

Abstract
Chromosome 17p13.3 is a region of genomic instability that is linked to different rare neurodevelopmental genetic diseases, depending on whether a deletion or duplication of the region has occurred. Chromosome microdeletions within 17p13.3 can result in either isolated lissencephaly sequence (ILS) or Miller-Dieker syndrome (MDS). Both conditions are associated with a smooth cerebral cortex, or lissencephaly, which leads to developmental delay, intellectual disability, and seizures. However, patients with MDS have larger deletions than patients with ILS, resulting in additional symptoms such as poor muscle tone, congenital anomalies, abnormal spasticity, and craniofacial dysmorphisms. In contrast to microdeletions in 17p13.3, recent studies have attracted considerable attention to a condition known as a 17p13.3 microduplication syndrome. Depending on the genes involved in their microduplication, patients with 17p13.3 microduplication syndrome may be categorized into either class I or class II. Individuals in class I have microduplications of the YWHAE gene encoding 14-3-3ε, as well as other genes in the region. However, the PAFAH1B1 gene encoding LIS1 is never duplicated in these patients. Class I microduplications generally result in learning disabilities, autism, and developmental delays, among other disorders. Individuals in class II always have microduplications of the PAFAH1B1 gene, which may include YWHAE and other genetic microduplications. Class II microduplications generally result in smaller body size, developmental delays, microcephaly, and other brain malformations. Here, we review the phenotypes associated with copy number variations (CNVs) of chromosome 17p13.3 and detail their developmental connection to particular microdeletions or microduplications. We also focus on existing single and double knockout mouse models that have been used to study human phenotypes, since the highly limited number of patients makes a study of these conditions difficult in humans. These models are also crucial for the study of brain development at a mechanistic level since this cannot be accomplished in humans. Finally, we emphasize the usefulness of the CRISPR/Cas9 system and next generation sequencing in the study of neurodevelopmental diseases.
Keywords: 17p13.3, microdeletion, microduplication, lissencephaly, autism spectrum disorder, neurodevelopmental disorder, CRISPR, next generation sequence
General introduction
Chromosome 17 boasts the third highest density of segmental duplications among human chromosomes and has the second-highest gene density. More than 23% of the short arm of chromosome 17 consists of low-copy repeats (LCRs), creating the opportunity for non-allelic homologous recombination (NAHR) to occur (Komoike et al., 2010; Shimojima et al., 2010). This differs from allelic homologous recombination (AHR), which serves to separate haplotypes and results in the interchange of homologous sequences to contribute to non-pathologic genetic variation. NAHR results in the recombination of sequences on different chromosomes, different positions of homologous chromosomes, or between two sequences on the same chromosome. This can lead to deletions and duplications (Clancy and Shaw, 2008). The high density of LCRs found within chromosome 17p13.3 makes it a “recombination hotspot.” LCRs are made up of the same or very similar repetitive sequences and are ~95% identical (Stankiewicz et al., 2003). Many deletion breakpoints have been identified within LCRs (Stankiewicz et al., 2003). All of these factors contribute to the association of chromosome 17p13.3 with rare genetic diseases caused by haploinsufficiency or duplication.
The instability seen in chromosome 17 contributes to the development of a wide variety of diseases including morphological brain disorders, mental illnesses, epilepsy, and tumors (De Smaele et al., 2004; Shimojima et al., 2010; Schnaiter and Stilgenbauer, 2013; Gazzellone et al., 2014). In this review, the four major phenotypes we will discuss are Miller-Dieker syndrome (MDS), isolated lissencephaly sequence (ILS), class I 17p13.3 microduplication syndrome, and class II 17p13.3 microduplication syndrome. The CRK, PAFAH1B1, and YWHAE genes located at chromosome 17p13.3 all have crucial roles in neuronal migration and contribute to each of these genetic disorders when microdeletions or microduplications arise. MDS and ILS are associated with 17p13.3 microdeletions. Earl Walker first described cases of lissencephaly in 1942 in reference to the “smooth brain” observed. Miller and Dieker contributed to the identification of MDS in 1963 and 1969 respectively, resulting in the nomenclature of the disease (Walker, 1942; Miller, 1963; Pilz and Quarrell, 1996). MDS results from contiguous gene deletion within 17p13.3 and mainly features cerebral agyria (absence of gyri), cerebral pachygyria (broad gyri), craniofacial deformities, and seizures (Barros Fontes et al., 2017). ILS phenotypes lack the craniofacial deformities and the most severe grade of lissencephaly only observed in MDS phenotypes. These features of MDS patients result from larger deletions within chromosome 17p13.3 as compared to ILS patients. MDS is caused by microdeletions containing PAFAH1B1 and YWHAE, at minimum, while ILS can result from heterozygous mutation or deletion of PAFAH1B1 (Dobyns et al., 1993; Reiner et al., 1993). The thickening and simplification of cortical layers associated with lissencephaly seen in both ILS and MDS are due to a defect of neural migration during cortical development. This results in a variety of defects, including mental retardation (Dobyns et al., 1993; Cardoso et al., 2003).
Patients with microduplications of 17p13.3 were first reported in 2009, and this condition is now referred to as 17p13.3 microduplication syndrome (Bi et al., 2009; Roos et al., 2009). Although there are currently only about 40 reported cases, the number of 17p13.3 microduplication syndrome patients has been increasing. The affected can be categorized into two classes based on the size of chromosome duplication (Bruno et al., 2010). Patients with class I microduplications never have PAFAH1B1 duplications. They typically display autistic and other behavioral symptoms, delay in speech and motor abilities, craniofacial deformities, hand and foot deformities, and postnatal overgrowth (Bruno et al., 2010). In contrast, PAFAH1B1 is always duplicated in patients with class II, who may also have duplications of CRK and YWHAE (Bruno et al., 2010). Microduplications in class II result in psychomotor and developmental delay, as well as hypotonia (Bruno et al., 2010). Microdeletions and microduplications around the MDS critical region, which is the region of chromosome 17 spanning from PAFAH1B1 to YWHAE, have distinct phenotypes. Even so, both share similarities and affect the same genes (Table 1).
Table 1.
Comparison of major phenotypes and genotypes associated with microdeletions and microduplications.
| Microdeletions | Microduplications | |||
|---|---|---|---|---|
| ILS | MDS | Class I | Class II | |
| Major phenotypes | Lissencephaly (grade 2–4) Epilepsy |
Lissencephaly (grade 1) Epilepsy Craniofacial dysmorphisms Congenital anomalies |
Learning disabilities Autism spectrum disorder Elevated growth factors Dysmorphic facial features Developmental delay Behavioral problems (severe) |
Developmental delay Psychomotor delay hypotonia Mild brain malformations Behavioral problems (mild) |
| Major genotypes | Deletion of PAFAH1B1 | Deletion of PAFAH1B1, YWHAE, and/or CRK | Duplication of YWHAE and/or CRK | Duplication of PAFAH1B1, and/or YWHAE, and/or CRK |
| References | Cardoso et al., 2003; Nagamani et al., 2009; Takahashi et al., 2015 | Bi et al., 2009; Bruno et al., 2010; Curry et al., 2013 | ||
It is difficult to study these conditions with human patients due to the small number of affected individuals. Fortunately, the areas of clinical interest within the MDS region are largely conserved between the human and mouse genome (Figure 1). The short arm of chromosome 17 in humans coincides approximately with the center of chromosome 11 in mice and the 26 human genes (Table 2) known in 17p13.3 have homologs in the mouse chromosome. The genes in the MDS critical region are in the same order in humans and mice; however, some neighboring genes are in a different order. These genetic similarities and the parallels between human and mouse development make the mouse model an excellent analog (Yingling et al., 2003). Previous studies have used knockout and transgenic mice to study genes within the MDS critical region. There are currently some single and double knockout mice available, which will be discussed. However, mouse models for the complete deletion or duplication of chromosome 17p13.3 have yet to be developed. In this review, we discuss microdeletions and microduplications of chromosome 17p13.3 with a focus on patient phenotypes, phenotypes seen in single and double knockout and transgenic mice, and limitations on current studies. We also propose a combined CRISPR/Cas9-Cre-loxP approach to create mouse models with microdeletions and microduplications of the complete chromosome 17p13.3 region, which may advance current studies by providing a more clinically relevant model. We then discuss the potential for applied analysis of next generation sequencing to improve identification of copy number variants.
Figure 1.

Outline of the location of the MDS critical region in humans and mice. In humans, it lies in region 17p13.3 and in mice it lies on chromosome 11 in region B5. The gene order of the MDS critical region in mice is defined above. Figure not to scale. The order and position of genes were obtained from the Human Genome Project Ensembl Database (release 90, accessed on 10/23/2017).
Table 2.
Summary of genes involved in microdeletions and microduplications of human chromosome 17p13.3.
| Gene | Functional relevance | References |
|---|---|---|
| BHLHA9* | Encodes a protein from the basic helix-loop-helix family; transcription factor involved in limb development | Klopocki et al., 2012 |
| YWHAE | Encodes protein 14-3-3ε which is a phosphoserine/threonine-binding protein and a critical regulator of neurogenesis and neuronal differentiation | Cornell et al., 2016 |
| CRK | Encodes a signaling protein downstream of Reelin; functions in cell proliferation, differentiation, migration, axonal growth | Huang et al., 2004; Matsuki et al., 2008; Park and Curran, 2008; Barros Fontes et al., 2017 |
| MYO1C | Encodes a member of the unconventional myosin family (myosin class 1); actin-based molecular motor with ATPase activity; nuclear isoform associates with RNA polymerase I and II to initiate transcription | Desh et al., 2014; Sarshad et al., 2014; Arif et al., 2016 |
| INPP5K | Encodes a protein that is a potential negative regulator of the actin cytoskeleton | Rahman et al., 2011 |
| PITPNA | Encodes a protein member of a family of lipid-binding proteins that transfer molecules of phosphatidylinositol/choline between membrane surfaces; implicated in phospholipase C signaling; implicated in production of phosphatidylinositol 3,4,5-triphosphate | Kular et al., 1997; Morgan et al., 2004 |
| SLC43A2 | Encodes a member of SLC43 (L-amino acid transporter 3) transporter protein family; mediates transport of L-isomers of neutral amino acids in a pH-, sodium-, and chloride- independent manner; implicated in the transfer of amino acids across the placenta to the fetus | Bodoy et al., 2005, 2013 |
| SCARF1 | Encodes scavenger receptor protein that is expressed in endothelial cells; regulates uptake of chemically-modified low-density lipoproteins | Dhaliwal and Steinbrecher, 1999; Ishii et al., 2007 |
| RILP | Encodes a lysosomal protein; potentially a downstream regulator of RAB7; RAB7 and RILP potentially interact to regulate endocytic traffic; potential regulator of lysosomal morphology | Cantalupo et al., 2001; Jordens et al., 2001; Harrison et al., 2003; van der Kant et al., 2013 |
| PRPF8 | Encodes a protein that is a part of the U2- and U12- dependent spliceosomal machinery; essential for step II of pre-mRNA splicing | Grainger and Beggs, 2005; Boesler et al., 2015; Wickramasinghe et al., 2015 |
| TLCD2 | Encodes TLC domain containing two proteins; unknown function | Zody et al., 2006 |
| WDR81 | Encodes a multi-domain transmembrane protein; predominantly expressed in brain; potential role in endolysosomal trafficking | Liu et al., 2017; Shaheen et al., 2017 |
| SERPINF2 | Encodes a member of the serpin family of serine protease inhibitors; major inhibitor of plasmin, which degrades fibrin and various other proteins | Abdul et al., 2016 |
| SERPINF1 | Encodes a member of the serpin family (no protease inhibitory activity, secreted protein); strongly inhibits angiogenesis, neurotrophic factor | Johnen et al., 2015 |
| SMYD4 | Encodes a protein with a SET domain, which often has lysine methyltransferase activity for histone modification; potential tumor suppressor gene | Hu et al., 2009 |
| RPA1 | Encodes the largest subunit of the heterotrimeric Replication Protein A (RPA) complex; role in recruiting molecules to DNA | Dubois et al., 2017; Zhang et al., 2017 |
| RTN4RL1 | Encodes a protein with a GPI-anchored domain and LRR motif; also known as Nogo receptor 3 (NgR3), a paralog of NgR1 | Zhang et al., 2011 |
| DPH1/OVCA1 | Encodes an enzyme involved in the biosynthesis of diphthamide; defects associated with autosomal recessive intellectual and craniofacial dysmorphisms | Loucks et al., 2015 |
| OVCA2 | Encodes a protein that is related to a variety of α-β hydrolases including esterases, lipases, and other enzymes; highly conserved gene | Prowse et al., 2002 |
| HIC1 | Tumor repressor gene, cell cycle regulator | Kumar, 2014; Janeckova et al., 2015; Hao et al., 2017 |
| SMG6 | Encodes a component of the telomerase ribonucleoprotein complex; role in nonsense-mediated mRNA decay (NMD) pathway | Chakrabarti et al., 2014; Nicholson et al., 2014; Schmidt et al., 2015; Colombo et al., 2017; Ottens et al., 2017 |
| SRR | Encodes serine racemase, which produces the NMDA co-agonist D-serine that is implicated in Alzheimer's disease, ALS, ischemic brain damage, and schizophrenia | Nelson et al., 2017 |
| TSR1 | Encodes a serine-arginine-rich protein; splicing factor, affects mRNA stability and rRNA processing | Gupta et al., 2014 |
| SGSM2 | Encodes a GTPase activator; regulates enzyme trafficking | Marubashi et al., 2016 |
| MNT | Encodes a protein in the Myc/Max/Mad network that has a basic-Helix-Loop-Helix-zipper domain; represses transcription | Link and Hurlin, 2015 |
| METTL16 | Encodes a methyltransferase; required for MAT2A splicing (MAT2A encodes the SAM synthetase expressed in most cells) | Pendleton et al., 2017 |
| PAFAH1B1 | Encodes non-catalytic α subunit of the intracellular Ib isoform of platelet-activating factor acetylhydrolase, which catalyzes the removal of an acetyl group on platelet-activating factor; deletion results in classical lissencephaly | Nagamani et al., 2009 |
Genes are listed in the approximate order they are found in the sequence. NCBI Gene Database: Gene [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004—[cited 2017 10 19] (Available from: https://www.ncbi.nlm.nih.gov/gene/).
Located within chromosome 17p13.3, but immediately outside the MDS critical region.
Chromosome 17p13.3 deletions
Introduction
Deletion of the 17p13.3 chromosome region results in an array of phenotypes in humans, depending on the severity of the deletion. Well-characterized phenotypes that may result from such a deletion include isolated lissencephaly sequence (ILS) and Miller-Dieker syndrome (MDS) (Ledbetter et al., 1992; Cardoso et al., 2003; Bruno et al., 2010; Barros Fontes et al., 2017). A region of clinical interest within chromosome 17p13.3 is book-ended by two genes: PAFAH1B1 and YWHAE. Deletion of PAFAH1B1 results in classical lissencephaly (LIS), while larger deletions between PAFAH1B1 and YWHAE, or in other words the MDS critical region, result in MDS (Cardoso et al., 2003; Nagamani et al., 2009). Deletions within the 17p13.3 region are associated with features such as intellectual disability, craniofacial defects, epilepsy, and a few rare symptoms.
Although both ILS and MDS are associated with LIS, they are different conditions. Patients with ILS lack many of the facial dysmorphic features seen with MDS (Ledbetter et al., 1992; Cardoso et al., 2003; Kim et al., 2011). Other clinical features not found in ILS patients, such as cardiac defects, cystic kidney, and polydactyly, may also occur in MDS patients (Cardoso et al., 2003). MDS is associated with the most severe grade of LIS (grade 1), while individuals with ILS have less severe LIS (grades 2–4). The difference between the MDS and ILS phenotypes results from the deletion of specific genes within the 17p13.3 region. Deletion of PAFAH1B1 along with YWHAE and/or CRK may cause the more severe grade of LIS seen in MDS patients (Cardoso et al., 2003). YWHAE encodes 14-3-3ε, while CRK encodes a signaling protein that functions downstream of Reelin and is involved in cell proliferation, differentiation, migration, and axonal growth (Huang et al., 2004; Matsuki et al., 2008; Park and Curran, 2008; Barros Fontes et al., 2017).
Previously, MDS was incorrectly thought to be an autosomal recessive disorder because it was occasionally reported in individuals within the same family (Ledbetter et al., 1989). It has been shown that MDS results from a de novo microdeletion of chromosome 17p13.3 that may have a paternal bias in origin (Ledbetter et al., 1989). It is generally accepted that MDS is a contiguous-gene deletion syndrome, meaning that it results from the deletion of multiple genes found in close proximity to each other on the chromosome. This was challenged by a study involving five MDS patients using polymerase chain reaction (PCR) and fluorescence in situ hybridization (FISH) to check for microdeletions in chromosome 17p13.3. A deletion was only found in three of the patients. Because all five patients had nearly identical clinical symptoms, the results suggest that MDS is not a contiguous gene-deletion syndrome (Kohler et al., 1995). These data contradict the widely accepted belief that MDS is a contiguous gene-deletion syndrome, but should not be ignored. It is estimated that there is a visible or submicroscopic deletion of chromosome 17p13.3 in 90% of MDS patients, but the cause of the other 10% of cases is unclear (Ledbetter et al., 1992).
ILS may occur when a smaller deletion of chromosome 17p13.3 occurs. About 60% of ILS cases are associated with a deletion or intragenic variation of PAFAH1B1 (Takahashi et al., 2015). One study found 17p13.3 deletions in only 6 out of 45 ILS patients, which suggests that intragenic variations such as point mutations may account for the majority of cases (Ledbetter et al., 1992).
Deletions of chromosome 17p13.3 result in LIS, which is associated with mental retardation and epilepsy seen in both ILS and MDS patients. Individuals with deletions of PAFAH1B1 often have ILS, while individuals with larger deletions of the MDS critical region have MDS. More severe lissencephaly and craniofacial dysmorphic features are seen in MDS patients. The relationship between critical genes and human phenotypes will be described next.
Phenotypes and genotypes in human patients
Almost all of our knowledge about the various phenotypes seen in patients with MDS comes from clinical case studies. Lissencephaly and craniofacial dysmorphism are well-characterized features of MDS. Epilepsy is another major feature of MDS, though this is also seen in ILS. Deletions of certain genes are strongly associated with some phenotypes we will describe.
Phenotypes: lissencephaly
Classical lissencephaly (LIS) largely results from arrest or defect of neuronal migration occurring between 10 and 14 weeks of gestation, causing abnormal cortical layering and a “smooth brain”, meaning that patients with LIS have an absence or reduction of gyri and sulci in the cerebral cortex (Ledbetter et al., 1992; Wynshaw-Boris and Gambello, 2001; Wynshaw-Boris et al., 2010; Moon and Wynshaw-Boris, 2013). LIS is typically associated with mental retardation, intractable epilepsy, spasticity, and reduced longevity (Dobyns et al., 1993). The severity of LIS ranges from agyria (LIS grade 1), mixed agyria-pachygyria (LIS grades 2 and 3), and pachygyria (LIS grade 4). Clinical symptoms correlate with the degree of agyria (Dobyns et al., 1999). LIS may also be caused by variations of the DCX gene on chromosome Xq23, but these cases may be distinguished from those associated with PAFAH1B1 because of differences in the gyral patterns and more common hypoplasia of the cerebellar vermis in individuals with DCX variations (Dobyns et al., 1999; Takahashi et al., 2015; Ayanlaja et al., 2017). Variations of PAFAH1B1 are linked to a posterior-to-anterior gradient of lissencephaly, while mutations of DCX are associated with an anterior-to-posterior gradient (Dobyns et al., 1999). Additionally, DCX mutations are associated with lissencephaly in males and a subcortical band heterotopia pattern in females, since DCX is located on the X chromosome (Gleeson et al., 1998; Poduri et al., 2013).
It has been established that LIS results from a defect in radial migration. However, previous reports indicate the involvement of interneurons, which have been shown to migrate non-radially (Pancoast et al., 2005). LIS is associated with mental retardation and epilepsy, which can often be linked to interneuron defects (Pancoast et al., 2005). A study involving two fetuses and two children with MDS found a non-radial migration defect in the calretinin-expressing interneuron subpopulation (Pancoast et al., 2005). Pancoast et al. hypothesizes that the clinically distinct forms of LIS have differences in interneurons, which may be useful for diagnosis and treatment once better understood (Pancoast et al., 2005). The idea that the migration of different cell types may be defective in different forms of LIS is supported by work from Marcorelles et al. (2010). A defect in tangential migration of GABAergic neurons has been associated with MDS, while defects in other aspects of tangential migration were reported for two other types of LIS (Marcorelles et al., 2010). Clearly identifying patterns in structural differences, like variations of gyral patterns and migration patterns, will aid in distinguishing between the types of LIS and could have some clinical applications.
While abnormal neuronal migration has been shown to contribute significantly to the lissencephaly phenotype, defects in neurogenesis may also play a role. LIS1, which is encoded by PAFAH1B1, influences cell proliferation at various neurodevelopmental stages (Tsai et al., 2005; Bi et al., 2009; Pramparo et al., 2010, 2011; Reiner and Sapir, 2013). A study where LIS1 levels were reduced throughout cortical development showed that LIS1 is involved in generating neuroblasts and postmitotic neurons and also has an influence on cell survival (Gambello et al., 2003). Lissencephaly in patients is analogous to the cortical disorganization observed in LIS1 deficient mice, which can be attributed to a combination of migration defects and LIS1-mediated reduction of cell numbers in the ventricular zone via modulation of cell proliferation and neuroblast survival (Gambello et al., 2003). It seems logical that defects in neurogenesis would impact subsequent stages of neurodevelopment since neurogenesis precedes and could potentially influence many events in development. Additionally, an increased time period in which neurogenesis occurs in a species has been evolutionarily correlated with the degree of convolution observed in the cortex. For example, neurogenesis is three times longer in ferrets and ten times longer in primates, as compared with rodents (Poluch and Juliano, 2015). A longer period of neurogenesis is also correlated with a slower depletion of the cortical progenitor pool (Kornack and Rakic, 1998; Calegari et al., 2005). This provides further support for neurogenesis as a contributing factor in the LIS phenotype, in addition to neuronal migration arrests or defects.
Phenotypes: epilepsy
Lissencephaly is closely linked with epilepsy. Since LIS occurs in patients with both ILS and MDS, it is a symptom common to both conditions. As with lissencephaly, YWHAE, CRK, and PAFAH1B1 have complicated roles in epilepsy. Two patients with deletions of YWHAE and CRK, but not PAFAH1B1, were described as having macrocephaly, epilepsy, and non-specific changes in white matter, among other clinical features (Tenney et al., 2011). Tenney et al. suggests that deletions of YWHAE and/or CRK indicate that a patient should be monitored carefully for seizure development (Tenney et al., 2011). Shimojima et al. supports this with similar findings regarding one of the three patients involved in the study (Shimojima et al., 2010). A different patient involved in this case study had a partial deletion of PAFAH1B1 resulting in isolated grade 3 lissencephaly and epilepsy (Shimojima et al., 2010). Thus, YWHAE, CRK, and PAFAH1B1 are critical genes involved in epilepsy.
Phenotypes: craniofacial dysmorphisms
Individuals with MDS have a characteristic facial appearance including tall square forehead, flattened midface, low-set posteriorly rotated ear, short upturned nose, and prominent lateral nasal folds (Cardoso et al., 2003; Nagamani et al., 2009; Bruno et al., 2010; Yu et al., 2014). One study involved a patient with a 284 kb deletion within the MDS critical region where CRK and MYO1C were deleted, but not YWHAE (Ostergaard et al., 2012). The patient had mental retardation in addition to slight facial and limb abnormalities, suggesting that CRK, but not YWHAE, may have a role in craniofacial defects and limb malformations (Ostergaard et al., 2012).
Additionally, deletion of the HIC1 and OVCA1 genes has a role in producing these craniofacial dysmorphisms (Barros Fontes et al., 2017). A study involving 19 patients with a 17p13.3 microdeletion reported that 13 of those individuals had haploinsufficiency of HIC1 and OVCA1 (Tenney et al., 2011). Cleft palate was observed in 4 of these patients, while craniofacial dysmorphisms were found in 11. These data implicate HIC1 and OVCA1 in craniofacial defects and provide evidence for their role in palatogenesis (Barros Fontes et al., 2017). Cardoso et al. identified a 400 kb critical region that differentiates ILS from MDS (Cardoso et al., 2003). Since craniofacial dysmorphisms are unique to MDS, the study of this critical region could help identify more genes involved in these characteristic craniofacial defects.
Rare and miscellaneous characteristics of MDS
Approximately 90% of MDS patients have a visible or submicroscopic deletion of chromosome 17p13.3 (Ledbetter et al., 1992). About 1 in 50,000–100,000 patients has a ring chromosome disorder (Kim et al., 2014). It is possible for deletions to occur during the formation of ring chromosomes, since they result from the fusion of the short and long arms (Kim et al., 2014). Ring chromosomes have been linked to birth defects and mental disabilities (Kim et al., 2014). Several individuals have been described as having a ring chromosome 17 (Ono et al., 1974; Qazi et al., 1979; Chudley et al., 1982; Sharief et al., 1991). Of those patients, a few have had clinical features consistent with MDS (Sharief et al., 1991). One case report described a MDS patient who had 46 chromosomes with a ring structure taking the place of one of the chromosomes 17. There was also a deletion equivalent to what is typically seen in MDS (Sharief et al., 1991). Recent developments may eventually lead to therapeutics for such individuals. When fibroblast cells derived from patients with ring chromosomes are reprogrammed into induced pluripotent stem cells (iPSCs), the cells lose the ring chromosome and duplicate the wild-type homolog through compensatory uniparental disomy (UPD) (Bershteyn et al., 2014). These results could have important clinical implications and may eventually lead to the development of an approach to correct large-scale chromosomal aberrations (Kim et al., 2017).
While lissencephaly, craniofacial dysmorphism, and epilepsy are seen in the vast majority of MDS patients, there are also some less common and minor symptoms associated with MDS. An example is spinal manifestations. A 6-month-old infant with MDS was reported to have a tethered spinal cord, with the conus medullaris terminating abnormally low at the upper level of L4 (Hsieh et al., 2013). An inflamed lumbosacral sinus dermal tract was also described (Hsieh et al., 2013). The low incidence of MDS makes it difficult to determine if the rare and minor symptoms reported from an individual case study are a direct result of MDS or if they are unrelated. Mouse models with gene mutations will help overcome this difficulty and provide a better understanding of the relationship between specific genes and minor symptoms.
Mouse models
The region of chromosome 17p13.3 that is frequently deleted in patients with MDS is homologous to a region within the mouse chromosome 11B5. Mnt, Hic1, and Ovca1 are localized at this region and have been studied in mice (Carter et al., 2000; Toyo-oka et al., 2004; Yu et al., 2014). Mnt is essential for embryonic development and survival and may play a role in craniofacial defects associated with MDS, since Mnt knockout mice have cleft palate and retardation of skull development (Toyo-oka et al., 2004). Hic1 knockout mice show no neuronal migration defects or disorganization of the cerebral cortex, but do show gross developmental defects ranging from exencephaly to limb abnormalities (Carter et al., 2000). Ovca1 encodes a protein involved in diphthamide biosynthesis, which is critical for craniofacial development. Ovca1 deficiency in tissue derived from the neural crest has been shown to contribute to the craniofacial defects associated with MDS (Yu et al., 2014). Aberrant craniofacial development has been best observed in Mnt, Hic1, and Ovca1 knockout mice (Yu et al., 2014), so the study of the facial dysmorphic features associated with MDS currently relies on these models.
Heterozygous Mnt+/−, Hic1+/−, and Ovca1+/− mice do not have distinctive developmental phenotypes (Carter et al., 2000; Yu et al., 2014). This raises a question about the representativeness of these models to the human population with MDS, since these individuals only have deletions in one of their chromosome 17p13.3 regions. In most cases, we are currently analyzing craniofacial dysmorphisms resulting from haploinsufficiency of a relatively large region by use of homozygous mouse model knockouts of individual genes. If deletion of these genes results in phenotypes in a dose-dependent manner, developmental phenotypes not seen in the heterozygous single knockout models may emerge when heterozygous knockouts of multiple genes are created. Study of the craniofacial dysmorphisms associated with MDS could be improved by creating Mnt/Hic1, Hic1/Ovca1, and/or Mnt/Ovca1 double heterozygous knockout mice. A Mnt/Hic1/Ovca1 triple heterozygous knockout model may show a more dramatic phenotype.
There are Pafah1b1, Ywhae, and Crk single knockout models and a Pafah1b1/Ywhae double knockout model available to study the neurodevelopmental defects associated with deletion of chromosome 17p13.3. Pafah1b1 encodes Lis1, which is involved in neuronal migration and has a role in dendritic filopodia dynamics and spine turnover (Hirotsune et al., 1998; Yamada et al., 2009; Sudarov et al., 2013). Ywhae encodes 14-3-3ε and is always deleted in patients with MDS. 14-3-3ε binds Ndel1 (also known as Nudel), which is phosphorylated by CDK5/p35, so Ndel1 remains phosphorylated (Toyo-oka et al., 2003). Single knockout models for Pafah1b1 and Ywhae are associated with similar defects in brain development and neuronal migration, while a double knockout of Pafah1b1 and Ywhae leads to a more severe phenotype that may be attributed to the non-sustained phosphorylation of Ndel1 (Toyo-oka et al., 2003).
The roles of Crk and Crk-like (Crkl) genes have been studied using mouse models. Crk encodes a signaling protein that functions downstream of Reelin and has roles in cell proliferation, differentiation, migration, and axonal growth (Huang et al., 2004; Tanaka et al., 2004; Matsuki et al., 2008; Park and Curran, 2008; Barros Fontes et al., 2017). Mutation of only Crk or Crkl does not compromise Reelin signaling and does not produce any obvious anatomical abnormalities (Park and Curran, 2008). However, double knockout of Crk and Crkl resulted in mice with grossly abnormal brain appearance (Park and Curran, 2008). This suggests overlapping functions for Crk and Crkl in Reelin signaling. Deletion of Crk, along with Ywhae, is thought to contribute to the more severe phenotype seen in MDS, as opposed to ILS (Tenney et al., 2011; Barros Fontes et al., 2017). At this time, Crk/Pafah1b1 and Crk/Ywhae double heterozygous knockout mice are not available. Creation of a Crk/Pafah1b1 double knockout could help determine if deletion of these genes contributes to the lissencephaly phenotype in a dose-dependent manner, as this seems to be the case when CRK and YWHAE are deleted together in humans (Tenney et al., 2011; Barros Fontes et al., 2017). There is a need for a Crk/Ywhae double knockout to be created, since combined deletion of these two genes is clinically important and has yet to be analyzed with a mouse model.
Limitations to current studies and future directions
Conventional cytogenetic analysis, or karyotyping, is not sufficient to detect microdeletion syndromes. Several case studies have used FISH to identify microdeletions of chromosome 17p13.3 in MDS and ILS patients (Kuwano et al., 1991; Kohler et al., 1995; Pilz et al., 1995; Cho et al., 2009). A relatively new alternative method is mental retardation syndrome multiplex ligation-dependent probe amplification (MRS-MLPA), which is capable of testing for multiple microdeletions at once and is less time consuming and less technically complicated than FISH. Another advantage of MRS-MLPA is its ability to detect smaller deletions that could be missed using FISH (Cho et al., 2009). The importance of this is illustrated by a case involving a MDS patient who had a partial deletion of the PAFAH1B1 locus (Izumi et al., 2007). A commercially available PAFAH1B1 FISH probe was used in an attempt to diagnose this patient, but due to the nature of the deletion, the patient's test results were normal (Izumi et al., 2007). The partial deletion was later discovered using a smaller sized FISH probe, drawing attention to the inability of FISH to detect relatively small deletions (Izumi et al., 2007). Other case studies have reported similar incidents (Shimojima et al., 2010). Use of FISH for diagnostic purposes may result in misdiagnosis for individuals with partial deletions of PAFAH1B1. Thus, the method that is considered the standard laboratory diagnostic tool for MDS has some shortcomings (Izumi et al., 2007). Unfortunately, the same is true of the mouse models currently being used to study MDS.
To date, there is no murine model for deletion of the complete MDS critical region. The lack of such a model is problematic because single or double knockout mice are not representative of the full range of phenotypes seen in MDS patients, who have relatively large deletions. While it would provide a more clinically relevant model, there are technical challenges in creating a mouse model for deletion of the complete MDS critical region. Later, in the conclusion of this paper, we describe these challenges and propose a strategy to address them.
Chromosome 17p13.3 duplications
Introduction
Chromosome 17p13.3 is a region of genomic instability that is prone to submicroscopic rearrangements due to a high density of LCRs (Roos et al., 2009; Capra et al., 2012). These submicroscopic rearrangements can lead to microduplications (Roos et al., 2009; Capra et al., 2012). Duplications that cause 17p13.3 microduplication syndrome have diverse mechanisms, come in various sizes, and include a variety of genes. It follows that these microduplications are associated with a diverse array of phenotypes. The phenotypes generally associated with microduplications of 17p13.3 include developmental and psychomotor delay, behavioral problems and autism spectrum disorder (ASD), structural brain abnormalities and malformations, and distinct physical features (Curry et al., 2013). Microduplications have also been associated with limb malformations and cleft lip and palate (Curry et al., 2013). Microduplications in 17p13.3 occur in the same gene region that when deleted causes MDS, and therefore is sometimes referred to as the MDS critical region. The microduplication minimal region has been defined as a 72kb region exclusively containing gene YWHAE, which encodes protein 14-3-3ε and is strongly associated with ASD (Bruno et al., 2010; Curry et al., 2013).
17p13.3 microduplication syndrome diagnosis splits duplications into two categories: class I and class II (Bruno et al., 2010). Class I duplications are categorized as containing the gene YWHAE, and not PAFAH1B1, which encodes protein LIS1 (Bi et al., 2009). Class I duplications can also involve other genes, such as CRK. Patients with class I duplications usually display phenotypes characterized by learning disabilities and ASD (Bi et al., 2009; Curry et al., 2013). These individuals may have congenital defects, macrosomia, dysmorphic facial features, mild to severe developmental delay, and behavioral problems such as increased aggression (Bi et al., 2009; Curry et al., 2013). Class II microduplications are any duplication in chromosome 17p13.3 that contains PAFAH1B1. Class II duplications can also involve other genes such as YWHAE and CRK (Bi et al., 2009; Roos et al., 2009; Bruno et al., 2010). Phenotypes generally associated with class II duplications are mild to severe developmental and/or psychomotor delay, hypotonia, and mild brain malformations including microcephaly (Bi et al., 2009; Bruno et al., 2010). In contrast to infants with class I microduplications, infants with class II microduplications may also have small body size at birth. There are some reports of major internal organ abnormalities, such as structural congenital heart disease, in patients with class II duplications (Bi et al., 2009). Seizures were also noted, but were not common phenotypes among both classes of duplications (Bi et al., 2009; Curry et al., 2013; Gazzellone et al., 2014). Overall, there are overlapping but distinct phenotypes observed in class I and class II microduplication syndrome patients.
Popular techniques for diagnosing and classifying 17p13.3 microduplications in patients are FISH, array comparative genomic hybridization (Array CGH), multiplex ligation-dependent probe amplification (MLPA), RT-PCR analysis, gene expression analysis, and chromosomal microarray (Bi et al., 2009; Roos et al., 2009; Bruno et al., 2010; Luk et al., 2014; Nagata et al., 2014; Petit et al., 2014; Eriksson et al., 2015). Array CGH has been used to detect gain of copy number, whereas FISH has been used to confirm duplications and regions (Bi et al., 2009). Case studies have analyzed the parental genomes of patients with 17p13.3 microduplication syndrome to determine potential inheritance patterns. In case studies where both parents were present and available for sequencing, duplications were largely classified as de novo. Only a few reported cases were maternally inherited (Bi et al., 2009; Curry et al., 2013). Simple and complex rearrangements have been observed. Suggested mechanisms for these rearrangements include non-homologous end joining (NHEJ) and replication fork stalling and template switching (Gu et al., 2008; Bi et al., 2009; Bruno et al., 2010). Bruno et al. also identified an individual with a microduplication that seemed to be caused by NAHR due to the observation of breakpoints lying within repetitive elements (Bruno et al., 2010). DNA sequencing of junction points was used to assess these hypothesized mechanisms (Bi et al., 2009; Bruno et al., 2010). It has been suggested that the microduplications present in 17p13.3 microduplication syndrome do not arise from stochastic events, but instead from intrinsic architectural features of the genome (Vissers et al., 2007).
Phenotypes: developmental/psychomotor delay and behavior problems
All patients present in case studies experienced some form of developmental delay, developmental disorder, cognitive impairment, speech abnormality, and/or behavior problems. Developmental delay is seen in both class I and class II duplications, whereas psychomotor delay is generally a characteristic unique to class II duplications (Bruno et al., 2010). Severity of the delay is highly diverse and variable. Case studies have shown developmental delay to vary greatly both between and within families (Curry et al., 2013). There was no correlation between size and location of duplication and degree of developmental delay or intellectual impairment. Curry et al. observed intellectual disability in 66% of patients who took part in their large-scale case studies (Curry et al., 2013). Cases consisted of individuals ranging from those who completed a high school education, to others who had average IQs accompanied by significant behavioral abnormalities, to those who were severely impaired (Curry et al., 2013). Delayed developmental milestones have been observed across patient populations. In a series of case studies performed by Roos et al. one patient with mild delays was observed to sit independently at 11 months, walk at 27 months, and have a vocabulary containing four words at 24 months (Roos et al., 2009). Other patients showed similar delays, with some more severe than others. Specifically, one more severely delayed patient was assessed at 22 months old and was just beginning to sit independently, had no recorded ability to walk, and had no meaningful vocabulary (Roos et al., 2009). In contrast, Bi et al. reported on one patient whose only impairment was fine motor delays by age 15 (Bi et al., 2009). Curry et al. observed behavioral issues in 100% of cases, but failed to observe a consistent behavioral phenotype for the patient population (Curry et al., 2013). Attention deficit hyperactivity disorder (ADHD) was observed in all individuals with duplications involving YWHAE (Bi et al., 2009). Behavioral phenotypes observed across case studies and classes of duplications included: poor social relationships, communication impairment, persistent/repetitive behaviors, attention deficit disorder, obsessive compulsive disorder or tendencies, obsessive food seeking, and mild to significant depression in older patients (Bi et al., 2009; Bruno et al., 2010; Hyon et al., 2011; Curry et al., 2013). As with developmental and psychomotor delay phenotypes, behavioral phenotypes were also variable and spanned duplication classes. Mechanistic etiological studies can help shed light on different microduplication region contributions to different variable delays and behavioral phenotypes.
Phenotypes: autism spectrum disorder (ASD)
Studies have found rare penetrative genetic copy number variations (CNVs) to account for between 5 and 15% of ASD cases. Copy number variations have been found in 8.6% of children diagnosed with autism, all of which were de novo (Gazzellone et al., 2014; Eriksson et al., 2015). It is important to note that <1% of autistic patients studied by Eriksson et al. presented with microduplications in 17p13.3, but when looking specifically at case studies of patients with 17p13.3 microduplication syndrome, ~32% presented with some form of ASD diagnosis or tendencies (Curry et al., 2013; Eriksson et al., 2015). Although this specific duplication is rare in the ASD patient population as a whole, ASD comorbidity is somewhat common in the 17p13.3 microduplication patient population. According to Curry et al., there are one or more autism loci in the 17p13.3 region (Curry et al., 2013). YWHAE and CRK are seen as candidate genes for ASD that arises in patients with 17p13.3 microduplication syndrome. This is due to the observance of autistic-like behaviors and diagnosis of ASD primarily in patients with class I duplications, and overlap of the same genes being duplicated in multiple patients (Curry et al., 2013).
Phenotypes: brain abnormalities and characteristic physical features
Brain abnormalities and characteristic physical features vary slightly across classes of microduplications, but are subtle across groups. Brain abnormalities have been assessed using MRI technology on few patients involved in case studies (Bi et al., 2009; Roos et al., 2009; Curry et al., 2013). Brain abnormalities are generally observed in individuals with class II duplications, which result in LIS1 overexpression, but the abnormality phenotype is usually minimal (Bi et al., 2009). One patient in a case study performed by Bi et al. had a triplication of PAFAH1B1 and showed mild volume loss in the cerebellum, dysgenesis of the corpus callosum, and cerebellar atrophy. The brain also appeared smaller, especially in the occipital cortex (Bi et al., 2009). Another subject with a duplication of PAFAH1B1 showed mild cerebellar volume loss, thinning of the splenium of the corpus callosum, and a smaller brain, once again most notably in the occipital cortex (Bi et al., 2009). Other case studies performed MRI imaging of six patients with 17p13.3 microduplication syndrome and found that three patients showed variable abnormalities of the corpus callosum and cerebellum, and the remaining three patients had normal MRI scans (Curry et al., 2013). Roos et al. performed case studies of patients with 17p13.3 microduplication syndrome and provided brain imaging data for two patients (Roos et al., 2009). MRI imaging of one patient revealed hypoplasia of the corpus callosum and dilation of lateral ventricles. Thinning of white matter may have occurred. The MRI showed a possible increase of the signal intensity paraventricularly. A small pituitary gland and enlargement of the cisterna magna were also observed (Roos et al., 2009). A brain axial CT scan was performed on one patient at two years old and revealed potential delayed myelination, but in a second axial CT scan at age four myelination appeared normal (Roos et al., 2009). A novel inverted 1.4 Mb microduplication that disrupted PAFAH1B1 in a patient has been reported (Classen et al., 2013). An MRI was performed on the patient presenting with this duplication and results showed diffuse pachygyria with a moderately thick cortex, smooth white-gray border and no microgyri, and mild-posterior predominate lissencephaly (Classen et al., 2013). Results also showed a thin, short corpus callosum and a mildly thin, flat brainstem (Classen et al., 2013). Brain abnormalities observed in this patient are more characteristic of 17p13.3 microdeletions than microduplications, and authors hypothesized that this phenotype was due to insertion of the 1.4 Mb duplication into intron 1 of PAFAH1B1, which disrupted normal splicing and effectively inactivated one copy of PAFAH1B1 (Classen et al., 2013). One case study observed a patient with a novel class I microduplication with slight brain malformations (Capra et al., 2012). An MRI was performed at 5 years of age and revealed a reduction in the volume and thickness of the isthmus of the cingulate gyrus and the splenium of the corpus callosum (Capra et al., 2012). A dysmorphic aspect of the rostrum of the corpus callosum was also observed (Capra et al., 2012). This is a somewhat rare case in which a patient with a class I microduplication also presented with brain malformations. Patients with class I microduplications usually present with dysmorphic facial features, as opposed to brain malformations. Facial features include asymmetric cranium, flat occipital region, dysmorphic appearance, frontal bossing, low set ears, broad nasal bridge, small nose, and hypertelorism (Bi et al., 2009; Roos et al., 2009; Bruno et al., 2010; Curry et al., 2013). Throughout the rest of the body, other general phenotypes have been observed across duplication classes such as abnormal body proportions, long limbs, and anisomelia (Roos et al., 2009; Bruno et al., 2010).
Phenotypes: overgrowth and related growth factors
Case studies analyzed by Bi et al. observed macrosomia in all but one patient with YWHAE duplication, which contrasts the severe growth restriction observed in individuals with duplications in PAFAH1B1 (Bi et al., 2009). Patients with class I duplications and macrosomia had duplications expanding into CRK, which is known to be involved in regulation of growth and cell differentiation. One case study reports a patient with a 1.58 Mb terminal gain of 17p13.3 (Henry et al., 2016). This gain is a class I duplication including YWHAE and CRK. The patient showed increased growth factors, pathologic tall stature, and entered puberty at 7 years old. This case study is unique in that it provided a detailed endocrinologic evaluation and reported involvement of the anterior pituitary gland. This suggests a potential hormonal mechanism for overgrowth associated with 17p13.3 microduplication syndrome (Henry et al., 2016). Future case studies of patients with class I microduplications in addition to macrosomia, accelerated growth, and/or elevated growth factors should aim to perform detailed endocrinologic analysis of the patient. This will further elucidate the hypothesized hormonal mechanism and allow for a better understanding of the consequences of CRK duplication.
Phenotypes: limb malformations
The BHLAH9 gene lies in the 17p13.3 microduplication syndrome region, just outside of the MDS critical region in humans, and has been associated with limb malformations (Nagata et al., 2014). Curry et al. hypothesized that duplication of BHLAH9 was necessary, but not sufficient, for limb malformation and that a complex mechanism including disruption or separation of nearby regulatory elements underlies the development of split hand/foot malformation with long bone deficiency (SHFLD) (Curry et al., 2013). Class I microduplications have been observed in patients with limb malformations such as SHFLD (Curry et al., 2013). For example, a case study reported a triplication involving BHLAH9 and a segment of YWHAE (Luk et al., 2014). A fetus was observed with bilateral split-hand malformation, and the triplication was maternally inherited. In a study of 51 Japanese families, BHLAH9 duplications were found to be the largest predictor of a range of different limb malformations (Nagata et al., 2014). A dosage effect was also observed in this sample, with the larger duplication, or in some cases triplication, correlating to more dramatic limb malformations. The limb malformations seen in this cohort were SHFLD, split hand/foot malformation (SHFM), and Gollop-Wolfgang complex (GWC) (Nagata et al., 2014). Like Curry et al., Nagata et al. also suggested that BHLAH9 copy number gains were necessary, but not sufficient, to cause limb malformations (Curry et al., 2013; Nagata et al., 2014). Case studies on 13 families with both BHLAH9 copy number gains and limb malformations found hand malformations in 75% of patients, foot malformations in 38% of patients, and long bone deficiency in 43% of patients (Petit et al., 2014). It has been hypothesized that duplications involving a 173 kb critical region correlate with SHFLD, due to the presence of overlapping microduplications in 17p13.3 in three patients involved in a case study (Armour et al., 2011). This 173 kb critical region contains gene BHLAH9 and exons 1-3 of ABR, another gene that lies just outside the MDS critical region (Armour et al., 2011). It has been proposed that duplication of this 173 kb region could potentially alter the dosage of a regulatory element involved in limb development or disrupt the interaction between a nearby regulatory element and its gene target or targets (Armour et al., 2011). These hypothesized mechanisms support the previously suggested mechanism that disruption or separation of nearby regulatory elements from their gene targets in the 17p13.3 microduplication region underlies the development of SHFLD (Curry et al., 2013).
Phenotypes: cleft lip and palate
Syndromic and non-syndromic cleft lip and palate have been observed in patients with both class I and class II 17p13.3 microduplication syndrome. YWHAE has been implicated in the etiology of this phenotype, because it is specifically evidenced in the development of midline craniofacial structures (Tucker and Escobar, 2014). It has been hypothesized that interactions between other genes in the duplication region and YWHAE contribute to the variations seen in this phenotype (Tucker and Escobar, 2014). Tucker and Escobar report on two cases of 17p13.3 class I microduplications involving YWHAE with cleft lip and palate phenotypes. These cases were the first reported cases with a strictly class I microduplication categorization (Tucker and Escobar, 2014). It was hypothesized that YWHAE duplications played a potentially causative role in the cleft lip and palate phenotype (Tucker and Escobar, 2014). A case study also reported a patient with 17p13.3 microduplication syndrome with a non-syndromic cleft lip and palate phenotype (Ibitoye et al., 2015). Curry et al. reported on two families with multiple members who had the 17p13.3 microduplication syndrome and cleft lip and palate with or without accompanying intellectual disability (Curry et al., 2013). Overall, cleft lip and palate is generally accepted as a rare phenotype observed in 17p13.3 microduplication syndrome patients (Curry et al., 2013).
Limitations to current studies and future directions
The majority of information about 17p13.3 microduplication syndrome has been obtained through case studies in human populations. Because 17p13.3 microduplication syndrome is classified as a rare disease, there are a very limited number of patients available for study. The largest review of case studies was authored by Curry et al. in collaboration with researchers, clinicians, and hospitals across the country to review 21 families (Curry et al., 2013). There are also no studies to date that have analyzed brains post-mortem due to the rarity of the disease and difficulty to obtain samples, so we must rely on techniques such as MRI to observe whole brain malformations. Additionally, gene expression patterns specific to the brain tissue of 17p13.3 microduplication syndrome patients has not been studied due to the limitations of obtaining post-mortem samples. Because participant numbers are small and phenotypes are incredibly variable, it is difficult to find distinct patterns between genotype and phenotype as well as disease etiology. Case studies allow for determining the presence and location of duplications, but difficulties arise when trying to determine mechanistic information.
A transgenic mouse model to conditionally overexpress Lis1 in the developing brain has been created (Bi et al., 2009). This model utilized the Cre-loxP system to increase expression of Pafah1b1, which caused a ~20% increase of Lis1 protein levels (Bi et al., 2009). Results showed that increased Lis1 expression in the developing brain may lead to neuronal migration defects as well as smaller brain size (Bi et al., 2009). They observed disorganization in the ventricular zone and disrupted cell polarity, which is critical for migration. Bi et al. also observed both radial and tangential migration disruptions in the cortex and an increase of apoptotic cells (Bi et al., 2009). This Lis1 overexpression mouse model was effective for investigating the effects of Lis1 overexpression in the developing mouse brain. However, in order to create a disease model for the 17p13.3 microduplication syndrome, other genes and proteins must also be considered, as almost all human patients have duplications that span multiple genes.
Cornell et al. used the technique of in utero electroporation to observe the effects of 14-3-3ε overexpression in the developing murine cortex (Cornell et al., 2016). Plasmids containing 14-3-3ε overexpression vectors were injected into the lateral ventricle of E14.5 or E16.5 embryonic brains. Electrodes were then positioned to direct plasmids into cells near the ventricular zone, which later migrate to their positions in the cortex. Embryos were then placed back into the uterus and allowed to develop uninterrupted. Brains were harvested and analyzed at P15. Results show that overexpression of 14-3-3ε results in a decrease of neurite formation through interactions with doublecortin (Dcx) (Cornell et al., 2016). This interaction decreases Dcx degradation and results in the failure of microtubule invasion into lamellipodia, which is a key step in neurite formation. Failure of microtubule invasion into the lamellipodia resulted in severe neuronal morphological defects. This was a useful technique for analyzing phenotypic differences in the developing mouse brain between 14-3-3ε overexpressing mice and control mice, but this system does not provide an exact mimicry of 14-3-3ε overexpression in human patients with the 17p13.3 microduplication syndrome. As was the case with Bi et al., this 14-3-3ε overexpression model proved effective for analyzing the effects of a single protein overexpression on neuronal development (Bi et al., 2009; Cornell et al., 2016). However, because both class I and class II duplications involve an interaction between multiple duplicated genes to create a phenotype, a mouse model must be created that encompasses multiple genes in chromosome 17p13.3 that have been identified as critical regions in humans.
Similar difficulties arise when studying deletions in 17p13.3. Models have been created to assess specific genes, but a successful mouse model has not yet been created to observe either 17p13.3 microdeletions or microduplications as they exist in human populations. Future directions include modification of available transgenic technology to create mice expressing certain 17p13.3 microdeletions or microduplications that correlate with specific known human phenotypes for mechanistic and/or etiological studies.
Mouse model with 17p13.3 deletion or duplication
Proposed Cre-LoxP and CRISPR/Cas9 combinatorial strategy for creating 17p13.3 deletion or duplication mouse models
To better understand both 17p13.3 microduplications and microdeletions, a new disease model must be created that encompasses critical regions containing both Pafah1b1 and Ywhae. There is a noted interaction between LIS1 and 14-3-3ε in all discussed disorders and both proteins are essential for correct neuronal migration (Toyo-oka et al., 2003; Bi et al., 2009; Bruno et al., 2010). Complications arise when trying to create models containing deletions or duplications of both Pafah1b1 and Ywhae because of the relatively large distance of ~1.3 Mbps between the two genes. This region is too large to either delete or duplicate using traditional Cre-loxP methods or the CRISPR/Cas9 system alone. We propose a methodology to create double transgenic mouse models using both Cre-loxP and CRISPR/Cas9 in combination to delete or duplicate a region of 17p13.3 containing both Pafah1b1 and Ywhae.
In theory, traditional Cre-loxP methods could be used to make a mouse model for deletion of the complete MDS critical region, but in practice this process would be quite laborious and likely require multiple attempts before mice with the correct genotype could be generated. Mice with loxP sites on the same DNA strand at both Pafah1b1 and Ywhae would need to be produced. A wide range of Cre transgenic mice could then be used to spatially and temporally delete the MDS critical region in mice. It would be possible to produce the Pafah1b1+/flox;Ywhae+/flox (in cis) mice by crossing Pafah1b1+/flox;Ywhae+/flox (in trans) mice with wild-type mice (Figure 2). This approach would rely on a meiotic crossover between alleles to place both loxP sites on the same chromosome (in cis). If there is no recombination, offspring would be positive for a loxP site at either the Pafah1b1 or Ywhae alleles. If crossover does occur, then the offspring will be positive for both alleles. The crossover could be confirmed by mating the Pafah1b1+/flox;Ywhae+/flox (in cis) mice to wild-type mice. We would expect ~1% embryonic recombinants from trans to cis (Merscher et al., 2001). This approach would require genotyping of hundreds of pups and relies heavily on chance, but has been used successfully in the past (Merscher et al., 2001). An additional concern is the Cre-mediated deletion would likely have an extremely low efficiency. The farther apart the loxP sites are, the more inefficient the deletion (Coppoolse et al., 2005).
Figure 2.

Traditional Cre-loxP approach for generating a mouse model for deletion of the complete MDS critical region. To produce Pafah1b1+/flox;Ywhae+/flox (in cis) mice, and create the opportunity for Cre-mediated deletion to occur, Pafah1b1+/flox;Ywhae+/flox (in trans) mice should be crossed with wild-type mice.
The approach for creating a mouse model for duplication of chromosome 17p13.3 is similar to that for creating the deletion model, with a few crucial differences. To create the duplication, the loxP sites should be on opposite DNA strands (Figure 3). Pafah1b1+/flox;Ywhae+/flox (in trans) mice would be crossed with Cre transgenic mice. This will result in the production of two alleles at the same time. One allele will have a duplication of the MDS critical region, while the other will have a deletion of the MDS critical region. This technique may also be used as an alternative approach for producing the deletion model. The two alleles can be segregated by crossing the mice with the deletion and duplication alleles with wild-type mice. However, a concern is that the recombination efficiency by Cre in trans is extremely low (Liu et al., 1998; Zheng et al., 2000). Therefore, this technique is interesting, but requires hard work.
Figure 3.

Workflow to simultaneously generate chromosome 17p13.3 deletion and duplication mouse models using a combinatorial CRISPR/Cas9—Cre-loxP approach.
Use of the CRISPR/Cas9 system alone would only allow the deletion of a single gene for each gRNA construct. If two gRNAs were designed to target both Pafah1b1 and Ywhae, mutation would likely occur close to the target sites, leaving most of the genes in the MDS critical region intact. A combined approach using CRISPR/Cas9 to insert the loxP sites near Pafah1b1 and Ywhae would be the most efficient way to create a mouse model with this relatively large deletion (Figure 4). This would allow researchers to bypass the laborious task described above of crossing the Pafah1b1+/flox;Ywhae+/flox (in trans) mice with wild-type mice. The loxP sites inserted by CRISPR/Cas9 should have the same directional orientations, so that the floxed sequence will be deleted. One concern is ensuring that both the loxP sites are inserted in the same chromosome and on the same DNA strand. To partially address this, a mutated nickase version of the Cas9 enzyme could be used. This would create a single-stranded break, as opposed to the double-stranded break created by the wild-type enzyme, which would make it more likely that the loxP sites would be inserted in the same DNA strand to create cis mice. Multiple attempts may be necessary before the loxP sites are inserted, since the efficiency of DNA break by the mutated nickase Cas9 is lower than wild-type Cas9.
Figure 4.
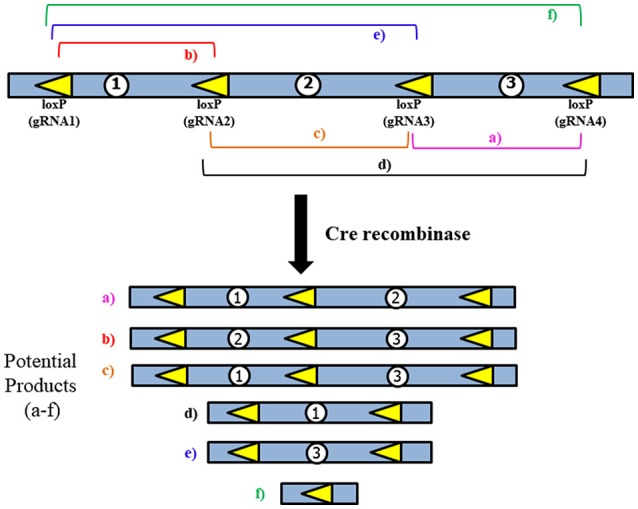
General strategy for use of multiple loxP sites to increase Cre-mediated deletion efficiency. LoxP sites would be inserted using the CRISPR/Cas9 system. The multiple products that could potentially be generated may aid in creating diverse models of multiple types of deletions, which would be similar to what is observed in human patients.
While the use of the CRISPR/Cas9 system would make insertion of the loxP sites significantly easier, the issue of extremely low Cre-mediated deletion efficiency remains due to the large size of the region we are proposing to delete. To address this, several loxP sites could be inserted to allow for a serial deletion of the complete MDS critical region (Figure 4). This would be done according to the method proposed above, but instead of only inserting loxP sites near Pafah1b1 and Ywhae multiple loxP sites would span the ~1.3 Mbps region. This should increase the efficiency of the Cre-mediated deletion.
Embryonic stem (ES) cells or fertilized eggs may be used to employ our strategy which combines Cre-loxP and CRISPR/Cas9 techniques. Use of ES cells is preferred because it will be simpler to inject the gRNA constructs in cells in culture and confirm that the loxP sites have been incorporated as desired. Once ES cells of the appropriate genotype have been generated, they may be used to create chimera mice. The chimeras should then be used for testing the germline transmission. These mice with the multiple pairs of loxP sites would finally be crossed with Cre transgenic mice. There are a few considerations that should be made when selecting which of the several varieties of Cre recombinase to use. EIIa-Cre causes recombination very early in development (Lakso et al., 1996). This would be most representative of human patients. However, this phenotype may be embryonic lethal. Nestin-Cre is neuron-specific and would cause recombination later in development (Giusti et al., 2014). This would be less representative of what occurs in human development, but may be the better option if inducing recombination at earlier time points proves to be lethal. Although numerous technical aspects would need to be considered throughout the process, generation of mouse models that have either a deletion or duplication of the region of 17p13.3 that contains both Pafah1b1 and Ywhae would likely provide the most clinically relevant models of the associated diseases to date.
Advantages and disadvantages of the proposed mouse model
Creation of mouse models for deletion and duplication of the MDS critical region will present unique technical challenges. There are many advantages associated with these models, but they are not without some disadvantages. Mouse models are a classical method for the study of neurodevelopmental disorders, since ethical concerns prevent the study of neurodevelopment using human subjects. Post-mortem analysis of brains and other techniques traditionally used to study phenotypes that emerge later in life do not allow developmental defects to be studied in real time, so these methods will not greatly advance our understanding of neurodevelopmental diseases. For these reasons, use of a mouse model that allows for the observation of neurodevelopment is highly advantageous. With recent advances in the use of iPSCs and organoids, it is possible to observe and analyze cellular, molecular, and simple morphogenic and migrational phenotypes associated with disease. Use of a mouse model would also allow for observation of these phenomena, but would additionally allow for analysis of accompanying behavioral phenotypes and craniofacial defects, which are homologous to those seen in human patients.
Although mice and humans differ in brain structure, chromosome structure, and behavior, creation of a mouse model is still useful to draw conclusions about brain and behavioral abnormalities in humans (Lui et al., 2011; Watson and Platt, 2012; Geschwind and Rakic, 2013; Florio and Huttner, 2014). There is a slight variation in the arrangement of genes surrounding the MDS critical region. The BHLHA9 gene that is located immediately outside the MDS critical region in humans is separated from the MDS critical region in mice by several other genes. This means that a frequently duplicated gene in humans may not factor into the mouse model for the 17p13.3 microduplication syndrome. Although this slight difference may cause variation, the entire MDS critical region does exist in the same order in humans and mice.
The most relevant behavioral difference between mice and humans is that it is impossible to draw conclusions regarding language development in mice, which would be of interest since language development is delayed in autistic human patients with 17p13.3 microduplication syndrome and developmentally delayed patients with 17p13.3 microdeletion syndrome (Silverman et al., 2010; Watson and Platt, 2012). However, there are other established measures of autistic behavior in mice that are homologous to humans such as stereotypic and repetitive behaviors, social behavior, and social communications (Crawley, 2004; Silverman et al., 2010; Watson and Platt, 2012). Social behavior is an especially important consideration for the study of ASD and related phenotypes in which patients may have characteristically poor social relationships.
Mouse models are also useful for analyzing typical phenotypes observed in patients with 17p13.3 microdeletion syndrome such as spasticity, epileptic seizures, decreased longevity, and intellectual disability (Dobyns et al., 1993). In addition, the craniofacial defects observed in human populations with 17p13.3 microdeletion syndrome are manifested in mice (Carter et al., 2000; Toyo-oka et al., 2004; Yu et al., 2014). Use of a mouse model with a large deletion or duplication, as opposed to single gene deletions and duplications, will be much more representative of the genotypes that occur in patients with ILS, MDS, and 17p13.3 microduplication syndrome. While there are drawbacks associated with any animal model, the creation of the proposed mouse models would advance the study of the rare neurodevelopmental diseases discussed in this review and is a goal worthy of effort.
Next generation sequencing and rare disease
Although the incidence of each individual rare disease is low, rare diseases collectively affect ~30 million people in the United States (Shen et al., 2015). Next generation sequencing (NGS) has accelerated the rate at which genes responsible for rare monogenic diseases are being identified (Boycott et al., 2013). Whole-exome sequencing (WES) is a popular technique that is currently favored over whole-genome sequencing (WGS) due to its lower cost and complexity for detecting genetic variations (Boycott et al., 2013). WES is an NGS technique that may be applied to identify novel rare disease causing genes. NGS techniques are not yet being used as a typical method for diagnosis, but this may become a common practice once more gene-phenotype relationships are identified (Shen et al., 2015). NGS has had a significant positive impact on rare disease research. However, this impact is rather limited to diseases that are caused by a single gene.
Progress in research has not been as rapid on rare diseases that involve large regions of the genome and multiple genes, such as MDS and 17p13.3 microduplication syndrome, due to the focus of previous research on identifying mutations in single genes. The NGS techniques that have been used to identify about half of the genes responsible for the approximated 7,000 known rare monogenic diseases need to be modified before they may be applied to the study of rare diseases involving multiple genes (Boycott et al., 2013). Specifically, data interpretation should be modified. To help identify the specific genes that have been deleted or duplicated in patients with MDS or 17p13.3 microduplication syndrome, respectively, studies should be looking at CNVs of genes within critical regions for these diseases. Such an approach may help identify additional genes of importance for these conditions. Applying this strategy to other diseases of unknown genetic origin may help determine if multiple genes are deleted or duplicated in other syndromes.
Study of CNVs using targeted NGS sequencing data, such as WES, is challenging because deletions and duplications may not begin or end within the exome, making them difficult to identify. However, a statistical method known as SeqCNV has recently been developed that can robustly identify CNVs using capture NGS data (Chen et al., 2017). First, a dataset is generated using WES. Then, the analysis is done using SeqCNV to identify the copy number ratio and CNV boundary through use of read depth information and maximum penalized likelihood estimation (MPLE) (Chen et al., 2017). CNVs can also be identified from targeted NGS data using a popular software package called ExomeDepth. The sensitivity and specificity of ExomeDepth v1.1.16 were determined to be 100% and 99.8%, respectively, validating this technique as an appropriate method for detection of CNVs (Ellingford et al., 2017). Methods such as SeqCNV and ExomeDepth are also advantageous because they can identify CNVs anywhere in the genome by using WES data, unlike other methods, such as FISH, where a region of interest must be identified to carry out the technique. Development of NGS techniques has already greatly improved our knowledge of rare monogenic diseases. A shift in attention to more complex genotypes involving the deletion or duplication of multiple genes may result in better approaches for identifying CNVs.
Conclusions
Microdeletions and microduplications of chromosome 17p13.3 lead to rare and complex diseases. The advent of NGS techniques and refinement of CRISPR/Cas9 mouse genetics opens new possibilities for the study of rare diseases such as the creation of new mouse models that previously would have been difficult or impossible to create, advanced analysis of WES data to better identify CNVs, and the potential for more accurate diagnostic tools. These techniques also offer alternate tools to study a rare disease that does not heavily rely on case study data, which is difficult to obtain. Study of neurodevelopmental disorders can also advance the field of neurodevelopmental research in general by contributing to our knowledge about fundamental processes involved in normal brain development including neurogenesis, neuronal migration, and neurite formation.
Author contributions
SMB, SAB, and TS wrote the initial draft of the manuscript. SMB and SAB: created the figures. KT edited and finalized it.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the National Institute of Neurological Disorders and Stroke (NINDS) and Rare Genomics Institute for their support. We also thank Dr. Anthony Wynshaw-Boris, Case Western Reserve University, for critical reading and his comments and Dr. Masahito Ikawa, Osaka University, for his comments.
Footnotes
Funding. This review has been supported by a research grant from the NINDS (NS096098) and BeHEARD Technology Prizes.
References
- Abdul S., Leebeek F. W., Rijken D. C., Uitte de Willige S. (2016). Natural heterogeneity of alpha2-antiplasmin: functional and clinical consequences. Blood 127, 538–545. 10.1182/blood-2015-09-670117 [DOI] [PubMed] [Google Scholar]
- Arif E., Sharma P., Solanki A., Mallik L., Rathore Y. S., Twal W. O., et al. (2016). Structural analysis of the Myo1c and Neph1 complex provides insight into the intracellular movement of Neph1. Mol. Cell. Biol. 36, 1639–1654. 10.1128/MCB.00020-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour C. M., Bulman D. E., Jarinova O., Rogers R. C., Clarkson K. B., DuPont B. R., et al. (2011). 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD). Eur. J. Hum. Genet. 19, 1144–1151. 10.1038/ejhg.2011.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayanlaja A. A., Xiong Y., Gao Y., Ji G., Tang C., Abdikani Abdullah Z., et al. (2017). Distinct features of doublecortin as a marker of neuronal migration and its implications in cancer cell mobility. Front. Mol. Neurosci. 10:199. 10.3389/fnmol.2017.00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros Fontes M. I., dos Santos A. P., Rossi Torres F., Lopes-Cendes I., Cendes F., Appenzeller S., et al. (2017). 17p13.3 microdeletion: insights on genotype-phenotype correlation. Mol. Syndromol. 8, 36–41. 10.1159/000452753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M., Hayashi Y., Desachy G., Hsiao E. C., Sami S., Tsang K. M., et al. (2014). Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature 507, 99–103. 10.1038/nature12923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Sapir T., Shchelochkov O. A., Zhang F., Withers M. A., Hunter J. V., et al. (2009). Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 41, 168–177. 10.1038/ng.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodoy S., Fotiadis D., Stoeger C., Kanai Y., Palacin M. (2013). The small SLC43 family: facilitator system l amino acid transporters and the orphan EEG1. Mol. Aspects Med. 34, 638–645. 10.1016/j.mam.2012.12.006 [DOI] [PubMed] [Google Scholar]
- Bodoy S., Martin L., Zorzano A., Palacin M., Estevez R., Bertran J. (2005). Identification of LAT4, a novel amino acid transporter with system L activity. J. Biol. Chem. 280, 12002–12011. 10.1074/jbc.M408638200 [DOI] [PubMed] [Google Scholar]
- Boesler C., Rigo N., Agafonov D. E., Kastner B., Urlaub H., Will C. L., et al. (2015). Stable tri-snRNP integration is accompanied by a major structural rearrangement of the spliceosome that is dependent on Prp8 interaction with the 5′ splice site. RNA 21, 1993–2005. 10.1261/rna.053991.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott K. M., Vanstone M. R., Bulman D. E., MacKenzie A. E. (2013). Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 14, 681–691. 10.1038/nrg3555 [DOI] [PubMed] [Google Scholar]
- Bruno D. L., Anderlid B. M., Lindstrand A., van Ravenswaaij-Arts C., Ganesamoorthy D., Lundin J., et al. (2010). Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J. Med. Genet. 47, 299–311. 10.1136/jmg.2009.069906 [DOI] [PubMed] [Google Scholar]
- Calegari F., Haubensak W., Haffner C., Huttner W. B. (2005). Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J. Neurosci. 25, 6533–6538. 10.1523/JNEUROSCI.0778-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G., Alifano P., Roberti V., Bruni C. B., Bucci C. (2001). Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 20, 683–693. 10.1093/emboj/20.4.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra V., Mirabelli-Badenier M., Stagnaro M., Rossi A., Tassano E., Gimelli S., et al. (2012). Identification of a rare 17p13.3 duplication including the BHLHA9 and YWHAE genes in a family with developmental delay and behavioural problems. BMC Med. Genet. 13:93. 10.1186/1471-2350-13-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso C., Leventer R. J., Ward H. L., Toyo-Oka K., Chung J., Gross A., et al. (2003). Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am. J. Hum. Genet. 72, 918–930. 10.1086/374320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M. G., Johns M. A., Zeng X., Zhou L., Zink M. C., Mankowski J. L., et al. (2000). Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum. Mol. Genet. 9, 413–419. 10.1093/hmg/9.3.413 [DOI] [PubMed] [Google Scholar]
- Chakrabarti S., Bonneau F., Schussler S., Eppinger E., Conti E. (2014). Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5-SMG7 and SMG6. Nucleic Acids Res. 42, 9447–9460. 10.1093/nar/gku578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zhao L., Wang Y., Cao M., Gelowani V., Xu M., et al. (2017). SeqCNV: a novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinformatics 18:147. 10.1186/s12859-017-1566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E. H., Park B. Y., Cho J. H., Kang Y. S. (2009). Comparing two diagnostic laboratory tests for several microdeletions causing mental retardation syndromes: multiplex ligation-dependent amplification vs fluorescent in situ hybridization. Korean J. Lab. Med. 29, 71–76. 10.3343/kjlm.2009.29.1.71 [DOI] [PubMed] [Google Scholar]
- Chudley A. E., Pabello P. D., McAlpine P. J., Nickel B. E., Shokeir M. H. (1982). Brief clinical report: ring chromosome 17 in a mentally retarded young man - clinical, cytogenetic, and biochemical investigations. Am. J. Med. Genet. 12, 219–225. 10.1002/ajmg.1320120212 [DOI] [PubMed] [Google Scholar]
- Clancy S., Shaw K. (2008). DNA deletion and duplication and the associated genetic disorders. Nat. Edu. 1:23. [Google Scholar]
- Classen S., Goecke T., Drechsler M., Betz B., Nickel N., Beier M., et al. (2013). A novel inverted 17p13.3 microduplication disrupting PAFAH1B1 (LIS1) in a girl with syndromic lissencephaly. Am. J. Med. Genet. A 161A, 1453–1458. 10.1002/ajmg.a.35904 [DOI] [PubMed] [Google Scholar]
- Colombo M., Karousis E. D., Bourquin J., Bruggmann R., Muhlemann O. (2017). Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 23, 189–201. 10.1261/rna.059055.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppoolse E. R., de Vroomen M. J., van Gennip F., Hersmus B. J., van Haaren M. J. (2005). Size does matter: cre-mediated somatic deletion efficiency depends on the distance between the target lox-sites. Plant Mol. Biol. 58, 687–698. 10.1007/s11103-005-7705-7 [DOI] [PubMed] [Google Scholar]
- Cornell B., Wachi T., Zhukarev V., Toyo-Oka K. (2016). Regulation of neuronal morphogenesis by 14-3-3epsilon (Ywhae) via the microtubule binding protein, doublecortin. Hum. Mol. Genet. 25, 4610 10.1093/hmg/ddx023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment. Retard. Dev. Disabil. Res. Rev. 10, 248–258. 10.1002/mrdd.20039 [DOI] [PubMed] [Google Scholar]
- Curry C. J., Rosenfeld J. A., Grant E., Gripp K. W., Anderson C., Aylsworth A. S., et al. (2013). The duplication 17p13.3 phenotype: analysis of 21 families delineates developmental, behavioral and brain abnormalities, and rare variant phenotypes. Am. J. Med. Genet. A 161A, 1833–1852. 10.1002/ajmg.a.35996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smaele E., Di Marcotullio L., Ferretti E., Screpanti I., Alesse E., Gulino A. (2004). Chromosome 17p deletion in human medulloblastoma: a missing checkpoint in the Hedgehog pathway. Cell Cycle 3, 1263–1266. 10.4161/cc.3.10.1200 [DOI] [PubMed] [Google Scholar]
- Desh H., Gray S. L., Horton M. J., Raoul G., Rowlerson A. M., Ferri J., et al. (2014). Molecular motor MYO1C, acetyltransferase KAT6B and osteogenetic transcription factor RUNX2 expression in human masseter muscle contributes to development of malocclusion. Arch. Oral Biol. 59, 601–607. 10.1016/j.archoralbio.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaliwal B. S., Steinbrecher U. P. (1999). Scavenger receptors and oxidized low density lipoproteins. Clin. Chim. Acta 286, 191–205. 10.1016/S0009-8981(99)00101-1 [DOI] [PubMed] [Google Scholar]
- Dobyns W. B., Reiner O., Carrozzo R., Ledbetter D. H. (1993). Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 270, 2838–2842. 10.1001/jama.1993.03510230076039 [DOI] [PubMed] [Google Scholar]
- Dobyns W. B., Truwit C. L., Ross M. E., Matsumoto N., Pilz D. T., Ledbetter D. H., et al. (1999). Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology 53, 270–277. 10.1212/WNL.53.2.270 [DOI] [PubMed] [Google Scholar]
- Dubois J. C., Yates M., Gaudreau-Lapierre A., Clement G., Cappadocia L., Gaudreau L., et al. (2017). A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res. 45, 8859–8872. 10.1093/nar/gkx571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford J. M., Campbell C., Barton S., Bhaskar S., Gupta S., Taylor R. L., et al. (2017). Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur. J. Hum. Genet. 25, 719–724. 10.1038/ejhg.2017.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M. A., Lieden A., Westerlund J., Bremer A., Wincent J., Sahlin E., et al. (2015). Rare copy number variants are common in young children with autism spectrum disorder. Acta Paediatr. 104, 610–618. 10.1111/apa.12969 [DOI] [PubMed] [Google Scholar]
- Florio M., Huttner W. B. (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. 10.1242/dev.090571 [DOI] [PubMed] [Google Scholar]
- Gambello M. J., Darling D. L., Yingling J., Tanaka T., Gleeson J. G., Wynshaw-Boris A. (2003). Multiple dose-dependent effects of Lis1 on cerebral cortical development. J. Neurosci. 23, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzellone M. J., Zhou X., Lionel A. C., Uddin M., Thiruvahindrapuram B., Liang S., et al. (2014). Copy number variation in Han Chinese individuals with autism spectrum disorder. J. Neurodev. Disord. 6:34. 10.1186/1866-1955-6-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind D. H., Rakic P. (2013). Cortical evolution: judge the brain by its cover. Neuron 80, 633–647. 10.1016/j.neuron.2013.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giusti S. A., Vercelli C. A., Vogl A. M., Kolarz A. W., Pino N. S., Deussing J. M., et al. (2014). Behavioral phenotyping of Nestin-Cre mice: implications for genetic mouse models of psychiatric disorders. J. Psychiatr. Res. 55, 87–95. 10.1016/j.jpsychires.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Gleeson J. G., Allen K. M., Fox J. W., Lamperti E. D., Berkovic S., Scheffer I., et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72. 10.1016/S0092-8674(00)80899-5 [DOI] [PubMed] [Google Scholar]
- Grainger R. J., Beggs J. D. (2005). Prp8 protein: at the heart of the spliceosome. RNA 11, 533–557. 10.1261/rna.2220705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Zhang F., Lupski J. R. (2008). Mechanisms for human genomic rearrangements. Pathogenetics 1:4. 10.1186/1755-8417-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. K., Chikne V., Eliaz D., Tkacz I. D., Naboishchikov I., Carmi S., et al. (2014). Two splicing factors carrying serine-arginine motifs, TSR1 and TSR1IP, regulate splicing, mRNA stability, and rRNA processing in Trypanosoma brucei. RNA Biol. 11, 715–731. 10.4161/rna.29143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M., Li Y., Wang J., Qin J., Wang Y., Ding Y., et al. (2017). HIC1 loss promotes prostate cancer metastasis by triggering epithelial-mesenchymal transition. J. Pathol. 242, 409–420. 10.1002/path.4913 [DOI] [PubMed] [Google Scholar]
- Harrison R. E., Bucci C., Vieira O. V., Schroer T. A., Grinstein S. (2003). Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol. Cell. Biol. 23, 6494–6506. 10.1128/MCB.23.18.6494-6506.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry R. K., Astbury C., Stratakis C. A., Hickey S. E. (2016). 17p13.3 microduplication including CRK leads to overgrowth and elevated growth factors: a case report. Eur. J. Med. Genet. 59, 512–516. 10.1016/j.ejmg.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S., Fleck M. W., Gambello M. J., Bix G. J., Chen A., Clark G. D., et al. (1998). Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat. Genet. 19, 333–339. 10.1038/1221 [DOI] [PubMed] [Google Scholar]
- Hsieh D. T., Jennesson M. M., Thiele E. A., Caruso P. A., Masiakos P. T., Duhaime A. C. (2013). Brain and spinal manifestations of Miller-Dieker syndrome. Neurol. Clin. Pract. 3, 82–83. 10.1212/CPJ.0b013e318278be63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Zhu Y. T., Qi C., Zhu Y. J. (2009). Identification of Smyd4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 69, 4067–4072. 10.1158/0008-5472.CAN-08-4097 [DOI] [PubMed] [Google Scholar]
- Huang Y., Magdaleno S., Hopkins R., Slaughter C., Curran T., Keshvara L. (2004). Tyrosine phosphorylated Disabled 1 recruits Crk family adapter proteins. Biochem. Biophys. Res. Commun. 318, 204–212. 10.1016/j.bbrc.2004.04.023 [DOI] [PubMed] [Google Scholar]
- Hyon C., Marlin S., Chantot-Bastaraud S., Mabboux P., Beaujard M. P., Al Ageeli E., et al. (2011). A new 17p13.3 microduplication including the PAFAH1B1 and YWHAE genes resulting from an unbalanced X;17 translocation. Eur. J. Med. Genet. 54, 287–291. 10.1016/j.ejmg.2010.12.006 [DOI] [PubMed] [Google Scholar]
- Ibitoye R. M., Roberts J., Goodacre T., Kini U. (2015). 17p13.3 microduplication, a potential novel genetic locus for nonsyndromic bilateral cleft lip and palate. Cleft Palate Craniofac. J. 52, 359–362. 10.1597/13-113 [DOI] [PubMed] [Google Scholar]
- Ishii J., Adachi H., Shibata N., Arai H., Tsujimoto M. (2007). Scavenger receptor expressed by endothelial cells (SREC)-I interacts with protein phosphatase 1alpha in L cells to induce neurite-like outgrowth. Biochem. Biophys. Res. Commun. 360, 269–274. 10.1016/j.bbrc.2007.06.047 [DOI] [PubMed] [Google Scholar]
- Izumi K., Kuratsuji G., Ikeda K., Takahashi T., Kosaki K. (2007). Partial deletion of LIS1: a pitfall in molecular diagnosis of Miller-Dieker syndrome. Pediatr. Neurol. 36, 258–260. 10.1016/j.pediatrneurol.2006.11.015 [DOI] [PubMed] [Google Scholar]
- Janeckova L., Pospichalova V., Fafilek B., Vojtechova M., Tureckova J., Dobes J., et al. (2015). HIC1 tumor suppressor loss potentiates TLR2/NF-kappaB signaling and promotes tissue damage-associated tumorigenesis. Mol. Cancer Res. 13, 1139–1148. 10.1158/1541-7786.MCR-15-0033 [DOI] [PubMed] [Google Scholar]
- Johnen S., Djalali-Talab Y., Kazanskaya O., Moller T., Harmening N., Kropp M., et al. (2015). Antiangiogenic and neurogenic activities of sleeping beauty-mediated PEDF-transfected RPE cells in vitro and in vivo. Biomed Res. Int. 2015:863845. 10.1155/2015/863845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens I., Fernandez-Borja M., Marsman M., Dusseljee S., Janssen L., Calafat J., et al. (2001). The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 11, 1680–1685. 10.1016/S0960-9822(01)00531-0 [DOI] [PubMed] [Google Scholar]
- Kim T., Bershteyn M., Wynshaw-Boris A. (2014). Chromosome therapy. Correction of large chromosomal aberrations by inducing ring chromosomes in induced pluripotent stem cells (iPSCs). Nucleus 5, 391–395. 10.4161/nucl.36300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T., Plona K., Wynshaw-Boris A. (2017). A novel system for correcting large-scale chromosomal aberrations: ring chromosome correction via reprogramming into induced pluripotent stem cell (iPSC). Chromosoma 126, 457–463. 10.1007/s00412-016-0621-6 [DOI] [PubMed] [Google Scholar]
- Kim Y. J., Byun S. Y., Jo S. A., Shin Y. B., Cho E. H., Lee E. Y., et al. (2011). Miller-Dieker syndrome with der(17)t(12;17)(q24.33;p13.3)pat presenting with a potential risk of mis-identification as a de novo submicroscopic deletion of 17p13.3. Korean J. Lab. Med. 31, 49–53. 10.3343/kjlm.2011.31.1.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopocki E., Lohan S., Doelken S. C., Stricker S., Ockeloen C. W., Soares Thiele de Aguiar R., et al. (2012). Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J. Med. Genet. 49, 119–125. 10.1136/jmedgenet-2011-100409 [DOI] [PubMed] [Google Scholar]
- Kohler A., Hain J., Muller U. (1995). Clinical and molecular genetic findings in five patients with Miller-Dieker syndrome. Clin. Genet. 47, 161–164. 10.1111/j.1399-0004.1995.tb03951.x [DOI] [PubMed] [Google Scholar]
- Komoike Y., Shimojima K., Liang J. S., Fujii H., Maegaki Y., Osawa M., et al. (2010). A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J. Hum. Genet. 55, 155–162. 10.1038/jhg.2010.1 [DOI] [PubMed] [Google Scholar]
- Kornack D. R., Rakic P. (1998). Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc. Natl. Acad. Sci. U.S.A. 95, 1242–1246. 10.1073/pnas.95.3.1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kular G., Loubtchenkov M., Swigart P., Whatmore J., Ball A., Cockcroft S., et al. (1997). Co-operation of phosphatidylinositol transfer protein with phosphoinositide 3-kinase gamma in the formylmethionyl-leucylphenylalanine-dependent production of phosphatidylinositol 3,4,5-trisphosphate in human neutrophils. Biochem. J. 325 (Pt 2), 299–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. (2014). P53 induction accompanying G2/M arrest upon knockdown of tumor suppressor HIC1 in U87MG glioma cells. Mol. Cell. Biochem. 395, 281–290. 10.1007/s11010-014-2137-9 [DOI] [PubMed] [Google Scholar]
- Kuwano A., Ledbetter S. A., Dobyns W. B., Emanuel B. S., Ledbetter D. H. (1991). Detection of deletions and cryptic translocations in Miller-Dieker syndrome by in situ hybridization. Am. J. Hum. Genet. 49, 707–714. [PMC free article] [PubMed] [Google Scholar]
- Lakso M., Pichel J. G., Gorman J. R., Sauer B., Okamoto Y., Lee E., et al. (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U.S.A. 93, 5860–5865. 10.1073/pnas.93.12.5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledbetter D. H., Ledbetter S. A., vanTuinen P., Summers K. M., Robinson T. J., Nakamura Y., et al. (1989). Molecular dissection of a contiguous gene syndrome: frequent submicroscopic deletions, evolutionarily conserved sequences, and a hypomethylated “island” in the Miller-Dieker chromosome region. Proc. Natl. Acad. Sci. U.S.A. 86, 5136–5140. 10.1073/pnas.86.13.5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledbetter S. A., Kuwano A., Dobyns W. B., Ledbetter D. H. (1992). Microdeletions of chromosome 17p13 as a cause of isolated lissencephaly. Am. J. Hum. Genet. 50, 182–189. [PMC free article] [PubMed] [Google Scholar]
- Link J. M., Hurlin P. J. (2015). The activities of MYC, MNT and the MAX-interactome in lymphocyte proliferation and oncogenesis. Biochim. Biophys. Acta 1849, 554–562. 10.1016/j.bbagrm.2014.04.004 [DOI] [PubMed] [Google Scholar]
- Liu P., Zhang H., McLellan A., Vogel H., Bradley A. (1998). Embryonic lethality and tumorigenesis caused by segmental aneuploidy on mouse chromosome 11. Genetics 150, 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Li Y., Wang X., Xing R., Liu K., Gan Q., et al. (2017). The BEACH-containing protein WDR81 coordinates p62 and LC3C to promote aggrephagy. J. Cell Biol. 216, 1301–1320. 10.1083/jcb.201608039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loucks C. M., Parboosingh J. S., Shaheen R., Bernier F. P., McLeod D. R., Seidahmed M. Z., et al. (2015). Matching two independent cohorts validates DPH1 as a gene responsible for autosomal recessive intellectual disability with short stature, craniofacial, and ectodermal anomalies. Hum. Mutat. 36, 1015–1019. 10.1002/humu.22843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui J. H., Hansen D. V., Kriegstein A. R. (2011). Development and evolution of the human neocortex. Cell 146, 18–36. 10.1016/j.cell.2011.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk H. M., Wong V. C. H., Lo I. F. M., Chan K. Y. K., Lau E. T., Kan A. S. Y., et al. (2014). A prenatal case of split-hand malformation associated with 17p13.3 triplication - a dilemma in genetic counseling. Eur. J. Med. Genet. 57, 81–84. 10.1016/j.ejmg.2013.12.005 [DOI] [PubMed] [Google Scholar]
- Marcorelles P., Laquerrière A., Adde-Michel C., Marret S., Saugier-Veber P., Beldjord C., et al. (2010). Evidence for tangential migration disturbances in human lissencephaly resulting from a defect in LIS1, DCX and ARX genes. Acta Neuropathol. 120, 503–515. 10.1007/s00401-010-0692-z [DOI] [PubMed] [Google Scholar]
- Marubashi S., Shimada H., Fukuda M., Ohbayashi N. (2016). RUTBC1 functions as a GTPase-activating protein for Rab32/38 and regulates melanogenic enzyme trafficking in melanocytes. J. Biol. Chem. 291, 1427–1440. 10.1074/jbc.M115.684043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T., Pramatarova A., Howell B. W. (2008). Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 121, 1869–1875. 10.1242/jcs.027334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S., Funke B., Epstein J. A., Heyer J., Puech A., Lu M. M., et al. (2001). TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104, 619–629. 10.1016/S0092-8674(01)00247-1 [DOI] [PubMed] [Google Scholar]
- Miller J. Q. (1963). LISSENCEPHALY IN 2 SIBLINGS. Neurology 13, 841–850. 10.1212/WNL.13.10.841 [DOI] [PubMed] [Google Scholar]
- Moon H. M., Wynshaw-Boris A. (2013). Cytoskeleton in action: lissencephaly, a neuronal migration disorder. Wiley Interdiscip. Rev. Dev. Biol. 2, 229–245. 10.1002/wdev.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C. P., Skippen A., Segui B., Ball A., Allen-Baume V., Larijani B., et al. (2004). Phosphorylation of a distinct structural form of phosphatidylinositol transfer protein alpha at Ser166 by protein kinase C disrupts receptor-mediated phospholipase C signaling by inhibiting delivery of phosphatidylinositol to membranes. J. Biol. Chem. 279, 47159–47171. 10.1074/jbc.M405827200 [DOI] [PubMed] [Google Scholar]
- Nagamani S. C., Zhang F., Shchelochkov O. A., Bi W., Ou Z., Scaglia F., et al. (2009). Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J. Med. Genet. 46, 825–833. 10.1136/jmg.2009.067637 [DOI] [PubMed] [Google Scholar]
- Nagata E., Kano H., Kato F., Yamaguchi R., Nakashima S., Takayama S., et al. (2014). Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex. Orphanet J. Rare Dis. 9:125. 10.1186/s13023-014-0125-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D. L., Applegate G. A., Beio M. L., Graham D. L., Berkowitz D. B. (2017). Human serine racemase structure/activity relationship studies provide mechanistic insight and point to position 84 as a hot spot for beta-elimination function. J. Biol. Chem. 292, 13986–14002. 10.1074/jbc.M117.777904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P., Josi C., Kurosawa H., Yamashita A., Muhlemann O. (2014). A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res. 42, 9217–9235. 10.1093/nar/gku645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K., Suzuki Y., Fujii I., Takeshita K., Arima M. (1974). [A case of ring chromosome E 17: 46, XX, r(17) (p13 yields q25) (author's transl)]. Jinrui Idengaku Zasshi 19, 235–242. [PubMed] [Google Scholar]
- Ostergaard J. R., Graakjaer J., Brandt C., Birkebaek N. H. (2012). Further delineation of 17p13.3 microdeletion involving CRK. The effect of growth hormone treatment. Eur. J. Med. Genet. 55, 22–26. 10.1016/j.ejmg.2011.09.004 [DOI] [PubMed] [Google Scholar]
- Ottens F., Boehm V., Sibley C. R., Ule J., Gehring N. H. (2017). Transcript-specific characteristics determine the contribution of endo- and exonucleolytic decay pathways during the degradation of nonsense-mediated decay substrates. RNA 23, 1224–1236. 10.1261/rna.059659.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancoast M., Dobyns W., Golden J. A. (2005). Interneuron deficits in patients with the Miller-Dieker syndrome. Acta Neuropathol. 109, 400–404. 10.1007/s00401-004-0979-z [DOI] [PubMed] [Google Scholar]
- Park T. J., Curran T. (2008). Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J. Neurosci. 28, 13551–13562. 10.1523/JNEUROSCI.4323-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton K. E., Chen B., Liu K., Hunter O. V., Xie Y., Tu B. P., et al. (2017). The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169, 824.e814–835.e814. 10.1016/j.cell.2017.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit F., Jourdain A. S., Andrieux J., Baujat G., Baumann C., Beneteau C., et al. (2014). Split hand/foot malformation with long-bone deficiency and BHLHA9 duplication: report of 13 new families. Clin. Genet. 85, 464–469. 10.1111/cge.12219 [DOI] [PubMed] [Google Scholar]
- Pilz D. T., Quarrell O. W. (1996). Syndromes with lissencephaly. J. Med. Genet. 33, 319–323. 10.1136/jmg.33.4.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilz D. T., Dalton A., Long A., Jaspan T., Maltby E. L., Quarrell O. W. (1995). Detecting deletions in the critical region for lissencephaly on 17p13.3 using fluorescent in situ hybridisation and a PCR assay identifying a dinucleotide repeat polymorphism. J. Med. Genet. 32, 275–278. 10.1136/jmg.32.4.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A., Evrony G. D., Cai X., Walsh C. A. (2013). Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758–1237758. 10.1126/science.1237758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poluch S., Juliano S. L. (2015). Fine-tuning of neurogenesis is essential for the evolutionary expansion of the cerebral cortex. Cereb. Cortex 25, 346–364. 10.1093/cercor/bht232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramparo T., Libiger O., Jain S., Li H., Youn Y. H., Hirotsune S., et al. (2011). Global developmental gene expression and pathway analysis of normal brain development and mouse models of human neuronal migration defects. PLoS Genet. 7:e1001331. 10.1371/journal.pgen.1001331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramparo T., Youn Y. H., Yingling J., Hirotsune S., Wynshaw-Boris A. (2010). Novel embryonic neuronal migration and proliferation defects in Dcx mutant mice are exacerbated by Lis1 reduction. J. Neurosci. 30, 3002–3012. 10.1523/JNEUROSCI.4851-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowse A. H., Vanderveer L., Milling S. W., Pan Z. Z., Dunbrack R. L., Xu X. X., et al. (2002). OVCA2 is downregulated and degraded during retinoid-induced apoptosis. Int. J. Cancer 99, 185–192. 10.1002/ijc.10334 [DOI] [PubMed] [Google Scholar]
- Qazi O. H., Madahar C., Kanchanapoomi R., Giridharan R., Beller E. (1979). Ring chromosome 17 in a mentally retarded boy. Ann. Genet. 22, 234–238. [PubMed] [Google Scholar]
- Rahman P., Huysmans R. D., Wiradjaja F., Gurung R., Ooms L. M., Sheffield D. A., et al. (2011). Silencer of death domains (SODD) inhibits skeletal muscle and kidney enriched inositol 5-phosphatase (SKIP) and regulates phosphoinositide 3-kinase (PI3K)/Akt signaling to the actin cytoskeleton. J. Biol. Chem. 286, 29758–29770. 10.1074/jbc.M111.263103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O., Sapir T. (2013). LIS1 functions in normal development and disease. Curr. Opin. Neurobiol. 23, 951–956. 10.1016/j.conb.2013.08.001 [DOI] [PubMed] [Google Scholar]
- Reiner O., Carrozzo R., Shen Y., Wehnert M., Faustinella F., Dobyns W. B., et al. (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364, 717–721. 10.1038/364717a0 [DOI] [PubMed] [Google Scholar]
- Roos L., Jonch A. E., Kjaergaard S., Taudorf K., Simonsen H., Hamborg-Petersen B., et al. (2009). A new microduplication syndrome encompassing the region of the Miller-Dieker (17p13 deletion) syndrome. J. Med. Genet. 46, 703–710. 10.1136/jmg.2008.065094 [DOI] [PubMed] [Google Scholar]
- Sarshad A. A., Corcoran M., Al-Muzzaini B., Borgonovo-Brandter L., Von Euler A., Lamont D., et al. (2014). Glycogen synthase kinase (GSK) 3beta phosphorylates and protects nuclear myosin 1c from proteasome-mediated degradation to activate rDNA transcription in early G1 cells. PLoS Genet. 10:e1004390. 10.1371/journal.pgen.1004390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S. A., Foley P. L., Jeong D. H., Rymarquis L. A., Doyle F., Tenenbaum S. A., et al. (2015). Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res. 43, 309–323. 10.1093/nar/gku1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaiter A., Stilgenbauer S. (2013). 17p deletion in chronic lymphocytic leukemia: risk stratification and therapeutic approach. Hematol. Oncol. Clin. North Am. 27, 289–301. 10.1016/j.hoc.2013.01.008 [DOI] [PubMed] [Google Scholar]
- Shaheen R., Sebai M. A., Patel N., Ewida N., Kurdi W., Altweijri I., et al. (2017). The genetic landscape of familial congenital hydrocephalus. Ann. Neurol. 81, 890–897. 10.1002/ana.24964 [DOI] [PubMed] [Google Scholar]
- Sharief N., Craze J., Summers D., Butler L., Wood C. B. (1991). Miller-Dieker syndrome with ring chromosome 17. Arch. Dis. Child. 66, 710–712. 10.1136/adc.66.6.710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T., Lee A., Shen C., Lin C. J. (2015). The long tail and rare disease research: the impact of next-generation sequencing for rare Mendelian disorders. Genet. Res. 97, e15. 10.1017/S0016672315000166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima K., Sugiura C., Takahashi H., Ikegami M., Takahashi Y., Ohno K., et al. (2010). Genomic copy number variations at 17p13.3 and epileptogenesis. Epilepsy Res. 89, 303–309. 10.1016/j.eplepsyres.2010.02.002 [DOI] [PubMed] [Google Scholar]
- Silverman J. L., Yang M., Lord C., Crawley J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P., Shaw C. J., Dapper J. D., Wakui K., Shaffer L. G., Withers M., et al. (2003). Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am. J. Hum. Genet. 72, 1101–1116. 10.1086/374385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarov A., Gooden F., Tseng D., Gan W. B., Ross M. E. (2013). Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol. Med. 5, 591–607. 10.1002/emmm.201202106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Tanaka R., Okano S., Okayama A., Suzuki N., Azuma H. (2015). Characterization of intragenic tandem duplication in the PAFAH1B1 gene leading to isolated lissencephaly sequence. Mol. Cytogenet. 8, 84. 10.1186/s13039-015-0186-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Serneo F. F., Higgins C., Gambello M. J., Wynshaw-Boris A., Gleeson J. G. (2004). Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709–721. 10.1083/jcb.200309025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney J. R., Hopkin R. J., Schapiro M. B. (2011). Deletion of 14-3-3{varepsilon} and CRK: a clinical syndrome with macrocephaly, developmental delay, and generalized epilepsy. J. Child Neurol. 26, 223–227. 10.1177/0883073810379638 [DOI] [PubMed] [Google Scholar]
- Toyo-oka K., Hirotsune S., Gambello M. J., Zhou Z. Q., Olson L., Rosenfeld M. G., et al. (2004). Loss of the Max-interacting protein Mnt in mice results in decreased viability, defective embryonic growth and craniofacial defects: relevance to Miller-Dieker syndrome. Hum. Mol. Genet. 13, 1057–1067. 10.1093/hmg/ddh116 [DOI] [PubMed] [Google Scholar]
- Toyo-oka K., Shionoya A., Gambello M. J., Cardoso C., Leventer R., Ward H. L., et al. (2003). 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat. Genet. 34, 274–285. 10.1038/ng1169 [DOI] [PubMed] [Google Scholar]
- Tsai J. W., Chen Y., Kriegstein A. R., Vallee R. B. (2005). LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J. Cell Biol. 170, 935–945. 10.1083/jcb.200505166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. E., Escobar L. F. (2014). Cleft lip/palate associated with 17p13.3 duplication involving a single candidate gene (YWHAE). Clin. Genet. 85, 600–601. 10.1111/cge.12237 [DOI] [PubMed] [Google Scholar]
- van der Kant R., Fish A., Janssen L., Janssen H., Krom S., Ho N., et al. (2013). Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 126(Pt 15), 3462–3474. 10.1242/jcs.129270 [DOI] [PubMed] [Google Scholar]
- Vissers L. E., Stankiewicz P., Yatsenko S. A., Crawford E., Creswick H., Proud V. K., et al. (2007). Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum. Genet. 121, 697–709. 10.1007/s00439-007-0359-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. (1942). Lissencephaly. Arch. Neurol. Psychiatry 48, 13–29. 10.1001/archneurpsyc.1942.02290070023002 [DOI] [Google Scholar]
- Watson K. K., Platt M. L. (2012). Of mice and monkeys: using non-human primate models to bridge mouse- and human-based investigations of autism spectrum disorders. J. Neurodev. Disord. 4, 21–21. 10.1186/1866-1955-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe V. O., Gonzalez-Porta M., Perera D., Bartolozzi A. R., Sibley C. R., Hallegger M., et al. (2015). Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5' splice site strength. Genome Biol. 16, 201. 10.1186/s13059-015-0749-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynshaw-Boris A., Gambello M. J. (2001). LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 15, 639–651. 10.1101/gad.886801 [DOI] [PubMed] [Google Scholar]
- Wynshaw-Boris A., Pramparo T., Youn Y. H., Hirotsune S. (2010). Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin. Cell Dev. Biol. 21, 823–830. 10.1016/j.semcdb.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M., Yoshida Y., Mori D., Takitoh T., Kengaku M., Umeshima H., et al. (2009). Inhibition of calpain increases LIS1(PAFAH1B1) and partially rescues in vivo phenotypes in a mouse model of lissencephaly. Nat. Med. 15, 1202–1207. 10.1038/nm.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling J., Toyo-Oka K., Wynshaw-Boris A. (2003). Miller-Dieker syndrome: analysis of a human contiguous gene syndrome in the mouse. Am. J. Hum. Genet. 73, 475–488. 10.1086/378096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. R., You L. R., Yan Y. T., Chen C. M. (2014). Role of OVCA1/DPH1 in craniofacial abnormalities of Miller-Dieker syndrome. Hum. Mol. Genet. 23, 5579–5596. 10.1093/hmg/ddu273 [DOI] [PubMed] [Google Scholar]
- Zhang H., Gan H., Wang Z., Lee J. H., Zhou H., Ordog T., et al. (2017). RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol. Cell 65, 272–284. 10.1016/j.molcel.2016.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Kuang X., Zhang J. (2011). Nogo receptor 3, a paralog of Nogo-66 receptor 1 (NgR1), may function as a NgR1 co-receptor for Nogo-66. J. Genet. Genomics 38, 515–523. 10.1016/j.jgg.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Zheng B., Sage M., Sheppeard E. A., Jurecic V., Bradley A. (2000). Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Mol. Cell. Biol. 20, 648–655. 10.1128/MCB.20.2.648-655.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody M. C., Garber M., Adams D. J., Sharpe T., Harrow J., Lupski J. R., et al. (2006). DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature 440, 1045–1049. 10.1038/nature04689 [DOI] [PMC free article] [PubMed] [Google Scholar]