

Abstract
Haploinsufficiency of the SHANK3 gene is causally linked to autism spectrum disorder (ASD), and ASD-associated genes are also enriched for chromatin remodelers. Here, we found that brief treatment with romidepsin, a highly potent class I histone deacetylase (HDAC) inhibitor, alleviated social deficits in Shank3-deficient mice, which persisted for ~3 weeks. HDAC2 transcription was upregulated in these mice, and knockdown of HDAC2 in prefrontal cortex also rescued their social deficits. Nuclear localization of β-catenin, a Shank3-binding protein that regulates cell adhesion and transcription, was increased in Shank3-deficient mice, which induced HDAC2 upregulation and social deficits. At the downstream molecular level, romidepsin treatment elevated the expression and histone acetylation of Grin2a and actin regulatory genes, and restored NMDAR function and actin filaments in Shank3-deficient mice. Taken together, these findings highlight an epigenetic mechanism underlying social deficits linked to Shank3 deficiency, which may suggest potential therapeutic strategies for ASD patients bearing SHANK3 mutations.
Introduction
Mouse models that reflect the core symptoms of autism spectrum disorder (ASD), including deficits in sociability and repetitive behaviors, are crucial for pre-clinical investigations of pathophysiological mechanisms and novel treatment avenues1. Haploinsufficiency of the SHANK3 gene, which encodes a master scaffolding protein in the postsynaptic density of glutamatergic synapses2, is causally linked to 22q13.3 deletion syndrome (also known as Phelan-McDermid Syndrome), which conveys a high risk for ASD3–6. Deletion or loss-of-function mutations of one copy of SHANK3 account for 0.5%-2.0% of ASD and intellectual disability cases5,6. Human genetic studies of ASD have found that enriched mutations are located at SHANK3 C-terminal region (exon 21)4, which contains binding sites for actin/Cortactin and mGluR/Homer and plays a crucial role in the synaptic targeting and postsynaptic assembly of Shank3 complex.
Mouse models with Shank3 deficiency exhibit autism-associated behaviors to different extents7–16. Male heterozygous mice with C-terminal (exon 21) deleted Shank3 (Shank3+/ΔC), which exhibit reduced full-length Shank3 expression11,12 and the loss of synaptic localization of the truncated Shank3 protein12, recapitulate social preference deficits and repetitive behaviors12. These behavioral deficits in Shank3+/ΔC mice are attributable to the loss of NMDAR function and synaptic trafficking due to actin dysregulation in pyramidal neurons of prefrontal cortex (PFC)12, a brain region that plays an essential role in mediating social cognition17.
Large scale genetic studies have revealed that many ASD-associated genes are involved in synaptic homeostasis, transcriptional regulation, and chromatin remodeling pathways18–21. This prompted us to speculate that targeting epigenetic enzymes to normalize gene expression and ameliorate synaptic defects may be a potential strategy to relieve social deficits in ASD. Here, we targeted histone deacetylase (HDAC) family proteins, which generally cause silencing of gene expression via condensing the chromatin architecture. HDACs play a key role in cognitive processes22,23, and altered histone acetylation and transcriptional dysfunction have been implicated in psychiatric disorders24, however, the efficacy of HDAC-targeting agents in models of ASD-associated mutations is unknown. Here we have identified an HDAC inhibitor that persistently alleviates social deficits in Shank3-deficient mice.
Results
Treatment with the HDAC inhibitor romidepsin lastingly relieves social deficits in Shank3-deficient mice
The level of global H3 acetylation (Fig. 1a) in the frontal cortex of Shank3+/ΔC mice was significantly lower than that from wild-type (WT) mice. A systemic administration of the low dose romidepsin (0.25 mg/kg, i.p., once daily for 3 days), a highly potent and brain-permeable class I-specific HDAC inhibitor (with nanomolar in vitro potency25) approved by FDA for cancer treatment26–28, significantly elevated the level of acetylated H3 in Shank3+/ΔC mice, while it had little effect in WT mice. These data suggest that Shank3-deficient mice have an abnormally low level of histone acetylation, which can be restored by romidepsin treatment.
Figure 1. Treatment with the HDAC inhibitor romidepsin induces the robust and prolonged rescue of autism-like social deficits in Shank3-deficient mice, while a variety of drugs for psychiatric disorders fail to do so.
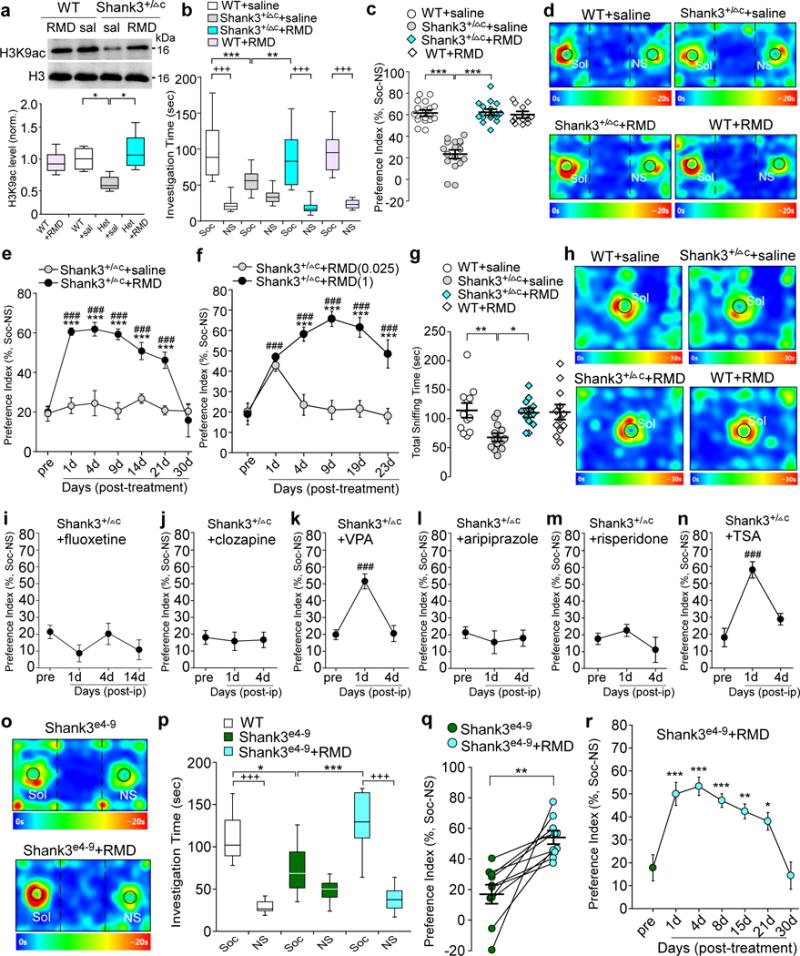
(a) Immunoblots and quantification analysis of the level of acetylated H3 and total H3 in the nuclear fraction of cortical slices from wild-type (WT) or Shank3+/ΔC (Het) mice injected (i.p.) with saline or romidepsin (RMD, 0.25 mg/kg, 3×). Immunoblotting was performed at 4-5 days post-injection. F1,20=11.3, P=0.0031; * P<0.05, two-way ANOVA, n=6 each group. (b, c) Plots showing the time spent investigating either the social (Soc) or nonsocial (NS) stimulus (b) and the social preference index (c) during 3-chamber sociability testing of saline-injected WT (n=18), saline-injected Shank3+/ΔC (n=17), RMD-treated Shank3+/ΔC (n=17) and RMD-treated WT (n=12) mice. In (b), F3,120=11.4, P<0.0001; +++ P<0.001 (Soc vs. NS), ** P<0.01, *** P<0.001, two-way ANOVA. In (c), F1,60=58.5, P<0.0001; *** P<0.001, two-way ANOVA. (d) Representative heat maps illustrating the time spent in different locations of the 3 chambers from the social preference tests of all groups. Locations of Sol and NS stimuli are labeled with the circles. (e) Plots of social preference index in Shank3+/ΔC mice treated with romidepsin (n=10) or saline (n=10) at different time points. F1,18(treatment)=124.3, P<0.0001; *** P<0.001 (saline vs. romidepsin), ### P<0.001 (pre- vs. post-injection), two-way rmANOVA. (f) Plots of social preference index in Shank3+/ΔC mice treated with different doses of romidepsin (0.025 mg/kg, n=9; 1 mg/kg, n=8) at different time points. F1,15(treatment)=51.3, P<0.0001; *** P<0.001 (1 mg/kg vs. 0.025 mg/kg RMD); ### P<0.001 (pre- vs. post-injection), two-way rmANOVA. (g) Scatter plots showing total sniffing time in social approach tests of saline-injected WT (n=12), saline-injected Shank3+/ΔC (n=14), RMD-treated Shank3+/ΔC (n=12) and RMD-treated WT (n=12) mice. F1,46=6.0, P=0.018; * P<0.05, ** P<0.01, two-way ANOVA. (h) Representative heat maps illustrating the time spent in different locations of the apparatus from the social approach tests of all groups. Locations of social stimuli are labeled with the circles. (i-n) Plots of social preference index in Shank3+/ΔC mice treated with fluoxetine (10 mg/kg, i.p., 14×, i, n=9), clozapine (5 mg/kg, i.p., 3×, j, n=11), valproic acid (VPA, 100 mg/kg, i.p., 3×, k, n=11), aripiprazole (1 mg/kg, i.p., 3×, l, n=9), risperidone (0.1 mg/kg, i.p., 3×, m, n=10), or Trichostatin A (TSA, 0.5 mg/kg, i.p., 3×, n, n=8). F2,30=19.2 (VPA), P<0.0001; F2,21=19.7, P<0.0001 (TSA); ### P<0.001 (pre- vs. post-injection), one-way ANOVA. (o) Representative heat maps illustrating the time spent in different locations of the 3 chambers from the social preference tests of a Shank3e4-9 mouse before and after romidepsin treatment (0.25 mg/kg, i.p., 3×). (p) Box plots showing the time spent investigating either Soc or NS stimulus during sociability testing in WT (n=8) or homozygous Shank3e4-9 mice (n=10) before and after romidepsin treatment. F2,50=9.2, P=0.0003; +++ P<0.001 (Soc vs. NS), * P<0.05, *** P<0.001, two-way ANOVA. (q) Scatter plots showing the preference index of the sociability testing in individual Shank3e4-9 mice before and after romidepsin treatment (n=10). t9=4.36, ** P=0.0018, paired two-tailed t-test. (r) Plots of social preference index in Shank3e4-9 mice (n=10) treated with romidepsin at different time points. F6,63=11.8, P<0.0001; * P<0.05, ** P<0.01, *** P<0.001 (pre- vs. post-injection), one-way ANOVA. Shank3+/ΔC mice (a-n) and WT mice (a-n,p) are all males (5-6 weeks old); Shank3e4-9 mice (o-r) are 6 males and 4 females (5-6 weeks old). Data are presented as median with interquartile range (a,b,p) or mean ± SEM (c,e-g,i-n,q,r). Each set of the experiments was replicated for at least 3 times. See Supplementary Fig. 8 for blot source data.
Next, we examined the impact of romidepsin on social deficits in young (5-6 weeks old) male Shank3+/ΔC mice, which exhibit loss of social preference in the three-chamber social interaction assay12. As shown in Fig. 1b and 1d, during the presentation of both a social (Soc) and a non-social (NS) stimuli, romidepsin (0.25 mg/kg, i.p, 3×)-treated Shank3+/ΔC mice spent significantly more time exploring the social stimulus over the non-social object, similar to WT mice, while saline-injected Shank3+/ΔC mice showed a significant loss of the preference for the social stimulus. WT mice treated with romidepsin had unchanged social preference. The significantly elevated social preference index in Shank3+/ΔC mice after romidepsin treatment (Fig. 1c) suggests that romidepsin alleviates the observed social deficits.
We then examined how long the effect of a brief romidepsin treatment could last. The social preference assays were performed in Shank3+/ΔC mice prior to and at various time points after drug treatment. As shown in Fig. 1e, the significantly increased social preference index in Shank3+/ΔC mice persisted for at least 21 days post-injection of romidepsin (0.25 mg/kg, i.p, 3×), while no improvement in social preference was found with repeated measurements of saline-injected Shank3+/ΔC mice. Romidepsin treatment not only persistently improved social preference, but also induced a sustained increase in the time interacting with the social stimulus in young Shank3+/ΔC mice (Supplementary Fig. 1a and 1b). The prolonged effect of romidepsin is consistent with the long duration of human response to romidepsin (13.7 months) revealed by pharmacokenetics analyses26,27. Romidepsin treatment of adult Shank3+/ΔC mice gave a less potent and more transient effect on social behaviors (Supplementary Fig. 1c and 1d), suggesting that the therapeutic efficacy of the drug is influenced by developmental processes.
The effective dose of romidepsin in rescuing social deficits (0.25 mg/kg, i.e. 0.75 mg/m2, i.p., once daily for 3 days) is equivalent to ~5% of the clinical anti-cancer dose in humans (14 mg/m2, i.v., once a week for 3 weeks). Dose response studies (Fig. 1f) indicated that a higher dose of romidepsin (1 mg/kg, i.p., 3×) gave a similar long-lasting rescuing effects on the social preference behavior in Shank3+/ΔC mice, while a lower dose of romidepsin (0.025 mg/kg, i.p., 3×) was ineffective in improving the social deficits persistently. Thus, romidepsin (0.25 mg/kg) was selected in following studies.
We further examined the impact of romidepsin on social approach behaviors in young Shank3+/ΔC mice. As shown in Fig. 1g and 1h, compared to WT mice, Shank3+/ΔC mice spent significantly less time approaching and interacting with social stimuli, which was almost completely reversed by romidepsin treatment. Taken together, these results suggest that a brief treatment with the low dose romidepsin can lead to prolonged rescue of autism-like social deficits in the Shank3-deficient model.
Moreover, we examined whether other pharmacological agents currently used for various psychiatric disorders were able to rescue social deficits in Shank3+/ΔC mice. As shown in Fig. 1i, treatment with the SSRI antidepressant fluoxetine (once daily for 14 days) failed to induce any improvement of the social preference in the three-chamber social interaction assay. The atypical antipsychotics clozapine (once daily for 3 days) was also ineffective in rescuing the social deficits (Fig. 1j). The mood stabilizer valproic acid (VPA, once daily for 3 days), a low affinity class I HDAC inhibitor (with millimolar in vitro potency25), only transiently increased the social preference index, which disappeared within a few days after treatment (Fig. 1k). No improvement on social preference was observed with aripiprazole (once daily for 3 days, Fig. 1l) or risperidone (once daily for 3 days, Fig. 1m), the only drugs approved by FDA for ASD treatment, which is consistent with the lack of effect of these drugs on the social communication deficits observed in ASD patients despite their utility in managing irritability and repetitive behaviors29. All these antipsychotics failed to increase the time interacting with the social stimulus in Shank3+/ΔC mice (Supplementary Fig. 2a–e). Treatment with the pan-HDAC inhibitor Trichostatin A (TSA, once daily for 3 days) significantly but transiently increased social preference index (Fig. 1n) and social interaction time (Supplementary Fig. 2f and 2g) in Shank3+/ΔC mice. These data suggest that the potent and prolonged effectiveness of romidepsin to alleviate social deficits is unique.
To examine the generalizability of the rescuing effect of romidepsin on social deficits, we examined another Shank3-deficient model, Shank3e4-9 mice, which exhibit the loss of major Shank3 isoforms because of the deletion of N-terminal exon 4-910. As shown in Fig. 1o–q, in the 3-chamber sociability test, homozygous Shank3e4-9 mice had significantly impaired social preference, consistent with previous findings10. After romidepsin treatment (0.25 mg/kg, i.p, 3×), the social interaction time and social preference index were significantly elevated. Moreover, the rescuing effect of romidepsin in Shank3e4-9 mice sustained for ~3 weeks (Fig. 1r), similar to what was found in Shank3+/ΔC mice. These results suggest that romidepsin is generally efficacious in mouse models of Shank3 deficiency.
A variety of other behaviors were also examined in romidepsin-treated young Shank3+/ΔC mice. Compared to wild-type mice or saline-injected Shank3+/ΔC mice, no differences were observed with romidepsin treatment in locomotion tests (Fig. 2a). Rotarod tests also found no difference with romidepsin treatment (Fig. 2b). Romidepsin- or saline-treated Shank3+/ΔC mice spent similar time in the center in open-field tests, compared to WT mice (Fig. 2c). These data suggest that romidepsin does not alter motor coordination or anxiety-like behavior. Romidepsin failed to normalize the increased repetitive grooming in Shank3+/ΔC mice (Fig. 2d).
Figure 2. Romidepsin treatment does not affect locomotor or anxiety-like behaviors or neuronal survival in Shank3-deficient mice.
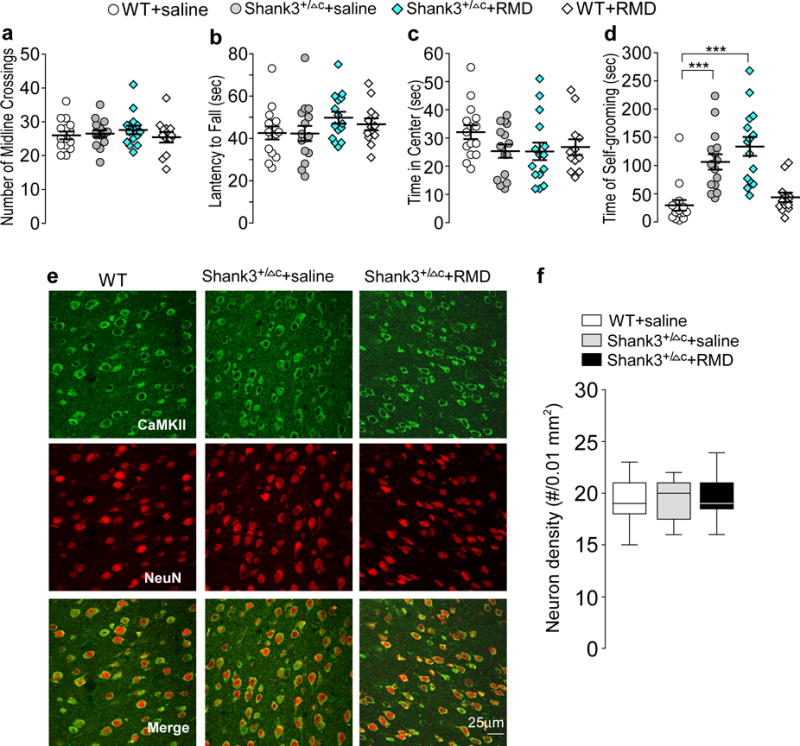
(a-d) Scatter plots showing a variety of behaviors in saline-injected WT (n=15), saline-injected Shank3+/ΔC (n=15), romidepsin (RMD, 0.25 mg/kg, 3×)-treated Shank3+/ΔC (n=15) and RMD-treated WT (n=12) mice, including the number of midline crossing in locomotion tests (a), the latency to fall during rotarod tests (b), the time spent in the center during open-field tests (c), and the time spent self-grooming (d). In (d), F1,53(treatment)=2.55, P=0.117; *** P<0.001, two-way ANOVA. (e, f) Confocal images (e) and quantification (f) of layer V prefrontal cortical neurons (immunostained with α-CaMKII and NeuN) in WT or Shank3+/ΔC mice treated with saline or romidepsin. Slices were collected for immunostaining at 4-5 days post-injection. n=27 images/3 mice each group. All animals used are males (5-6 weeks old). Data are presented as mean ± SEM (a-d) or median with interquartile range (f). Each set of the experiments was replicated for at least 3 times.
Romidepsin treatment did not cause non-specific behavioral effects, suggesting that it is generally safe, consistent with comprehensive safety studies showing that this FDA-approved anti-cancer drug is safe and well tolerated26,28. To further examine the potential neuronal toxicity of low dose romidepsin treatment in young Shank3+/ΔC mice, we performed immunocytochemical experiments to compare the survival neurons. Pyramidal neurons were immunostained with α-CaMKII and NeuN (Neuronal Nuclei), and cell density was counted in the PFC region. As shown in Fig. 2e and 2f, neuronal morphology and density were not altered by romidepsin treatment.
HDAC2 is upregulated in Shank3-deficient mice via a β-catenin-mediated transcriptional mechanism
To find out which HDAC family member may mediate the therapeutic effect of romidepsin, we first examined the expression of class I HDACs. Quantitative PCR analyses indicated that the level of HDAC2 mRNA was significantly higher in PFC lysates from Shank3+/ΔC mice, while the level of HDAC1, HDAC3 and HDAC8 mRNA was largely unchanged (Fig. 3a). Western blot analyses showed that the protein level of HDAC2, but not HDAC1, in the nucleus fraction of PFC was significantly higher in Shank3+/ΔC mice (Fig. 3b). The significantly elevated HDAC2 mRNA and HDAC2 protein were also detected in the PFC of Shank3e4-9 mice (Fig. 3c). It suggests that the upregulated HDAC2 may be responsible for the abnormally low histone acetylation in Shank3-deficient mice.
Figure 3. Shank3 deficiency induces HDAC2 upregulation, and HDAC2 knockdown in PFC rescues autism-like social deficits.
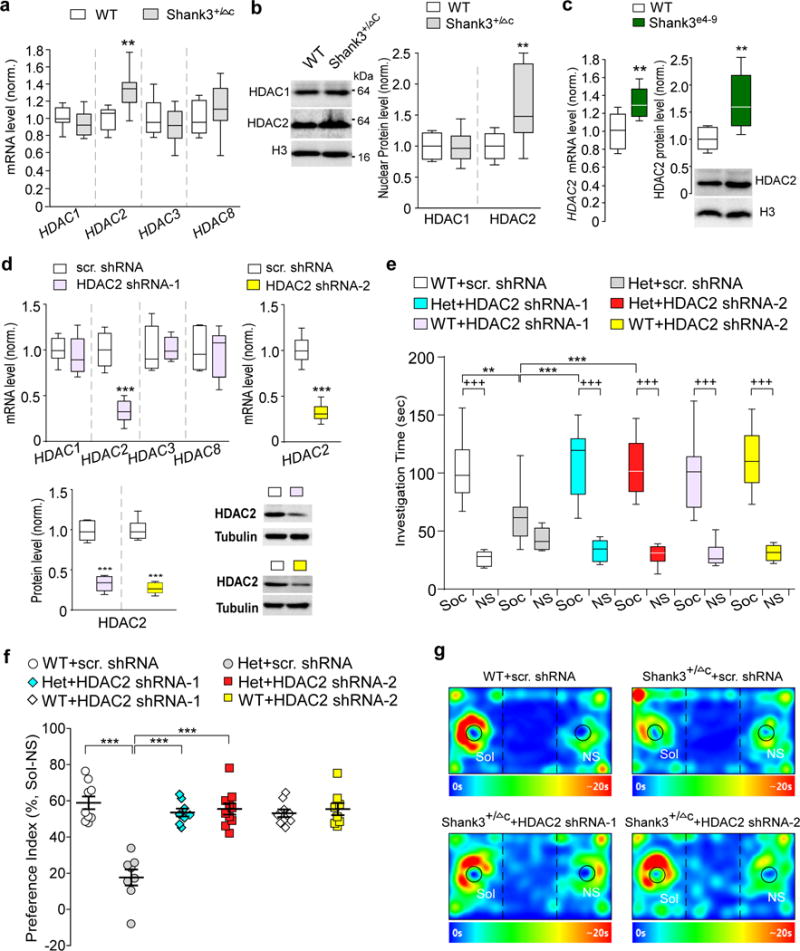
(a) Quantitative real-time RT-PCR data on the mRNA level of class I HDAC family members (HDAC1, 2, 3, 8) in PFC from WT and Shank3+/ΔC mice. t16=3.75, ** P=0.0017 (HDAC2), n=9 each group, two-tailed t-test. (b) Immunoblots and quantification analysis of the protein level of HDAC1 and HDAC2 in the nuclear fraction of PFC neurons from WT vs. Shank3+/ΔC mice. t16=3.00, ** P=0.0085 (HDAC2), n=9 each group, two-tailed t-test. (c) qPCR and Western blot data showing HDAC2 mRNA and protein levels in PFC from WT and Shank3e4-9 mice. t14=3.24, ** P=0.006 (mRNA), n=8 each group; t16=3.79, ** P=0.0016 (protein), n=9 each group, two-tailed t-test. (d) qPCR and Western blot data showing HDAC2 mRNA and protein levels in PFC infected with one of the two different HDAC2 shRNA lentiviruses or a scrambled shRNA lentivirus. shRNA-1: t10=7.28, *** P<0.0001 (HDAC2 mRNA), n=6 each group; t10=10.84, *** P<0.0001 (HDAC2 protein), n=6 each group; shRNA-2: t14=11.33, *** P<0.0001 (HDAC2 mRNA), n=8 each group; t10=12.47, *** P<0.0001 (HDAC2 protein), n=6 each group, two-tailed t-test. (e, f) Plots showing the time spent investigating either the social (Soc) or nonsocial (NS) stimulus (e) and the social preference index (f) during 3-chamber sociability testing of WT or Shank3+/ΔC mice with PFC injection of one of the two different HDAC2 shRNA lentiviruses or a scrambled shRNA lentivirus (n=8-10 each group). In (e), F5,92=5.1, P=0.0004; In (f), F2,46=26.2, P<0.0001; +++ P<0.001 (Soc vs. NS), ** P<0.01, *** P<0.001, two-way ANOVA. (g) Representative heat maps of the 3-chamber sociability tests of WT or Shank3+/ΔC mice injected with different viruses. All animals used are males (5-8 weeks old). Data are presented as median with interquartile range (a,b,c,d,e) or mean ± SEM (f). Each set of the experiments was replicated for at least 3 times. See Supplementary Fig. 8 for blot source data.
Next, we tested whether knockdown of HDAC2 by a stereotaxic injection of HDAC2 shRNA lentivirus to medial PFC of Shank3+/ΔC mice might rescue the social deficits. To prevent potential off-target effects, two shRNA lentiviruses targeting different sequences of the mouse HDAC2 gene (HDAC2 shRNA-1 and HDAC2 shRNA-2) were used. As shown in Fig. 3d, compared to a scrambled control shRNA, HDAC2 shRNA-1 specifically knocked down the mRNA level of HDAC2, but not other class I HDAC family members. HDAC2 shRNA-2 was also highly effective in suppressing HDAC2 mRNA. The level of HDAC2 protein was significantly diminished by injecting these HDAC2 shRNA lentiviruses in vivo. Behavioral assays indicated that Shank3+/ΔC mice with PFC injection of either HDAC2 shRNA-1 or HDAC2 shRNA-2 lentivirus exhibited the significantly increased social interaction time and improved preference for the social stimulus over the non-social object in 3-chamber sociability tests (Fig. 3e–g). These data suggest that knockdown of HDAC2 in PFC induces a similar rescuing effect as romidepsin treatment on social deficits.
To understand how the loss of Shank3 at synapses leads to the upregulation of HDAC2 transcription in the nucleus, we turned our attention to β-catenin, a nucleocytoplasmic shuttle protein involved in both cell adhesion and transcriptional regulation30. In the default state, β-catenin is phosphorylated and retained in the cytoplasm, where it is constitutively targeted for proteasomal degradation. Activation of Wnt signaling results in the nuclear translocation of β-catenin, where it activates target genes in a complex with LEF/TCF transcription factors. Neural activity induces the redistribution of β-catenin into spines, where it interacts with the adhesion molecule cadherin to influence synaptic size and strength31. β-catenin has also been identified as a critical mediator of dendritic morphogenesis via interacting with cadherin to stabilize actin cytoskeleton32. Our co-immunoprecipitation experiments revealed that β-catenin bound to Shank3 in WT mice (Fig. 4a). In PFC of Shank3+/ΔC mice, β-catenin at the synaptic membrane fraction was significantly reduced, while the level of active (non-phosphorylated) β-catenin in the cytosol and the level of β-catenin in the nucleus were significantly elevated (Fig. 4b). In PFC of Shank3e4-9 mice, the decreased synaptic β-catenin and increased nuclear β-catenin were also observed (Fig. 4c). These results suggest that Shank3 deficiency induces the redistribution of β-catenin from synapses to nucleus, probably due to the attenuated association of β-catenin with synaptic scaffolding proteins and adhesion molecules.
Figure 4. β-catenin is nuclear-translocated to activate HDAC2 transcription in Shank3-deficient mice, and manipulation of β-catenin directly affects HDAC2 transcription and social behaviors.
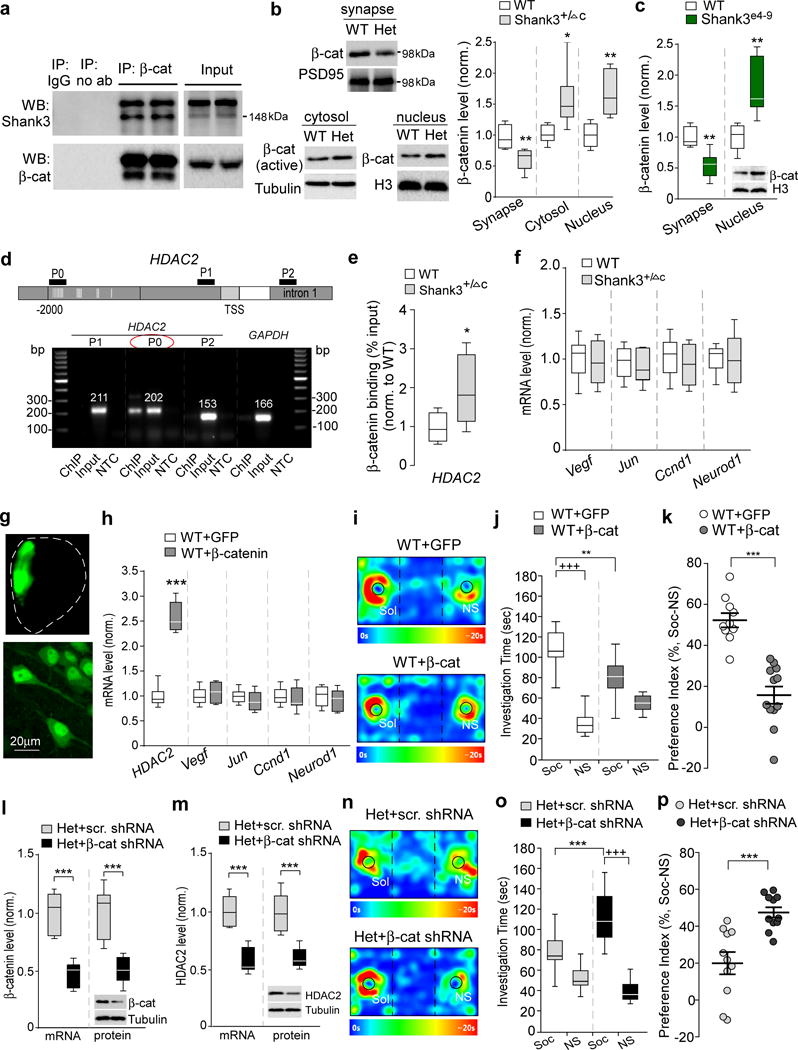
(a) Co-immunoprecipitation data from cortical lysates of WT mice showing the specific binding of β-catenin to Shank3. (b) Immunoblots and quantification analysis of the level of β-catenin in synaptic membrane, cytosol or nuclear fractions of cortical slices from WT and Shank3+/ΔC (Het) mice. t10=3.35, ** P=0.0074 (synapse), t10=2.78, * P=0.0194 (cytosol), t10=4.01, ** P=0.0025 (nucleus), n=6 each group, two-tailed t-test. (c) Quantification analysis of synaptic and nuclear β-catenin levels in cortical slices from WT and Shank3e4-9 mice. t10=4.33, ** P=0.0015 (synapse), t10=3.82, ** P=0.0034 (nucleus), n=6 each group, two-tailed t-test. (d) PCR images showing the ChIP (β-catenin-occupied DNA), input (total DNA) and no-template control (NTC) signals with 3 primers (P0, P1, P2) designed against different regions of the HDAC2 gene, including the promoter region containing (P0) or lacking (P1) the TCF/LEF binding motif (labeled with vertical lines) and a down-stream intron (P2). TSS, transcriptional start site. GAPDH was used as a control. (e) ChIP assay data showing the binding of β-catenin at HDAC2 promoter region (containing TCF/LEF binding motif) in PFC lysates from WT and Shank3+/ΔC mice. t12=2.6, * P=0.02, n=7 each group, two-tailed t-test. (f) Quantitative real-time RT-PCR data on the mRNA level of other β-catenin target genes (Vegf, Jun, Ccnd1, and Neurod1) in PFC from WT and Shank3+/ΔC mice (n=8 each group). (g) Images showing the β-catenin (GFP-tagged) adenovirus-infected medial PFC region (top) and PFC neurons (bottom). (h) qPCR data on the mRNA level of HDAC2 and other β-catenin target genes in PFC of WT mice with the overexpression of GFP-tagged β-catenin (n=10) or GFP control (n=8). t16=13.44, *** P<0.0001 (HDAC2), two-tailed t-test. (i-k) Representative heat maps (i), plots of social interaction time (j) and social preference (k) in 3-chamber sociability tests of WT mice with the overexpression of β-catenin (n=12) or GFP control (n=10) in PFC. j: F1,40=20.5, P<0.0001; +++ P<0.001 (Soc vs. NS), ** P<0.01 (GFP vs. β-catenin), two-way ANOVA. k: t20=6.4, *** P<0.0001, two-tailed t-test. (l, m) qPCR and Western blot data showing the mRNA and protein level of β-catenin (l) and HDAC2 (m) in Shank3+/ΔC mice with the stereotaxic injection of β-catenin shRNA or a scrambled shRNA lentivirus into the PFC. In (I), t10=6.47, *** P<0.0001 (β-catenin mRNA); t10=4.88, *** P=0.0006 (β-catenin protein). In (m), t10=6.20, *** P=0.0001 (HDAC2 mRNA); t10=5.05, *** P=0.0005 (HDAC2 protein), n=6 each group, two-tailed t-test. (n-p) Representative heat maps (n), plots of social interaction time (o) and social preference (p) in 3-chamber sociability tests of Shank3+/ΔC mice injected with β-catenin shRNA (n=11) or a scrambled shRNA (n=11) lentivirus into the PFC. o: F1,40=15.0, P=0.0004; +++ P<0.001 (Soc vs. NS), *** P<0.001 (β-catenin shRNA vs. scrambled shRNA), two-way ANOVA. p: t20=4.36, *** P=0.0003, two-tailed t-test. All animals used are males (5-8 weeks old). Data are presented as median with interquartile range (b,c,e,f,h,j,l,m,o) or mean ± SEM (k,p). Each set of the experiments was replicated for at least 3 times. See Supplementary Fig. 8 for blot source data.
The increased nuclear translocation of β-catenin in Shank3-deficient mice suggests that it can activate the TCF/LEF transcriptional machinery to regulate target genes. Our ChIP assays indicated that β-catenin specifically associated with the HDAC2 promoter region containing TCF/LEF binding elements (Fig. 4d). β-catenin enrichment on HDAC2 promoter was significantly increased in PFC of Shank3+/ΔC mice (Fig. 4e), implicating β-catenin in the upregulated transcription of HDAC2. Other β-catenin target genes found in cancer cells or neural stem cells, such as Vegf, Jun (encoding c-jun), Ccnd1 (encoding Cyclin D1), and Neurod1, were not significantly altered in PFC of Shank3+/ΔC mice (Fig. 4f).
To further reveal the involvement of β-catenin in the regulation of genes and behaviors, we elevated β-catenin expression by injecting β-catenin adenovirus into the PFC of wild-type mice. A high level of β-catenin was found in the nucleus of PFC pyramidal neurons (Fig. 4g). HDAC2 mRNA was significantly and specifically upregulated by β-catenin overexpression in PFC of wild-type mice (Fig. 4h), similar to what was found in Shank3+/ΔC mice. Moreover, the social preference deficits were induced in wild-type mice with elevated β-catenin expression in PFC (Fig. 4i–k), reminiscent to the behavioral consequence of the upregulated nuclear β-catenin in Shank3-deficient mice.
Next, we diminished β-catenin expression by injecting β-catenin shRNA lentivirus into the PFC of Shank3-deficient mice. Effective knockdown of β-catenin expression in vivo was achieved with the β-catenin shRNA (Fig. 4l). HDAC2 expression in PFC was significantly reduced in β-catenin shRNA-injected Shank3+/ΔC mice, compared to those injected with a scrambled control shRNA (Fig. 4m). Furthermore, Shank3+/ΔC mice with β-catenin knockdown in PFC exhibited the significantly increased social interaction time and social preference in 3-chamber sociability tests (Fig. 4n–p). Taken together, these results have provided direct evidence on the sufficiency and necessity of β-catenin in the regulation of HDAC2 and social behavior.
Romidepsin treatment elevates NMDAR transcription and restores NMDAR function in Shank3-deficient mice
We then sought to find out molecular mechanisms downstream of HDAC inhibition that may underlie the amelioration of social deficits. Diminished synaptic signals at glutamatergic synapses are strongly linked to autism-like phenotypes, including social deficits and repetitive behaviors7–16, making glutamate receptors a potential key target of romidepsin. Quantitative PCR analyses (Fig. 5a) indicated that in Shank3+/ΔC mice treated with romidepsin, the mRNA level of Grin2a (encoding NMDAR NR2A subunit) was significantly elevated, while the transcripts encoding other NMDA receptor subunits (Grin1, Grin2b) or AMPA receptor subunits (Gria1, Gria2) were not significantly changed. Consistently, romidepsin significantly increased the protein level of NR2A subunit, but not other glutamate receptor subunits. The reduced Shank3 mRNA and protein levels in Shank3+/ΔC mice were not restored by romidepsin treatment (Fig. 5b and 5c).
Figure 5. Romidepsin treatment increases Grin2a transcription and histone acetylation, and restores NMDAR synaptic function in PFC of Shank3-deficient mice.
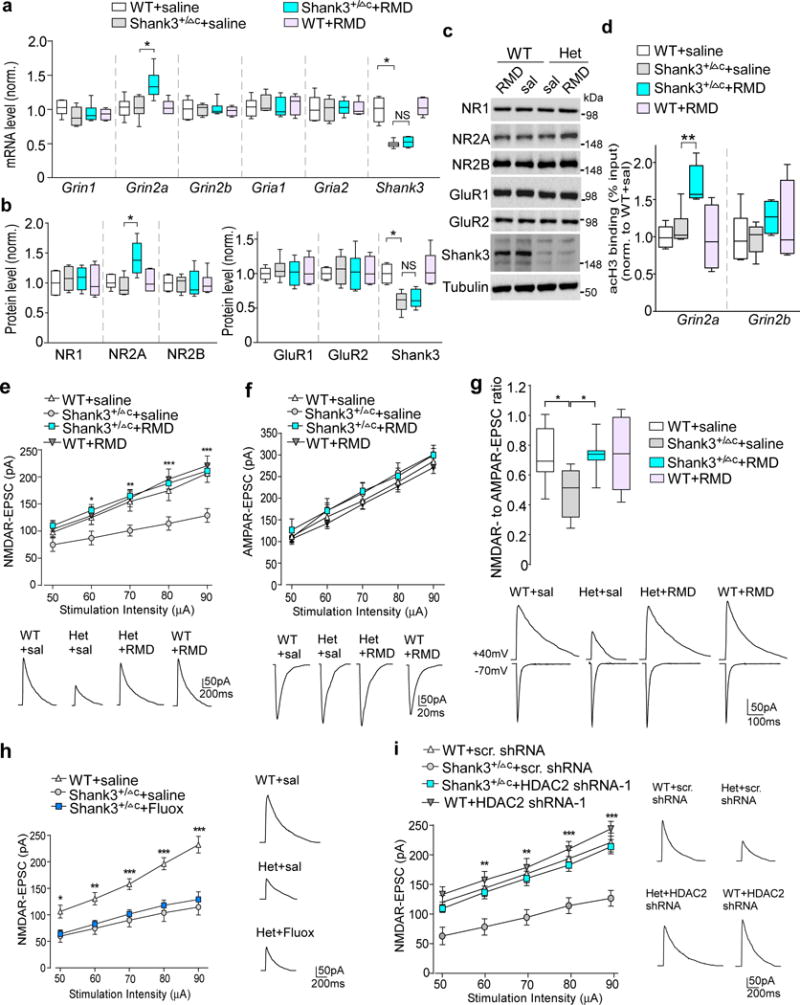
(a) Quantitative real-time RT-PCR data on the mRNA level of NMDAR and AMPAR subunits and Shank3 in PFC slices from saline-injected WT (n=8), saline-injected Shank3+/ΔC (n=8), romidepsin (RMD, 0.25 mg/kg, 3×)-treated Shank3+/ΔC (n=10) and RMD-treated WT (n=6) mice. F1,28(treatment)=5.96, P=0.021 (Grin2a); F1,28(treatment)=0.52, P=0.48 (Shank3); * P<0.05; ns, not significant, two-way ANOVA. (b, c) Quantification analysis and representative immunoblots of the protein level of NMDAR and AMPAR subunits and Shank3 in PFC slices from WT or Shank3+/ΔC mice injected with saline or romidepsin. F1,20(treatment)=5.58, P=0.030 (Grin2a); F1,20(treatment)=0.62, P=0.44 (Shank3); * P<0.05, two-way ANOVA, n=6 each group. (d) ChIP assay data showing the acetylated histone H3 level at Grin2a and Grin2b promoter regions in PFC lysates from saline-injected WT (n=6), saline-injected Shank3+/ΔC (n=6), RMD-treated Shank3+/ΔC (n=6) and RMD-treated WT (n=4) mice. F1,18(treatment)=5.88, P=0.026 (Grin2a); ** P<0.01, two-way ANOVA. (e, f) Input-output curves of NMDAR-EPSC (e) and AMPAR-EPSC (f) in PFC pyramidal neurons from WT vs Shank3+/ΔC mice treated with romidepsin or saline (n=16 cells/4 mice each group). Recordings were performed at 4-5 days post-injection. In (e), * P<0.05, ** P<0.01, *** P<0.001 (Shank3+/ΔC+RMD vs. Shank3+/ΔC+saline), two-way rmANOVA. Inset: representative NMDAR-EPSC and AMPAR-EPSC traces. (g) Box plots showing the NMDAR-EPSC to AMPAR-EPSC ratio in PFC pyramidal neurons from WT vs Shank3+/ΔC mice treated with romidepsin or saline. Inset: representative EPSC traces. F1,36=4.8, P=0.036; * P<0.05, two-way ANOVA, n=10 cells/3 mice each group. (h) Input-output curves of NMDAR-EPSC in Shank3+/ΔC mice treated with fluoxetine (5 mg/kg, i.p., 14×). Inset: representative NMDAR-EPSC traces. * P<0.05, ** P<0.01, *** P<0.001 (Shank3+/ΔC+fluoxetine vs. WT+saline), two-way rmANOVA, n=12 cells/3 mice each group. (i) Input-output curves of NMDAR-EPSC in PFC pyramidal neurons of WT or Shank3+/ΔC mice with the PFC injection of a HDAC2 shRNA or a scrambled control shRNA lentivirus. ** P<0.01, *** P<0.001 (Shank3+/ΔC+HDAC2 shRNA vs. Shank3+/ΔC+scrambled shRNA), two-way rmANOVA, n=12 cells/3 mice each group. All animals used are males (5-6 weeks old). Data are presented as median with interquartile range (a,b,c,g) or mean ± SEM (e,f,h,i). Each set of the experiments was replicated for at least 3 times. See Supplementary Fig. 9 for blot source data.
The transcriptional increase of Grin2a by romidepsin prompted us to find out whether histone acetylation of NMDA receptors was altered by this HDAC inhibitor. To test this, we performed ChIP assays to examine the histone acetylation at Grin2a and Grin2b promoters in PFC neurons. H3 acetylation (Supplementary Fig. 3) was identified at the Grin2a proximal promoter region (~200 bp from TSS) and Grin2b promoter region (~700 bp from TSS). As shown in Fig. 5d, romidepsin treatment significantly increased H3 acetylation at Grin2a promoter in Shank3+/ΔC mice. No significant effect of romidepsin on H3 acetylation at Grin2b promoter was observed. These data suggest that romidepsin treatment of Shank3+/ΔC mice elevates Grin2a gene expression by enhancing its histone acetylation.
The upregulation of Grin2a transcription by romidepsin could lead to elevated NMDAR function in Shank3+/ΔC mice. Thus, we performed electrophysiological experiments to record NMDAR-mediated excitatory postsynaptic currents (EPSC) at 3-5 days after romidepsin treatment in layer V PFC pyramidal neurons, which exhibit the clearest deficits in the brains of autistic children33. As shown in Fig. 5e, the amplitudes of NMDAR-EPSC induced by a series of stimulus intensities were markedly reduced in saline-injected Shank3+/ΔC mice, compared to saline-injected WT mice. A strong recovery of the input/output curves of NMDAR-EPSC was observed in PFC neurons of Shank3+/ΔC mice with romidepsin treatment. AMPAR-ESPC was unchanged in Shank3+/ΔC mice treated with romidepsin or saline (Fig. 5f). NMDAR- to AMPAR-EPSC ratio was significantly reduced in Shank3+/ΔC mice, which was also restored by romidepsin treatment (Fig. 5g). In contrast, fluoxetine treatment (10 mg/kg, i.p. 14×) failed to elevate NMDAR-EPSC in Shank3+/ΔC mice (Fig. 5h). Moreover, in PFC pyramidal neurons of HDAC2 shRNA-injected Shank3+/ΔC mice, the diminished NMDAR-EPSC was significantly restored (Fig. 5i). The romidepsin-induced rescue of NMDAR-EPSC, as well as the romidepsin-induced normalization of global H3 acetylation, could be observed at 16-18 days post-treatment, but not 30-32 days post-treatment (Supplementary Fig. 4), consistent with the therapeutic time window of romidepsin on behaviors (Fig. 1e). These results suggest that the recovery of NMDAR function in PFC may underlie the rescue of social deficits in Shank3-deficient mice with HDAC2 inhibition or knockdown.
Romidepsin treatment elevates the transcription of actin regulators and restores F-actin in Shank3-deficient mice
To find out the potential existence of other molecular targets of romidepsin, we focused on various actin regulators in the “Shank3 interactome”34 that could regulate synaptic structure and NMDAR trafficking12. Enriched mutations in genes regulating actin filament network were identified in genetic analyses of ASD patients19,35, suggesting that actin dysregulation at glutamatergic synapses is a potential pathophysiological mechanism. It has been found that the Rac1-LIMK-PAK-cofilin signaling is disrupted in Shank3-deficient mice, leading to F-actin dysregulation12, so we first examined the mRNA level of these genes. As shown in Fig. 6a, Arhgef7, which encodes βPIX, the guanine nucleotide exchange factor (GEF) for Rac1 and a Shank-interacting protein at excitatory synaptic sites36, was significantly lost in Shank3+/ΔC mice, and romidepsin treatment rescued Arhgef7 gene transcription. The significantly decreased Limk1, which encodes a major Rac1 downstream target controlling cofilin activity, was also restored by romidepsin treatment of Shank3-deficient mice. The mRNA level of Rac1, Pak1/2/3 and Cfl1 (encoding cofilin) was largely unchanged.
Figure 6. Romidepsin treatment elevates the expression of actin regulators and normalizes actin filaments in PFC of Shank3-deficient mice.
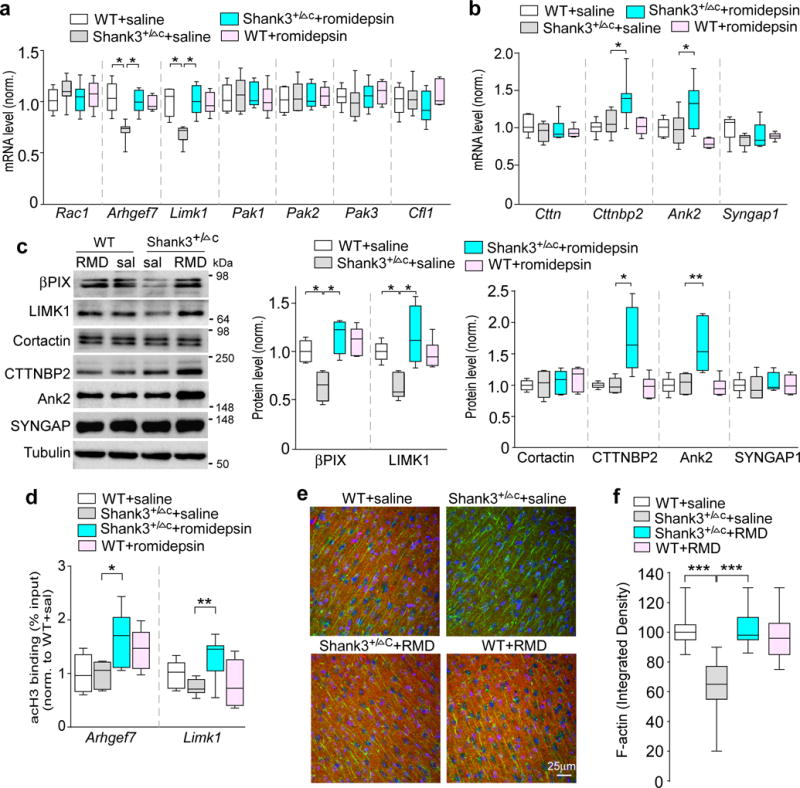
(a, b) Quantitative RT-PCR data on the mRNA level of actin regulators in the Rac1 signaling pathway (a) and those identified as ASD risk factors (b) in PFC slices from saline-injected WT (n=8), saline-injected Shank3+/ΔC (n=8), romidepsin (RMD, 0.25 mg/kg, 3×)-treated Shank3+/ΔC (n=10) and RMD-treated WT (n=6) mice. F1,28=6.42, P=0.017 (Arhgef7); F1,28=8.04, P=0.0086 (Limk1); F1,28(tretment)=5.93, P=0.022 (Cttnbp2); F1,28(treatment)=7.71, P=0.01 (Ank2); * P<0.05, two-way ANOVA. (c) Immunoblots and quantification analysis of the protein level of selective actin regulators in PFC slices from WT or Shank3+/ΔC mice injected with saline or romidepsin. F1,20=6.87, P=0.016 (βPIX); F1,20=10.87, P=0.0036 (LIMK1); F1,20(treatment)=4.97, P=0.038 (CTTNBP2); F1,20(treatment)=9.12, P=0.0068 (ANK2); * P<0.05, ** P<0.01, two-way ANOVA, n=6 each group. (d) ChIP assay data showing the acetylated histone H3 level at Arhgef7 and Limk1 promoter regions in PFC lysates from saline-injected WT (n=8), saline-injected Shank3+/ΔC (n=7), RMD-treated Shank3+/ΔC (n=9) and RMD-treated WT (n=6) mice. F1,26(treatment)=14.6, P=0.0007 (Arhgef7); F1,26=10.0, P=0.004 (Limk1); * P<0.05, ** P<0.01, two-way ANOVA. (e) Merged confocal images (40×) of F-actin (phalloidin, red) co-stained with PSD-95 (green) and DAPI (blue) in PFC slices of WT and Shank3+/ΔC mice treated with romidepsin or saline. Immunohistochemistry was performed at 4-5 days post-injection. (f) Quantification of F-actin (integrated densities) in PFC slices of different animal groups. F1,104=54.49, P<0.0001; *** P<0.001, two-way ANOVA, n=27 images/3 mice each group. All animals used are males (5-6 weeks old). Data are presented as median with interquartile range (a,b,c,d,f). Each set of the experiments was replicated for at least 3 times. See Supplementary Fig. 9 for blot source data.
The mRNA levels of several other Shank3-interacting actin regulators that have been identified as ASD risk factors19 were also examined. As shown in Fig. 6b, Cttnbp2, which encodes a protein binding to Cortactin (an F-actin and Shank3-interacting protein), was upregulated in Shank3+/ΔC mice treated with romidepsin. A similar upregulation was found in Ank2 (encoding Ankyrin2), while Cttn (encoding Cortactin) and Syngap1 mRNAs showed no change.
We further examined the alteration of protein levels of these actin regulators. As shown in Fig. 6c, the diminished expression of βPIX and LIMK1 in Shank3-deficient mice was restored by romidepsin treatment. Moreover, romidepsin significantly increased the protein level of CTTNBP2 and Ank2 without altering Cortactin or SYNGAP1.
To test whether the transcriptional increase of actin regulators by romidepsin could be due to the increased histone acetylation, we performed ChIP assays to examine the histone acetylation at Arhgef7 and Limk1 promoters in PFC neurons. H3 acetylation (Supplementary Fig. 5) was identified at the Arhgef7 promoter region (~800 bp from TSS) and Limk1 promoter region (~500 bp from TSS). As shown in Fig. 6d, romidepsin treatment of Shank3+/ΔC mice significantly increased the enrichment of H3 acetylation at Arhgef7 promoter and Limk1 promoter.
The romidepsin-induced upregulation of key actin regulators in “Shank3 interactome” may help to restore and maintain the integrity of actin cytoskeleton at synapses. To test this, we performed immunostaining with phalloidin to directly visualize F-actin. As shown in Fig. 6e, 6f and Supplementary Fig. 6, compared to WT mice, Shank3+/ΔC mice (saline-injected) had a significant decrease of F-actin (red) expression in PFC slices, and romidepsin treatment restored cortical actin filaments to the normal level. The expression of PSD-95 (green) in PFC of Shank3+/ΔC mice was unchanged by romidepsin treatment. The normalization of actin filaments may facilitate the actin-based NMDAR membrane delivery.
To find out the region specificity of romidepsin-induced gene expression changes, we performed qPCR profiling in several other brain regions and peripheral organs. In contrast to what was found in PFC, the levels of mRNA encoding NMDAR subunits (Grin1, Grin2a, and Grin2b) and actin regulators (Arhgef7 and Limk1) in dorsal striatum, ventral tegmental area (VTA), kidney and heart of Shank3+/ΔC mice were not significantly altered by romidepsin treatment (Supplementary Fig. 7).
Romidepsin treatment induces genome-wide restoration or elevation of genes involved in neural signaling in PFC of Shank3-deficient mice
Finally, to determine whether HDAC inhibition has genome-wide effects on gene expression, we used RNA sequencing to analyze the mRNA profile in PFC of WT and Shank3+/ΔC mice treated with romidespin or saline control. We mapped the sequences to 24,420 mouse genes. Compared to WT, 365 genes in saline-treated Shank3+/ΔC mice showed a change in expression values of at least 1.2 fold and p ≤ 0.01 (213 downregulated, 152 upregulated). In romidepsin-treated Shank3+/ΔC mice, ~88% of the downregulated genes (n=187 genes) were normalized to control values (Supplementary Table 1). The heat map generated with the expression values for the 187 restored genes showed that the saline-treated Shank3+/ΔC samples clustered together and separated from the WT samples. Moreover, romidepsin-treated Shank3+/ΔC samples were closer to those from WT than the saline-treated Shank3+/ΔC samples (Fig. 7a). Further analysis of the romidepsin-restored genes revealed enrichment in actin cytoskeleton-mediated transport, signal transduction pathways, and developmental processes (Fig. 7b). In addition, romidepsin treatment increased the expression of 41 genes that were unchanged in Shank3+/ΔC samples (Supplementary Table 2), which were enriched in cell-cell signaling, synaptic transmission and neurotransmitter secretion (Fig. 7c). These results suggest that romidepsin treatment can rescue or elevate the expression of many genes involved in neural signaling in Shank3-deficient mice, which may collectively contribute to its therapeutic effects on behavioral deficits (Fig. 7d).
Figure 7. Romidepsin treatment induces genome-wide restoration or elevation of genes involved in neural signaling in PFC of Shank3-deficient mice.
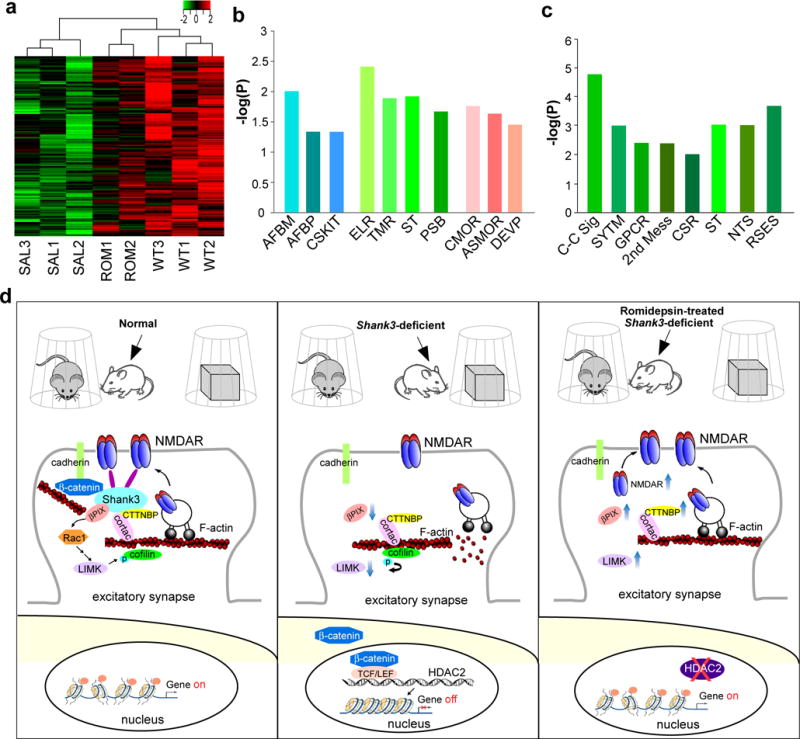
(a) Heat map representing expression (row z-score) of 187 genes that were downregulated in saline-treated Shank3+/ΔC mice (SAL, n=3) and normalized in romidepsin (RMD, 0.25 mg/kg, 3×)-treated Shank3+/ΔC mice (ROM, n=2), compared to wild-type mice (WT, n=3). All animals used are males (5-6 weeks old). (b) Enrichment analysis using gene sets derived from the Biological Process Ontology for the 187 genes that were suppressed in Shank3+/ΔC mice and restored by romidepsin treatment. AFBM: Actin filament-based movement; AFBP: Actin filament-based process; CSKIT: Cytoskeleton-dependent intracellular transport; ELR: Enzyme-linked receptor protein signaling pathway; TMR: Transmembrane receptor protein signaling pathway; ST: Signal transduction; PSB: Regulation of protein stability; CMOR: Regulation of cell morphogenesis; ASMOR: Anatomical structure morphogenesis; DEVP: Regulation of developmental process. (c) Enrichment analysis for the 41 genes that were unchanged in Shank3+/ΔC mice but were elevated by romidepsin treatment. C-C Sig: Cell-cell signaling; SYTM: Synaptic transmission; GPCR: G protein-coupled receptor protein signaling pathway; 2nd Mess: 2nd messenger signaling; CSR: Cell surface receptor-linked signal transduction; ST: Signal transduction; NTS: Neurotransmitter secretion; RSES: Response to external stimulus. (d) A schematic model showing the potential mechanism underlying the therapeutic effect of romidepsin in Shank3-deficient mice. Normally, Shank3 crosslinks NMDARs to actin cytoskeleton, and binds to the adhesive junction-associated protein β-catenin. Loss of Shank3 leads to the translocation of β-catenin from synapses to nucleus, inducing the upregulation of HDAC2 and chromatin remodeling. The ensuing transcriptional suppression of actin regulators results in the disruption of actin filaments and the synaptic delivery of NMDARs. Treatment with the HDAC inhibitor romidepsin restores or elevates many target genes, including NMDAR subunits, key actin regulators and others involved in neuronal signaling, leading to the normalization of actin cytoskeleton and NMDAR synaptic function. Consequently, the autism-like social deficits are rescued.
Discussion
Here, we show that Shank3-deficient mice exhibit an abnormally low level of histone acetylation resulting from HDAC2 upregulation in the PFC. A brief treatment with a low dose of romidepsin, a potent class I HDAC inhibitor, leads to robust and long-lasting rescue of social deficits, a core symptom of ASD. This effect of romidepsin was unique, as many of the pharmacological agents currently used in psychiatric practice, including fluoxetine, clozapine, valproic acid, risperidone and aripiprazole, all fail to profoundly and persistently improve social behaviors in the Shank3 model. This low dose romidepsin treatment does not induce locomotor deficits, changes in anxiety-like behavior, or neuronal loss in young animals. The most effective therapeutic window of romidpesin is the juvenile to late adolescent period, which is a critical developmental stage for social and communication skills. The sustained rescuing effect on social deficits of a single administration of romidepsin in young Shank3-deficient mice is promising, however it is limited by the lack of chronic effects at adulthood with repeated administrations. Using romidepsin as a tool compound to interrogate the biology of Shank3 deficiency, we have demonstrated how aberrations in synaptic, transcriptional, and epigenetic pathways, all of which have been implicated in autism19–21, are interconnected.
One important question is the molecular mechanism underlying the epigenetic aberrations in Shank3 deficiency models. We have found that Shank3 deficiency induces the nuclear translocation of its binding partner at synapses, β-catenin. HDAC2 transcription is upregulated in Shank3-deficient mice, which is associated with the increased enrichment of β-catenin at the HDAC2 promoter region containing TCF/LEF binding elements. Direct overexpression of β-catenin in PFC of wild-type mice leads to the upregulation of HDAC2 transcription, and knockdown of β-catenin in PFC of Shank3-deficient mice leads to the reduction of HDAC2 transcription, further suggesting that HDAC2 is a target gene of β-catenin. Moreover, the social deficits are rescued by inhibition of HDAC2, knockdown of HDAC2, or knockdown of β-catenin in PFC of Shank3-deficient mice, and are induced by elevating β-catenin expression in PFC of wild-type mice. These bi-directional lines of evidence strongly suggest that β-catenin/HDAC2 plays a causal role in social deficits of Shank3-deficiency models and the therapeutic effect of romidepsin.
Finding potential targets downstream of HDAC inhibition involved in the amelioration of social deficits is another challenge. Aberrant synaptic function resulting from genetic mutations are thought to be a major pathogenic factor in ASD18–20,37,38. The behavioral abnormalities exhibited in Shank3-deficient mice have been attributed to the alteration of glutamatergic synapses7–16. Particularly, aberrations in NMDA receptor, a Shank3-associated key synaptic protein2 controlling neural development and synaptic plasticity underlying cognitive processes, are strongly linked to social deficits12,39–41. Previous studies have demonstrated that the Shank3 deficiency-induced NMDAR hypofunction is due to the loss of actin-based synaptic delivery of NMDARs because of the reduced Rac1-PAK-cofilin signaling12,42. Romidepsin treatment of Shank3-deficient mice normalizes the aberrant histone acetylation, elevates the transcription of many downstream target genes, including NMDAR subunits and key actin regulators, leading to the restoration of synaptic NMDAR function and F-actin in PFC neurons. These results are also supported by our RNA sequencing data. The majority (~88%) of the 213 downregulated genes in Shank3-deficient mice is restored by romidepsin treatment, and these genes are highly enriched in actin cytoskeleton-mediated transport, signal transduction pathways, and developmental processes. The reversal of the haploinsufficiency of a number of genes identified as key ASD risk factors provides a potential mechanism for the romidepsin-induced rescue of social deficits in Shank3-deficient mouse models.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Online Methods
The mice expressing C-terminal (exon 21) deleted Shank3 (Jackson Labs, Bar Harbor, ME), which exhibited the significant loss of full-length Shank3 expression, were generated, genotyped and maintained as previously described11,12. Since only the hemizygous deletion or loss-of-function mutation in the Shank3 gene has been linked to human autism and intellectual disability3,4, heterozygous Shank3+/ΔC mice (5-8 weeks old, male) and age-matched wild-type littermates (male) were used in this study. Female Shank3+/ΔC mice lack the autism-like social deficits, so they were not used.
Shank3e4-9 mice with the loss of major isoforms of the Shank3 gene resulting from the deletion of N-terminal exons 4-9 (a kind gift from Dr. Yong-Hui Jiang at Duke University School of Medicine) were generated, genotyped and maintained as previously described10. Both male and female homozygous Shank3e4-9 mice, which exhibited prominent autism-like social preference deficits10, were used. Similar results were obtained, so they were pooled together.
Mice of different genotypes were randomly assigned to drug/saline groups. Experiments were carried out by investigators in a blind fashion (with no prior knowledge about the genotypes and treatments). All experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the State University of New York at Buffalo. We have complied with all relevant ethical regulations.
Romidepsin (Selleckchem), Fluoxetine (NIH), Valproic acid (Tocris), aripiprazole (Tocris), risperidone (Tocris), or Trichostatin A (Sigma) was prepared by dissolving in DMSO to make the stock solution and diluting with 0.9% saline before injections (DMSO concentration of the working solution: <0.2%). All the tests were performed at various days as indicated in the paper after the last injection of the compound.
Behavioral Testing
Social Preference Test
A three-chamber social interaction assay was performed to assess social deficits12. Briefly, an apparatus (L: 101.6 cm, W: 50.8 cm, H: 50.8 cm) containing 3 chambers with retractable doorways allowing for access to side chambers was used. The test was composed of two phases with different stimulus in each of two side chambers. Each stimulus was placed inside a capsule (an inverted pencil cup, D: 10.2 cm, H: 10.5 cm). An upright plastic drinking cup was placed on top of each capsule to prevent the subject mouse from climbing on top. The 1st phase contained two identical nonsocial stimuli (folded black papers), the 2nd phase contained a nonsocial (NS) stimulus (a wood block) and a social (Sol) stimulus (age- and sex-matched mouse of the same strain). Locations of the NS and Sol stimuli were counterbalanced. Animals were habituated in the 3-chamber apparatus at 1 day before the initial testing. During the habituation, two empty capsules were placed at side chambers, and animals were allowed to explore for 10 min. During the sociability measurement, different stimuli were placed inside capsules at side chambers, and the test animal was placed in the center chamber, and was free to explore the apparatus for 10 min in each phase, while it was returned to home cage during the 5-min intervals between test phases. The chamber was cleaned with 75% Ethanol after each phase. Interaction time was counted based on “investigating” behaviors of the test animal to each stimulus. In earlier experiments, an experimentalist measured the time of the test animal spent on actively seeking and sniffing the stimulus. In later experiments, a computer running the Any-maze behavior tracking software (Stoelting, Wood Dale, IL) measured the time of the test animal spent at the close proximity of the capsule (distance of animal head to cup edge: ≤3.5 cm). Data generated manually and automatically were consistent, so they were pooled together. Preference scores were calculated, where time spent with one stimulus was subtracted from the time spent with the other stimulus and divided by the total time spent exploring both stimuli.
Social Approach Test
The test animal was habituated in an apparatus (L: 67.7 cm, W: 50.8 cm, H: 50.8 cm) containing a capsule (an inverted pencil cup, placed in the center area) for 10 min, then was returned to the home cage. The apparatus was cleaned and a social stimulus (an age- and sex-matched mouse) was placed inside the capsule. The test animal was put back into the apparatus to explore for 10 min. The time spent on interacting with the social stimulus was measured.
Locomotion Test
Animals were placed in a cage (L: 42 cm, W: 20 cm, H: 20 cm) devoid of any bedding materials for 5 minutes and the number of times crossing a midline was counted.
Rotarod Test
To assess motor coordination and balance, an accelerating rotarod (SD instruments, San Diego CA) was used. Mice were placed on a cylinder, which slowly accelerated from 4 to 40 revolutions per minute over a 5-min test session. The task requires mice to walk forward in order to remain on top of the rotating cylinder rod.
Self-Grooming
Mice were scored for spontaneous grooming behaviors when placed individually in a clean cage. The cage was lined with a thin layer of bedding (~1 cm) in order to reduce neophobia, but prevent digging, a potentially competing behavior. Prior to the testing period, animals were allowed to habituate to the novel environment for 10 minutes. Each mouse was rated for 10 minutes on cumulative time spent grooming.
Open Field Test
Animals were placed in a rectangular arena (60 cm × 80 cm); the amount of time the animal spent in the center (25 cm × 25 cm) was counted. Anxious animals spend less time in the center and more time in the corner of the field.
Western Blotting of Nuclear, Synaptic, and Total Proteins
Nuclear extracts from mouse brains was prepared according to the manufacturer’s instructions (Life Technologies) with modifications. Briefly, ten PFC punches (diameter: 2 mm) from fresh mouse slices (300 μm) per animal were collected, and then homogenized with 500 μl homogenization buffer (20 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 1 mM PMSF, with cocktail protease inhibitor). The homogenate was incubated on ice for 15 min and followed by centrifugation at 3,000 × g, 4°C for 10 min. The nuclear pellet was resuspended in 50 μl nuclear extract buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 10% glycerol, 1 mM PMSF, with cocktail protease inhibitor) and incubated on ice for 30 min with vortex periodically to re-suspend the pellet. After centrifugation, the supernatant for nuclear fractions was collected, boiled in 2 × SDS loading buffer for 5 min and then separated on 10% SDS-polyacrylamide gels. Western blotting experiments for nuclear proteins were performed with antibodies against acetylated H3K9 (1:1000, Cell Signaling, 9649), H3 (1:500, Cell Signaling Technology, 4499), HDAC1 (1:500, Santa Cruz Biotech., sc-7872), HDAC2 (1:500, Santa Cruz Biotech., sc-7899), and β-catenin (1:1000, Cell Signaling, 8480) as previously described43,44.
Synaptic membrane and cytosolic fractions were prepared as described previously12. In brief, blocks of frontal cortex were cut out, weighed, and homogenized in ice-cold lysis buffer (10 ml/g, 15 mM Tris, pH 7.6, 0.25 M sucrose, 1 mM PMSF, 2 mM EDTA, 1 mM EGTA, 10 mM Na3VO4, 25 mM NaF, 10 mM sodium pyrophosphate, and protease inhibitor tablet). After centrifugation at 800 × g for 5 min to remove nuclei and large debris, the remaining supernatant was subjected to 10,000 × g centrifugation for 10 min. The crude synaptosome fraction (pellet) was suspended in lysis buffer containing 1% Triton X-100 and 300 mM NaCl, homogenized again, and centrifuged at 16,000 × g for 15 min. Triton insoluble fraction which mainly includes membrane-associated proteins from synpatosomes was dissolved in 1% SDS. Samples were boiled in 2× SDS loading buffer for 5 min, and separated on 7.5% SDS-PAGE. Anti-β-catenin (1:1000, Cell Signaling, 8480) was used to detect β-catenin in the synaptic membrane fraction, and anti-non-phosphorylated β-catenin (1:1000, Cell Signaling, 8814) was used to detect the active form of β-catenin in the cytosol.
Western blotting of total proteins12,43 were performed using antibodies against NR1 (1:500, NeuroMab, 75-272), NR2A (1:500, Millipore, 07-632), NR2B (1:500, Millipore, 06-600), GluR1 (1:500, Millipore, AB1504), GluR2 (1:500, NeuroMab 75-002), Shank3 (1:500, NeuroMab, 75-344, clone N367/62), tubulin (Sigma, T9026), βPIX (1:500, Santa Cruz Biotechnology, sc393814), LIMK (1:1000, BD Biosciences, 611748), Cortactin (1:1000, Cell Signaling, #3502), CTTNBP2 (1:1000, Proteintech Group, 17893), Ank2 (1:500, NeuroMab clone N105/17), and SYNGAP (1:1000, Cell Signaling, #9479).
Co-Immunoprecipitation (Co-IP)
Frontal cortical slices from WT mice were collected and homogenized in lysis buffer (in mM: 50 NaCl, 30 sodium pyrophosphate, 50 NaF, 10 Tris, 5 EDTA, 0.1 Na3VO4, 1 PMSF, with 1% Triton X-100 and protease inhibitor tablet). Lysates were centrifuged (12,000 × g) at 4°C for 15 min. Supernatant fraction was incubated with the antibody against β-catenin (8 μl, Cell Signaling, 2698) for overnight at 4°C (negative controls used the antibody against mouse IgG (Millipore, 12-371) or omitted antibody), followed by incubation with 50 μl of protein A/G plus agarose (Santa Cruz Biotech., sc-2003) for 1 hr atantibodyin5 4°C. Immunoprecipitates were washed 3 times with lysis buffer, then boiled in 2×SDS loading buffer for 5 min, and separated on 7.5% SDS-polyacrylamide gels. Western blotting experiments were performed with anti-Shank3 (1:1000, Abcam, ab101919) and anti-β-catenin (1:1000, Cell Signaling, 2698).
Chromatin Immunoprecipitation (ChIP)
Briefly, ten PFC punches from mouse slices per two animals were collected, cross-linked with 1% formaldehyde for 12 min and quenched by the addition of glycine at a final concentration of 0.125 M for 5 min before freezing at −80°C. The chromatin was extracted by the 1% SDS lysis buffer, followed by the sheering using a Fisher Scientific Sonic Dismembrator Model 300 at 28% of power (ten 15-s pulses with 30 s pause between pulses). This procedure resulted in DNA fragment sizes of 200-500 bp. After centrifugation, ~10% of the supernatant were saved to serve as input controls. To reduce nonspecific background, the supernatant were diluted in ChIP dilution buffer and pre-cleared with 80 μl of salmon sperm DNA/protein A agarose-50% slurry (Millipore, 16-157) for 30 min at 4°C with agitation. The pre-cleared supernatant was incubated with antibodies against β-catenin (Cell Signaling, 8480, 20 μl per reaction) or pan-acetylated H3 (Millipore, 06-599, 7 μg per reaction) overnight at 4°C under constant rotation, following by incubation with 60 μl of Salmon Sperm DNA/Protein A agarose-50% Slurry for 1 hr at 4°C. Rabbit IgG was used as a non-specific control for immunoprecipitation assays. After 5 times of washing, bound complex was eluted from the beads by incubating with 150 μl elution buffer for twice at room temperature. After reversing crosslinks in 65°C for 4 hours, proteins and RNA were removed using proteinase K (Invitrogen) and RNase (Roche), respectively. Then immunoprecipitated DNA and input DNA were purified by phenol/chlorofrom extraction. Quantification of ChIP signals was calculated as percent input.
Primers from multiple sites relative to TSS (within 2000 bp) were designed and pre-tested in both input and ChIP samples. Only those with solid signals in both input and ChIP samples were chosen for the experiment. Purified DNA was subjected to qPCR reactions with primers against mouse HDAC2 promoter (Forward, −1748 bp ~ −1728 bp relative to TSS, 5′-GGGTTGCAGACCCCTTTAGT-3′; Reverse, −1566 bp ~ −1546 bp relative to TSS, 5′-TCACAAGAGGACAAGCCGAC-3′); GAPDH promoter (Forward, −441 bp ~ −422 bp relative to TSS, 5′-GCATTCAGGTCTCTGGGTCC-3′; Reverse, −295 bp ~ −276 bp relative to TSS, 5′-GTGGGCTCCGAACTGATAGG -3′); Grin2a promoter (Forward, −112 bp ~ −93 bp relative to TSS, 5′-CCCGCTCTGAGAGTCGAATG-3′; Reverse, −233 bp ~ −214 bp relative to TSS, 5′-GGCAAAATAGCTCGGCTTGG-3′), Grin2b promoter (Forward, −733 bp ~ −712 bp relative to TSS, 5′-AGATTTGAGGACTTTGGGGTTC-3′; Reverse, −833 bp ~ −813 bp relative to TSS, 5′-GCTGGGTTAGTCAGTGCTGTA-3′), Arhgef7 promoter (Forward, −950 bp ~ −932 bp relative to TSS, 5′-AAACTGTCGAGGAGTGCCAG-3′; Reverse, −749 bp ~ −730 bp relative to TSS, 5′- TCAAGAGCACATCGCAACCT-3′), and Limk1 promoter (Forward, −644 bp ~ −625 bp relative to TSS, 5′-TTCCGCTGTTCACAGACTCC-3′; Reverse, −447 bp ~ −428 bp relative to TSS, 5′-ATAACCACGCCCATCAAGCA-3′).
Quantitative Real-time RT-PCR
To compare the mRNA levels, quantitative RT-PCR was used. Total RNA was isolated from mouse PFC punches using Trizol reagent (Invitrogen) and treated with DNase I (Invitrogen) to remove genomic DNA. Then SuperScript III first-strand synthesis system for RT-PCR (Invitrogen) was used to obtain cDNA from the tissue mRNA, followed by the treatment with RNase H (2 U/l) for 20 min at 37°C. Quantitative real-time RT-PCR was carried out using the iCycler iQ™ Real-Time PCR Detection System and iQ™ Supermix (Bio-Rad) according to the manufacturer’s instructions. In brief, GAPDH was used as the housekeeping gene for quantitation of the expression of target genes in samples from WT and Shank3+/ΔC mice treated with romidepsin or saline control. Fold changes in the target genes were determined by: Fold change = 2-Δ(ΔCT), where ΔCT = CT(target) – CT(GAPDH), and Δ(ΔCT) = ΔCT(treated group) - ΔCT(WT+saline). CT (threshold cycle) is defined as the fractional cycle number at which the fluorescence reaches 10× the standard deviation of the baseline. A total reaction mixture of 25 μl was amplified in a 96-well thin-wall PCR plate (Bio-Rad) using the following PCR cycling parameters: 95°C for 5 min followed by 40 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 60 sec. Primers for all the target genes are listed in Supplementary Table 3.
Immunohistochemistry
Mice were anesthetized and transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA) prior to brain removal. Brains were post-fixed in 4% PFA for 2 days and cut into 100 μm slices. Slices were cut coronally and permeabilized using 0.5% Triton in PBS for 1 hour, washed, and blocked for 1 hour in PBS containing 3% BSA and 0.3% Triton. After washing, slices were incubated with the primary antibody against CaMKII (1:500, Santa Cruz Biotechnology, sc-9035), NeuN (1:1000, Millipore, MAB377) or PSD-95 (Neuromab, 75-028, 1:1000) for 48 hours at 4°C. After washing 3 times (1/2 hours, 1 hour, and 1 hour with gentle shaking) in PBS, slices were incubated with one or two secondary antibody (Alex488, Invitrogen, A21202, 1:1000; Alex594, Invitrogen A11032) for 1 hour at room temperature, followed by 3 washes with PBS. For F-actin staining, slices were then incubated with DAPI antibody (1:10,000) for 10 minutes at room temperature, followed by 3 washes with PBS. Slices were mounted on slides with VECTASHIELD mounting media (Vector Laboratories). Images were acquired using a 40× objective on a Zeiss LSM 510 Confocal Microscope. All specimens were imaged under identical conditions and analyzed with identical parameters using Image J software. Z-axis stacked confocal images (10) of 9 prefrontal cortical areas (225 μm × 225 μm) from 3-4 slices per animal were quantified.
Electrophysiological Recordings
Whole-cell voltage-clamp recording technique was used to measure synaptic currents in layer V pyramidal neurons of prefrontal cortical slices, as previously described12,43. Mouse slices (300 μm) were positioned in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated ACSF (in mM: 130 NaCl, 26 NaHCO3, 1 CaCl2, 5 MgCl2, 3 KCl, 1.25 NaH2PO4, 10 glucose, pH 7.4, 300 mOsm). Bicuculline (20 μM) and CNQX (20 μM) were added in NMDAR-EPSC recordings. Bicuculline and D-APV (50 μM) were added in AMPAR-EPSC recordings. Patch electrodes contained internal solution (in mM): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2 QX-314, 12 phosphocreatine, 5 MgATP, 0.2 Na3GTP, 0.1 leupeptin, pH 7.2-7.3, 265-270 mOsm. Layer V mPFC pyramidal neurons were visualized with a 40× water-immersion lens and recorded with the Multiclamp 700A amplifier (Molecular Devices, Sunnyvale, CA). Evoked EPSC were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Grass Technologies, West Warwick, RI). A bipolar stimulating electrode (FHC, Bowdoinham, ME) was placed ~100 μm from the neuron under recording. For NMDAR-EPSC, the cell (clamped at −70 mV) was depolarized to +40 mV for 3 s before stimulation to fully relieve the voltage-dependent Mg2+ block. Membrane potential was maintained at −70 mV for AMPAR-EPSC recordings. For input-output responses, EPSC was elicited by a series of pulses with different stimulation intensities (50-90 μA) delivered at 0.033 Hz. To obtain NMDAR- to AMPAR-EPSC ratio, AMPAR‐EPSC was first recorded at −70 mV in ACSF solution (containing bicuculline). Then the mixture of AMPAR-EPSC and NMDAR-EPSC was recorded at +40 mV with the same stimulation pulse (0.4 ms, 70 μA). The peak of NMDAR-EPSC was calculated at 40 ms from the onset of the EPSC mixture.
Virus Generation and Delivery
The shRNA oligonucleotide targeting mouse HDAC2 sequence (CCCAATGAGTTGCCATATAAT, Open Biosystem) was inserted to the lentiviral vector pLKO.3G (Addgene), which contains an eGFP marker. For the production of lentiviral particles, a mixture containing the pLKO.3G shRNA plasmid (against HDAC2), psPAX2 packaging plasmid and pMD2.G envelope plasmid (Addgene) was transfected to HEK-293FT cells using lipofectamine 2000. The transfection reagent was removed 12-15 hours later, and cells were incubated in fresh DMEM (containing 10% FBS + penicillin/streptomycin) for 24 hrs. The media harvested from the cells, which contained lentiviral particles, was concentrated by centrifugation (2,000 × g, 20 min) with Amicon Ultra Centrifugal Filter (Ultracel-100K, Millipore). The concentrated virus was stored at −80°C. Another shRNA lentivirus targeting different sequences of the mouse HDAC2 gene (CATGAGAGATGGTATAGAT, GCTTGGTTGTTTCAATCTA, ACATGCACCTGGTGTTCAA) was obtained from Santa Cruz Biotech (sc-29346-V). The GFP-tagged β-catenin adenovirus was obtained from Vector Biolabs (1182). The shRNA lentivirus targeting mouse β-catenin gene (GCCTTCAGATCTTAGCTTA, CAGCTGGAATTCTCTCTAA, CTGCAGAACTCCAGAAAGA, GTCGAGGAGTAACAATACA) was obtained from Santa Cruz Biotech (sc-29210-V). In vivo delivery of the viral suspension (1 μl each side) was achieved by stereotaxic injection bilaterally into medial PFC (2.0 mm anterior to bregma; 0.25 mm lateral) with a Hamilton syringe (needle gauge 31, extended to a depth of 2.0 mm below the tissue surface) as previously described12,44.
RNA Sequencing and Bioinformatic Analysis
PFC samples were obtained from three WT mice, three saline-treated Shank3+/ΔC mice and two romidepsin-treated Shank3+/ΔC mice. We generated strand-specific RNA libraries from 1 μg purified RNA using TruSeq stranded total RNA plus Ribo-zero kits (Illumina). The sequencing was performed at the Genomics and Bioinformatics Core of State University of New York at Buffalo. Single end reads per sample were obtained using the HiSeq 2500 platform from Illumina. Reads were first trimmed using Cutadapt45 to remove the 3′ end adapters and trailing sequences, followed by aligning to mouse RefSeq mRNAs using TopHat246. Transcript counts were estimated using HTSeq47. Differences in gene expression levels between samples were assessed with edgeR48 and calculated as log2 fold change. Functional enrichment analyses of differentially expressed genes were undertaken using gene sets derived from the Biological Process Ontology from GSEA (http://software.broadinstitute.org/gsea/msigdb/index.jsp).
Statistics
Data analyses were performed with Clampfit (Molecular Devices, Sunnyvale, CA), Kaleidagraph (Synergy Software, Reading, PA) and GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA). For statistical significance, experiments with two groups were analyzed using two-tailed Student’s t-tests. Eexperiments with more than two groups were subjected to one-way ANOVA, two-way ANOVA, or two-way repeated measure ANOVA (rmANOVA), followed by post hoc Bonferroni tests for multiple comparisons. Measurements were taken from distinct samples. No sample was excluded from the analysis. The sample size was based on power analyses and was similar to those reported in previous works12,43,44. We have tested the normality and variance of data distribution between groups being statistically compared. Data in box plots are presented as: center line, median; box limits, upper and lower quartiles; whiskers, minimal and maximum values. Data in all other formats are presented as mean ± SEM. F values, degrees of freedom and P values for all ANOVAs (unless otherwise stated, they represent statistics of main interaction effects and post hoc comparisons), as well as t-values and degrees of freedom for t-tests, are included in figure legends.
Life Science Reporting Summary
Further information on experimental design and reagents is available in the Life Sciences Reporting Summary.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank X.Q. Chen for excellent technical support. This work was supported by Nancy Lurie Marks Family Foundation and National Institutes of Health (MH112237, MH108842 and DA037618) to Z. Y. We also thank E. F. Trachtman and Varanasi family for their donations.
Footnotes
Accession Codes
RNA sequencing data have been deposited to GEO (accession #: GSE109328), which is publically available (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109328).
Author Contributions
L.Q. performed immunocytochemical and electrophysiological experiments and analyzed data. K.M. performed behavioral tests and analyzed data. L.Q., Z-J.W., E.M., and J.W. performed biochemical and molecular biological experiments and analyzed data. Z.H. performed bioinformatic analysis. Z.Y. designed experiments, supervised the project and wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 3.Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffard O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism. 2013;4:17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10:e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je S, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, et al. Loss of predominant shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J Neurosci. 2013;33:18448–68. doi: 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duffney LJ, Zhong P, Matas E, Wei J, Cheng J, Dietz DM, Kajiwara Y, Buxbaum JD, Yan Z. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Reports. 2015;11:1400–1413. doi: 10.1016/j.celrep.2015.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, Dutta N, Hayrapetyan V, Yu C, Gaidis E, Zhao S, Ding JD, Xu Q, Chung L, Rodriguiz RM, Wang F, Weinberg RJ, Wetsel WC, Dzirasa K, Yin H, Jiang YH. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun. 2016;7:11459. doi: 10.1038/ncomms11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bidinosti M, Botta P, Krüttner S, Proenca CC, Stoehr N, Bernhard M, Fruh I, Mueller M, Bonenfant D, Voshol H, Carbone W, Neal SJ, McTighe SM, Roma G, Dolmetsch RE, Porter JA, Caroni P, Bouwmeester T, Lüthi A, Galimberti I. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science. 2016;351:1199–203. doi: 10.1126/science.aad5487. [DOI] [PubMed] [Google Scholar]
- 15.Speed HE, Kouser M, Xuan Z, Reimers JM, Ochoa CF, Gupta N, Liu S, Powell CM. Autism-Associated Insertion Mutation (InsG) of Shank3 Exon 21 Causes Impaired Synaptic Transmission and Behavioral Deficits. J Neurosci. 2015;35:9648–65. doi: 10.1523/JNEUROSCI.3125-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S, Powell CM. Altered Striatal Synaptic Function and Abnormal Behaviour in Shank3 Exon4-9 Deletion Mouse Model of Autism. Autism Res. 2016;9:350–75. doi: 10.1002/aur.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006;7:268–77. doi: 10.1038/nrn1884. [DOI] [PubMed] [Google Scholar]
- 18.Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med. 2013;19:685–94. doi: 10.1038/nm.3193. [DOI] [PubMed] [Google Scholar]
- 19.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: genetics and pathology. Annu Rev Pathol. 2015;10:111–44. doi: 10.1146/annurev-pathol-012414-040405. [DOI] [PubMed] [Google Scholar]
- 21.Crawley JN, Heyer WD, LaSalle JM. Autism and Cancer Share Risk Genes, Pathways, and Drug Targets. Trends Genet. 2016;32:139–46. doi: 10.1016/j.tig.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–17. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–67. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 25.Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126–35. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- 26.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, Panageas K, Wright JJ, Pandolfi PP, Nimer SD. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826–32. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 27.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, Reeder C, Joske D, Figg WD, Gardner ER, Steinberg SM, Jaffe ES, Stetler-Stevenson M, Lade S, Fojo AT, Bates SE. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–7. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fraczek J, Vanhaecke T, Rogiers V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert Opin Drug Metab Toxicol. 2013;9:441–57. doi: 10.1517/17425255.2013.754011. [DOI] [PubMed] [Google Scholar]
- 29.McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J. A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics. 2011;127:e1312–e1321. doi: 10.1542/peds.2011-0427. [DOI] [PubMed] [Google Scholar]
- 30.Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31:2714–36. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murase S, Mosser E, Schuman EM. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron. 2002;35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- 32.Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–77. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- 33.Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–19. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han K, Holder JL, Jr, Schaaf CP, Lu H, Chen H, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503:72–7. doi: 10.1038/nature12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–9. doi: 10.1074/jbc.M301052200. [DOI] [PubMed] [Google Scholar]
- 37.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–71. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. 2015;28:91–102. doi: 10.1097/WCO.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 39.Carlson GC. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol Biochem Behav. 2012;100:850–854. doi: 10.1016/j.pbb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Won H, Lee HR, Gee HY, Mah W, Kim JI, et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- 41.Chung W, Choi SY, Lee E, Park H, Kang J, Park H, Choi Y, Lee D, Park SG, Kim R, Cho YS, Choi J, Kim MH, Lee JW, Lee S, Rhim I, Jung MW, Kim D, Bae YC, Kim E. Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nature neuroscience. 2015;18:435–443. doi: 10.1038/nn.3927. [DOI] [PubMed] [Google Scholar]
- 42.Duffney LJ, Wei J, Cheng J, Liu W, Smith KR, Kittler JT, Yan Z. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J Neurosci. 2013;33:15767–78. doi: 10.1523/JNEUROSCI.1175-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–77. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei J, Xiong Z, Lee JB, Cheng J, Duffney LJ, Matas E, Yan Z. Histone modification of Nedd4 ubiquitin ligase controls the loss of AMPA receptors and cognitive impairment induced by repeated stress. J Neurosci. 2016;36:2119–30. doi: 10.1523/JNEUROSCI.3056-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcel M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 46.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.