

Summary
Rare neurological diseases shed light onto universal neurobiological processes. However, molecular mechanisms connecting genetic defects to their disease phenotypes are elusive. Here, we obtain mechanistic information by comparing proteomes of cells from individuals with rare disorders with proteomes from their disease-free consanguineous relatives. We use triple-SILAC mass spectrometry to quantify proteomes from human pedigrees affected by mutations in ATP7A, which cause Menkes disease, a rare neurodegenerative and neurodevelopmental disorder stemming from systemic copper depletion. We identified 214 proteins whose expression was altered in ATP7A−/y fibroblasts. Bioinformatic analysis of ATP7-Amutant proteomes identified known phenotypes and processes affected in rare genetic diseases causing copper dyshomeostasis, including altered mitochondrial function. We found connections between copper dyshomeostasis and the UCHL1/PARK5 pathway of Parkinson’s disease, which we validated with mitochondrial respiration and Drosophila genetics assays. We propose that our genealogical ‘omics’ strategy can be broadly applied to identify mechanisms linking a genomic locus to its phenotypes.
eTOC
Rare genetic diseases provide fundamental insight into important biological questions and common diseases. Zlatic et al. present a strategy, termed genealogical proteomics, to investigate the molecular manifestations and mechanism of disease by comparing within a family samples from normal and affected subjects. Using this approach, the authors study a rare copper metabolism genetic disorder and find tantalizing connections with molecules and mechanisms known in Parkinson’s disease. Thus, genealogical proteomics is a promising approach for biological discovery and precision medicine.
Introduction
The study of rare hereditary disorders provides unparalleled insight into the inner workings of normal biological processes (Garrod, 1928; McKusick, 2007). Presently the number of human genes with phenotype-causing mutations is ~3,700, most of them correspond to rare and novel disorders (https://omim.org/statistics/geneMap) (Amberger et al., 2015). It is estimated that 7,000 to 15,000 human genes could be affected by phenotype-causing mutations (Cooper et al., 2010; Kohler et al., 2017; McKusick, 2007). Nearly half of all rare diseases diagnosed are neurological diseases, frequently of pediatric onset. These rare neurological diseases offer an unprecedented resource to shed light into normal and abnormal neurodevelopmental and neurological processes in humans (Gahl et al., 2012). However, once a mutation is identified, the challenge is to uncover molecular mechanisms by which a genetic defect expresses its phenotypes. Here we address this aspect focusing on rare mutations in genes required for copper homeostasis.
One of these afflictions is Menkes disease, an X-linked neurological childhood disorder caused by genetic defects in the copper transporter ATP7A (OMIM 309400). Menkes disease is caused by systemic copper deficiency due to defective gut metal absorption (Kaler, 2011). In contrast, cultured cells lacking ATP7A accumulate copper due to defective metal Golgi sequestration and plasma membrane efflux. Thus, Menkes cells in vitro resemble in part the systemic phenotype of Wilson disease (OMIM 277900), another rare disease caused by systemic copper accumulation. Wilson disease affects multiple organs in particular the liver and brain and it is caused by mutations in the ATP7B copper transporter. ATP7B is required for biliary copper excretion (Kaler, 2011).
Despite significant advances in establishing mutation-to-phenotype cause and effect relationships, there is a vast gap between a genomic defect and the molecular pathways connecting a mutation to its disease phenotype (Boycott et al., 2013). Filling this gap is particularly challenging in rare monogenic disorders when few patients are available or the disease is the result of coincidental rare Mendelian diseases (Gahl et al., 2016). Introduction of mutations in model genetic organisms as well as genetic, and biochemical interactomes of mutated genes have been used to bridge the gap between gene and phenotypes. Proteomes have the distinctive advantage of being executors of phenotypic programs in cells and tissues. Therefore, proteomes are causally closer to the identity of phenotypes and disease mechanisms than genomes and transcriptomes (Mullin et al., 2013). However, the abundance of each protein within a proteome is subject to the regulation of multiple genomic loci and their allelic variants (Chick et al., 2016; Foss et al., 2007; Ghazalpour et al., 2011; Klose et al., 2002; Wu et al., 2013). Therefore, quantitative proteomes are prone to interindividual variability, thus limiting the study of proteomes to high prevalence yet genetically heterogeneous diseases (Seyfried et al., 2017).
We present an approach that minimizes the genomic variability that influences proteomes by comparing within a pedigree proteomes of rare disease-affected subjects with their unaffected kin. We termed this approach genealogical proteomics. We predicted that whole proteome profiling of biological samples from subjects within a pedigree should identify protein expression traits that cosegregate with the mutation. We reasoned that rare-disease proteome datasets defined through genealogical proteomics should enrich information to bioinformatically predict diagnostic disease phenotypes from the bottom-up, as well as known and novel mechanisms of disease.
We applied genealogical proteomics to Menkes patient fibroblasts and assessed the predictive power of genealogical proteomic-generated datasets to identify mechanisms of dyshomeostatic copper accumulation. We chose Menkes disease cells because of abundant literature precisely defining the pathogenesis of systemic Menkes diseases phenotypes including cutis laxa, cerebral vascular tortuosity, aneurisms, and hypopigmentation. These phenotypes have been attributed to the reduced activity of cuproenzymes involved in extracellular matrix and melanin synthesis. In contrast, the pathogenesis of Menkes neurodevelopmental and neurodegeneration symptoms is still unknown (Zlatic et al., 2015). Additionally, in vitro cultured Menkes disease cells mimic the systemic cellular consequences of noxious copper accumulation similar to those that characterize Wilson disease, a disorder that includes psychiatric and Parkinsonian symptomatology (Bandmann et al., 2015; Davies et al., 2016; Dusek et al., 2015; Kaler, 2011). The precise mechanisms of these neurological, neurodevelopmental, and psychiatric phenotypes caused by copper depletion, in Menkes disease, and copper accumulation, in Wilson disease, remains mostly unexplored. It has been suggested that the source of Menkes disease brain symptoms result from decreased enzymatic activity in cuproenzymes necessary for the synthesis of neurotransmitters, neuropeptides, and mitochondrial respiration (Kaler, 2011). However, Menkes neurodevelopmental and neurodegeneration mechanisms are likely to involve factors other than cuproenzymes as suggested by the ATP7A interactome that enriches neurodevelopmental disorder and neurodegeneration causative genes (Comstra et al., 2017; Zlatic et al., 2015).
Here we report that genealogical proteomics and bioinformatic analysis of copper overloaded Menkes disease cells in vitro predicts, from the bottom-up, characteristic phenotypes and known metabolites associated to the copper dyshomeostasis found in Menkes and Wilson diseases. Importantly, this approach identified previously unreported mechanisms of neurodegeneration due to copper dyshomeostasis that intersect with ubiquitin metabolism through the UCHL1/PARK5 pathway of Parkinson’s disease (Bilguvar et al., 2013; Corti et al., 2011; Leroy et al., 1998; Saigoh et al., 1999). We propose genealogical ‘omics’ as a conceptual approach to garner mechanistic information from genetic diseases of limited sample size.
Results
Protein expression traits identified by genealogical proteomics of Menkes patient fibroblasts
We used Menkes disease fibroblasts from two human pedigrees (Fig. 1A) to test the capacity of genealogical proteomics to reveal known and novel copper homeostatic mechanisms. We confirmed the absence of ATP7A polypeptide in Menkes patient fibroblasts (Fig. 1A) and their increased cellular copper content by inductive-coupled mass spectrometry (Fig. 1B). ATP7A null cells increased their total cellular copper by 3 to 5-fold as compared to non-affected relatives (Fig. 1B), thus validating expected cell autonomous Menkes cellular phenotypes and providing a platform for genealogical proteomic studies.
Fig. 1. Copper Phenotypes of Menkes Diseases Fibroblasts.
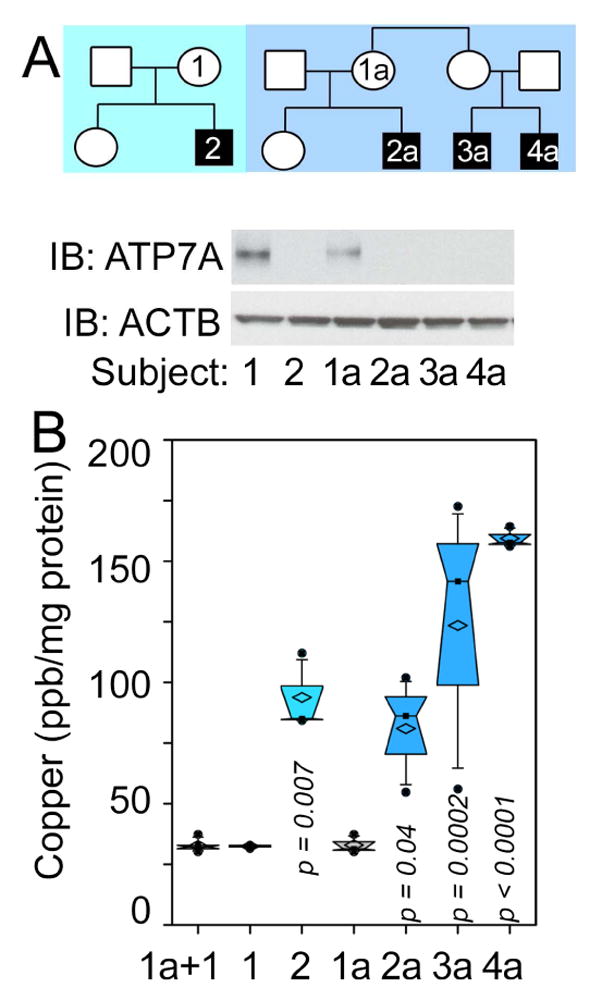
A) Pedigrees of Menkes disease families and immunoblot confirmation for ATP7A null status. B) shows copper content of fibroblasts from all subjects measured by inductively coupled plasma MS (One Way ANOVA followed by Dunnett’s multiple comparisons, n=3).
We quantified whole-cell detergent soluble proteomes of ATP7A wild type and mutant individuals within each family using triple-SILAC (Fig. 2) (Ong and Mann, 2006). Wild type ATP7A cells were labeled to equilibrium with 12C and 14N arginine and lysine (R0K0) while ATP7A null cells were incubated with either 13C and 2H (R6K4) or 13C and 15N-tagged arginine- and lysine-containing media (R10K8). Incorporation of isotopically labeled aminoacids into proteomes was >98% in all cells. We performed 6,645 mass spectrometry protein quantitations across two Menkes pedigrees (see examples in Figs. 2, 2A1, and 2A2; 2B–2B1, and 2C–2C1). Among all these protein quantitations, 450 total and 214 non-redundant proteins increased >2-fold or decreased <0.5-fold their expression in ATP7A−/y subjects as compared to unaffected female relatives within each pedigree. These 214 non-redundant and ATP7A−/y sensitive proteins, henceforth referred as the copper dyshomeostasis proteome, were absent in a proteome dataset comparing two unaffected and unrelated females (Figs. 2D–E). Eighteen proteins were modified in all ATP7A−/y subjects studied. These 18 proteins include isoforms of known cuproenzymes that require ATP7A activity for copper loading in the Golgi complex (LOXL2, MOXD1), ATP7A interactors (GLRX) (Lim et al., 2006; Singleton et al., 2010), enzymes involved in retinol, glutathione and eicosanoid metabolism (RBP4, RETSAT, PTGIS, EPHX1 see Fig. S2 and Sup. Table 1), and eight proteins implicated in neurodegenerative diseases (ATP7A, UCHL1/PARK5, PTGIS, GLRX, DPP4, ADD3, EPHX1, and QPRT, see Fig. S2 and Sup. Table 1).
Fig. 2. Genealogical Proteomics of Menkes Pedigree Cells Using Triple-SILAC Labeling.

A) Experimental design for Menkes Pedigree subject cells 1a–3a. A1 depict all mass spectrometry quantifications where the color code denotes individuals being compared (arrows). A2, presents all significant protein expression changes (>2 or <0.5, areas outside the grey box) contained in A1 (average ± SD). Venn diagram summarizes proteins with significant expression changes in A2. B–B1 and C–C1 present additional triple SILAC experiments. D) Presents all significant protein expression changes when comparing two control females. E) Venn diagram summarizing overlap of significant protein expression changes identified in B to D. F) Summary of overlap in all triple-SILAC experiments, 18 proteins were affected in all individuals. UCHL1 is depicted in A2-B1 as a red dot. MS/MS data can be found in Supplementary Tables 2–4.
We validated the genealogical proteomics triple-SILAC data focusing on these 18 proteins and other polypeptides affected in more than two Menkes subjects. We confirmed their expression changes by semiquantitative immunoblot (Fig. 3A). The most pronounced changes in protein expression affected the neurodegeneration factors UCHL1/PARK5, a ubiquitin C-terminal hydrolase mutated in Parkinson disease 5 (OMIM: 613643) (Bilguvar et al., 2013; Corti et al., 2011; Leroy et al., 1998; Saigoh et al., 1999), and QPRT, a quinolinate phosphoribosyltransferase required for catabolism of the excitotoxin quinolinate (el-Defrawy et al., 1986; Fukuoka et al., 2012; Guidetti et al., 2006). Both proteins increased by 4-fold in Menkes subjects as compared to unaffected female relatives within each pedigree (Fig. 3A–B). UCHL1/PARK5 and QPRT protein expression modifications were even more pronounced than expression changes in two factors known to require or interact with ATP7A, LOX and GLRX (Fig. 3A–B) (Lim et al., 2006; Singleton et al., 2010; Zlatic et al., 2015). We further confirmed the changes in UCHL1/PARK5 and QPRT protein expression measuring their transcripts, which increase 6–8-fold in ATP7A mutant cells (Fig. 3B–C). The correspondence between protein and transcript expression for other gene products in the copper dyshomeostasis proteome was partial with a correlation of 50%. This correlation is well within the range of protein-transcript gene expression studies in isogenic model systems (Sup Fig. 1, r=0.5, n=29 protein-transcript measurement pairs) (Maier et al., 2009). We further analyzed the reliability of transcripts to identify copper overload expression traits in mouse ileum from Atp7abr/y mice (Fig. 3D). We chose ileum since its copper content is increased in Atp7abr/y mice, much like the case of copper accumulation in Menkes patient cells in culture (Camakaris et al., 1979). UCHL1/PARK5 transcript expression was increased in Atp7abr/y ileum, yet other transcripts did not change their expression despite modifications in their mRNA in Menkes patient fibroblasts (Fig. 3D). These results indicate that UCHL1/PARK5 is a robust molecular phenotype in diverse cells and species to genetically model copper dyshomeostasis. Moreover, our findings show that quantitative genealogical proteomics reliably identifies expression traits associated to copper metabolism defects.
Fig. 3. ATP7A null cells overexpress UCHL1 and other proteins.

A–A1) Human pedigrees of families affected by Menkes disease and studied in B–C. Immunoblots of ATP7A, UCHL1, and antigens identified by genealogical proteomics of human Menkes fibroblasts. Actin (ACTB) and clathrin (CLTC) were used as loading controls. B) Quantitation of immunoblots as a ratio of Menkes patient to the carrier female within pedigree. Color code of the bars depict the pedigree in A1. Average ± SE comparisons were done against beta actin (ACTB). Kruskal-Wallis test followed by pairwise comparisons with Mann-Whitney Rank Sum Test, for n=3–8. C) mRNA quantification by qRT-PCR of antigens in B expressed as a ratio of Menkes patient to the carrier female in the pedigree. Color code of the bars depict the pedigree in A1. Average ± SE comparisons were done between diseased and non-diseased relative (ACTB, VAMP2 and 3 mRNAs are reference housekeeping genes). EPHX1 mRNA was not measured. D) Ileum mRNA quantification of mouse orthologues from of antigens presented in B. qRT-PCR data expressed as a ratio of Atp7abr/y to Atp7abr/+. GSTM3 was not measured. (VAMP2 and 3 mRNAs are reference housekeeping genes and VIL1, villin is a marker of ileal epithelial cells). C–D Benjamini and Hochberg FDR corrected two-tails t-test, n=3. All changes in expression are significant unless marked by NS.
The proteome predicts phenotypes of genetic copper metabolism disorders
We reasoned that if genealogical proteomics were to offer insight into rare genetic defects, then ontologies generated from genealogical proteomic datasets by bioinformatics should identify known and novel traits as well as mechanisms of disease. We tested this hypothesis feeding the copper dyshomeostasis proteome dataset into diverse bioinformatic tools to unbiasedly assess traits, cell, and tissue pathways significantly enriched within this copper dyshomeostasis dataset.
We interrogated annotated mouse phenotypes, KEGG pathways, and metabolite ontology databases with the copper dyshomeostasis proteome. We fed each individual pedigree triple-SILAC dataset (Fig. 4A–C) or all pedigrees datasets combined into the copper dyshomeostasis proteome (Fig. 4C) to the ENRICH mouse phenotypes (Fig. 4) and KEGG pathways prediction engines (Sup Fig. 2) (Chen et al., 2013). The copper dyshomeostasis proteome outperformed each individual pedigree triple-SILAC dataset in predicting copper dyshomeostasis disease traits in the mouse phenotype, KEGG pathway, and metabolite ontologies (Fig. 4 and Sup Fig. 2). Mouse phenotype categories predicted by the copper dyshomeostasis proteome dataset identified Atp7a loss-of-function mouse alleles in the Mouse Genomic Informatics engine even though the copper dyshomeostasis proteome is defined in cells overloaded with copper (Fig. 4F). These phenotypic ontological categories describe cardinal traits of Menkes disease, such as defects in skin tensile strength (MP0005275) and collagen (MP0008438) both related to the vascular alterations and cutis laxa found in Menkes patients (Zlatic et al., 2015). Mouse phenotypes predicted from the copper dyshomeostasis proteome poorly overlapped with phenotypes predicted with a proteome dataset obtained by comparing two unrelated disease-free subjects (Fig. 4A–E Overlay row). The predictive value of the copper dyshomeostasis proteome extended to KEGG pathways and metabolite ontologies identifying copper, glutathione, as well as oxidized and reduced forms of nicotinamide adenine dinucleotide species (Fig. 4G and Sup Fig. 2A–E). All of these metabolites have been either associated directly or indirectly with genetic forms of copper dyshomeostasis and are central to the diagnosis of these diseases, as is the case of copper (Fig. 4G, p-value corrected with Bonferroni = 0.003396) (Bhattacharjee et al., 2016; Kunz et al., 1999; Kuznetsov et al., 1996; Mercer et al., 2016; Singleton et al., 2010; Zlatic et al., 2015). We confirmed these bioinformatic findings using three additional bioinformatic engines GeneTerm Linker, ClueGo, and Gene set Disease Association GDA (Fig. S2F–I) (Bindea et al., 2009; Fontanillo et al., 2011; Park et al., 2014). The GDA engine predicted a significant enrichment of neurodegeneration and brain vascular pathology (Fig. S2I). Neurodegeneration and cerebral arterial tortuosity are phenotypes present in all patients affected by Menkes disease (Kaler, 2013; Manara et al., 2017a; Manara et al., 2017b). These bioinformatic results demonstrate that genealogical proteomics datasets contain sufficient information to predict clinical traits which are diagnostic of genetic diseases of copper metabolism.
Fig. 4. Bioinformatic Analysis of Genealogical Proteomics from Menkes Disease Pedigrees.

A–E) MGI mouse phenotype ontology analyzed with ENRICHR. Data are depicted as canvases where coordinates are occupied by an individual ontology category. First canvas row depicts SILAC comparing diseased and non-diseased pairs, second row in red depicts non-diseased pair comparison. Third row is their overlay. F) MGI mouse phenotype ontology associated to the copper dyshomeostasis proteome analyzed with ENRICHR depicted as combined score (z-score x −log(p-value)) (Chen et al., 2013; Kuleshov et al., 2016). Red depicts categories found under Atp7a mouse mutations in MGI. G) Human Metabolome Database (HMDB) metabolites associated to the copper dyshomeostasis proteome analyzed with ENRICHR engine depicted as combined score. Individual family and collective bioinformatics data can be found in Supplementary Tables 1–4.
Mitochondrial function alterations are caused by genetic defects of copper metabolism
A common theme from our bioinformatic studies are cellular processes converging on mitochondria including glutathione, purine and pyrimidine metabolism, and neurodegenerative diseases such as Parkinson’s. However, only 15 proteins of the 214 proteins in the copper dyshomeostasis proteome (7%) belong to the human mitochondrial proteome (Mitocarta 2.0, Fig. 5A–B). In fact, the mitoproteome measured in copper overloaded Menkes patient cells is mostly unchanged (Fig. S3). Mitochondrial MGST1 or microsomal glutathione S-transferase 1 was the only gene whose expression detected by triple-SILAC was affected in all Menkes pedigree cells examined, a finding we confirmed by qRT-PCR (Fig. 5C).
Fig. 5. Mitochondrial Respiration is Increased in Copper Overloaded ATP7A null fibroblasts.

A) Venn diagram depicting the overlap of the copper dyshomeostasis proteome and the human Mitoproteome Mitocarta 2.0. B) List of 15 proteins common to the Menkes and Mitocarta Proteomes in A. MS/MS data can be found in Supplementary Tables 2–4. C) Quantitation of the mitochondrial MGST1 antigen as per SILAC fold of change and its mRNA by qRT-PCR in Menkes fibroblasts. Benjamini and Hochberg FDR corrected two-tails t-test. Depicted is average ± SE, n=3. Asterisks mark significant changes in message. D) Oxygen consumption rates in non-diseased mother 1 (grey) and Menkes son fibroblasts 2 (cyan). Arrows (a) to (c) indicate injection of oligomycin (a), FCCP (b), and rotenone plus antimycin A (c) for stress test. Depicted is average ± SD (n=6). E–G) Rescued and non-diseased mother 1 (grey, columns 1 and 3) and Menkes patient fibroblasts (cyan, columns 2 and 4,) basal, maximal, and ATP-dependent respiratory rates. Cells in columns 1 and 2 are immortalized fibroblasts from the same Menkes patient either rescued by transfection of ATP7A in 1 (ATP7AR/y) or empty vector in 2 (ATP7A−/y). Cells in column 3 and 4 are primary fibroblast cultures of non-diseased mother 1 (ATP7A+/y) and Menkes son (ATP7A−/y). Each point is an independent experiment. Menkes patient fibroblasts possess significantly increased respiration (asterisks p<0.0001, Mann-Whitney Rank Sum Test). E) Menkes cell respiration is sensitive to increasing extracellular copper challenge. Figure depicts the change in basal respiration (acute response) at different copper concentrations. Average ± SE (n=6). One Way ANOVA followed by Bonferroni test compares wild type and Menkes patient fibroblasts at different copper concentrations.
To address the dichotomy between bioinformatics and proteomics, we analyzed mitochondrial respiration in Menkes patient fibroblasts by Seahorse technology (Divakaruni et al., 2014). We measured mitochondrial respiration in human Menkes pedigree cells as well as immortalized Menkes fibroblasts rescued by recombinant expression of ATP7A employing extracellular flux analysis (Fig. 5D–H). Menkes disease fibroblasts from human pedigrees had an increased basal rate of oxygen consumption (Fig. 5D–E), increased maximal respiration after FCCP protonophore addition (Fig. 5D and F columns 3–4), and increased oxygen consumption sensitivity to oligomycin (Fig. 5D and G columns 3–4). These mitochondrial phenotypes associated with copper accumulation due to ATP7A genetic defects were rescued by recombinant expression of ATP7A in Menkes immortalized fibroblasts (Fig. 5E–G, compared rescued cells in column 1, ATP7AR/y, with Menkes patient fibroblasts in column 2, ATP7A−/y). The increased mitochondrial respiration phenotype did not associate with enhanced content of total free radicals in Menkes patient fibroblasts, as measured by flow cytometry with diverse probes sensitive to free radical species (Dickinson and Chang, 2008; Marullo et al., 2013) (Fig. S3E). However, the increased respiration in Menkes patient fibroblasts did associate with an enhanced sensitivity of basal mitochondrial respiration to excess extracellular copper (Fig. 5H–I). In contrast, basal mitochondrial respiration was minimally sensitive to excess extracellular copper in control ATP7A+/− cells (Fig. 5H–I). These data demonstrate that ATP7A mutations increase mitochondrial respiration conferring rapid mitochondrial sensitivity to added extracellular copper.
Pharmacological and genetic Inhibition of UCHL1/PARK5 ameliorates copper dyshomeostasis phenotypes
Mutations in the familial Parkinson’s genes PARK6, 7, 9 17, 23 increase basal and/or maximal mitochondrial oxygen consumption in diverse cell types including fibroblasts and excitable cells (Cooper et al., 2012; Grunewald et al., 2012; Lesage et al., 2016; Shi et al., 2015; Tang et al., 2015). This evidence prompted us to ask whether mitochondrial dysfunction caused by copper accumulation due to ATP7A genetic defects is modulated by pharmacological or genetic manipulation of UCHL1/PARK5 activity. We focused on UCHL1/PARK5 because it is a Parkinson’s causative gene and UCHL1/PARK5 is the top hit among the upregulated proteins in the copper dyshomeostasis proteome (Fig. 2). We sought to discriminate whether increased UCHL1/PARK5 expression observed in copper overloaded Menkes fibroblasts is a pathogenic mechanism or, alternatively, a compensatory adaptive mechanism to dyshomeostatic copper overload.
We used the UCHL1/PARK5 inhibitor LDN57444 to assess whether Menkes patient fibroblast mitochondrial phenotypes were sensitive to UCHL1/PARK5 activity (Liu et al., 2003). Cells from wild type unrelated males (ATP7A+/y, Fig. 6A), carrier mother (ATP7A−/+, Fig. 6B) and Menkes affected son (ATP7A-/y, Fig. 6C) were incubated in the presence of vehicle (Fig. 6, gray symbols) or LDN57444 (Fig. 6, blue symbols) followed by a mitochondrial stress test (Fig. 6A, arrows a–c). We first confirmed in blind ATP7A immunofluorescence experiments that all ATP7A+/y cells expressed ATP7A, while transporter expression occurred in half of the ATP7A−/+ mother’s cells, and none of the Menkes disease son’s fibroblasts (Fig. S3). Addition of LDN57444 decreased basal and maximal respiration in all fibroblasts, yet effects were significantly more pronounced in Menkes patient fibroblasts (Fig. 6D, compare columns 2 and 6). In fact, the inhibitory effect of LDN57444 upon mitochondrial respiration was proportional to ATP7A gene dosage reduction as determined by the changes in basal respiration before and after LDN57444 incubation (Fig. 6D, compare even columns). To test the specificity of LDN57444, we assessed drug effect on wild type and Uchl1 mutant mouse fibroblasts mitochondrial respiration (Fig. S5) (Saigoh et al., 1999). Uchl1 genetic defect did not affect mitochondrial respiration, Atp7a expression, and copper sensitivity (Fig. S6). These results indicate that UCHL1/PARK5 is downstream or parallel to ATP7A and suggest that UCHL1/PARK5 inhibition could protect ATP7A mutant cells from copper dyshomeostasis.
Fig. 6. Mitochondrial Respiration in Cultured Menkes patient fibroblasts is Restored by a UCHL1 inhibitor.

A–C Oxygen consumption rates in wild type (ATP7A+/y), non-diseased mother (ATP7A−/+) and Menkes son (ATP7A−/y) fibroblasts treated with vehicle (grey) and the UCHL1 inhibitor LDN57444 (cyan) for 250 min while performing Seahorse analysis. Average ± SE, n=5. D) Box plot presents the acute responses per genotype expressed as the O2 consumption rate change as compared to pre-vehicle or drug-addition. Note that the inhibitor effect on respiration increases with the gene dosage reduction of ATP7A. Mann-Whitney Rank Sum Test, n1-n6= 21, 22, 50, 58, 56, 60 respectively. Box plots depict percentiles fifth and 95th. Box line represents sample median and diamonds sample mean and notches mark the half-width.
To determine whether reduced activity of UCHL1/PARK5 is protective, we genetically modified the expression of Drosophila ATP7A (Atp7, CG1886) and tested the effects of UCHL1 (Uchl, CG4265). We used the GAL4-UAS system to up- and down-regulate ATP7 expression. Cell autonomous ATP7 overexpression mimics the systemic effects of Menkes disease in tissues by decreasing cellular copper. In contrast, ATP7 RNAi mimics the systemic Wilson disease copper accumulation and the increased copper content observed in cultured ATP7A-deficient cells due to decreased copper efflux from the cytoplasm (Hwang et al., 2014; Lye et al., 2011).
Pan neuronal or thoracic dorsal midline overexpression of ATP7 with the drivers GAL4-elav C155 and GAL4-Pannier (Pnr) (Heitzler et al., 1996; Lin and Goodman, 1994), respectively, induced high adult mortality (Fig. 7A and D, compare row 1 with 2). Death due to ATP7 overexpression requires copper transport activity as demonstrated by the reduced effect of ATP7 mutant transgenes carrying mutations in the metal binding (MBD) and ATP binding domains of ATP7 (TAP, Fig. 7A and D, compare row 1 with 3–4) (Norgate et al., 2006). Similarly, ATP7 or Uchl RNAi increased mortality, though to a lesser extent (Fig. 7A and D, rows 5–6).
Fig. 7. ATP7A and UCHL1 Drosophila Orthologues Genetically Interact.

A) Mortality of transgenic animals expressing UAS-ATP7, UAS-ATP7 RNAi, and UAS-Uchl RNAi in all neurons using the C155-GAL4 driver. UAS-ATP7 transgenes carrying mutations that impair copper transport (mbs or tap) decrease mortality. N represents total number of animals used in these experiments. B) UAS-ATP7A and UAS-Uchl RNAi were expressed in dopaminergic neurons using the Ddc-GAL4 driver. P values represent difference between two independent proportions for the indicated rows. C) UAS-ATP7A RNAi and UAS-Uchl RNAi were expressed in dopaminergic neurons using the Ddc-GAL4 driver. B and C) Mortality was induced by copper feeding for three days. In the absence of copper in the diet there is no mortality phenotype. n=24 experiments with 10 animals each, total 240 animals per genotype). Data are presented for females and male flies. P values calculated with Kruskal-Wallis test followed by pairwise comparisons with Mann-Whitney Rank Sum test. D) Mortality of transgenic animals expressing UAS-ATP7, UAS-ATP7 RNAi, and UAS-Uchl RNAi in the mid thoracic segment using the Pnr-GAL4 driver. UAS-ATP7 transgenes carrying mutations that impair copper transport (mbs or tap) decrease mortality. N represents total number of animals used in these experiments. P values represent difference between two independent proportions for the indicated rows. E) Images of animals in D. Arrows point to scutellum bristles.
We tested the effect of down-regulation of Uchl on mortality induced by ATP7 up- and down-regulation using the pan-neuronal C155 driver. The mortality induced by ATP7 overexpression or downregulation remained unaffected by Uchl RNAi (Fig. 7A compare row 2 and 7, 5 and 8). The C155 driver expresses from the neuroblast stage in all neurons, thus ATP7-dependent mortality phenotypes could be plateaued early in development. Therefore, we used dopa decarboxylase (Ddc)-GAL4 driver (Feany and Bender, 2000), which drives the expression of UAS-ATP7 reagents and Uchl RNAi selectively in dopaminergic and serotoninergic neurons. Changes in ATP7 expression with the Ddc-GAL4 driver makes animals susceptible to death induced by copper feeding thus allowing environmental control of ATP7 expression-dependent phenotypes. Uchl RNAi in dopaminergic neurons was protective of the copper feeding-induced mortality in wild type animals (Fig. 7B rows 5–6 and Fig. 7C rows 1 and 2). Moreover, this Uchl RNAi protective effect was evident even when dopaminergic neurons where sensitized by ATP7 up-regulation, a condition that adapt cells to low cellular copper content and mimics in part the systemic effects of cytoplasmic copper depletion in Menkes disease (Fig. 7B, rows 7 and 8) or when ATP7 was down-regulated, a condition that adapts cells to increased cellular copper similar to the copper overload observed in Wilson disease tissues (Fig. 7C, rows 3 and 4).
We further explored protective effects of Uchl down-regulation in other fly tissues. We focused on the thoracic dorsal midline marked by the pannier GAL4 driver because ATP7 up-regulation causes adult mortality, hypopigmentation, midline developmental defects, and loss of sensory bristles. Mortality induced by ATP7 up-regulation in the paneer domain of the thorax was ameliorated by Uchl RNAi both in males and females (Fig. 7D compare rows 2 and 7). In contrast, mortality induced by ATP7 RNAi in the thoracic dorsal midline was not significantly modified by Uchl down-regulation (Fig. 7D compare rows 5 and 8) yet scutellum bristle phenotypes in ATP7 RNAi animals were rescued by down-regulation of Uchl (Fig. 7E).
We determined whether Uchl down-regulation protects synapses from ATP7 up-regulation, a condition that depletes copper from the cytoplasm of cells (Fig. S6). Up-regulation of ATP7 expression in neurons decreases the footprint of presynaptic terminals at the neuromuscular junction of third instar larvae, which we quantified as an increased number of boutons per branch length (Comstra et al., 2017). We reproduced this increase number of boutons per branch length phenotype by expressing ATP7 with the pan-neuronal driver C155 (Fig. S6A–B, C155>ATP7-wt). In contrast, neuronal down-regulation of Uchl increased the complexity and footprint of presynaptic terminals by increasing the number of branches, their length, and number of boutons yet without affecting the number of boutons per branch length (Fig. S6A–B, C155> Uch RNAi). Combination of ATP7 up-regulation with down-regulation of Uchl in the same neuron resembled the complexity of C155> Uch RNAi terminals indicating that the effects of ATP7 overexpression were rescued by decreasing Uchl expression (Fig. S6A–B, C155> ATP7-wt, Uch RNAi). These results demonstrate that reduced Uchl expression protects cells, synapses, and tissues from copper dyshomeostasis. We propose that the increased expression of UCHL1 observed in Menkes patient fibroblasts is a copper dyshomeostasis pathogenesis mechanism rather than a compensatory adaptation.
Discussion
Genealogical proteomics is a paradigm to extract mechanistic information from low frequency/small population biological specimens such as rare disorders. We can infer from a discrete proteome dataset, the copper dyshomeostasis proteome, cardinal clinical phenotypes and known processes affected in genetic diseases that affect copper metabolism. Furthermore, we identified putative novel pathways affected by copper dyshomeostasis, such as mitochondrial function and pathways affected by genetic forms Parkinson’s disease. These findings demonstrate that inborn copper metabolism defects share mechanisms and molecules with Parkinson’s disease.
Mitochondrial respiration in copper overloaded ATP7A null fibroblasts resembles mitochondrial phenotypes in cells from familial Parkinson’s caused by mutations in either PARK6, 7, 9, 17 and 23. These mutations increase basal and/or maximal mitochondrial oxygen consumption in fibroblasts, neurons and other excitable tissues (Cooper et al., 2012; Grunewald et al., 2012; Lesage et al., 2016; Shi et al., 2015; Tang et al., 2015). However, this mitochondrial phenotype in Parkinson gene mutant cells is likely to be neither universal across different Parkinson’s gene defects nor cell types as indicated by Uchl1 null mouse fibroblasts, which lack this mitochondrial respiration phenotype (Fig. S5). Despite the limitations of fibroblast studies to infer neuronal mechanisms, it is intriguing to consider that the most sensitive neurons to Parkinson’s disease reside in the substantia nigra compacta and these cells have increased mitochondrial oxygen consumption as compared to other neurons (Pacelli et al., 2015). Pacelli has proposed that increased mitochondrial respiration may predispose these neurons to cell death (Pacelli et al., 2015). Thus, amelioration of the increased mitochondrial respiration observed in copper overloaded fibroblast could protect cells from copper imbalances. The increased UCHL1 expression in copper overloaded fibroblasts and the effects of UCHL1 pharmacological inhibition on mitochondrial respiration suggests that UCHL1 increased expression is part of a pathogenic pathway of copper dyshomeostasis.
UCHL1 is a ubiquitin-protein hydrolase that cleaves the C-terminal glycine of ubiquitin during the processing of ubiquitin precursors and of ubiquitinated proteins, thus restoring free ubiquitin pools (Bishop et al., 2016). Loss-of-function UCHL1 mutations causes several neurodegenerative diseases that result in increased mortality including familial Parkinson’s disease (PARK5), childhood neurodegeneration, and spinal gracile axonal dystrophy. Interestingly, the latter two phenotypes are also observed in hypomorphic ATP7A mutations in humans (Bilguvar et al., 2013; Graham and Liu, 2017; Saigoh et al., 1999; Wintermeyer et al., 2000). We reproduced the increased mortality effects of UCHL1 loss-of-function in Drosophila by pan-neuronal RNAi of the fly UCHL1 orthologue (Uchl). We tested whether Uchl participates directly in copper chaperoning or ATP7 stability. We discriminated these models by changing the expression of ATP7. Surprisingly, Uchl down-regulation in dopaminergic neurons is protective after an acute copper exposure irrespective of whether ATP7 expression is maintained at wild type levels, or is either up- or down-regulated Drosophila dopaminergic neurons. Cell autonomous overexpression of ATP7A reduces cellular copper levels while ATP7 RNAi increases copper in cells (Hwang et al., 2014; Lye et al., 2011). Thus, these findings exclude a direct role of UCHL1 in copper chaperoning or ATP7A expression as we demonstrate in Uchl1 null cells.
We propose that the target of ATP7A-dependent copper metabolism imbalances and UCHL1 activity is the mitochondria respiratory chain. We favor this model because of: a) the increased mitochondrial respiration observed in copper overloaded Menkes fibroblast which is ameliorated by pharmacological inhibition of UCHL1, b) the decreased mitochondrial glutathione levels reported in cultured ATP7A null cells mitochondria (Bhattacharjee et al., 2016), and c) because UCHL1 belongs to a network of Parkinson’s disease causing genes (PARK2, PARK7), which includes mitochondrial ATPase subunits and peroxiredoxins, and diverse SLC25A inner membrane mitochondrial transporters (Davison et al., 2009; Havugimana et al., 2012; McKeon et al., 2015; Wan et al., 2015; Zanon et al., 2013). Our finding that UCHL1 downregulation is protective of neurodegeneration is in line with the report that parkin-dependent neuronal death requires UCHL1 expression (Corsetti et al., 2015) and the observation that UCHL1 gain-of-function mutations cause neurodegeneration in the form of hereditary spastic paraplegia (SPG79, OMIM: 615491) (Rydning et al., 2017). However, the role of UCHL1 in neuronal viability is likely more nuanced because of the neuroprotective role that UCHL1 gain-of-function can also play in chronic neurodegenerative mouse models of Parkinson’s and Alzheimer’s disease (Gong et al., 2006; Kyratzi et al., 2008; Liu et al., 2002; Lombardino et al., 2005; Yasuda et al., 2009). We speculate that normal turnover of damaged mitochondria requires the activity of UCHL1, however excessive turnover of globally damaged mitochondria due to an acute noxious agent such as copper feeding in the Drosophila model, may compromise cells unless turnover is slowed down by inhibition of the UCHL1 pathway.
We used fibroblasts to inform mechanisms in neuronal cells, a practice that has received some support (Kalman et al., 2016) despite its inability to comprehensively reveal neuronal-specific mechanisms. Even after this limitation of our study, we still gained mechanistic insight from the 214 proteins of the copper dyshomeostasis proteome by three strategies. First, focusing on the most pronounced protein expression changes, such as the case of UCHL1 that was also prioritized by bioinformatics. Second, by testing genetic interactions between proteins belonging to the copper dyshomeostasis proteome in Drosophila neurons. Finally, by inferring ontological categories from the entire copper dyshomeostasis proteome using bioinformatic tools. The ontological category of mitochondrial function (MP0006036) raised above a statistical threshold in two of the three SILAC studies. Similarly, copper (HMDB00657) was a significant bioinformatic finding in only one family SILAC study with 66 ATP7A mutation-sensitive proteins (corrected p-value with Bonferroni = 0.003396). The addition of all three SILAC proteomic datasets into the copper dyshomeostasis proteome allowed a robust recognition of copper and mitochondrial function as significant bioinformatic hits (corrected p-value with Bonferroni of 0.003396 and 0.0005486, respective). These results argue that a single patient pedigree may not be sufficient to uncover disease mechanisms unless a disease-specific proteome dataset is expanded by increasing either the number of families with affected probands or the depth coverage of the quantified cellular proteome. Alternatively, a disease-specific genealogical proteome dataset could be combined with hits obtained from proteomes gathered from animal or cellular model of the disease, from genealogical transcriptomes or genealogic metabolomes. We propose that the concept of genealogical ‘omics’ can be applied to genetically heterogeneous populations in order to identify expression traits and ontological categories cosegregating with genomic loci and phenotypes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Dr. Victor Faundez MD, PhD (vfaunde@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Tissue
Mouse tissues from Atp7abr/y and Atp7abr/+ were collected following University of Minnesota IACUC approved protocols. C57BL/6-Atp7aMo-br/J are available from The Jackson Laboratory (Stock No: 002566 (RRID:IMSR_JAX:002566)).
Tissue from UCHL1−/− and UCHL1+/+ were collected following Emory University IACUC approved protocols. B6:129P2-Uchl1tm1Dgen/Mmnc mice are available from the Mutant Mouse Resource and Research Center (MMRRC stock number MMRRC:011642-UNC; RRID:MMRRC_011642-UNC).
Tissue Culture Cell Lines
Pedigrees of Menkes Fibroblasts were obtained from Coriell cell repository (Cat#. GM01983\(RRID:CVCL_F591), GM01981 (RRID:CVCL_F589), GM04068 (RRID:CVCL_1L28), GM00245 (RRID:CVCL_W623), GM00220 (RRID:CVCL_H963), GM01057 (RRID:CVCL_H964)). An immortalized Menkes fibroblast line and the ATP7A rescue cell line, ME32 (ATP7A−/y) and ME344 (ATP7AR/y), were previously described (Comstra et al., 2017; La Fontaine et al., 1998). All cells were cultured in DMEM (Corning 10-013-CV) media supplemented with 10–15% fetal bovine serum (FBS) (Atlanta Biologicals S12450) and 100 μg/ml penicillin and streptomycin (Hyclone SV30010) at 37°C in 5% CO2. Media was supplemented with CuCl2 (Sigma 203149) as described in the text.
Drosophila Husbandry and Tissue
P{UAS-ATP7.FLAG}, P{UAS-ATP7.MBS1.FLAG}, and P{UAS-ATP7.TAP.FLAG}, were gifts from R. Burke (Norgate et al., 2006). w[1118] (#3605 RRID:BDSC_3605), C155-GAL4 (P{w[+mW.hs]=GawB}elav[C155] (#458 RRID:BDSC_458), Ddc-GAL4 (w[1118]; P{w[+mC]=Ddc-GAL4.L}Lmpt[4.36] (#7009 RRID:BDSC_7009), and pnr-GAL4 (y[1] w[1118]; P{w[+mW.hs]=GawB}pnr[MD237] (#3039 RRID:BDSC_3039) came from the Bloomington Drosophila Stock Center. UAS-Uchl RNAi w[1118];P{GD11265}v26468 (RRID:FlyBase_FBst0456429) and UAS-ATP7 RNAi (P{KK100396}VIE-260B v108159 (RRID:FlyBase_FBst0479971) came from the Vienna Drosophila Stock Center. Flies were reared in a 12hr:12hr light:dark cycle at 25°C incubator on standard molasses food.
METHODS DETAILS
Trio SILAC Labeling Mass Spectrometry
Fibroblasts were labeled using the protocol described below and as cited (Gokhale et al., 2016; Gokhale et al., 2012; Gokhale et al., 2015; Ong et al., 2002; Ong and Mann, 2006; Ryder et al., 2013). Cells were grown in DMEM with either “light” unlabeled 12C- and 14N- arginine and lysine amino acids (R0K0). “medium” 13C- labeled arginine, and 2D-labeled lysine amino acids (R6K4) or “heavy” 13C- and 15N-labeled arginine, and 13C- and 15N-labeled lysine amino acids (R10K8) supplemented with 15% FBS and 100 μg/ml penicillin and streptomycin. Cells were grown for a minimum of seven passages ensuring maximum incorporation (97.5%) of the amino acids into the cellular proteins. All reagents for stable isotope labeling by amino acids in cell culture (SILAC) labeling were obtained from Dundee Cell Products. Cell lysates were prepared, as described below. Mass spectrometry was performed using the services of MS Bioworks (Gokhale et al., 2016; Gokhale et al., 2015; Perez-Cornejo et al., 2012; Ryder et al., 2013). The clarified SILAC labeled supernatants were pooled 1:1:1 and 20μg of this mix was separated on a 4–12% Bis-Tris Novex mini-gel (Invitrogen) using the MOPS buffer system. The gel was stained with Coomassie and the lanes excised into 40 equal segments using a grid. Gel pieces were robotically processed (ProGest, DigiLab) by first washing with 25mM ammonium bicarbonate followed by acetonitrile, followed by reduction with 10mM dithiothreitol at 60°C, alkylation with 50mM iodoacetamide at room temperature. Pieces were digested with trypsin (Promega) at 37°C for 4h and quenched with formic acid. The supernatant was analyzed directly without further processing.
Gel digests were analyzed by nano LC/MS/MS with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher Q Exactive. Peptides were loaded on a trapping column and eluted over a 75μm analytical column at 350nL/min; both columns were packed with Jupiter Proteo resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with MS and MS/MS performed in the Orbitrap at 70,000 FWHM resolution and 17,500 FWHM resolution, respectively. The fifteen most abundant ions were selected for MS/MS. Data were processed through the MaxQuant software 1.4.1.2 (www.maxquant.org) which served the following functions: 1. Recalibration of MS data. 2. Filtering of database search results at the 1% protein and peptide false discovery rate (FDR). 3. Calculation of SILAC heavy:light ratios. Data were searched using a local copy of Andromeda with the following parameters: Enzyme: Trypsin. Database: Swissprot (concatenated forward and reverse plus common contaminants). Fixed modification: Carbamidomethyl (C). Variable modifications: Oxidation (M), Acetyl (Protein N-term), 13C6/15N2 (K), 13C6/15N4 (R), 4H2 (K), 13C6 (R). Fragment Mass Tolerance: 20ppm. Pertinent MaxQuant settings were: Peptide FDR 0.01. Protein FDR 0.01. Min. peptide Length 7. Min. unique peptides 0. Min. ratio count 2. Re-quantify TRUE. Second Peptide TRUE.
SILAC Proteomic Analysis and Bioinformatics
Bioinformatic analyses were performed with the ENRICHR engine, GeneTerm Linker, ClueGo, and Gene set Disease Association GDA algorithms (Bindea et al., 2009; Chen et al., 2013; Fontanillo et al., 2011; Kuleshov et al., 2016; Park et al., 2014). These are described in our previous work (Comstra et al., 2017; Gokhale et al., 2016; Larimore et al., 2017). Bioinformatic statistics are reported in the pertinent supplementary tables 1–3.
Cell Lysis
Cells were washed twice in phosphate buffered saline (PBS Corning 21-040-CV) containing 0.1mM CaCl2 and 1.0mM MgCl2. Cells were then scraped up in lysis buffer (10mM HEPES, 150mM NaCl, 1mM EGTA, 0.1mM MgCl2, 0.5% Triton X-100) containing Complete (Roche 11-836-145-001) protease inhibitor, incubated at 4°C with periodic vortexing for 30min, and spun at 16,100 × g for 15 min at 4°C. SILAC samples were scraped up and sonicated prior to the 4°C incubation above. The cleared lysate was collected for analysis. Protein concentration was determined using the Bradford Assay (BioRad 5000006).
SDS-PAGE and Western Blotting
For western blot, lysate was reduced and denatured in sample buffer containing SDS and 2-mercaptoethanol and heated for 5 minutes at 75°C. Equal concentrations of sample were loaded into Criterion gels (BioRad 5671094) for electrophoresis and transferred to PVDF (Millipore IPFL00010) using the semi-dry transfer method. Membranes were blocked with 5% non-fat milk in Tris buffered saline containing 0.05% Triton X-100 (TBST), rinsed and incubated overnight with primary antibody diluted in antibody base solution (PBS with 3% Bovine Serum Albumin, 0.2% Sodium Azide). Membranes were then washed in TBST and incubated in HRP conjugated secondary antibody diluted 1:5000 in the blocking solution above. Washed membranes were then exposed to Amersham Hyperfilm ECL (GE Healthcare 28906839) with Western Lightning Plus ECL reagent (Perkin Elmer NEL105001EA). Densitometry was performed and quantified with Fiji Image J 1.51n software.
Antibodies
Antibodies for western blot analysis are as follows: ATP7A (Santa Cruz Biotechnology Cat# sc-376467 RRID:AB_11150485), beta Actin (Sigma-Aldrich Cat# A5441 RRID:AB_476744), Complex II Abcam Cat# ab14715 RRID:AB_301433, Complex IV Abcam Cat# ab14705 RRID:AB_2084810, SOD2 Santa Cruz Biotechnology Cat# sc-30080 RRID:AB_661470, TOMM20 Santa Cruz Biotechnology Cat# sc-11415 RRID:AB_2207533, TRX-2 Santa Cruz Biotechnology Cat# sc-50336 RRID:AB_2212130, UCHL1 (McKeon et al., 2015), QPRT (Santa Cruz Biotechnology Cat# sc-100809 RRID:AB_1128879), LOX (Proteintech Group Cat# 17958-1-AP RRID:AB_2234541), EPHX1 (BD Biosciences Cat# 611372 RRID:AB_398894), CD9 (Abcam Cat# ab92726 RRID:AB_10561589), GSTM3 (Proteintech Group Cat# 15214-1-AP RRID:AB_2116070), GSTT1 (Proteintech Group Cat# 15838-1-AP RRID:AB_2116344), GLRX (Abcam Cat# ab45953 RRID:AB_880242), STON2 (a gift from the lab of Lena von Oertzen, pAB rabbit aff. Purified 2424.5), HSP90 BD Biosciences Cat# 610418 RRID:AB_397798, SV2 DSHB Cat# SV2 also AB_528480 RRID:AB_2315385, CLTC, BD Biosciences Cat# 610499 RRID:AB_397865. Secondary antibodies were against mouse or rabbit conjugated to HRP (A10668; RRID:AB_2534058 and G21234; RRID:AB_2536530).
Antibodies for mammalian immunofluorescence are as follows: ATP7A, a custom affinity purified antibody was produced from Bethyl Laboratories against the mATP7A peptide sequence SEPDKHSLLVGDFREDDDTT, CLTC Thermo Fisher Scientific Cat# MA1-065 RRID:AB_2083179. Secondary antibodies are Alexa fluor conjugated anti-mouse 555 (A21127; RRID:AB_2535769) and anti-rabbit 488 (A11034; RRID:AB_2576217).
Immunofluorescence and Microscopy
Immunofluorescence and confocal microscopy were performed as described (Ryder et al., 2013; Zlatic et al., 2013). Cells were seated onto Matrigel (Corning 356237) coated glass coverslips as per manufactures protocol. Cells were then washed twice on ice with ice cold phosphate buffered saline (PBS Corning 21-040-CV) containing 0.1mM CaCl2 and 1.0mM MgCl2, fixed with 4% paraformaldehyde in PBS/Mg2+/Ca2+ at 4°C. Cells were then washed twice again in PBS/Mg2+/Ca2+ and permeablized and blocked in blocking solution containing 2% bovine serum albumin, 1% fish gelatin, 0.02% sapponin, and 15% horse serum in phosphate buffered saline. Cells were then incubated with primary antibody at 37°C, washed three times in blocking solution, incubated in Alexa Fluor conjugated secondary antibody at 37°C, washed three more times in blocking solution and once in PBS/Mg2+/Ca2+. All immunofluorescent antibodies were diluted in blocking solution. Coverslips were mounted to slides with gelvatol and sealed. Confocal imaging was done on an inverted 510 Zeiss LSM microspope run with Zen imaging Software. Images were captured with a 60X oil immersion objective and sequential GFP and rhodamine filter sets.
RNA Isolation and RT-PCR
RNA extraction for cells and tissues was done using Trizol Reagent (Invitrogen 15596026) following manufactures protocol. Concentration and purity of RNA were determined using the BioRad SmartSpec Plus Spectrophotometer. First strand synthesis was performed using the Superscript III First Strand Synthesis System Kit (Invitrogen 18080-051) with 5 ♣g total RNA per reaction and random hexamer primers following manufactures protocol. RT-PCR was done with 1♣l cDNA from first strand synthesis in LightCycler 480 SYBR Green I Master (Roche 04707516001) according to manufactures protocol on a LightCycler 480 Instrument with 96-well format. RT-PCR run consisted of an initial denaturation at 95° C for 5min, followed by 45 cycles of amplification with a 5 second hold at 95°C ramped at 4.4°C/s to 55°C. Temperature was then held for 10 seconds at 55°C and ramped up to 72°C at 2.2°C/s. Temperature was held at 72°C for 20 seconds were a single acquisition point was collected and then ramped at 4.4°C/s to begin the cycle anew. A melting curve was collected following amplification. Here, temperature was held at 65° for 1 min and ramped to 97°C at a rate of 0.11°C/s. Five acquisition points were collected per °C. Primers were designed with either the Roche Universal Probe Library Assay Design Center or the IDT Real Time qPCR Assay Entry site using site recommended parameters. Primers were purchased through Sigma Custom DNA Oligo service. Melting curves confirmed primer specificity to single transcripts. A primer list is provided in supplementary table 3. For quantification, standard curves for each primer were applied to all samples using LightCycler 480 software. Ratios of experimental to control samples, normalized to reference genes, are reported. Primers are described in Supplementary Table 5.
Copper Determinations
Copper determinations were made at the University of Georgia Center for Applied Isotype Studies with inductively coupled plasma mass spectroscopy (ICP-MS). Samples were prepared from human fibroblasts cultured in 10% FBS culture media. Cells were then washed with PBS, lifted in PBS supplemented with 10mM EDTA, pelleted and sent for digestion and analysis at the University of Georgia Center for Applied Isotope Studies.
Seahorse Flux Analysis
Cells were cultured in 10% FBS culture media, lifted with trypsin and plated to 96-well Seahorse culture plates (Agilent 101085-004) approximately 24 hours prior to oxygen consumption analysis on a Seahorse XFe96 Analyzer (Agilent). ME32 and ME344 cells were plated at 40,000 cells/well. Human and mouse fibroblasts were plated at 20,000 and 16,000 cells/well respectively. Seahorse XFe96 FluxPaks probe and ports (Agilent 102416-100) were hydrated and loaded according to manufactures recommendations. On the day of analysis, cells were washed twice in Seahorse analysis medium (Seahorse XF Base Medium (Agilent 102353), 1mM Sodium Pyruvate (Sigma S8636), 10mM D-Glucose (Sigma G8769), 2mM L-Glutamine (Hyclone SH30034.01), pH 7.4 with NaOH). A final volume of 180 μl was added to wells and incubated at 37°C in a non-CO2 injected incubator for 1 hour prior to the mitochondrial stress test. Basal oxygen consumption rates were collected for 4 hours following the addition of CuCl2, LDN57444 (Tocris 3998), or vehicle control. Stress tests were performed following the 4 hour basal drug or vehicle control incubations. Final well concentrations for human fibroblast stress test consisted of 1 μM Oligomycin A (Sigma 75351), 1 μM Carbonyl cyanide 4- (trifluoromethoxy)phenylhydrazone, FCCP (Sigma C2920), and 0.5 μM Rotenone (Sigma R8875) and Antimycin A (Sigma A8674) at injections a, b, and c respectively. For mouse fibroblasts, stress tests were conducted with 1 μM Oligomycin, 0.5 μM FCCP, and 0.5 μM Rotenone and Antymicin A. ME32 and ME344 cell stress test used 2μM Oligomycin, 0.125 μM FCCP, and 0.5 μM Rotenone and Antimycin A. Change in oxygen levels was acquired based on recommended Seahorse parameters with cycled rate recordings of a 3 minute mix followed by a 3 minute read of oxygen change over time. Seahorse Wave Software version 2.2.0.276 was used for data analysis.
Flow Cytometry of Reactive Oxygen Species
Fibroblast were plated in 6 well plates the day before the experiment. Cells were incubated in the well with either 4 μg/ml 2,7-Dichlorodihydrofluorescein diacetate (Sigma D6883,) diluted in PBS, 8 μg/ml MitoPy (Tocris cat#4428), 3 μg/ml Dihydroethidium (Invitrogen D11347), 1 μg/ml MitoSox (InvitrogenM36008) in media for 30 min at 37°C. Fluorescence of 10,000 cells was acquired in a LSRII Cytometer (BD, Franklin Lakes, NY, USA) and analyzed using FlowJo (TreeStar Inc. Ashland, OR, USA). Each determination was performed in triplicate wells. To calculate the fold of induction, the average of readings in unaffected female family members was defined as 1.
Drosophila Neuromuscular Microscopy
Larvae were dissected in Ca2+ free HL3 ringer’s solution at room temperature, fixed in 4% paraformaldehyde, stained overnight at 4 degrees with anti-HRP antibody conjugated with FITC. Imaging was performed on an inverted 510 Zeiss LSM microscope. Samples were imaged and analyzed blind. Confocal settings such as black level (offset/contrast), gain, pixel dwell time, pinhole, and the number of iterative samplings (Kalman averaging) were kept constant. Quantification of bouton and branch parameters were done using FIJI (https://imagej.net/Fiji). (Comstra et al., 2017; Gokhale et al., 2016).
Drosophila Viability and Copper Toxicity Assay
The number of male and female flies were counted upon eclosion from a minimum of three replicate vials. Flies were aged 8–2 days and then starved for 3 hours. Age matched flies were placed in a vial with either control food of 5% glucose or food containing 100mM CuSO4 in 5% glucose on a filter disc. Survival was monitored every 24hrs. Flies were imaged using LAS and Zerene Stacker.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental conditions were compared using Synergy Kaleida-Graph, version 4.1.3 (Reading, PA) or Aabel NG2 v5 x64 by Gigawiz as specified in each figure.
DATA AND SOFTWARE AVAILABILITY
All Trio SILAC Labeled Mass Spectrometry raw data were deposited in the public repository PeptideAtlas, which can be reached at: http://www.peptideatlas.org/PASS/PASS01115
Supplementary Material
Fig. S1, related to Fig. 3. Correlation of Transcripts and Protein Expression in the Copper Dyshomeostasis Proteome.
A) Menkes disease pedigrees used for studies in B) where qRT-PCR quantitation of indicated copper dyshomeostasis proteome gene products was performed. Data are represented as heat map where the minimum and maximum changes within a column is depicted. Each determination was performed in triplicate. C) Correlation between mRNA expression and SILAC fold of change expressed as diseased/non-diseased ration. N represents the number of mRNA-protein expression changes measured.
Fig. S2, related to Fig. 4. Additional Bioinformatic Analyses of Genealogical Proteomics from Menkes Disease Pedigrees.
A–E) Kyoto Encyclopedia of Genes and Genomes (KEEG) pathways ontology analyzed with ENRICHR engine, the GeneTerm Linker (F–G), the ClueGo (H), and Gene set Disease Association GDA (I) algorithms (Bindea et al., 2009; Chen et al., 2013; Fontanillo et al., 2011; Kuleshov et al., 2016; Park et al., 2014). A–E) Data are depicted as canvases where every coordinate is occupied by an individual KEGG category whose p value significance is depicted by color intensity. First canvas row depicts SILAC experiments comparing diseased and nondiseased pairs, second row in red depicts non-diseased pair comparison. Third row is the overlay. G) Presents KEGG Term clusters depicted in F. H) ClueGo node size depicts the enrichment significance of the terms. In (I) Gene set Disease Association GDA indicates diseases represented in the copper dyshomeostasis proteome dataset. Individual family and collective bioinformatics data can be found in Supplementary Tables 1–4.
Fig. S3, related to Fig. 5 and 6. Mitoproteome and Free Radical Species in Menkes Disease Fibroblasts.
A) Human pedigrees of families affected by Menkes disease and studied in B–C. B) depict all mass spectrometry quantitations where the color code denotes individuals being compared with pedigrees in A. Changes >2 or <0.5 fall in areas outside the grey box. MS/MS data can be found in Supplementary Tables 2–4. C) Immunoblots of ATP7A and mitochondrial antigens identified in the mitoproteome of human Menkes fibroblasts. Actin was used as loading control (ACTB). D) Quantitation of immunoblots as a ratio of Menkes patient to the carrier female within its own pedigree. Color code of the bars depict the pedigree in A. Average ± SE comparisons were done against beta actin (ACTB). All changes in antigen expression were non-significant, n=3. E) Flow cytometry analysis of human fibroblasts of the specified genotype with reactive oxygen species-sensitive probes 2′,7′-dichlorodihydrofluorescein diacetate (DCF), dihydroethidium (ETH), MitoSox (MSox), and mitoPY (MPTY) (Marullo et al., 2013) (Dickinson and Chang, 2008). Average ± SE n=3 independent experiments done in triplicate except for MPTY n=1 in triplicate.
Fig. S4, related to Fig. 6. Immunodetection of ATP7A Antigen in Fibroblasts with ATP7A Gene Reduction.
A–B) Two wild type (ATP7A+/y), carrier mother (ATP7A+/−, subject 1) and Menkes son (ATP7A−/y, subject 2) fibroblasts were stained with antibodies against ATP7A and clathrin heavy chain (CHC) and imaged by confocal microscopy. Cells were scored blind of their genotype as positive or negative for ATP7A immunoreactivity yet positive for clathrin in B. A) presents two cells from carrier mother ATP7A+/− one positive and another negative for ATP7A immunoreactivity. B) Percent of cells positive for ATP7A. P values represent difference between two independent proportions for the indicated rows.
Fig. S5, related to Fig. 6. Mitochondrial Respiration in Uchl1 null fibroblasts.
A) Oxygen consumption rates in wild type (grey) and Uchl1 null fibroblasts (cyan). Arrows (a) to (c) indicate injection of oligomycin (a), FCCP (b), and rotenone plus antimycin A (c) for stress test. Depicted is average ± SD (n=6). B–D) Wild type (grey) and Uchl1 null fibroblasts (cyan) basal, maximal, and ATP-dependent respiratory rates. Each point is an independent experiment performed in two wild type and two Uchl1 null fibroblast isolates. E) Uchl1 null cell respiration is insensitive to increasing extracellular copper challenge. Figure depicts the change in basal respiration (acute response) at different copper concentrations. Average ± SE (n=6). F) Oxygen consumption rates in wild type and Uchl1 null fibroblasts incubated with vehicle (grey) or LDN57444 (cyan). Arrows (a) to (c) indicate injection of oligomycin (a), FCCP (b), and rotenone plus antimycin A (c) for stress test. Depicted is average ± SD (n=6). G) Immunoblot analysis of wild type and Uchl1 null hippocampus. Hsp90 and the synaptic vesicle SV2 antigen were used as loading controls. H) Immunoblot analysis of wild type and Uchl1 null fibroblasts. The mitochondrial transporter Slc25a1 was used as a loading control antigen. I) Quantitation of blots shown in G–I. Fibroblast data are marked as fib, n are provided by italic numbers. Average ± SE.
Fig. S6, related to Fig. 7. Drosophila ATP7 and Uchl genetically interact to specify Drosophila melanogaster synapse development.
Third instar larvae neuromuscular junction synapses stained with anti-HRP antibodies (A). B. Scoring was done blind to the animal genotype. Control animals (C155 outcross or UAS-Uchl outcross, columns 1–2), animals expressing either UAS-Uchl RNAi or UAS-ATP7A-wt driven by the C155 GAL4 driver (column 3–4), and flies overexpressing ATP7A plus Uchl in neuronal cells (c155>UAS-ATP7A, UAS-Uchl; column 5) were analyzed. Kruskal-Wallis Test followed by pairwise Mann-Whitney U Test, n1-n5= 11, 8, 11, 12, and 9 respectively.
Supplementary Table 1, related to Fig. 2–3. Combined Copper Dyshomeostasis Proteome Hits Summary and Bioinformatic Analysis of Triple SILAC Experiments.
Supplementary Table 2, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 1.
Supplementary Table 3, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 2.
Supplementary Table 4, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 3.
Supplemental Table 5, related to STAR Methods. Primers Used in these Studies.
Highlights.
Genealogical proteomics identifies phenotypes and mechanisms of genetic disease.
Copper dyshomeostasis, due to defects in ATP7A, increases the expression of UCHL1.
UCHL1 expression is required for the pathomechanism of copper dyshomeostasis.
Copper dyshomeostasis mechanisms intersect with Parkinson’s causative genes.
Acknowledgments
This work was supported by grants from the National Institutes of Health NS088503 and R56 MH111459 to VF, DK093386 to MP, and NS092343 to LL. SRR was supported by the Goizueta Foundation STEM Success Initiative, the Frances Marx Shillinglaw Women in Science Fund, and the SURE Scholars Program at Emory University. JBD was supported by the Mattel Initiative. We are indebted to the Faundez lab members for their comments. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study.
Footnotes
Declaration of Interests
There are no interests to declare by all authors
Author Contributions
Conceptualization, V.F., A.V-M., A.G., C.H. and S.Z. Methodology, V.F., A.V-M., A.G., C.H., and S.Z.; Investigation, A.V-M., A.G., C.H., S.Z., L.J.C; E.S., R.B., S.R-R., J.B.D., M.M. M, and E.W; Writing – Original Draft, V.F; Writing – Review & Editing, V.F., A.V-M., A.G., C.H. and S.Z.; Resources, M.P. and L.L.; Funding Acquisition, Supervision, Formal Analysis, Data curation, and Visualization, V.F.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic acids research. 2015;43:D789–798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14:103–113. doi: 10.1016/S1474-4422(14)70190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Yang H, Duffy M, Robinson E, Conrad-Antoville A, Lu YW, Capps T, Braiterman L, Wolfgang M, Murphy MP, et al. The Activity of Menkes Disease Protein ATP7A Is Essential for Redox Balance in Mitochondria. The Journal of biological chemistry. 2016;291:16644–16658. doi: 10.1074/jbc.M116.727248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3489–3494. doi: 10.1073/pnas.1222732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop P, Rocca D, Henley JM. Ubiquitin C-terminal hydrolase L1 (UCH-L1): structure, distribution and roles in brain function and dysfunction. The Biochemical journal. 2016;473:2453–2462. doi: 10.1042/BCJ20160082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- Camakaris J, Mann JR, Danks DM. Copper metabolism in mottled mouse mutants: copper concentrations in tissues during development. The Biochemical journal. 1979;180:597–604. doi: 10.1042/bj1800597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, Gygi SP. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstra HS, McArthy J, Rudin-Rush S, Hartwig C, Gokhale A, Zlatic SA, Blackburn JB, Werner E, Petris M, D’Souza P, et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. Elife. 2017;6 doi: 10.7554/eLife.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Chen JM, Ball EV, Howells K, Mort M, Phillips AD, Chuzhanova N, Krawczak M, Kehrer-Sawatzki H, Stenson PD. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Human mutation. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Science translational medicine. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, Della Valle F, Borreca A, Manca A, Meli G, et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Human molecular genetics. 2015;24:3058–3081. doi: 10.1093/hmg/ddv059. [DOI] [PubMed] [Google Scholar]
- Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiological reviews. 2011;91:1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- Davies KM, Mercer JF, Chen N, Double KL. Copper dyshomoeostasis in Parkinson’s disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci (Lond) 2016;130:565–574. doi: 10.1042/CS20150153. [DOI] [PubMed] [Google Scholar]
- Davison EJ, Pennington K, Hung CC, Peng J, Rafiq R, Ostareck-Lederer A, Ostareck DH, Ardley HC, Banks RE, Robinson PA. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics. 2009;9:4284–4297. doi: 10.1002/pmic.200900126. [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods in enzymology. 2014;547:309–354. doi: 10.1016/B978-0-12-801415-8.00016-3. [DOI] [PubMed] [Google Scholar]
- Dusek P, Litwin T, Czlonkowska A. Wilson disease and other neurodegenerations with metal accumulations. Neurol Clin. 2015;33:175–204. doi: 10.1016/j.ncl.2014.09.006. [DOI] [PubMed] [Google Scholar]
- el-Defrawy SR, Boegman RJ, Jhamandas K, Beninger RJ. The neurotoxic actions of quinolinic acid in the central nervous system. Can J Physiol Pharmacol. 1986;64:369–375. doi: 10.1139/y86-060. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Fontanillo C, Nogales-Cadenas R, Pascual-Montano A, De las Rivas J. Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PloS one. 2011;6:e24289. doi: 10.1371/journal.pone.0024289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, Kruglyak L. Genetic basis of proteome variation in yeast. Nature genetics. 2007;39:1369–1375. doi: 10.1038/ng.2007.22. [DOI] [PubMed] [Google Scholar]
- Fukuoka S, Kawashima R, Asuma E, Shibata K, Fukuwatari T. Quinolinate Accumulation in the Brains of the Quinolinate Phosphoribosyltransferase (QPRT) Knockout Mice. In: Tunali NE, editor. Huntington’s Disease - Core Concepts and Current Advances. INTECH; 2012. [Google Scholar]
- Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, Tifft CJ, Udn The NIH Undiagnosed Diseases Program and Network: Applications to modern medicine. Mol Genet Metab. 2016;117:393–400. doi: 10.1016/j.ymgme.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod A. The lessons of rare maladies. Lancet. 1928;211:1055–1061. [Google Scholar]
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS genetics. 2011;7:e1001393. doi: 10.1371/journal.pgen.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Hartwig C, Freeman AH, Das R, Zlatic SA, Vistein R, Burch A, Carrot G, Lewis AF, Nelms S, et al. The Proteome of BLOC-1 Genetic Defects Identifies the Arp2/3 Actin Polymerization Complex to Function Downstream of the Schizophrenia Susceptibility Factor Dysbindin at the Synapse. J Neurosci. 2016;36:12393–12411. doi: 10.1523/JNEUROSCI.1321-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C, Lupashin VV, Smith Y, Faundez V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci. 2012;32:3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Mullin AP, Zlatic S, Easley CA, Merritt ME, Raj N, Larimore J, Gordon DE, Peden AA, Sanyal S, et al. The N-Ethylmaleimide Sensitive Factor (NSF) and Dysbindin Interact to Modulate Synaptic Plasticity. J Neurosci. 2015;35:7643–7653. doi: 10.1523/JNEUROSCI.4724-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloidinduced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Graham SH, Liu H. Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res Rev. 2017;34:30–38. doi: 10.1016/j.arr.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiology of aging. 2012;33:1843e1841–1847. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiology of disease. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzler P, Haenlin M, Ramain P, Calleja M, Simpson P. A genetic analysis of pannier, a gene necessary for viability of dorsal tissues and bristle positioning in Drosophila. Genetics. 1996;143:1271–1286. doi: 10.1093/genetics/143.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JE, de Bruyne M, Warr CG, Burke R. Copper overload and deficiency both adversely affect the central nervous system of Drosophila. Metallomics: integrated biometal science. 2014;6:2223–2229. doi: 10.1039/c4mt00140k. [DOI] [PubMed] [Google Scholar]
- Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nature reviews Neurology. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG. Inborn errors of copper metabolism. Handbook of clinical neurology. 2013;113:1745–1754. doi: 10.1016/B978-0-444-59565-2.00045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman S, Garbett KA, Janka Z, Mirnics K. Human dermal fibroblasts in psychiatry research. Neuroscience. 2016;320:105–121. doi: 10.1016/j.neuroscience.2016.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose J, Nock C, Herrmann M, Stuhler K, Marcus K, Bluggel M, Krause E, Schalkwyk LC, Rastan S, Brown SD, et al. Genetic analysis of the mouse brain proteome. Nature genetics. 2002;30:385–393. doi: 10.1038/ng861. [DOI] [PubMed] [Google Scholar]
- Kohler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Ayme S, Baynam G, Bello SM, Boerkoel CF, Boycott KM, et al. The Human Phenotype Ontology in 2017. Nucleic acids research. 2017;45:D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz WS, Kuznetsov AV, Clark JF, Tracey I, Elger CE. Metabolic consequences of the cytochrome c oxidase deficiency in brain of copper-deficient Mo(vbr) mice. Journal of neurochemistry. 1999;72:1580–1585. doi: 10.1046/j.1471-4159.1999.721580.x. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Clark JF, Winkler K, Kunz WS. Increase of flux control of cytochrome c oxidase in copper-deficient mottled brindled mice. The Journal of biological chemistry. 1996;271:283–288. doi: 10.1074/jbc.271.1.283. [DOI] [PubMed] [Google Scholar]
- Kyratzi E, Pavlaki M, Stefanis L. The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Human molecular genetics. 2008;17:2160–2171. doi: 10.1093/hmg/ddn115. [DOI] [PubMed] [Google Scholar]
- La Fontaine SL, Firth SD, Camakaris J, Englezou A, Theophilos MB, Petris MJ, Howie M, Lockhart PJ, Greenough M, Brooks H, et al. Correction of the copper transport defect of Menkes patient fibroblasts by expression of the Menkes and Wilson ATPases. The Journal of biological chemistry. 1998;273:31375–31380. doi: 10.1074/jbc.273.47.31375. [DOI] [PubMed] [Google Scholar]
- Larimore J, Zlatic SA, Arnold M, Singleton KS, Cross R, Rudolph H, Bruegge MV, Sweetman A, Garza C, Whisnant E, et al. Dysbindin Deficiency Modifies the Expression of GABA Neuron and Ion Permeation Transcripts in the Developing Hippocampus. Front Genet. 2017;8:28. doi: 10.3389/fgene.2017.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. American journal of human genetics. 2016;98:500–513. doi: 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CM, Cater MA, Mercer JF, La Fontaine S. Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochemical and biophysical research communications. 2006;348:428–436. doi: 10.1016/j.bbrc.2006.07.067. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lashuel HA, Choi S, Xing X, Case A, Ni J, Yeh LA, Cuny GD, Stein RL, Lansbury PT., Jr Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem Biol. 2003;10:837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Lombardino AJ, Li XC, Hertel M, Nottebohm F. Replaceable neurons and neurodegenerative disease share depressed UCHL1 levels. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8036–8041. doi: 10.1073/pnas.0503239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye JC, Hwang JE, Paterson D, de Jonge MD, Howard DL, Burke R. Detection of genetically altered copper levels in Drosophila tissues by synchrotron x-ray fluorescence microscopy. PloS one. 2011;6:e26867. doi: 10.1371/journal.pone.0026867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Manara R, D’Agata L, Rocco MC, Cusmai R, Freri E, Pinelli L, Darra F, Procopio E, Mardari R, Zanus C, et al. Neuroimaging Changes in Menkes Disease, Part 1. AJNR Am J Neuroradiol. 2017a doi: 10.3174/ajnr.A5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manara R, Rocco MC, D’Agata L, Cusmai R, Freri E, Giordano L, Dara F, Procopio E, Toldo I, Peruzzi C, et al. Neuroimaging Changes in Menkes Disease, Part 2. AJNR Am J Neuroradiol. 2017b doi: 10.3174/ajnr.A5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS one. 2013;8:e81162. doi: 10.1371/journal.pone.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon JE, Sha D, Li L, Chin LS. Parkin-mediated K63-polyubiquitination targets ubiquitin C-terminal hydrolase L1 for degradation by the autophagy-lysosome system. Cell Mol Life Sci. 2015;72:1811–1824. doi: 10.1007/s00018-014-1781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. American journal of human genetics. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer SW, La Fontaine S, Warr CG, Burke R. Reduced glutathione biosynthesis in Drosophila melanogaster causes neuronal defects linked to copper deficiency. Journal of neurochemistry. 2016;137:360–370. doi: 10.1111/jnc.13567. [DOI] [PubMed] [Google Scholar]
- Mullin AP, Gokhale A, Moreno-De-Luca A, Sanyal S, Waddington JL, Faundez V. Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl Psychiatry. 2013;3:e329. doi: 10.1038/tp.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgate M, Lee E, Southon A, Farlow A, Batterham P, Camakaris J, Burke R. Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Molecular biology of the cell. 2006;17:475–484. doi: 10.1091/mbc.E05-06-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & cellular proteomics: MCP. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Current biology: CB. 2015;25:2349–2360. doi: 10.1016/j.cub.2015.07.050. [DOI] [PubMed] [Google Scholar]
- Park J, Wick HC, Kee DE, Noto K, Maron JL, Slonim DK. Finding novel molecular connections between developmental processes and disease. PLoS computational biology. 2014;10:e1003578. doi: 10.1371/journal.pcbi.1003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cornejo P, Gokhale A, Duran C, Cui Y, Xiao Q, Hartzell HC, Faundez V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10376–10381. doi: 10.1073/pnas.1200174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder PV, Vistein R, Gokhale A, Seaman MN, Puthenveedu M, Faundez V. The WASH Complex, an Endosomal Arp2/3 Activator, Interacts with the Hermansky-Pudlak Syndrome Complex BLOC-1 and its Cargo Phosphatidylinositol-4-kinase Type II Alpha. Molecular biology of the cell. 2013 doi: 10.1091/mbc.E13-02-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydning SL, Backe PH, Sousa MML, Iqbal Z, Oye AM, Sheng Y, Yang M, Lin X, Slupphaug G, Nordenmark TH, et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Human molecular genetics. 2017;26:1031–1040. doi: 10.1093/hmg/ddw391. [DOI] [PubMed] [Google Scholar]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nature genetics. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017;4:60–72. e64. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SY, Lu SY, Sivasubramaniyam T, Revelo XS, Cai EP, Luk CT, Schroer SA, Patel P, Kim RH, Bombardier E, et al. DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice. Nature communications. 2015;6:7415. doi: 10.1038/ncomms8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton WC, McInnes KT, Cater MA, Winnall WR, McKirdy R, Yu Y, Taylor PE, Ke BX, Richardson DR, Mercer JF, et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. The Journal of biological chemistry. 2010;285:27111–27121. doi: 10.1074/jbc.M110.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, Xiong WC. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell reports. 2015;12:1631–1643. doi: 10.1016/j.celrep.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. Panorama of ancient metazoan macromolecular complexes. Nature. 2015;525:339–344. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintermeyer P, Kruger R, Kuhn W, Muller T, Woitalla D, Berg D, Becker G, Leroy E, Polymeropoulos M, Berger K, et al. Mutation analysis and association studies of the UCHL1 gene in German Parkinson’s disease patients. Neuroreport. 2000;11:2079–2082. doi: 10.1097/00001756-200007140-00004. [DOI] [PubMed] [Google Scholar]
- Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Nihira T, Ren YR, Cao XQ, Wada K, Setsuie R, Kabuta T, Wada K, Hattori N, Mizuno Y, et al. Effects of UCH-L1 on alpha-synuclein over-expression mouse model of Parkinson’s disease. Journal of neurochemistry. 2009;108:932–944. doi: 10.1111/j.1471-4159.2008.05827.x. [DOI] [PubMed] [Google Scholar]
- Zanon A, Rakovic A, Blankenburg H, Doncheva NT, Schwienbacher C, Serafin A, Alexa A, Weichenberger CX, Albrecht M, Klein C, et al. Profiling of Parkin-binding partners using tandem affinity purification. PloS one. 2013;8:e78648. doi: 10.1371/journal.pone.0078648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic S, Comstra HS, Gokhale A, Petris MJ, Faundez V. Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease. Neurobiology of disease. 2015;81:154–161. doi: 10.1016/j.nbd.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatic SA, Grossniklaus EJ, Ryder PV, Salazar G, Mattheyses AL, Peden AA, Faundez V. Chemical-genetic disruption of clathrin function spares adaptor complex 3-dependent endosome vesicle biogenesis. Molecular biology of the cell. 2013;24:2378–2388. doi: 10.1091/mbc.E12-12-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1, related to Fig. 3. Correlation of Transcripts and Protein Expression in the Copper Dyshomeostasis Proteome.
A) Menkes disease pedigrees used for studies in B) where qRT-PCR quantitation of indicated copper dyshomeostasis proteome gene products was performed. Data are represented as heat map where the minimum and maximum changes within a column is depicted. Each determination was performed in triplicate. C) Correlation between mRNA expression and SILAC fold of change expressed as diseased/non-diseased ration. N represents the number of mRNA-protein expression changes measured.
Fig. S2, related to Fig. 4. Additional Bioinformatic Analyses of Genealogical Proteomics from Menkes Disease Pedigrees.
A–E) Kyoto Encyclopedia of Genes and Genomes (KEEG) pathways ontology analyzed with ENRICHR engine, the GeneTerm Linker (F–G), the ClueGo (H), and Gene set Disease Association GDA (I) algorithms (Bindea et al., 2009; Chen et al., 2013; Fontanillo et al., 2011; Kuleshov et al., 2016; Park et al., 2014). A–E) Data are depicted as canvases where every coordinate is occupied by an individual KEGG category whose p value significance is depicted by color intensity. First canvas row depicts SILAC experiments comparing diseased and nondiseased pairs, second row in red depicts non-diseased pair comparison. Third row is the overlay. G) Presents KEGG Term clusters depicted in F. H) ClueGo node size depicts the enrichment significance of the terms. In (I) Gene set Disease Association GDA indicates diseases represented in the copper dyshomeostasis proteome dataset. Individual family and collective bioinformatics data can be found in Supplementary Tables 1–4.
Fig. S3, related to Fig. 5 and 6. Mitoproteome and Free Radical Species in Menkes Disease Fibroblasts.
A) Human pedigrees of families affected by Menkes disease and studied in B–C. B) depict all mass spectrometry quantitations where the color code denotes individuals being compared with pedigrees in A. Changes >2 or <0.5 fall in areas outside the grey box. MS/MS data can be found in Supplementary Tables 2–4. C) Immunoblots of ATP7A and mitochondrial antigens identified in the mitoproteome of human Menkes fibroblasts. Actin was used as loading control (ACTB). D) Quantitation of immunoblots as a ratio of Menkes patient to the carrier female within its own pedigree. Color code of the bars depict the pedigree in A. Average ± SE comparisons were done against beta actin (ACTB). All changes in antigen expression were non-significant, n=3. E) Flow cytometry analysis of human fibroblasts of the specified genotype with reactive oxygen species-sensitive probes 2′,7′-dichlorodihydrofluorescein diacetate (DCF), dihydroethidium (ETH), MitoSox (MSox), and mitoPY (MPTY) (Marullo et al., 2013) (Dickinson and Chang, 2008). Average ± SE n=3 independent experiments done in triplicate except for MPTY n=1 in triplicate.
Fig. S4, related to Fig. 6. Immunodetection of ATP7A Antigen in Fibroblasts with ATP7A Gene Reduction.
A–B) Two wild type (ATP7A+/y), carrier mother (ATP7A+/−, subject 1) and Menkes son (ATP7A−/y, subject 2) fibroblasts were stained with antibodies against ATP7A and clathrin heavy chain (CHC) and imaged by confocal microscopy. Cells were scored blind of their genotype as positive or negative for ATP7A immunoreactivity yet positive for clathrin in B. A) presents two cells from carrier mother ATP7A+/− one positive and another negative for ATP7A immunoreactivity. B) Percent of cells positive for ATP7A. P values represent difference between two independent proportions for the indicated rows.
Fig. S5, related to Fig. 6. Mitochondrial Respiration in Uchl1 null fibroblasts.
A) Oxygen consumption rates in wild type (grey) and Uchl1 null fibroblasts (cyan). Arrows (a) to (c) indicate injection of oligomycin (a), FCCP (b), and rotenone plus antimycin A (c) for stress test. Depicted is average ± SD (n=6). B–D) Wild type (grey) and Uchl1 null fibroblasts (cyan) basal, maximal, and ATP-dependent respiratory rates. Each point is an independent experiment performed in two wild type and two Uchl1 null fibroblast isolates. E) Uchl1 null cell respiration is insensitive to increasing extracellular copper challenge. Figure depicts the change in basal respiration (acute response) at different copper concentrations. Average ± SE (n=6). F) Oxygen consumption rates in wild type and Uchl1 null fibroblasts incubated with vehicle (grey) or LDN57444 (cyan). Arrows (a) to (c) indicate injection of oligomycin (a), FCCP (b), and rotenone plus antimycin A (c) for stress test. Depicted is average ± SD (n=6). G) Immunoblot analysis of wild type and Uchl1 null hippocampus. Hsp90 and the synaptic vesicle SV2 antigen were used as loading controls. H) Immunoblot analysis of wild type and Uchl1 null fibroblasts. The mitochondrial transporter Slc25a1 was used as a loading control antigen. I) Quantitation of blots shown in G–I. Fibroblast data are marked as fib, n are provided by italic numbers. Average ± SE.
Fig. S6, related to Fig. 7. Drosophila ATP7 and Uchl genetically interact to specify Drosophila melanogaster synapse development.
Third instar larvae neuromuscular junction synapses stained with anti-HRP antibodies (A). B. Scoring was done blind to the animal genotype. Control animals (C155 outcross or UAS-Uchl outcross, columns 1–2), animals expressing either UAS-Uchl RNAi or UAS-ATP7A-wt driven by the C155 GAL4 driver (column 3–4), and flies overexpressing ATP7A plus Uchl in neuronal cells (c155>UAS-ATP7A, UAS-Uchl; column 5) were analyzed. Kruskal-Wallis Test followed by pairwise Mann-Whitney U Test, n1-n5= 11, 8, 11, 12, and 9 respectively.
Supplementary Table 1, related to Fig. 2–3. Combined Copper Dyshomeostasis Proteome Hits Summary and Bioinformatic Analysis of Triple SILAC Experiments.
Supplementary Table 2, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 1.
Supplementary Table 3, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 2.
Supplementary Table 4, related to Fig. 2–3. Copper Dyshomeostasis Proteome Hits and Bioinformatic Analyses of SILAC Experiment 3.
Supplemental Table 5, related to STAR Methods. Primers Used in these Studies.