is modified with a lipophilic moiety to form LC3b-II (or LC3-II). Membrane bound LC3b-II plays a role in forming the phagophore and promoting membrane fusion to form the autophagosome. Immunostaining for LC3b in a punctate pattern is commonly used to detect autophagosomes. Patient derived fibroblasts under basal culture conditions were probed with LC3b antibodies to detect autophagosomes. A dramatic increase in LC3b-immunolabled punctae per cell was observed (Fig. 3, wild type mean 1.4±0.54, TBCK mean 9.4±1.7, punctae/cell ± SEM (standard error of the mean)), consistent with autophagosomes accumulation in TBCK−/− fibroblasts. Since mTORC1 inhibition is known to increase autophagosome content in cells, this observation is consistent with our previous data suggesting that loss of TBCK results in inhibition of mTORC1 signaling2.
Figure 3. TBCK−/− fibroblasts show significant increase in LC3+ autophagosomes.
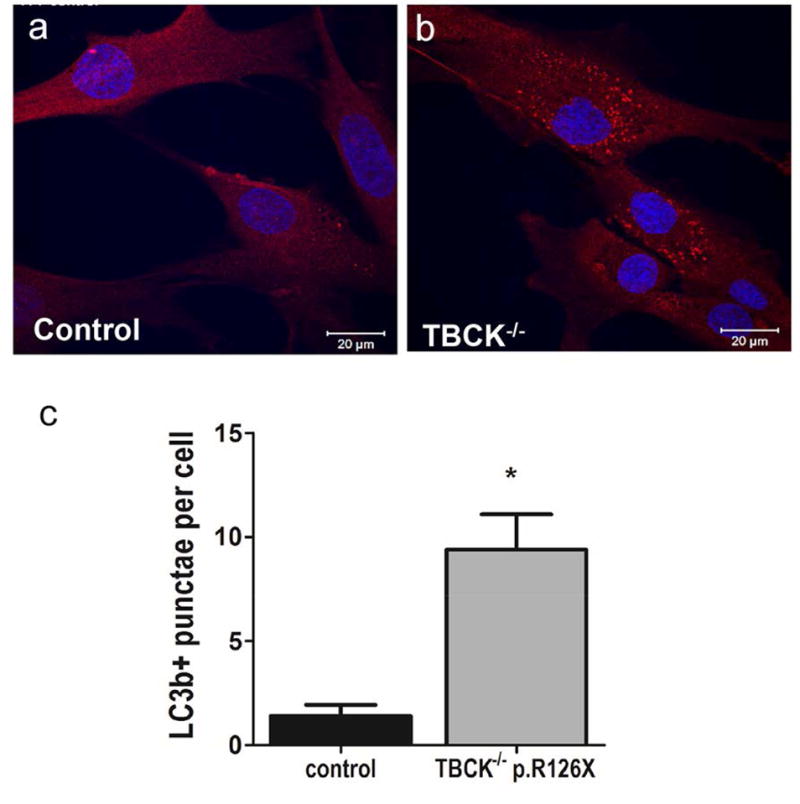
LC3b (red) immunostaining of TBCK-encephalopathy patients (b) compared to controls (a). Nuclei stained with DAPI (blue). Panel C shows automated punctae quantification using Zeiss Zen blue software (n=51 wild type and n=106 TBCK cells from 2 independent experiments); bars show SEM, Mann Whitney test p value .0044. TBCK p.R126X data pooled from 3 different patient lines (TBCK 126-1, 126-3, 126-5).
TBCK p.R126X fibroblasts exhibit increased autophagic flux at baseline but not under stress
Given the substantial increase in autophagosomes found during routine culture conditions, we assayed fibroblast for LC3-II/LC3-I ratio, which is indicative of autophagic flux20. Immunoblots (Figure 4a) showed that TBCKE patient fibroblasts have increased LC3-II/LC3-I ratio at baseline conditions compared to controls (Figure 4b). Increased autophagic flux is associated to increased p62 levels and TBCKE-fibroblasts showed a trend to increased p62, albeit not statistically significant (figure 4b). Under baseline conditions, Beclin-1 levels showed a small but statistically significant reduction (figure 4c, 4d) in TBCKE-fibroblasts relative to controls. To assess how TBCKE-fibroblasts respond to known stressors and autophagy modulators, we assayed serum-free cells, as well as serum-free cells treated with chloroquine and leucine. Under autophagy induction conditions (serum-free), there was no significant difference in autophagic flux between TBCK and wild type cells (figure 4e, 4f) Chloroquine is a lysosomal lumen alkalizer, blocking the autophagic progress by impairing lysosomal function,20 resulting in increased LC3-II/LC3-I ratio. As expected, chloroquine increased autophagic flux, although there was no difference in response between TBCK−/− and controls. Since L-leucine stimulates mTORC1 signaling, it is expected to inhibit autophagy under stress conditions. Our previous studies showed rescue of mTORC1 signaling deficits in TBCK-fibroblasts2 with similar treatment protocol, so we tested whether L-Leucine treatment could change autophagic flux. Under serum-free conditions, the 5 hour treatment with 600μg/mL did not result in significant changes in autophagic flux (Figure 4e).
Figure 4. Western Assays from TBCK−/− fibroblasts show increased autophagic flux at baseline but no difference in response to stress or autophagy modulators.
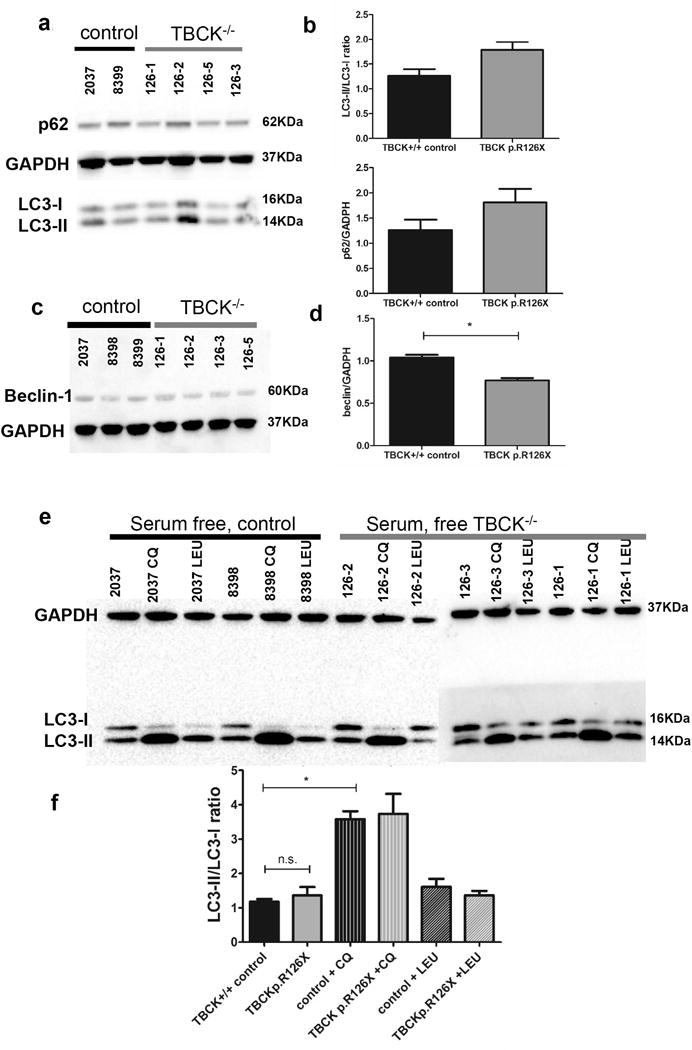
Immunoblots for TBCK p.R126X fibroblasts at baseline (complete media) conditions assaying for autophagy markers LC3-I, LC3-II and p62 (panel a) were quantified and normalized to GADPH (panel b). LC3-II/LC3-I ratios are reported as indicative of autophagic flux. Similarly, Beclin-1 levels were assayed (panel c) and quantified relative to GADPH (panel d) at baseline conditions. Quantification includes 3 independent experiments, each including at least 2 wild type and 3 TBCK−/− lines, each blot was normalized to GADPH and statistical significance was defined as two-tailed p value of <.05 using unpaired t-test. To determine if loss-of-function of TBCK affects autophagic response under stress, cells were assayed after 24 hours without serum. Cells were also treated with known autophagy modulators chloroquine (impairs lysosomal function, blocking progression of autophagy) and leucine (enhances mTORC1 signaling). Panel e shows results of immunoblots. As seen in panel f, there is no significant change in the response of controls versus TBCK p.R126X cells under stress or autophagy modulating conditions.
TBCK p.R126X leads to a deficiency in oligosaccharide degradation
Since the autophagic-lysosomal pathway is crucial for the degradation of glycosylated proteins, free sialylated oligosaccharide profiles can provide insight into this cellular process by assaying the byproducts of degradation. Previous studies have shown that cells with defects either initiating autophagy or with lysosomal dysfunction accumulate aberrant intracellular free sialylated oligosaccharide species. We therefore tested whether patient-derived fibroblasts with the homozygous TBCK p.R126X mutations exhibit aberrant oligosaccharides profiles. We found that the loss-of-function of TBCK leads to abnormal accumulation of sialylated oligosaccharide species in patient-derived fibroblasts (Figure 5c). Treatment of control cells with mTORC1 inhibitor rapamycin also resulted in accumulation of aberrant oligosaccharide species in control fibroblasts in a similar pattern (Figure 5a and b). Moreover, the aberrant oligosaccharide species in TBCK−/− fibroblasts can be partially rescued by treatment with the mTORC1 activator leucine (Figure 5d). All the sialylated oligosaccharides diminished after leucine treatment, while the elevation of GlcNAc1Man2-Man4 persists. It has been reported before that intracellular sialylated free oligosaccharides are specifically from lysosomal sialic acid transporters while GlcNAc1Man2-4 could also come from ER. Since free oligosaccharides can also be detected in urine, we tested if the oligosaccharide findings in fibroblasts of patients with p.R126X TBCK mutations were reflected in urine specimens. As shown in figure 6 we have found a similar aberrant oligosaccharide pattern in the urine of TBCKE patients. We detected a similar pattern of abnormalities in all TBCKE patients tested to date, 5 with the TBCK p.R126X mutations (ages 11-15 years old) and 3 with other genotypes (2 patients Het c.[2060-2A>G];[803_806delTGAA] p.[=];[Met268fsArg*26]; 1 patient with a homozygous deletion of exon 21). Although only one patient (126-3) had urine glycosaminoglycan (GAG) testing, this was also significantly abnormal for increased total GAG quantity as well as abnormal GAG fractionation pattern, further supporting reduced lysosomal degradation capacity.
Figure 5. Fibroblasts from TBCK p. R126X patients show aberrant oligosaccharide profiles.
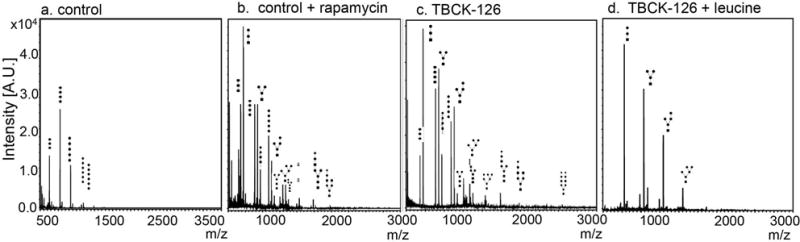
MALDI-TOF mass spectrometry profiles of control primary fibroblasts (a). Oligosaccharide species with higher molecular weight (right of profile) accumulate due to impaired autophagic-lysosomal degradation in control cells treated with mTORC1 inhibitor rapamycin (b) and in TBCK−/− R126X cells (c). Degradation defects in TBCK−/− p.R126X cells can be ameliorated by leucine (d), which activates mTORC1 signaling
Figure 6. A representative urine free oligosaccharide profile from a control and a TBCKE patient.
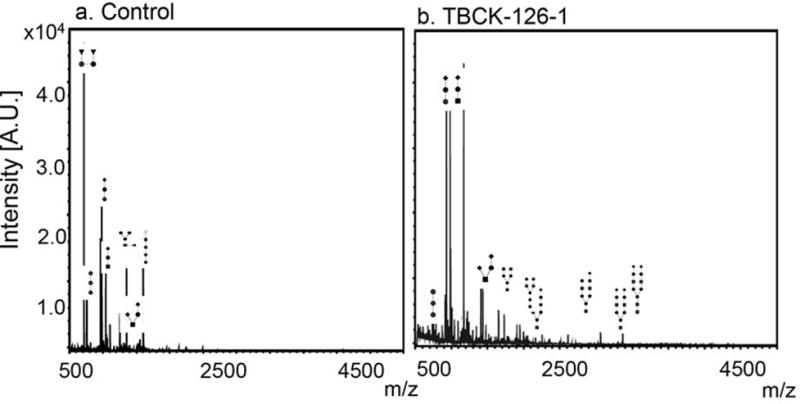
Moderate increase of sialylated complexed oligosaccharides were detected in urine from the TBCKE patient (panel b, 126-1) relative to healthy control (panel a), in a pattern consistent with a deficiency in lysosomal associated degradation of glycoproteins.
DISCUSSION
We describe the distinct neurological phenotype of a subset of patients with homozygous mutations in the TBCK gene which we called TBCK-Encephaloneuronopathy. All eight patients with the Boricua mutation (p.R126X) share a severe phenotype with age-dependent neurodegeneration, including features of motor neuronopathy, which has not been clearly or consistently characterized in previous reports of TBCK associated disease1–4. Common systemic clinical features include hypothyroidism, dyslipidemia and osteoporosis. We provide evidence that TBCK loss-of-function mutations result in inappropriate increase in autophagic flux at baseline conditions, but does not seem to impair autophagic response during stress. Therefore, we provide evidence that autophagic-lysosomal dysfunction may play a role in the pathophysiology of TBCKE.
The consistent neuromuscular phenotype seen in patients with the TBCK p.R126X genotype significantly contributes to disease morbidity, as the progressive neuromuscular weakness results in chronic respiratory insufficiency in all patients by teenage years. All patients who underwent neurophysiological studies (n=5) have evidence of motor neuronopathy although older patients might have additional myopathic features (table 2). Interestingly, especially in the setting of acute illness such as respiratory infections, patients exhibit prominent fasciculations, which were visualized in three acutely ill patients using muscle ultrasound (see supplemental video). We suspect the neuromuscular involvement in TBCK-associated disease is not exclusive to the Boricua mutation, and likely present in all children with truncating mutations leaving little functional protein activity. In patients who had serial neurophysiological studies, there is evidence that the neuromuscular abnormalities are progressive, consistent with their clinical course. Studies by EMG and muscle ultrasound are most consistent with active denervation as the result of motor neuronopathy. As we have no muscle biopsies from patients in their teenage years, when myopathic features were seen on EMG, it remains unclear whether myopathic changes are secondary to chronic denervation or the development of an additional myopathy. The latter is conceivable given the multisystemic nature of the disease.
In addition to progressive neuromuscular weakness, our TBCKE cohort clearly exhibits progressive brain degeneration and neurodevelopmental regression. This neurodegenerative aspect of the disease has not been consistently appreciated in previously reported patients with TBCK biallelic mutations. While all patients reported to date present with hypotonia and delayed milestones, some patients have a milder phenotype, achieving independent ambulation and expressive language without clinical evidence for regression2. In contrast, patients in our TBCKE cohort presented with profound hypotonia and global delays in the neonatal period, and subsequently experienced further developmental regression with loss of skills (such as purposeful reaching or attempting to crawl) later in childhood, typically before 10 years. We submit that cognitive regression is not due only to seizures, and more likely due to leukoencephalopathy, since typically at age of regression most children had not yet developed medication-refractory epilepsy. As shown in figure 2 neuroimaging shows progressive global cerebral atrophy and white matter abnormalities.
Figure 2.
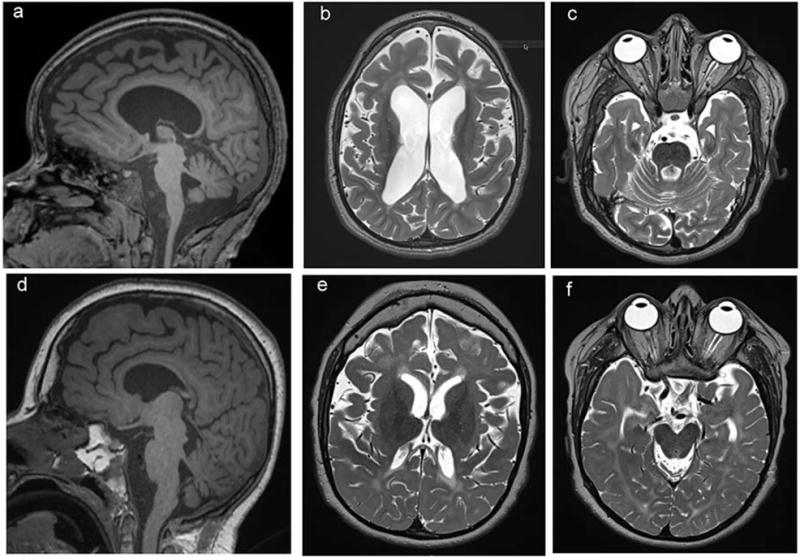
Neuroimaging features of patients with TBCK p.R126X demonstrate thinning of corpus callosum (a, d) and white matter with abnormal T2 signal hyperintensity, most evident in periventricular white matter (b, e) and prominent fourth ventricle due to cerebellar atrophy (c, f). Top panel (a-c): Neuroimaging corresponding to patient 126-3 at age 11years old, bottom panel (d-f), patient 126-2 at age 14 years old.
Consistent with an emerging genotype-phenotype correlation for TBCK-encephalopathy, we do have some evidence that patients with TBCK-encephalopathy presenting with a milder and non-progressive phenotype (siblings previously reported as subjects 4-1 and 4-22) have no clinical evidence of neuropathy/neuronopathy and normal EMG/NCS at an age when all Puerto Rican patients have profound weakness and abnormalities in neurophysiological studies.
Compromised autophagic function has emerged as a mechanistic common denominator in many neurodegenerative disorders17. mTORC1 inhibitors are well known to induce autophagy the process by which proteins, lipids and organelles are trafficked to lysosomes for degradation16, 17. Importantly, activation of mTORC1 is also required for autophagy associated lysosome reformation and loss of mTORC1 activation could lead to a reduction in lysosome content21. Our studies in TBCKE suggest that chronic mTORC1 inhibition may be detrimental, resulting in inappropriate increase in autophagic flux at baseline conditions. Consistently, we observed a significant increase in accumulation of autophagosomes in TBCK patient-derived fibroblasts (Fig. 3) in baseline conditions. Under autophagy inducing conditions, such as serum deprivation, or in response to autophagy inhibitors such as chloroquine, TBCKE-fibroblasts response did not differ from wild type. Treatment with L-leucine did not significantly alter autophagic flux in serum-free conditions, although it is possible that the short duration of treatment (5 hours) was insufficient to influence autophagic flux. Our past studies with mTORC1 signaling phosphorylation targets showed rescue of phosphorylation deficits at similar dose and exposure time, but under baseline conditions. Further studies should examine if longer exposure to L-leucine can alter autophagic flux at baseline conditions on TBCKE-fibroblasts.
However, the increase in autophagosome content does not necessarily imply more efficient degradation. On the contrary, our oligosaccharide profile data suggests that glycosylated proteins are not being properly degraded in TBCK null fibroblasts, implying a possible reduction in lysosome content or lysosomal degradation capacity. Therefore, it is possible that increased autophagosome content and autophagic flux ratios at baseline are indicative of a block in the autophagic-lysosomal cascade, reminiscent of cells treated with chloroquine. This could explain the increased autophagosome content, increased LC3II/LC3I ratios and impaired degradation of glycosylated proteins at baseline. Further studies should address lysosomal function in TBCKE cells and dissect the steps along the autophagic-lysosomal pathway to better understand the cellular effects of disease causing mutations in TBCK and their role in neurodegeneration. Crucially, future studies need to examine the physiological role of TBCK as it pertains to the regulation of mTORC1 signaling, either by interacting with proteins upstream of the mTORC1 complex or due to another yet undetermined mechanism. There are only two reports in the literature regarding TBCK prior to its disease association, and they are conflicting regarding its potential role in regulating cell proliferation7, 22. Future animal studies in TBCK knockout mice may be instrumental in determining the physiologic role of the TBCK protein and providing further insight into its role in the brain and peripheral nervous system.
Aberrant oligosaccharide species can accumulate in cells with defects along the molecular cascade spanning from initiating autophagy to lysosomal degradation23. Our free oligosaccharide profile data suggests impaired glycosylated proteins degradation as a consistent finding in patient fibroblasts as well as in urine specimens. Our data is consistent with aberrant autophagic/lysosomal activity resulting from impaired mTORC1 signaling, since abnormal oligosaccharides can be recapitulated by rapamycin treatment in wild type cells. Furthermore, the mTORC1 activator leucine partially rescues the oligosaccharide profile abnormalities in TBCK−/− fibroblasts. Although our data does not show differences in terms of autophagic flux when cells were treated with leucine, the discrepancy could be due to different times of treatment (5 hours for flux experiments versus 16 hours for oligosaccharide experiments). Future studies should examine if long term treatment with leucine alters autophagic flux, and most importantly to determine therapeutic potential, whether leucine can rescue the phenotype in neuronal models. Further studies will be needed to address if urine oligosaccharide profile abnormalities are present in all patients with TBCK-associated disease regardless of genotype. As patients with the Boricua mutation have a more severe phenotype, it would be interesting to measure profiles in milder phenotype patients to determine if the severity correlates with the degree of free oligosaccharide excretion. We propose that free oligosaccharide profiles could serve as a biomarker of autophagic-lysosomal dysfunction in this condition.
Our previous studies in TBCK-encephalopathy patient cells also showed that mTORC1 signaling activation with leucine could rescue PS6K phosphorylation deficits2. Here we show partial rescue of aberrant free oligosaccharide accumulation in fibroblasts from TBCK-encephalopathy patients treated with leucine, suggesting potential therapeutic relevance of leucine and similar mTORC1 signaling activators. Further studies are needed to determine whether inappropriate autophagy is also present in TBCK-null neurons or glia, and whether leucine can reverse their phenotype. Neuronal and animal models will be of particular relevance given recent reports that autophagy in hippocampal neurons may be independent of mTORC1 signaling.
The study of TBCK-associated encephalopathy reveals new insights into the role of over-inhibition of mTORC1 signaling in neurodevelopmental disabilities. Specifically the p.R126X mutation leads to a unique clinical phenotype and may provide additional insight into the link between autophagy and mTORC1 signaling, and links its dysregulation to autophagic-lysosomal pathway dysfunction, and progressive central and peripheral nervous system degeneration.
Supplementary Material
Acknowledgments
This work was supported by the Robert Wood Johnson Harold Amos Faculty Development Award (XOG) and the National Institute for Neurological Disorders and Stroke (5T32NS7413-14 and K12 NS049453-08, XOG), and R01-NS021328-30 (DCW); The National Institute for Mental Health (R01-MH108592-01, DCW); the National Institutes of Health Intramural Research Program Funding from the National Institute of Neurological Disorders and Stroke (CGB). Alex Petterson and James Peterson provided assistance to our genetic counselor (KK) during their training. We are grateful to Elizabeth Bhoj and Katherine Helbig assisted for referring patients to our cohort.
Footnotes
Author Contributions:
Study concept and design: XOG, CGB, MH, PC and DCW. XOG, JATH, MH and XL contributed to experimental data acquisition and analysis, while XOG, SY, DXBH, ARF, CGB, SKK and KK contributed clinical and neurophysiological data acquisition and analysis. XOG, JATH, MH, PC contributed to manuscript drafting and figure preparation.
Potentials Conflicts of Interest:
Nothing to disclose.
References
- 1.Chong JX, Caputo V, Phelps IG, et al. Recessive Inactivating Mutations in TBCK, Encoding a Rab GTPase-Activating Protein, Cause Severe Infantile Syndromic Encephalopathy. Am J Hum Genet. 2016 Apr;98(4):772–81. doi: 10.1016/j.ajhg.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhoj EJ, Li D, Harr M, et al. Mutations in TBCK, Encoding TBC1-Domain-Containing Kinase, Lead to a Recognizable Syndrome of Intellectual Disability and Hypotonia. Am J Hum Genet. 2016 Apr;98(4):782–8. doi: 10.1016/j.ajhg.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerreiro RJ, Brown R, Dian D, de Goede C, Bras J, Mole SE. Mutation of TBCK causes a rare recessive developmental disorder. Neurol Genet. 2016 Jun;2(3):e76. doi: 10.1212/NXG.0000000000000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandel H, Khayat M, Chervinsky E, Elpeleg O, Shalev S. TBCK-related intellectual disability syndrome: Case study of two patients. Am J Med Genet A. 2017 Feb;173(2):491–4. doi: 10.1002/ajmg.a.38019. [DOI] [PubMed] [Google Scholar]
- 5.Hartley T, Wagner JD, Warman-Chardon J, et al. Whole-exome sequencing is a valuable diagnostic tool for inherited peripheral neuropathies: outcomes from a cohort of 50 families. Clin Genet. 2017 Jul 14; doi: 10.1111/cge.13101. [DOI] [PubMed] [Google Scholar]
- 6.Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015 Jan 13;10(2):148–61. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Yan X, Zhou T. TBCK influences cell proliferation, cell size and mTOR signaling pathway. PLoS ONE. 2013;8(8):e71349-e. doi: 10.1371/journal.pone.0071349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012 Apr;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004 Aug 15;18(16):1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 10.Crino PB. mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb Perspect Med. 2015 Apr;5(4) doi: 10.1101/cshperspect.a022442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J Neurosci. 2015 Oct;35(41):13836–42. doi: 10.1523/JNEUROSCI.2656-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rangasamy S, Olfers S, Gerald B, Hilbert A, Svejda S, Narayanan V. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Res. 2016;5:2269. doi: 10.12688/f1000research.8156.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricciardi S, Boggio EM, Grosso S, et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011 Mar 15;20(6):1182–96. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- 14.Bidinosti M, Botta P, Kruttner S, et al. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science. 2016 Mar 11;351(6278):1199–203. doi: 10.1126/science.aad5487. [DOI] [PubMed] [Google Scholar]
- 15.Jinks RN, Puffenberger EG, Baple E, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain. 2015 Aug;138(Pt 8):2173–90. doi: 10.1093/brain/awv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993 Oct;333(1–2):169–74. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 17.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015 Jun;16(6):345–57. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 18.Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis. 2016 Jan;85:144–54. doi: 10.1016/j.nbd.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Finbraten AK, Syversen U, Skranes J, Andersen GL, Stevenson RD, Vik T. Bone mineral density and vitamin D status in ambulatory and non-ambulatory children with cerebral palsy. Osteoporos Int. 2015 Jan;26(1):141–50. doi: 10.1007/s00198-014-2840-0. [DOI] [PubMed] [Google Scholar]
- 20.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 2016 Aug 15;25(16):3432–45. doi: 10.1093/hmg/ddw185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Li Q, Li Y, et al. A long type of TBCK is a novel cytoplasmic and mitotic apparatus-associated protein likely suppressing cell proliferation. Journal of Genetics and Genomics. 2014;41(2):69–72. doi: 10.1016/j.jgg.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Xia B, Asif G, Arthur L, et al. Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin Chem. 2013 Sep;59(9):1357–68. doi: 10.1373/clinchem.2012.201053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.