

Abstract
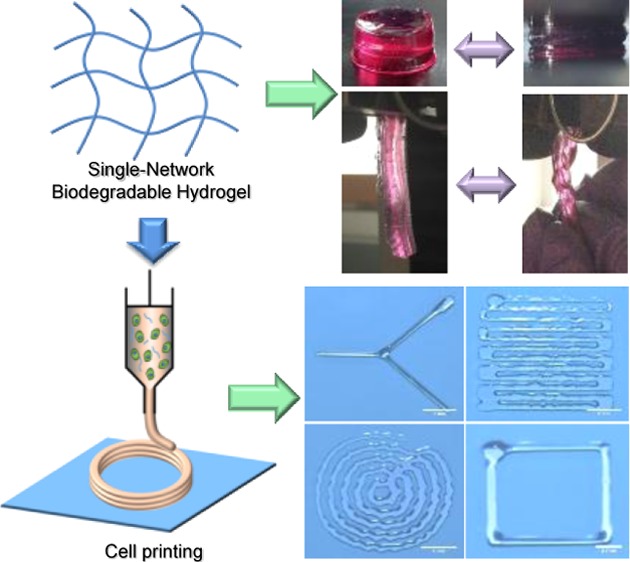
Cell printing is becoming a common technique to fabricate cellularized printed scaffold for biomedical application. There are still significant challenges in soft tissue bioprinting using hydrogels, which requires live cells inside the hydrogels. Moreover, the resilient mechanical properties from hydrogels are also required to mechanically mimic the native soft tissues. Herein, we developed a visible-light cross-linked, single-network, biodegradable hydrogel with high elasticity and flexibility for cell printing, which is different from previous highly elastic hydrogel with double-network and two components. The single-network hydrogel using only one stimulus (visible light) to trigger gelation can greatly simplify the cell printing process. The obtained hydrogels possessed high elasticity, and their mechanical properties can be tuned to match various native soft tissues. The hydrogels had good cell compatibility to support fibroblast growth in vitro. Various human cells were bioprinted with the hydrogels to form cell–gel constructs, in which the cells exhibited high viability after 7 days of culture. Complex patterns were printed by the hydrogels, suggesting the hydrogel feasibility for cell printing. We believe that this highly elastic, single-network hydrogel can be simply printed with different cell types, and it may provide a new material platform and a new way of thinking for hydrogel-based bioprinting research.
Keywords: cell printing, single network, biodegradable hydrogel, elasticity, tissue regeneration
Introduction
Cell printing has gained a surge of interest in biomedical engineering field because it combines biocompatible materials, cells, and supportive components into printed constructs.1 Today, nonbiological printing is very successful to fabricate stiff biomaterial scaffold (without cells), such as osteoinductive materials to match patients’ anatomy for bone repair.2−5 However, in soft tissue bioprinting, which requires live cells inside the scaffolds, there are still significant challenges. Current cell printing of soft tissue uses biodegradable hydrogel materials, which are naturally derived polymers, such as fibrin,6 gelatin,7 hyaluronic acid,8,9 alginate,10 and agarose;11 synthetic polymers, such as poly(ethylene glycol) (PEG)12,13 and methacrylated gelatin (GelMA);14−16 or natural–synthetic composites.17−19 However, these hydrogels are brittle and unstretchable due to lack of flexibility and elasticity, which cannot mimic the mechanical behavior of softness, stretchability, and elasticity of human soft tissues, such as skin, skeletal muscle, blood vessels, and heart muscles. To make stretchable hydrogels, some groups have developed dual cross-linking-network hydrogel system to achieve high elasticity and mechanical strength,20−22 but such dual-network system using two cross-linking mechanisms significantly increases the difficulty and complexity in cell printing control and handling. To overcome these challenges, we pursue a simple system using single cross-linking mechanism to achieve a highly elastic and robust, biodegradable, and biocompatible hydrogel for cell printing, which is our work reported in this paper. We found that a triblock biodegradable polymer of polycaprolactone–poly(ethylene glycol)–polycaprolactone (PCL–PEG–PCL) with two end groups of acrylates and visible-light water-soluble initiator can form a hydrogel with high elasticity and flexibility, which is also feasible for cell printing. Both PCL and PEG components are widely used in Food and Drug Administration (FDA)-approved devices and implants; thus our approach will facilitate quick translation of this material into preclinical and clinical trials in the future.
Our single-network elastic hydrogel system is simple and thus can be easily controlled during bioprinting process. In contrast, other methods are still quite complex and difficult to implement for cell printing. For example, a dual-network hydrogel consisted of PEG and sodium alginate was bioprinted into complex, cellularized structures with high stretchability and toughness.20 However, the use of two cross-linking mechanisms for cell printing encounters some challenges and limitations. First, the preparation of two or more precursors before cell printing is tedious and time consuming. Second, in many cases, there are more than one external stimulus involving in gelation process, such as temperature, light, ion concentration, and enzyme (thrombin), which will increase the difficulty of cell printing. For example, to load cells into a polycaprolactone (PCL)/poly(lactide-co-caprolactone) (PLCL)/fibrin hydrogel during the cell printing process, a cooling system was required to maintain the bioprinter chamber at 18 °C to avoid the cells contacting the high-temperature PCL and PLCL.17 Hence, single-component-based hydrogel system, which only need one external stimulus for gelation and can be simply prepared and bioprinted, is considered a better choice for cell printing. In addition, to mimic the resilient native soft tissues,23 biodegradable hydrogels with good mechanical strength and elasticity are desirable. Herein, a bioprintable, biodegradable hydrogel based on single component, possessing good mechanical strength and elasticity was designed in this study (Figure 1), which has been rarely reported.
Figure 1.

Preparation and characterization of highly elastic, visible-light cross-linked, single-network, biodegradable hydrogel for cell printing. An acrylated PCL–PEG–PCL triblock polymer was synthesized and then cross-linked using visible light to form a highly elastic single-network biodegradable hydrogel. The hydrogel has attractive mechanical properties, and it is stretchable, compressible, and twistable. The hydrogel can also be bioprinted with various human cells and form complex patterns upon visible-light exposure.
In this study, we designed a triblock copolymer, PCL–PEG–PCL diacrylate (DA) as the single-component precursor to form a cross-linked hydrogel network (Figure S1). Specifically, a PCL–PEG–PCL triblock copolymer diol containing hydrophilic PEG segments and hydrophobic PCL segments was first synthesized via ring-opening reaction. The acryloyl groups were then introduced to the two ends of the PCL–PEG–PCL diols to obtain the PEG–PCL-DA polymers, which formed hydrogel under visible-light exposure. The chemical structure, water absorption, mechanical properties, and elasticity of the PEG–PCL-DA hydrogels were characterized. The in vitro cytocompatibility of the PEG–PCL-DA hydrogels was evaluated by mouse 3T3 fibroblasts. Various human cells were bioprinted with the hydrogels, and the cell viabilities were studied after 7 days of culture. Basic geometric shapes were printed to prove the printability of the PEG–PCL-DA hydrogels.
Materials and Methods
Materials
Poly(ethylene glycol) (PEG, MW = 20 000, Sigma) and ε-caprolactone (CL, Sigma) were dried in a vacuum oven at 60 °C to remove residual water before use. Stannous octoate (Sn(Oct)2, Sigma), triethylamine (TEA, Sigma), acryloyl chloride (Sigma), dichloromethane (Sigma), dimethyl phenylphosphonite (Acros Organics), 2,4,6-trimethylbenzoyl chloride (Sigma), lithium bromide (Sigma), and 2-butanone (Sigma) were used as received.
Synthesis of PCL–PEG–PCL Copolymer Diols
The PCL–PEG–PCL diols were synthesized using PEG to initiate a ring-opening polymerization of CL at 120 °C for 24 h under N2 atmosphere (Figure S1).24 Sn(Oct)2 was used as a catalyst. Products were dissolved in dichloromethane, precipitated in cold diethyl ether, and then dried in a vacuum oven at 60 °C for 3 days.
Acrylation of PCL–PEG–PCL Copolymer Diols
Acrylation of the PCL–PEG–PCL copolymer diols was executed through acryloyl chloride (Figure S1). The copolymer diols were dissolved in 15 mL of dichloromethane in a three-neck flask under N2 protection, to which TEA was added dropwise under stirring for 30 min in an ice bath.25 Acryloyl chloride in 15 mL of dichloromethane was then added to the mixture dropwise. The molar ratio of the hydroxyl groups (−OH) in PCL–PEG–PCL diols/TEA/acryloyl chloride was set at 1:2:2. The reaction was first performed in an ice bath for 30 min and then heated to 40 °C for 24 h under N2 atmosphere. The reaction mixture was cooled to room temperature and then precipitated in cold diethyl ether. The precipitates were filtered, dried in a desiccator for 2 days, redissolved in deionized water, and dialyzed over 2 days. The synthesized PEG–PCL-DA was collected after lyophilization. The PEG–PCL-DA polymers were set as PEG–PCL(X)-DA, with X referring to the block length of the PCL–PEG–PCL diols. PEG-DA without PCL segments was set as the control group, which was synthesized from PEG (MW = 20 000) and acryloyl chloride via the same reaction process as described above.
Proton Nuclear Magnetic Resonance (1H NMR) Spectroscopy
The chemical structures of the PEG–PCL-DA and PEG-DA polymers were characterized by 1H NMR (JEOL ECX Instrument, 300 MHz) with D2O as solvent. The block length of the PCL–PEG–PCL copolymer diols and the degree of substitution (DS) of the acryloyl group on both ends of the PCL–PEG–PCL diols were both calculated from the 1H NMR spectra.
Elastic Hydrogel Formation
The PEG–PCL-DA hydrogel was formed by photopolymerization using lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (LAP) as the water-soluble, visible-light initiator which was synthesized according to the previous studies (Figure S1).26,27 The PEG–PCL-DA polymers were dissolved in deionized water and mixed with the LAP water solution to reach various final concentrations (10, 20, and 40% w/v). The mixed solution was then poured into a cylinder or strip mold, followed by exposure under an light-emitting diode (LED) splash lighter (395–405 nm, 5 W) for 2 min to irradiate the mixed solution. Then, the PEG–PCL-DA hydrogels were formed. Unless otherwise noted, the hydrogels used for all measurements were at a concentration of 40%.
Hydrogel Water Absorption
To determine the swelling ratio of the PEG–PCL-DA hydrogels, the cylinder samples (4 mm height; 9 mm diameter; n = 3) were immersed in phosphate-buffered saline (PBS) at 37 °C for 24 h.28 The hydrogels were then taken out, and the surface water of the hydrogels was removed gently with a filter paper; the hydrogels were weighed (Ws). The swollen hydrogels were rinsed by deionized water and lyophilized. The weight of dry hydrogel after freeze-drying was recorded as Wd. The swelling ratio was calculated as (Ws – Wd)/Wd × 100%.
Mechanical Property Measurement
The hydrogels used for all mechanical measurements were in wet state (immersion in PBS for 24 h) before testing. The compression testing of cylinder hydrogels (4 mm height; 9 mm diameter; n = 4) was performed on MTS Insight Testing System with a 500 N load cell and a cross-head rate of 1 mm/min.29 For uniaxial tensile testing, the strips of PEG–PCL-DA hydrogels with 50 mm length, 5 mm width, and 3 mm height (n = 3) were prepared and tested on MTS Insight Testing System with a 500 N load cell and a cross-head rate of 10 mm/min according to ASTM D638-03.30 The instant strain recovery of PEG–PCL-DA hydrogel strips (n = 4) were carried out and measured under the same conditions as described above.31 The hydrogel strip was stretched to 10% strain, held for 1 min, and then released. The stretching cycle was repeated three times. The original length (L0) and the length after stretching (L1) were measured by a caliper. The instant strain recovery was calculated as (1 – (L1 – L0)/L0) × 100%. For cyclic stretch, the PEG–PCL-DA hydrogel specimens (50 mm length, 5 mm width; 3 mm height; n = 3) were stretched to the maximum strain of 30% and released back to 0% strain for 10 cycles at a constant rate of 10 mm/min.32
Cell Viability in Hydrogels
PEG–PCL-DA polymers (1.2 g) and LAP (0.015 g) were sterilized under UV radiation for 1 h and dissolved in 1 mL of sterilized PBS to obtain 120% (w/v) PEG–PCL-DA/PBS solution and 1.5% (w/v) LAP/PBS solution, respectively. Mouse 3T3 fibroblasts (ATCC, Manassas, VA) in 1 mL cell culture medium (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) at a density of 1.5 × 107 cells/mL was first mixed with the PEG–PCL-DA/PBS solution and then subsequently blended with the LAP/PBS solution. The final PEG–PCL-DA hydrogel concentration was 40% (w/v), the final initiator concentration was 0.5% (w/v), and the final cell density in the obtained cell/hydrogel precursor was 5 × 106 cells/mL. The cell/hydrogel precursor was injected into a mold by a 1 mL syringe and exposed to the LED splash lighter to form cell/hydrogel construct as described above. Standard biopsy punches (6 mm, Miltex) were used to punch the cell/hydrogel construct to obtain cell/hydrogel disks (6 mm diameter), which were transferred to 24-well cell culture plates and incubated at 37 °C in a 5% CO2 environment. The cell culture medium was exchanged every 2 days. The cell viabilities after 1 and 3 days of incubation in PEG–PCL-DA hydrogels (n = 5) were detected by a mitochondrial activity assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Sigma) at 1 and 3 days. The MTT results were verified using a live and dead staining (live, SYTO 10 green fluorescent nucleic acid stain; dead, ethidium homodimer-1 nucleic acid stain, Life Technologies, Inc.) kit to visualize the 3T3 fibroblasts in the hydrogels. The images of live/dead stained 3T3 fibroblasts were taken on a fluorescence microscope.
Viscosity of PEG–PCL-DA Solution
PEG–PCL-DA polymers were dissolved in PBS to create solutions
ranging from 10 to 40% (w/v). The viscosity of the polymer solutions
was measured using a falling ball viscometer (size 3, Gilmont) under
manufacturer’s instructions. A glass ball was used to measure
the viscosity of polymer solutions that were less than 10% (w/v),
and a stainless steel ball was used for the other concentrations.
Three measurements were taken for each concentration. Shear stress
within the nozzle during bioprinting is estimated using the Hagen–Poiseuille
equation: 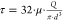 , where μ is the viscosity, Q is the mean volumetric flow rate, and d is the diameter of the needle.
, where μ is the viscosity, Q is the mean volumetric flow rate, and d is the diameter of the needle.
Cell Printing of PEG–PCL-DA
All cell printing experiments were performed using a modified printing platform reported in previous studies.8 The printer consists of a dispenser mounted onto a Cartesian robotic stage that moves in the x–y direction. Another motor-controlled stage that moves in the z direction acts as the printing substrate. The extrusion-based dispenser is driven by a syringe pump (Harvard Apparatus); the speed of extrusion can be controlled by programming the pump. In addition, printing resolution can be controlled by adjusting the speed of extruder movement, extrusion speed, and nozzle size. Syringe barrels containing the bioink and tubing attached to the dispenser were covered in aluminum foil to prevent premature cross-linking during the printing process. The printing apparatus was housed in a sterile laminar flow hood to prevent contamination. Basic geometric shapes were printed by extruding a 20% (w/v) PEG–PCL-DA solution through an 18G needle and a 21G needle.
Cell Viability under Printing Conditions
Sterilized PEG–PCL-DA polymers of varying weight (0.1–0.3 g) were dissolved in 500 μL of media to obtain polymer/medium solutions. Three types of media were used: fibroblast growth medium (FGM-2, Lonza) for neonatal human lung fibroblasts, endothelial growth medium (EGM-2, Lonza) for human umbilical vein endothelial cells, and smooth muscle growth medium (SmGM-2, Lonza) for human aortic smooth muscle cells. The cells were suspended in their respective media at a concentration of 2.5 × 106 cells/mL. The cell suspension (400 μL) was mixed with the polymer solution and then subsequently mixed with 100 μL of LAP/PBS solution to obtain a PEG–PCL-DA bioink with a final concentration of 10–30% (w/v), a final initiator concentration of 0.5% (w/v), and a final cell density of 1.0 × 106 cells/mL. The resulting bioink was wrapped in aluminum foil and kept on ice prior to printing. The bioink was loaded into sterile 10 mL Luer Lock syringes (Becton Dickinson) and mounted onto our printing apparatus. The bioink was extruded through nozzles of four different gauges (18G, 21G, 23G, and 25G) at a constant flow rate of 0.15 mL/min and photo-cross-linked for 30 s to create cylindrical cell/hydrogel constructs. Live/dead staining (live, calcein AM; dead, ethidium homodimer-1, Thermo Fisher Scientific) was used to determine cell viability of cells immediately after printing (n = 3). The constructs were immersed in media and incubated under 37 °C in a 5% CO2 environment. Media was changed every 2 days, and live/dead staining was performed again 7 days after printing.
Statistical Analysis
All results are presented as mean ± standard deviation. All data were analyzed by one-way analysis of variance, followed by a post hoc Tukey–Kramer test. Differences were considered statistically significant when p < 0.05.
Results and Discussion
PEG–PCL-DA Polymer Synthesis and Hydrogel Formation
The chemical structures of synthesized PEG-DA (Figure 2A) and PEG–PCL-DA (Figure 2B) were confirmed by 1H NMR spectra. The specific peaks of the ethylene oxide protons of the PEG segments in PEG-DA and PEG–PCL-DA polymers were located at 3.55 and 4.20 ppm, and 3.56 and 4.09 ppm, respectively (Figure 2). The methyl protons of the PCL blocks in the PEG–PCL-DA polymer were assigned to chemical shifts between 1.22 and 3.99 ppm (Figure 2B). The specific peaks of the acryloyl protons in the PEG–PCL-DA were located from 5.81 to 6.30 ppm (Figure 2). The block length of the PCL–PEG–PCL was calculated from the 1H NMR spectra and distributed as 1863–20 000–1863 for PEG–PCL(24k) and 1151–20000–1151 for PEG–PCL(22k) (Table 1). The degrees of substitution (DS) of the acryloyl group calculated from the 1H NMR spectra were 55, 59, and 60% for PEG-DA, PEG–PCL(22k)-DA, and PEG–PCL(24k)-DA, respectively. The DS of the acryloyl group for PEG-DA is comparable to the previous study,33 which verified the success of the acrylation.
Figure 2.

1H NMR spectra of (A) PEG-DA and (B) PEG–PCL(24k)-DA.
Table 1. Block Lengths of PCL–PEG–PCL Copolymer Diols.
| copolymer diols | theoretical block length of PCL–PEG–PCL | calculated block length of PCL–PEG–PCL |
|---|---|---|
| PEG–PCL(24k) | 2000–20 000–2000 | 1863–20 000–1863 |
| PEG–PCL(22k) | 1000–20 000–1000 | 1151–20 000–1151 |
The hydrogel was formed by covalent cross-linking though visible-light initiation. The formed hydrogel was transparent. The triblock PCL–PEG system was previously designed for thermosensitive biodegradable hydrogel, which depended on the hydrophobicity/hydrophilicity balance by changing the feeding ratio of PEG–PCL and total macromolecular weight. The thermosensitive PEG–PCL hydrogel was opaque and not elastic.34−36 In contrast, the acrylated PEG–PCL polymer in this work can form transparent and elastic hydrogel via photopolymerization under exposure to visible light. The selected photoinitiator, LAP, can maintain high cell viability during direct cellular encapsulation because it has very good cell compatibility in vitro and in vivo,26,37 and can trigger the photopolymerization under visible light (395–405 nm) exposure.26 The visible light for cross-linking is much safer than the common UV cross-linking (365 nm), and it would allow a longer duration and safer cell printing. In addition, light cross-linking technique is highly compatible with the current three-dimensional printing technique.
Hydrogel Water Absorption
Water absorption of PEG–PCL-DA hydrogel is summarized in Table 2. A decrease in water absorption was observed with an increase in the concentration of PEG–PCL-DA precursor solution. The water absorption of PEG–PCL(24k)-DA-10% was 1930 ± 140%; however, it decreased to 766 ± 18% when the concentration of PEG–PCL(24k)-DA precursor solution increased to 40% (Table 2). It was primarily because more polymer chains involved in cross-linking process would result in denser hydrogel mesh, which lead to lower water absorption rate.38
Table 2. PEG–PCL-DA Hydrogel Characterization*,#.
| polymers | concentrations (%) | compressive modulus (kPa) | compressive stress at 80% strain (kPa) | water absorption (%) |
|---|---|---|---|---|
| PEG–PCL(24k)-DA | 40 | 26.7 ± 4.5a | 356.8 ± 18.7a | 766 ± 18a |
| 20 | 14.9 ± 3.1b | 162.5 ± 14.5b | 1184 ± 43b | |
| 10 | 10.7 ± 2.5b | 78.6 ± 4.5c | 1930 ± 140c | |
| PEG–PCL(22k)-DA | 40 | 19.4 ± 1.5a | -## | 971 ± 48a |
| 20 | 8.9 ± 1.7b | 122.4 ± 2.3a | 1593 ± 51b | |
| 10 | 4.9 ± 0.4c | 45.0 ± 5.2b | 2504 ± 337c | |
| PEG-DA | 40 | 9.2 ± 1.8a | -## | 1692 ± 296a |
| 20 | 4.7 ± 0.5b | 24.1 ± 2.7a | 2819 ± 292b | |
| 10 | 1.1 ± 0.2c | 8.8 ± 1.8b | 3798 ± 276c |
a, b, c represent significantly different groups for each characteristic. #The compression testing of hydrogels was carried out after immersion in PBS for 24 h. ##The PEG-DA and PEG–PCL(22k)-DA hydrogels at 40% concentration were broken at 80% strain during the compressive test.
Furthermore, the incorporation of hydrophobic PCL segments into the network resulted in a decrease in water absorption rate. The PEG-DA without PCL segments had the highest water absorption rate at 1692 ± 296%. The PEG–PCL(24k)-DA with the highest PCL content had the lowest water absorption rate at 766 ± 18%. Additionally, there is a balance between the water absorption from the hydrophilic PEG moiety and the enhanced mechanical properties and elasticity majorly attributed to the hydrophobic PCL moiety.
The water absorption is important to be characterized to predict the swollen shape and size for a bioprinted construct,39 and it is also required to be optimal for cell culture. However, water adsorption range appropriate for both printing and cell survival has not been well defined yet. Some publications have reported that materials with water absorption from 78% (PEG-alginate hydrogel) to 1183% (GelMA/collagen hydrogel) were acceptable for printing.20,40−42 However, the optimal hydrogel water absorption rates for printing and cell growth are often opposing.43,44 No strong evidence exists to show that such water absorption ranges were good for cell culture. In our study, the water absorption rates of PEG–PCL(24k)-DA hydrogel (766–1930%) were comparable to the reported acceptably printable range (78–1183%). In addition, introducing low-molecular-weight cross-linker or polymers with low water absorption can assist in reducing the water absorption rate of the PEG–PCL-DA hydrogel.42,45,46
Mechanical Properties of the PEG–PCL-DA Hydrogel
It is a desired property that the hydrogel maintains its elasticity under the media culture condition (wet state). We therefore examine their mechanical properties after submerging the hydrogel in media overnight. As shown in Figure 3 and Video S1, the PEG–PCL(24k)-DA hydrogel exhibited high flexibility and elasticity by withstanding large deformations of stretching (Figure 3B), compression (Figure 3D), and twisting (Figure 3F) without any obvious breakage. Particularly, after removal of the applied force, the PEG–PCL(24k)-DA hydrogel could recover quickly from deformation. In contrast, the swollen PEG-DA hydrogel became broken after slightly stretching (Figure 3A), large deformation of compression (Figure 3C), and twisting (Figure 3E, Video S2). The brittle nature of the PEG-DA hydrogel is consistent with a previous study, which also showed the swollen PEG-DA hydrogel (Mn = 20 000 g/mol) broken upon high deformation of compression or slight stretching.33 As determined by the compressive testing (Figures 4A, S2A, and Table 2 in the Supporting Information), the initial modulus and compressive strength at 80% strain of the PEG–PCL-DA hydrogels increased with increasing PCL segments when the polymer concentration was fixed (p < 0.05). The PEG–PCL(24k)-DA hydrogel achieved a compressive stress of 356.8 ± 18.7 kPa and a compressive modulus of 26.7 ± 4.5 kPa at 80% strain, which was around 3 times higher than the compressive modulus of PEG-DA hydrogel (9.2 ± 1.8 kPa). The compressive stress at 80% strain of PEG-DA was not able to be collected due to the hydrogel collapsing at 80% deformation. The tensile properties (Figures 4B, S2B, and Table 3) of the PEG–PCL-DA hydrogels demonstrate similar trends with their compressive properties. The initial modulus and tensile stress of PEG–PCL(24k)-DA hydrogel were 37.7 ± 1.7 and 34.5 ± 2.5 kPa, which were 6.3 times and 8.2 times higher than the initial modulus (6.0 ± 1.2 kPa) and tensile stress (4.2 ± 1.2 kPa) of PEG-DA, respectively. There was no significant difference on the breaking strain between the PEG–PCL(24k)-DA (150 ± 14%), PEG–PCL(22k)-DA (145 ± 17%), and PEG-DA (187 ± 27%) (p > 0.05). The instant recovery of both PEG–PCL(22k)-DA and PEG–PCL(24k)-DA hydrogel was ≥99% for three cycles at 10% strain, which was larger than that of PEG-DA hydrogel (97 ± 1%) (p < 0.05; Table 3). The PEG–PCL(24k)-DA hydrogel had the highest suture retention strength at 0.32 ± 0.06 N/mm2, whereas the PEG-DA had the lowest suture retention strength at 0.15 ± 0.01 N/mm2 (p < 0.05; Table 3). Cyclic stretching was carried out to study the elasticity of the PEG–PCL-DA hydrogels at a maximum strain of 100% (Figure 4C). All of the hydrogels exhibited a large hysteresis loop in the first cycle and much smaller hysteresis loops in the next nine cycles. The irreversible deformations decreased with the increasing block length of the PCL segments in the PEG–PCL-DA polymer chain from PEG-DA (∼45%) to PEG–PCL(24k)-DA (∼20%). The PEG–PCL-DA hydrogel with increasing PCL amounts had an increasing capacity to hold their own weight when suspended on cantilever right after gelation, which can be proved by the increasing angles from 8° (PEG-DA) to 39° (PEG–PCL(24k)-DA) between the hydrogels and vertical lines (Figure S3). These results suggested that the incorporation of PCL segments enhanced the strength, toughness, flexibility, and elasticity of the single-component hydrogel network. These observations were mainly attributed to the hydrophobic interactions between the PCL moieties in the hydrogel network. The hydrophobic interactions may form a second physical network along with the chemical cross-linking network, which may dissipate the crack energy applied along the hydrogel sample due to the reversible dissociation of the hydrophobic interactions.47,48 The developed PEG–PCL-DA hydrogel with good mechanical strength and elasticity is promising for soft tissue engineering. For example, the initial moduli of PEG–PCL(24k)-DA-40% (37.7 ± 1.7 kPa), PEG–PCL(22k)-DA-40% (28.2 ± 2.4 kPa), PEG–PCL(24k)-DA-20% (16.4 ± 4.8 kPa), and PEG–PCL(22k)-DA-20% (10.7 ± 1.7 kPa) fall in the range of that of human myocardium (Emyocardium = 10–500 kPa).49 The initial moduli of PEG–PCL(24k)-DA-20% (16.4 ± 4.8 kPa), PEG–PCL(24k)-DA-10% (9.4 ± 1.9 kPa), and PEG–PCL(22k)-DA-20% (10.7 ± 1.7 kPa) are comparable to those of human muscle (Emuscle = 8–17 kPa).50
Figure 3.

Photographs demonstrating the attractive mechanical properties of the PEG–PCL(24k)-DA hydrogel under stretching, compression, and twisting. (A) PEG-DA hydrogel was broken under stretching. (B) PEG–PCL(24k)-DA hydrogel could be stretched and recoiled back to the original length. (C) PEG-DA hydrogel was broken into pieces under compression at 80% strain. (D) PEG–PCL(24k)-DA hydrogel deformed and recovered under compression. (E) PEG-DA hydrogel was broken after twisting for four cycles. (F) PEG–PCL(24k)-DA hydrogel could be twisted for four cycles and recovered after releasing.
Figure 4.

Mechanical properties of the PEG–PCL(24k)-DA hydrogel. (A) Compressive stress–strain curves of PEG–PCL-DA hydrogels. (B) Tensile stress–strain curves of PEG–PCL-DA hydrogels. (C) Cyclic stretching of PEG–PCL-DA hydrogels at 100% deformation for 10 cycles.
Table 3. Tensile Testing of PEG–PCL-DA Hydrogel*,#.
| polymers | concentrations (%) | initial modulus (kPa) | tensile strength (kPa) | breaking strain (%) | suture retention (N/mm2) | instant recovery (%) |
|---|---|---|---|---|---|---|
| PEG–PCL(24k)-DA | 40 | 37.7 ± 1.7a | 34.5 ± 2.5a | 150 ± 14a | 0.32 ± 0.06a | 100 ± 1a |
| 20 | 16.4 ± 4.8b | 18.7 ± 4.3b | 165 ± 25a,b | |||
| 10 | 9.4 ± 1.9c | 10.8 ± 2.6c | 202 ± 27b | |||
| PEG–PCL(22k)-DA | 40 | 28.2 ± 2.4a | 25.3 ± 2.1a | 145 ± 17 | 0.20 ± 0.02b | 99 ± 1a |
| 20 | 10.7 ± 1.3b | 12.3 ± 1.1b | 160 ± 12 | |||
| 10 | 5.5 ± 0.6c | 8.6 ± 1.0c | 176 ± 21 | |||
| PEG-DA | 40 | 6.0 ± 1.2a | 4.2 ± 1.2a | 187 ± 27a | 0.15 ± 0.01c | 97 ± 1b |
| 20 | 1.4 ± 0.3b | 1.2 ± 0.2b | 102 ± 15b | |||
| 10 | -## | -## | -## |
a, b, c represent significantly different groups for each characteristic. #The tensile testing of hydrogels was carried out after immersion in PBS for 24 h. The suture retention and instant recovery were measured for hydrogels at a concentration of 40%. ##The PEG-DA-10% hydrogel was too weak to be loaded on the MTS machine.
In Vitro Cytocompatibility of the PEG–PCL-DA Hydrogel
The two chemical components in the hydrogel, PEG and PCL, are widely known as biocompatible materials, and have been used in FDA-approved devices.31 Hence, we expected that the hydrogel consisting of the two moieties can possess good cytocompatibility. The ability of the PEG–PCL-DA hydrogel to support the encapsulated cell growth was evaluated using mouse 3T3 fibroblasts. Live/dead staining was used to determine the cell viability in the hydrogel over 3 days of culture. In Figure 5A, the 3T3 fibroblasts maintained a round morphology inside the PEG-DA hydrogel on day 1 and day 3. However, a portion of the 3T3 fibroblasts exhibited elongated cell morphology after 1 day culture inside the PEG–PCL(24k)-DA hydrogel, which suggested the ability of the PEG–PCL(24k)-DA hydrogel to support cell attachment. This might be attributed to the hydrophobic PCL moiety, which can promote the protein adsorption to the hydrogel and thus improve the cell–hydrogel interactions.33,51 Cells encapsulated in PEG-based synthetic hydrogels are difficult to spread out because the high hydrophilicity of the inert hydrogels resists protein adhesion and cannot support interactions between cells and the hydrogels.52 Incorporating bioactive components into the synthetic networks, such as proteins, peptides, and polysaccharides, can significantly improve cell growth.25,53,54 For example, cells with rounded morphology were observed in PEG hydrogel; however, with the addition of hyaluronic acid or fibrinogen, the cells exhibited spreading morphology inside the hydrogels.53,54 Furthermore, the cell survival rates of the 3T3 fibroblasts inside the PEG–PCL(24k)-DA and PEG-DA hydrogels were both over 90% from day 1 to day 3 (Figure 5B). There was no significant difference in the cell viability between PEG–PCL(24k)-DA and PEG-DA within 3 days of culture (p > 0.05) (Figure 5C). The results indicate that the PEG–PCL-DA hydrogel with good cell compatibility can support the growth of the photoencapsulated 3T3 fibroblasts in vitro.
Figure 5.

In vitro cytocompatibility of elastic hydrogels. (A) Live and dead stained 3T3 fibroblasts encapsulated in PEG-DA and PEG–PCL(24k)-DA hydrogels after 1 day and 3 days of culture. (B) Cell viability calculated as percentage of live cells (green) from the live and dead staining images. (C) Metabolic index of 3T3 fibroblasts encapsulated in PEG-DA and PEG–PCL(24k)-DA hydrogels.
Viscosity of PEG–PCL-DA Solution
The viscosity of the hydrogel precursors can be tuned for bioprinting. The viscosity of the PEG–PCL-DA precursors increased exponentially with their concentration (Figure 6A). In particular, precursors exceeding 30% (w/v) showed remarkable increase in viscosity with respect to their concentration, whereas 0–20% precursor solution has very low viscosity. We envision that different bioprinting techniques may be needed for hydrogel at different viscosities. For example, inkjet-based printer can be used for precursor solution below 20%. For high-concentration solutions, extrusion-based printer will be used, which is suitable for printing high viscous materials.
Figure 6.

Cytotoxicity and printability of elastic PEG–PCL(24k)-DA hydrogel using an extrusion bioprinter. (A) Viscosity curve of elastic PEG–PCL(24k)-DA precursor solution. (B, C) Cell viability of different cell types in printed 10% elastic PEG–PCL(24k)-DA hydrogel. Live/dead assay was performed immediately after gel polymerization and after 7 days in culture (scale bars represent 500 μm). (D) Effect of different needle sizes and precursor solution concentrations on viability of neonatal human lung fibroblasts. (E) Effect of shear stress on cell viability evaluated immediately after printing. (F, G) Sample shapes printed using a printer with different needle sizes (scale bars in (F) represent 2 mm and scale bars in (G) represent 5 mm).
Cellular Printing
We printed the hydrogel loaded with the cells to test whether this material can be applied toward bioprinting of cell–gel construct. As for the constructs that were printed with PEG–PCL(24k)-DA at 10% of polymer concentration, cell viability of over 83% were observed across three different human cell types: human umbilical vein endothelial cells, neonatal human lung fibroblasts, and human aortic smooth muscle cells. These results suggest that the PEG–PCL-DA could potentially be used to print a wide range of different human soft tissues. The cells within the constructs continued to exhibit high viability after 7 days in culture printing (Figure 6B,C). In addition, no significant difference in cell viability was observed with decreasing nozzle diameter immediately after printing (Figure 6D). Similarly, there was no significant difference in cell viability with increasing shear stress (Figure 6E). However, there was a significant decrease in cell viability after 7 days in culture (Figure 6D). This could be a result of a decrease in the rate of nutrient diffusion from the increased polymer concentration and/or cell damage experienced from the printing process.
The PEG–PCL-DA hydrogel can also be easily printed for various patterns. Basic geometric shapes were printed using the PEG–PCL-DA precursor, and their resolution can be adjusted by changing the nozzle diameter (Figure 6F). Complex patterns could also be achieved by modifying the printing pattern on our cell printing platform; however, the polymer solution spread as it contacted the substrate, limiting the minimum feature size (Figure 6G). The chemical composition of the polymer may need to be modified to create smaller features. There are two major factors affecting the hydrogel shape fidelity, viscosity and swelling behavior.43 For hydrogel swelling behavior, high levels of cross-linking extent and charge densities will result in low swelling ratio, which, however, will impair cell growth and migration due to reduced pore sizes.55 In addition, hydrogel fidelity can also be improved by increasing its viscosity and enhancing its shear-thinning properties. For example, micro/nanoparticles could be incorporated into the precursor to make it more suitable for bioprinting.20,56
Conclusions
In summary, we have developed a visible-light cross-linked, single-component, elastic, and biodegradable hydrogel system based on a triblock copolymer of PEG and PCL. It is biocompatible, biodegradable, and has tunable mechanical properties. The possessed elastic properties are much desired for soft tissue engineering based on the premise that the elastic material, capable of transducing the correct mechanical stimulation to cells, will improve tissue adaptation to biomechanical environment. Various cells can be incorporated with the hydrogel for bioprinting. The selected two components are FDA-approved, which is helpful for quickly translating our research into the preclinical and clinical trials. Ultimately, this elastic hydrogel may be fully compatible with many other recently developed biomaterial approaches, such as incorporating biomimetic peptides, proteins, growth factors, or other bioactive molecules, and thus allow easy optimization for further improvement for soft tissue engineering.
Acknowledgments
The work was supported by Beginning Grant-in-Aid 14BGIA20510066 (Y.H.) from the American Heart Association, Faculty Career Development (CAREER) award #1554835 (Y.H.) from the National Science Foundation, and R21HD090680 (G.D. and Y.H.) and R01HL118245 (G.D.) from the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b01294.
Synthetic schemes of PCL–PEG–PCL copolymer diols, acrylated PEG–PCL-DA and PEG-DA polymers, and PCL–PEG-DA hydrogel (Figure S1); comparison of compression and tensile properties of PEG–PCL-DA hydrogels (Figure S2); qualitatively depiction of hydrogel bending (Figure S3) (PDF)
PEG–PCL(24k)-DA-40% stretching and twisting (AVI)
PEG-DA-40% stretching and twisting (AVI)
Author Contributions
Y.H. and G.D. designed the research. C.X. conducted polymeric experiments, characterization, and data analysis. W.L. conducted the cell printability evaluations. C.X., W.L., G.D., and Y.H. wrote and edited the manuscript. All authors reviewed the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Murphy S. V.; Atala A. 3D Bioprinting of Tissues and Organs. Nat. Biotechnol. 2014, 32, 773–785. 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- Lin K.-F.; He S.; Song Y.; Wang C.-M.; Gao Y.; Li J.-Q.; Tang P.; Wang Z.; Bi L.; Pei G.-X. Low-Temperature Additive Manufacturing of Biomimic Three-Dimensional Hydroxyapatite/Collagen Scaffolds for Bone Regeneration. ACS Appl. Mater. Interfaces 2016, 8, 6905–6916. 10.1021/acsami.6b00815. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Hua S.; Yang M.; Fu Z.; Teng S.; Niu K.; Zhao Q.; Yi C. Fabrication and Characterization of electrospinning/3D Printing Bone Tissue Engineering Scaffold. RSC Adv. 2016, 6, 110557–110565. 10.1039/C6RA17718B. [DOI] [Google Scholar]
- Lee S. J.; Lee D.; Yoon T. R.; Kim H. K.; Jo H. H.; Park J. S.; Lee J. H.; Kim W. D.; Kwon I. K.; Park S. A. Surface Modification of 3D-Printed Porous Scaffolds via Mussel-Inspired Polydopamine and Effective Immobilization of rhBMP-2 to Promote Osteogenic Differentiation for Bone Tissue Engineering. Acta Biomater. 2016, 40, 182–191. 10.1016/j.actbio.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Inzana J. A.; Olvera D.; Fuller S. M.; Kelly J. P.; Graeve O. A.; Schwarz E. M.; Kates S. L.; Awad H. A. 3D Printing of Composite Calcium Phosphate and Collagen Scaffolds for Bone Regeneration. Biomaterials 2014, 35, 4026–4034. 10.1016/j.biomaterials.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X.; Boland T. Human Microvasculature Fabrication Using Thermal Inkjet Printing Technology. Biomaterials 2009, 30, 6221–6227. 10.1016/j.biomaterials.2009.07.056. [DOI] [PubMed] [Google Scholar]
- Wang X.; Yan Y.; Pan Y.; Xiong Z.; Liu H.; Cheng J.; Liu F.; Lin F.; Wu R.; Zhang R.; Lu Q. Generation of Three-Dimensional Hepatocyte/Gelatin Structures with Rapid Prototyping System. Tissue Eng. 2006, 12, 83–90. 10.1089/ten.2006.12.83. [DOI] [PubMed] [Google Scholar]
- Lee V.; Singh G.; Trasatti J. P.; Bjornsson C.; Xu X.; Tran T. N.; Yoo S.-S.; Dai G.; Karande P. Design and Fabrication of Human Skin by Three-Dimensional Bioprinting. Tissue Eng., Part C 2014, 20, 473–484. 10.1089/ten.tec.2013.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T.; Gregory C. A.; Molnar P.; Cui X.; Jalota S.; Bhaduri S. B.; Boland T. Viability and Electrophysiology of Neural Cell Structures Generated by the Inkjet Printing Method. Biomaterials 2006, 27, 3580–3588. 10.1016/j.biomaterials.2006.01.048. [DOI] [PubMed] [Google Scholar]
- Xu C.; Chai W.; Huang Y.; Markwald R. R. Scaffold-Free Inkjet Printing of Three-Dimensional Zigzag Cellular Tubes. Biotechnol. Bioeng. 2012, 109, 3152–3160. 10.1002/bit.24591. [DOI] [PubMed] [Google Scholar]
- Fedorovich N. E.; De Wijn J. R.; Verbout A. J.; Alblas J.; Dhert W. J. A. Three-Dimensional Fiber Deposition of Cell-Laden, Viable, Patterned Constructs for Bone Tissue Printing. Tissue Eng., Part A 2008, 14, 127–133. 10.1089/ten.a.2007.0158. [DOI] [PubMed] [Google Scholar]
- Hoffman A. S. Hydrogels for Biomedical Applications. Adv. Drug Delivery Rev. 2012, 64, 18–23. 10.1016/j.addr.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Arcaute K.; Mann B. K.; Wicker R. B. Stereolithography of Three-Dimensional Bioactive Poly(ethylene glycol) Constructs with Encapsulated Cells. Ann. Biomed. Eng. 2006, 34, 1429–1441. 10.1007/s10439-006-9156-y. [DOI] [PubMed] [Google Scholar]
- Billiet T.; Gevaert E.; De Schryver T.; Cornelissen M.; Dubruel P. The 3D Printing of Gelatin Methacrylamide Cell-Laden Tissue-Engineered Constructs with High Cell Viability. Biomaterials 2014, 35, 49–62. 10.1016/j.biomaterials.2013.09.078. [DOI] [PubMed] [Google Scholar]
- Bertassoni L. E.; Cardoso J. C.; Manoharan V.; Cristino A. L.; Bhise N. S.; Araujo W. A.; Zorlutuna P.; Vrana N. E.; Ghaemmaghami A. M.; Dokmeci M. R.; Khademhosseini A. Direct-Write Bioprinting of Cell-Laden Methacrylated Gelatin Hydrogels. Biofabrication 2014, 6, 024105 10.1088/1758-5082/6/2/024105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton S.; Yu C. H.; Ersoy F.; Emadi S.; Khademhosseini A.; Tasoglu S. 3D-Printed Microfluidic Chips with Patterned, Cell-Laden Hydrogel Constructs. Biofabrication 2016, 8, 025019 10.1088/1758-5090/8/2/025019. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Fu Q.; Yoo J.; Chen X.; Chandra P.; Mo X.; Song L.; Atala A.; Zhao W. 3D Bioprinting of Urethra with PCL/PLCL Blend and Dual Autologous Cells in Fibrin Hydrogel: an in vitro Evaluation of Biomimetic Mechanical Property and Cell Growth environment. Acta Biomater. 2017, 50, 154–164. 10.1016/j.actbio.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Gao G.; Schilling A. F.; Hubbell K.; Yonezawa T.; Truong D.; Hong Y.; Dai G.; Cui X. Improved Properties of Bone and Cartilage Tissue from 3D Inkjet-Bioprinted Human Mesenchymal Stem Cells by Simultaneous Deposition and Photocrosslinking in PEG-GelMA. Biotechnol. Lett. 2015, 37, 2349–2355. 10.1007/s10529-015-1921-2. [DOI] [PubMed] [Google Scholar]
- Xu T.; Binder K. W.; Albanna M. Z.; Dice D.; Zhao W.; Yoo J. J.; Atala A. Hybrid Printing of Mechanically and Biologically Improved Constructs for Cartilage Tissue Engineering Applications. Biofabrication 2013, 5, 015001 10.1088/1758-5082/5/1/015001. [DOI] [PubMed] [Google Scholar]
- Hong S.; Sycks D.; Chan H. F.; Lin S.; Lopez G. P.; Guilak F.; Leong K. W.; Zhao X. 3D Printing of Highly Stretchable and Tough Hydrogels into Complex, Cellularized Structures. Adv. Mater. 2015, 27, 4035–4040. 10.1002/adma.201501099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Tadepalli V.; Wiley B. J. 3D Printing of A Double Network Hydrogel with A Compression Strength and Elastic Modulus Greater than those of Cartilage. ACS Biomater. Sci. Eng. 2017, 3, 863–869. 10.1021/acsbiomaterials.7b00094. [DOI] [PubMed] [Google Scholar]
- Wei J.; Wang J.; Su S.; Wang S.; Qiu J.; Zhang Z.; Christopher G.; Ning F.; Cong W. 3D Printing of An Extremely Tough Hydrogel. RSC Adv. 2015, 5, 81324–81329. 10.1039/C5RA16362E. [DOI] [Google Scholar]
- Guilak F.; Butler D. L.; Goldstein S. A.; Baaijens F. P. T. Biomechanics and Mechanobiology in Functional Tissue Engineering. J. Biomech. 2014, 47, 1933–1940. 10.1016/j.jbiomech.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Huang Y.; Tang L.; Hong Y. Low-Initial-Modulus Biodegradable Polyurethane Elastomers for Soft Tissue Regeneration. ACS Appl. Mater. Interfaces 2017, 9, 2169–2180. 10.1021/acsami.6b15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hern D. L.; Hubbell J. A. Incorporation of Adhesion Peptides into Nonadhesive Hydrogels Useful for Tissue Resurfacing. J. Biomed. Mater. Res. 1998, 39, 266–276. . [DOI] [PubMed] [Google Scholar]
- Fairbanks B. D.; Schwartz M. P.; Bowman C. N.; Anseth K. S. Photoinitiated Polymerization of PEG-Diacrylate with Lithium Phenyl-2,4,6-Trimethylbenzoylphosphinate: Polymerization Rate and Cytocompatibility. Biomaterials 2009, 30, 6702–6707. 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majima T.; Schnabel W.; Weber W. Phenyl-2,4,6-Trimethylbenzoylphosphinates as Water-Soluble Photoinitiators. Generation and Reactivity of O=Ṗ(C6H5)(O–) Radical Anions. Macromol. Chem. Phys. 1991, 192, 2307–2315. 10.1002/macp.1991.021921010. [DOI] [Google Scholar]
- Park H.; Guo X.; Temenoff J. S.; Tabata Y.; Caplan A. I.; Kasper F. K.; Mikos A. G. Effect of Swelling Ratio of Injectable Hydrogel Composites on Chondrogenic Differentiation of Encapsulated Rabbit Marrow Mesenchymal Stem Cells in vitro. Biomacromolecules 2009, 10, 541–546. 10.1021/bm801197m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Ding Q.; Dutta A.; Wang Y.; Huang Y.-h.; Weng H.; Tang L.; Hong Y. An Injectable Extracellular Matrix Derived Hydrogel for Meniscus Repair and Regeneration. Acta Biomater. 2015, 16, 49–59. 10.1016/j.actbio.2015.01.027. [DOI] [PubMed] [Google Scholar]
- Chuang T. W.; Masters K. S. Regulation of Polyurethane Hemocompatibility and Endothelialization by Tethered Hyaluronic Acid Oligosaccharides. Biomaterials 2009, 30, 5341–5351. 10.1016/j.biomaterials.2009.06.029. [DOI] [PubMed] [Google Scholar]
- Xu C.; Huang Y.; Wu J.; Tang L.; Hong Y. Triggerable Degradation of Polyurethanes for Tissue Engineering Applications. ACS Appl. Mater. Interfaces 2015, 7, 20377–20388. 10.1021/acsami.5b06242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Huang Y.; Yepez G.; Wei Z.; Liu F.; Bugarin A.; Tang L.; Hong Y. Development of Dopant-Free Conductive Bioelastomers. Sci. Rep. 2016, 6, 34451 10.1038/srep34451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Aung A.; Liao L.; Varghese S. A Novel Single Precursor-Based Biodegradable Hydrogel with Enhanced Mechanical Properties. Soft Matter 2009, 5, 3831–3834. 10.1039/b912102a. [DOI] [Google Scholar]
- Gong C.; Shi S.; Dong P.; Kan B.; Gou M.; Wang X.; Li X.; Luo F.; Zhao X.; Wei Y.; Qian Z. Synthesis and Characterization of PEG–PCL–PEG Thermosensitive Hydrogel. Int. J. Pharm. 2009, 365, 89–99. 10.1016/j.ijpharm.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Ma G.; Miao B.; Song C. Thermosensitive PCL–PEG–PCL Hydrogels: Synthesis, Characterization, and Delivery of Proteins. J. Appl. Polym. Sci. 2010, 116, 1985–1993. 10.1002/app.31654. [DOI] [Google Scholar]
- Gong C. Y.; Dong P. W.; Shi S.; Fu S. Z.; Yang J. L.; Guo G.; Zhao X.; Wei Y. Q.; Qian Z. Y. Thermosensitive PEG–PCL–PEG Hydrogel Controlled Drug Delivery System: Sol–Gel–Sol Transition and in vitro Drug Release Study. J. Pharm. Sci. 2009, 98, 3707–3717. 10.1002/jps.21694. [DOI] [PubMed] [Google Scholar]
- Mariner P. D.; Wudel J. M.; Miller D. E.; Genova E. E.; Streubel S.-O.; Anseth K. S. Synthetic Hydrogel Scaffold is an Effective Vehicle for Delivery of INFUSE (rhBMP2) to Critical-Sized Calvaria Bone Defects in Rats. J. Orthop. Res. 2013, 31, 401–406. 10.1002/jor.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick J. A.; Chung C.; Jia X.; Randolph M. A.; Langer R. Controlled Degradation and Mechanical Behavior of Photopolymerized Hyaluronic Acid Networks. Biomacromolecules 2005, 6, 386–391. 10.1021/bm049508a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurman W.; Levett P. A.; Pot M. W.; van Weeren P. R.; Dhert W. J. A.; Hutmacher D. W.; Melchels F. P. W.; Klein T. J.; Malda J. Gelatin–Methacrylamide Hydrogels as Potential Biomaterials for Fabrication of Tissue-Engineered Cartilage Constructs. Macromol. Biosci. 2013, 13, 551–561. 10.1002/mabi.201200471. [DOI] [PubMed] [Google Scholar]
- Lim K. S.; Schon B. S.; Mekhileri N. V.; Brown G. C. J.; Chia C. M.; Prabakar S.; Hooper G. J.; Woodfield T. B. F. New Visible-Light Photoinitiating System for Improved Print Fidelity in Gelatin-Based Bioinks. ACS Biomater. Sci. Eng. 2016, 2, 1752–1762. 10.1021/acsbiomaterials.6b00149. [DOI] [PubMed] [Google Scholar]
- Suntornnond R.; Tan E. Y. S.; An J.; Chua C. K. A Highly Printable and Biocompatible Hydrogel Composite for Direct Printing of Soft and Perfusable Vasculature-like Structures. Sci. Rep. 2017, 7, 16902 10.1038/s41598-017-17198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakarich S. E.; Gorkin R. III; in het Panhuis M.; Spinks G. M. Three-Dimensional Printing Fiber Reinforced Hydrogel Composites. ACS Appl. Mater. Interfaces 2014, 6, 15998–16006. 10.1021/am503878d. [DOI] [PubMed] [Google Scholar]
- Hölzl K.; Lin S.; Tytgat L.; Van Vlierberghe S.; Gu L.; Ovsianikov A. Bioink Properties Before, During and After 3D Bioprinting. Biofabrication 2016, 8, 032002 10.1088/1758-5090/8/3/032002. [DOI] [PubMed] [Google Scholar]
- Malda J.; Visser J.; Melchels F. P.; Jüngst T.; Hennink W. E.; Dhert W. J. A.; Groll J.; Hutmacher D. W. 25th Anniversary Article: Engineering Hydrogels for Biofabrication. Adv. Mater. 2013, 25, 5011–5028. 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- Bukhari S. M. H.; Khan S.; Rehanullah M.; Ranjha N. M. Synthesis and Characterization of Chemically Cross-Linked Acrylic Acid/Gelatin Hydrogels: Effect of pH and Composition on Swelling and Drug Release. Int. J. Polym. Sci. 2015, 2015, 1–15. 10.1155/2015/187961. [DOI] [Google Scholar]
- Son K. H.; Lee J. W. Synthesis and Characterization of Poly(Ethylene Glycol) Based Thermo-Responsive Hydrogels for Cell Sheet Engineering. Materials 2016, 9, 854 10.3390/ma9100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncaboylu D. C.; Sari M.; Oppermann W.; Okay O. Tough and Self-Healing Hydrogels Formed via Hydrophobic Interactions. Macromolecules 2011, 44, 4997–5005. 10.1021/ma200579v. [DOI] [Google Scholar]
- Abdurrahmanoglu S.; Can V.; Okay O. Design of High-Toughness Polyacrylamide Hydrogels by Hydrophobic Modification. Polymer 2009, 50, 5449–5455. 10.1016/j.polymer.2009.09.042. [DOI] [Google Scholar]
- Venugopal J. R.; Prabhakaran M. P.; Mukherjee S.; Ravichandran R.; Dan K.; Ramakrishna S. Biomaterial Strategies for Alleviation of Myocardial Infarction. J. R. Soc. Interface 2012, 9, 1–19. 10.1098/rsif.2011.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Schiraldi C.; D’Agostino A.; Oliva A.; Flamma F.; De Rosa A.; Apicella A.; Aversa R.; De Rosa M. Development of Hybrid Materials Based on Hydroxyethylmethacrylate as Supports for Improving Cell Adhesion and Proliferation. Biomaterials 2004, 25, 3645–3653. 10.1016/j.biomaterials.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Nicodemus G. D.; Bryant S. J. Cell Encapsulation in Biodegradable Hydrogels for Tissue Engineering Applications. Tissue Eng., Part B 2008, 14, 149–165. 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almany L.; Seliktar D. Biosynthetic Hydrogel Scaffolds Made from Fibrinogen and Polyethylene Glycol for 3D Cell Cultures. Biomaterials 2005, 26, 2467–2477. 10.1016/j.biomaterials.2004.06.047. [DOI] [PubMed] [Google Scholar]
- Kutty J. K.; Cho E.; Lee J. S.; Vyavahare N. R.; Webb K. The Effect of Hyaluronic Acid Incorporation on Fibroblast Spreading and Proliferation within PEG-Diacrylate Based Semi-Interpenetrating Networks. Biomaterials 2007, 28, 4928–4938. 10.1016/j.biomaterials.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Bencherif S. A.; Srinivasan A.; Horkay F.; Hollinger J. O.; Matyjaszewski K.; Washburn N. R. Influence of the Degree of Methacrylation on Hyaluronic Acid Hydrogels Properties. Biomaterials 2008, 29, 1739–1749. 10.1016/j.biomaterials.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Markstedt K.; Mantas A.; Tournier I.; Ávila H. M.; Hägg D.; Gatenholm P. 3D Bioprinting Human Chondrocytes with Nanocellulose–Alginate Bioink for Cartilage Tissue Engineering Applications. Biomacromolecules 2015, 16, 1489–1496. 10.1021/acs.biomac.5b00188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.