

Abstract
Background
M6620 is a novel inhibitor of the DNA damage repair enzyme ATR, and has potentiated the activity of cisplatin and irinotecan in NSCLC and colon cancer xenografts, respectively.
Procedures
M6620 was tested in vitro at concentrations ranging from 1.0 nM to 10.0 μM and at 75 nM in combination with cisplatin or melphalan. M6620 was tested against 24 solid tumor xenografts alone and in combination with cisplatin. Cisplatin was administered intraperitoneally on day 1 and 8 at a dose of 5 mg/kg. M6620 was administered intravenously on days 2 and 9 at 20 mg/m2 approximately 16 hours after cisplatin.
Results
The median relative IC50 (rIC50) value for M6620 was 0.19 μM, (range 0.03–1.38 μM). M6620 reduced the mean IC50 of cisplatin and melphalan by 1.48- and 1.95-fold, respectively. M6620 as a single agent in vivo induced significant differences in EFS distribution in 5 of 24 (21%) solid tumor xenografts, but induced no objective responses. Cisplatin as a single agent induced significant differences in EFS distribution compared to control in 18 of 24 (75%) solid tumor xenografts. Three objective responses were observed to cisplatin. The M6620 and cisplatin combination induced significant differences in EFS distribution compared to control in 21 of 24 (88%), with four objective responses.
Conclusions
M6620 showed modest potentiation of cisplatin and melphalan activity for some cell lines. M6620 showed little single agent activity and the addition of M6620 to cisplatin significantly prolonged time to event for a minority of tested xenografts across several histologies.
Keywords: Preclinical Testing, Developmental Therapeutics, ATR inhibitor, Cisplatin, Pediatric Tumor Xenografts
INTRODUCTION
Agents that damage DNA such as cyclophosphamide, cisplatin, topoisomerase poisons and ionizing radiation remain the backbone of curative therapy for many childhood cancers. The cytotoxic effect of these agents is best correlated with induction of DNA strand breaks, particularly double strand breaks that trigger the cascade of events leading to apoptosis. The effectiveness of these agents is limited by repair of DNA damage (strand breaks, or DNA adducts) through a complex series of repair mechanisms that have evolved for all organisms to survive endogenous and exogenous genotoxins.
The DNA damage response (DDR) is regulated by two homologous protein kinases, ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR). ATM-CHK2 and ATR-CHK1 checkpoints are central genome surveillance systems that function to maximize cell survival while minimizing genome instability [1]. Activated CHK2 and CHK1 phosphorylate downstream effectors to amplify and relay the signals to engage DDR leading to cell cycle arrest, DNA damage repair, senescence or apoptosis [2,3]. The major functions of checkpoints are to facilitate DNA repair and promote recovery from replication stalling [4–6], thereby maintaining cell survival. DNA replication forks undergo frequent stalling during normal cell cycle progression when they encounter endogenous DNA lesions estimated to occur at a frequency of at least 2×104 per cell/day [7]. Stabilization of stalled replication forks is regulated by a highly conserved process involving ATR-CHK1, which makes the ATR-CHK1 checkpoint essential for cell survival in all eukaryotes [2]. Eukaryotes have a highly efficient DNA repair network where, under normal growth conditions, the baseline DNA damage incurred from extracellular and intracellular agents is rapidly repaired and there is no checkpoint activation. However, in response to massive DNA damage, DNA damage checkpoints will be activated to arrest cell cycle progression in order to provide time for repair machinery to repair DNA lesions. Thus, through checkpoint signaling genome integrity is maintained [3,8,9], and defects in DDR facilitate emergence of genetically unstable cancer cells.
It has long been recognized that ATM patients are hypersensitive to ionizing radiation, and defects in DDR predispose to cells becoming hypersensitive to DNA damaging agents. Emerging clinical data show that ovarian cancers deficient in homologous recomination as a consequence of BRCA 1/2 mutations respond better to cisplatin-based therapy than do cancers with wild type BRCA genes [10–12]. Similarly, responses to platinum-based therapy are reported to be better in patients with lung cancers with mutated or compromised ERCC1 function [13] raising the anticipation that many defects in DDR may predispose cancer cells to cytotoxic effects of chemotherapy. Indeed, hypersensitivity to the PARP inhibitor talazoparib (and to cisplatin) in a Wilms tumor xenograft model was associated with a mutation in PALB2, a protein that plays a critical role in homologous recombination repair (HRR) through its ability to recruit BRCA2 and RAD51 to DNA breaks [14].
It has been proposed that a combination of replication stress and defective ATM signaling (as a consequence of defects upstream such as loss of p53 function) may promote reliance on ATR signaling to maintain cell viability [15–17]. Thus, it has been proposed that ATR inhibition may increase the effectiveness of DNA damaging chemotherapeutics such as cisplatin [18], topoisomerase I poisons [19], and ionizing radiation [20]. M6620 is a potent and selective inhibitor of ATR that has entered clinical trials and is being studied as a single agent and in combination with radiation therapy and with agents that induce DNA damage (e.g., cisplatin/carboplatin, gemcitabine, irinotecan) [18,20,21]. In this report, we present evaluation of M6620 as a single agent, or in combination with cisplatin or melphalan in pediatric cancer cell lines and in combination with cisplatin in xenograft models derived from pediatric cancers.
MATERIALS AND METHODS
In vitro testing
Testing was performed using DIMSCAN, a semiautomatic fluorescence-based digital image microscopy system that quantifies viable (using fluorescein diacetate [FDA]) cell numbers in tissue culture multiwell plates using standard Pediatric Preclinical Testing Program (PPTP) procedures described previously [22]. Cells were incubated in the presence of drug for 96 hours. Mean fluorescence values were determined for each concentration tested and then normalized to the mean control fluorescence for the line to determine relative mean fluorescence values.
For analysis of in vitro testing results, a non-linear regression, sigmoidal dose-response model was fitted using GraphPad Prism 5.03 to the relative mean fluorescence values vs. the log-transformed concentration (X) for the in vitro PPTP study data:
The terms are defined as follows: rIC50 (relative IC50) is the concentration of agent that gives a response half way between Bottom and Top; HillSlope describes the steepness of the dose-response curve; and Top and Bottom are the plateaus in the Treatment/Control (T/C%) values at low and high concentrations, respectively. To compare activity between cell lines, the ratio of the median relative IC50 to individual cell line’s relative IC50 value is used (larger values connote greater sensitivity). Observed Ymin is the minimum survival fraction (Treated/Control %) at the range of concentrations of the drug employed.
The Relative In/Out (I/O)% values compare the relative difference in final cell number compared with the starting cell number for treated cells and for control cells calculated as follows: (Observed Ymin−Y0)/(100−Y0) if Observed Ymin>Y0; and (Observed Ymin−Y0)/(Y0) if Observed Ymin<Predicted Ymin). Y0 is an estimate of the starting cell number derived from determinations of the doubling time of the cell line.
Combination testing was performed using methods previously applied to testing cytotoxic agents with rapamycin [23]. Data obtained for the cytotoxic alone were normalized to controls with no M6620 present and data obtained for the combination were normalized to controls with 75 nM M6620 present. Using these normalized data, rIC50 values were determined for the agent in the presence and absence of M6620, and the ratio of the two values was used to evaluate for potentiation induced by M6620. Cisplatin was tested at concentrations from 3 nM to 10 μM, and melphalan was tested at concentrations from 10 nM to 30 μM.
In vivo testing
CB17SC scid−/− female mice (Taconic Farms, Germantown NY), were used to propagate subcutaneously implanted kidney/rhabdoid tumors, sarcomas (Ewing, osteosarcoma, rhabdomyosarcoma), neuroblastoma, and brain tumors. Female mice were used irrespective of the patient gender from which the original tumor was derived. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee at the Research Institute, Nationwide Children’s Hospital. Ten mice were used in each control or treatment group. Tumor volumes (cm3) were determined and responses were determined using three activity measures as previously described [24]. An in-depth description of the analysis methods is included in the Supplemental Response Definitions S1 section.
Statistical Methods
The exact log-rank test, as implemented using Proc StatXact for SAS®, was used to compare event-free survival distributions between treatment and control groups. P-values were two-sided and were not adjusted for multiple comparisons given the exploratory nature of the studies. P values equal to or less than 0.05 were considered as statistically significant. Objective response is defined as ≥ 50% tumor volume regression in treated animals. The Pearson correlation coefficient was used to explore the relationship between potentiation of cisplatin and melphalan by M6620 across the cell lines.
Drugs and Formulation
M6620 was provided to the Pediatric Preclinical Testing Program by Vertex Pharmaceuticals (Europe) Inc., through the Cancer Therapy Evaluation Program (NCI). M6620 was formulated as a 20 mg/ml suspension in 5% captisol/3% mannitol in sterile water with stirring (30 min) until a clear solution was prepared. Solutions were prepared fresh. M6620 was administered by intravenous (IV) injection (0.05 ml/10 g body weight) days 1, 8, or in combination with cisplatin days 2 and 9 approximately 16 hours after the dose of cisplatin. Cisplatin (5 mg/kg) was administered by intraperitoneal (IP) injection days 1 and 8. M6620 and cisplatin were provided in coded vials for blinded testing.
RESULTS
M6620 in vitro testing
M6620 was tested against the PPTP’s in vitro cell line panel at concentrations ranging from 1.0 nM to 10.0 μM using the standard 96 hour exposure period. The median rIC50 value for the 24 cell lines was 0.19 μM, with a range from 0.03 μM to 1.38 μM (Table 1). The most sensitive cell line, Karpas-299, is an anaplastic large cell lymphoma (ALCL) cell line.
Table 1.
In vitro activity of M6620 against PPTP cell lines.
| Cell Line | Histotype | rIC50 (μM) | Ymin (Observed) | Relative In/Out% |
|---|---|---|---|---|
| RD | Rhabdomyosarcoma | 0.53 | 0.03 | −100% |
| Rh41 | Rhabdomyosarcoma | 0.21 | 0.00 | −100% |
| Rh18 | Rhabdomyosarcoma | 0.87 | 0.01 | −100% |
| Rh30 | Rhabdomyosarcoma | 0.10 | 0.01 | −100% |
| BT-12 | Rhabdoid | 0.72 | 0.00 | −100% |
| CHLA-266 | Rhabdoid | 1.38 | 0.05 | −100% |
| TC-71 | Ewing sarcoma | 0.08 | 0.01 | −99% |
| CHLA-9 | Ewing sarcoma | 0.07 | 0.01 | −100% |
| CHLA-10 | Ewing sarcoma | 0.26 | 0.01 | −100% |
| CHLA-258 | Ewing sarcoma | 0.31 | 0.01 | −100% |
| GBM2 | Glioblastoma | 0.19 | 0.11 | −99% |
| NB-1643 | Neuroblastoma | 0.26 | 0.00 | −100% |
| NB-EBc1 | Neuroblastoma | 0.48 | 0.00 | −100% |
| CHLA-90 | Neuroblastoma | 0.93 | 0.02 | −100% |
| CHLA-136 | Neuroblastoma | 0.45 | 11.26 | −61% |
| NALM-6 | Acute lymphoblastic leukemia | 0.10 | 0.00 | −100% |
| COG-LL-317 | Acute lymphoblastic leukemia | 0.13 | 0.00 | −100% |
| RS4;11 | Acute lymphoblastic leukemia | 0.09 | 0.08 | −99% |
| MOLT-4 | Acute lymphoblastic leukemia | 0.12 | 0.00 | −100% |
| CCRF-CEM (1) | Acute lymphoblastic leukemia | 0.12 | 0.00 | −100% |
| CCRF-CEM (2) | Acute lymphoblastic leukemia | 0.13 | 0.01 | −100% |
| Kasumi-1 | Acute myelogenous leukemia | 0.17 | 0.00 | −100% |
| Karpas-299 | T cell Non-Hodgkins lymphoma | 0.03 | 0.00 | −100% |
| Ramos-RA1 | Non-Hodgkins lymphoma | 0.20 | 0.06 | −94% |
| Median | 0.19 | 0.01 | −100% | |
| Minimum | 0.03 | 0.00 | −100% | |
| Maximum | 1.38 | 11.26 | −61% |
Tumor Volume T/C value: Relative tumor volumes (RTV) for control (C) and treatment (T) mice were calculated at day 21 or when all mice in the control and treated groups still had measurable tumor volumes (if less than 21 days). The T/C value is the mean RTV for the treatment group divided by the mean RTV for the control group. High activity = T/C ≤ 0.15; Intermediate activity = T/C ≤ 0.45 but > 0.15; and Low activity = T/C > 0.45.
EFS T/C values = the ratio of the median time to event of the treatment group and the median time to event of the respective control group. High activity requires: a) an EFS T/C > 2; b) a significant difference in EFS distributions, and c) a net reduction in median tumor volume for animals in the treated group at the end of treatment as compared to at treatment initiation. Intermediate activity = criteria a) and b) above, but not having a net reduction in median tumor volume for treated animals at the end of the study. Low activity = EFS T/C < 2.
Objective response measures are described in detail in the Supplemental Response Definitions. PD1 = progressive disease with EFS T/C ≤ 1.5, and PD2 = progressive disease with EFS T/C > 1.5.
A metric used to compare the relative responsiveness of the PPTP cell lines to M6620 is the ratio of the median rIC50 of the entire panel to that of each cell line. Higher ratios are indicative of greater sensitivity to M6620 and are shown in Supplemental Figure S1 by bars to the right of the midpoint line. The median rIC50 for the neuroblastoma cell lines exceeded that of the non-neuroblastoma cell lines (0.47 μM vs 0.15 μM, respectively, p=0.05), while the median rIC50 for the ALL cell lines was less than that of the non-ALL cell lines (0.12 μM vs 0.26 μM, respectively, p=0.03).
The Relative In/Out (I/O)% values compare the relative difference in final cell number compared with the starting cell number for treated cells and for control cells. Relative I/O% values range between 100% (no treatment effect) to −100% (complete cytotoxic effect), with a Relative I/O% value of 0% being observed for a completely effective cytostatic agent. As shown in Table 1, all PPTP cell lines showed a pronounced cytotoxic effect at the higher concentrations tested, with Relative I/O% values approaching −100% for all cell lines.
M6620 in vitro combination testing
To test for the ability of M6620 to potentiate the cytotoxic activity of standard chemotherapy agents, it was tested at a fixed concentration of 75 nM in combination with cisplatin and melphalan. The M6620 concentration was selected based on its reported cellular IC50 for inhibition of ATR (19 nM) and on data developed by Vertex showing potentiation of cisplatin at concentrations of 62 nM to 125 nM. Table 2 shows the rIC50 values for cisplatin and melphalan in the presence and absence of 75 nM M6620. When considering the ratio of the rIC50 values in the absence and presence of M6620, there is evidence of potentiation for both cisplatin and melphalan, with the median ratio of the IC50 values being 1.48 and 1.95, respectively. For cisplatin, the degree of potentiation was greatest for MOLT-4 (15.1-fold). For melphalan, the ratio was greatest for Kasumi-1 (7.55-fold). There was overlap of only one cell line between the top quartile of lines showing potentiation of cisplatin (MOLT-4, CHLA-136, Kasumi-1, NALM-6, NB-EBc1, and Rh30) and those showing potentiation of melphalan (Kasumi-1, Rh18, Karpas-299, CHLA-266, CHLA-90, and CCRF-CEM).
Table 2.
Relative IC50 Values for cisplatin and melphalan in the presence and absence of M6620
| Line | rIC50 for Cisplatin | rIC50 for Cisplatin + M6620 (75nM) | Ratio | rIC50 for Melphalan | rIC50 for Melphalan + M6620 (75nM) | Ratio |
|---|---|---|---|---|---|---|
| RD | 1.84 μM | 2.10 μM | 0.88 | 5.34 μM | 2.48 μM | 2.15 |
| Rh41 | 5.56 μM | 3.48 μM | 1.60 | 8.37 μM | 2.32 μM | 3.61 |
| Rh18 | 1.22 μM | 0.72 μM | 1.70 | 15.59 μM | 3.17 μM | 4.92 |
| Rh30 | 2.18 μM | 0.97 μM | 2.24 | 3.35 μM | 2.22 μM | 1.51 |
| BT-12 | 0.73 μM | 0.69 μM | 1.05 | 3.37 μM | 3.41 μM | 0.99 |
| CHLA-266 | 2.92 μM | 3.44 μM | 0.85 | 16.18 μM | 3.69 μM | 4.38 |
| TC-71 | 1.52 μM | 1.07 μM | 1.42 | 11.29 μM | 9.49 μM | 1.19 |
| CHLA-9 | 0.25 μM | 0.28 μM | 0.89 | 0.69 μM | 0.52 μM | 1.33 |
| CHLA-10 | 1.39 μM | 0.76 μM | 1.83 | 3.23 μM | 2.01 μM | 1.61 |
| CHLA-258 | 7.96 μM | 9.64 μM | 0.83 | 5.61 μM | 1.72 μM | 3.26 |
| GBM2 | 0.74 μM | 0.88 μM | 0.84 | 4.53 μM | 4.98 μM | 0.91 |
| NB-1643 | 0.75 μM | 0.85 μM | 0.88 | 1.19 μM | 1.19 μM | 1.00 |
| NB-EBc1 | 1.55 μM | 0.67 μM | 2.31 | 4.29 μM | 2.90 μM | 1.48 |
| CHLA-90 | 7.94 μM | 5.16 μM | 1.54 | 51.68 μM | 12.79 μM | 4.04 |
| CHLA-136 | 5.25 μM | 0.75 μM | 6.99 | 3.16 μM | 1.73 μM | 1.83 |
| NALM-6 | 1.44 μM | 0.55 μM | 2.62 | 1.73 μM | 0.50 μM | 3.46 |
| COG-LL-317 | 1.37 μM | 1.41 μM | 0.97 | 0.73 μM | 0.48 μM | 1.53 |
| RS4-11 | 0.89 μM | 0.84 μM | 1.06 | 2.19 μM | 1.05 μM | 2.07 |
| MOLT-4 | 1.36 μM | 0.09 μM | 15.1 | 1.13 μM | 1.24 μM | 0.91 |
| CCRF-CEM | 2.33 μM | 2.55 μM | 0.91 | 3.49 μM | 0.91 μM | 3.82 |
| CCRF-CEM-2 | 1.76 μM | 1.31 μM | 1.34 | 3.20 μM | 1.29 μM | 2.48 |
| KASUMI-1 | 2.93 μM | 1.09 μM | 2.69 | 5.53 μM | 0.73 μM | 7.55 |
| KARPAS-299 | 0.50 μM | 0.30 μM | 1.67 | 3.43 μM | 0.72 μM | 4.79 |
| RAMOS | 3.40 μM | 1.82 μM | 1.87 | 3.65 μM | 3.95 μM | 0.92 |
| Median | 1.53 μM | 0.92 μM | 1.48 | 3.46 μM | 1.87 μM | 1.95 |
M6620 in vivo testing
M6620 was tested against 24 PPTP solid tumor xenografts alone and in combination with cisplatin. Cisplatin was administered IP on day 1 and 8 at a dose of 5 mg/kg. M6620 was administered IV on days 2 and 9 at a dose of 20 mg/kg. For combination testing, M6620 was administered 16 hours after the cisplatin treatments on days 1 and 8. The total planned treatment and observation period was up to 12 weeks.
Toxicity testing was performed prior to efficacy testing using a M6620 dose of 20 mg/kg and cisplatin doses of 5, 3, and 2 mg/kg. M6620 did not exacerbate weight loss when added to cisplatin, with no more than approximately 10% weight loss at 5 mg/kg cisplatin. The weight loss nadir (9%) occurred on day 3, with full recovery by day 6 (100% of pretreatment weight). Based on the toxicity testing, the cisplatin dose selected for efficacy testing was 5 mg/kg. During efficacy testing, M6620 and cisplatin were well tolerated with 2/237 (0.8%), and 3/325 (1.3%) deaths, respectively. The combination of M6620 with cisplatin was also well tolerated (5/327 deaths 2.1%).
All 24 tested xenograft models were considered evaluable for efficacy. Complete details of testing are provided including total numbers of mice, number of mice that died (or were otherwise excluded), numbers of mice with events and average times to event, tumor growth delay, as well as numbers of responses and T/C values (Supplemental Table S1).
M6620 as a single agent induced significant differences in EFS distribution compared to control in 5 of 24 (21%) of the solid tumor xenografts studied (Table 3). For those xenografts with a significant difference in EFS distribution between treated and control groups, the EFS T/C activity measure additionally requires an EFS T/C value of > 2.0 for intermediate activity and indicates a substantial agent effect in slowing tumor growth. High activity further requires a reduction in final tumor volume compared to the starting tumor volume. M6620 induced tumor growth inhibition meeting criteria for intermediate EFS T/C activity in 0 of 24 (0%) solid tumor xenografts studied. No objective responses were observed among the 24 solid tumor xenografts.
Table 3.
Summary of in Vivo Activity of M6620, Cisplatin, and the Combination of M6620 and cisplatin
| Line | Tumor Type | Treatment Group | Median Time to Event | P-value | EFS T/C | Median RTV/CD45 at End of Study | Tumor Volume T/C | EFS Activity | Response |
|---|---|---|---|---|---|---|---|---|---|
| BT-29* | Rhabdoid | Cisplatin | 23.5 | 0.004 | 1.5 | >4 | 0.69 | Low | PD1 |
| BT-29* | Rhabdoid | M6620 | 23.6 | 0.019 | 1.5 | >4 | 0.69 | Low | PD1 |
| BT-29* | Rhabdoid | Combo | 36.8 | <0.001 | 2.3 | >4 | 0.48 | Int | PD2 |
| KT-12 | Rhabdoid | Cisplatin | 20.4 | <0.001 | 1.7 | >4 | 0.48 | Low | PD2 |
| KT-12 | Rhabdoid | M6620 | 13.3 | 0.858 | 1.1 | >4 | 0.83 | Low | PD1 |
| KT-12 | Rhabdoid | Combo | 19.8 | 0.006 | 1.7 | >4 | 0.46 | Low | PD2 |
| KT-10 | Wilms | Cisplatin | > EP | <0.001 | > 9.1 | 0 | 0.01 | High | MCR |
| KT-10 | Wilms | M6620 | 9.3 | 0.141 | 0.9 | >4 | 1.12 | Low | PD1 |
| KT-10 | Wilms | Combo | > EP | <0.001 | > 9.1 | 0 | 0.01 | High | MCR |
| KT-13 | Wilms | Cisplatin | 18.6 | <0.001 | 1.7 | >4 | 0.29 | Low | PD2 |
| KT-13 | Wilms | M6620 | 10.6 | 0.595 | 1 | >4 | 0.97 | Low | PD1 |
| KT-13 | Wilms | Combo | 19.9 | <0.001 | 1.9 | >4 | 0.28 | Low | PD2 |
| KT-5* | Wilms | Cisplatin | 45.7 | <0.001 | 2.7 | >4 | 0.27 | Int | PD2 |
| KT-5* | Wilms | M6620 | 17.5 | 0.309 | 1 | >4 | 0.88 | Low | PD1 |
| KT-5* | Wilms | Combo | > EP | <0.001 | > 5.3 | 3.2 | 0.06 | Int | CR |
| SK-NEP-1 | Ewing | Cisplatin | > EP | <0.001 | > 9.0 | 0 | 0.01 | High | MCR |
| SK-NEP-1 | Ewing | M6620 | 13.4 | 0.016 | 1.3 | >4 | 0.78 | Low | PD1 |
| SK-NEP-1 | Ewing | Combo | > EP | <0.001 | > 9.0 | 0 | 0.01 | High | MCR |
| EW5 | Ewing | Cisplatin | 56.9 | <0.001 | 1.8 | >4 | 0.08 | Int | CR |
| EW5 | Ewing | M6620 | 15.3 | 0.495 | 0.5 | >4 | 1.06 | Low | PD1 |
| EW5 | Ewing | Combo | 39.5 | <0.001 | 1.2 | >4 | 0.2 | Low | CR |
| EW8 | Ewing | Cisplatin | 12.8 | 0.185 | 0.9 | >4 | 0.98 | Low | PD1 |
| EW8 | Ewing | M6620 | 12.2 | 0.605 | 0.9 | >4 | 1.01 | Low | PD1 |
| EW8 | Ewing | Combo | 12.5 | 0.714 | 0.9 | >4 | 1 | Low | PD1 |
| TC-71 | Ewing | Cisplatin | 16.1 | 0.002 | 1.3 | >4 | 0.53 | Low | PD1 |
| TC-71 | Ewing | M6620 | 11.2 | 0.927 | 0.9 | >4 | 1.02 | Low | PD1 |
| TC-71 | Ewing | Combo | 19.5 | <0.001 | 1.5 | >4 | 0.41 | Low | PD1 |
| Rh30 | Alveolar RMS | Cisplatin | 28.5 | 0.001 | 1.3 | >4 | 0.4 | Low | PD1 |
| Rh30 | Alveolar RMS | M6620 | 14.5 | 0.731 | 0.6 | >4 | 1.42 | Low | PD1 |
| Rh30 | Alveolar RMS | Combo | 28.2 | 0.005 | 1.3 | >4 | 0.36 | Low | PD1 |
| Rh30R | Alveolar RMS | Cisplatin | 19.9 | 0.015 | 1.4 | >4 | 0.68 | Low | PD1 |
| Rh30R | Alveolar RMS | M6620 | 14.3 | 0.99 | 1 | >4 | 0.94 | Low | PD1 |
| Rh30R | Alveolar RMS | Combo | 19.9 | 0.029 | 1.4 | >4 | 0.72 | Low | PD1 |
| Rh41 | Alveolar RMS | Cisplatin | 15.1 | <0.001 | 2.2 | >4 | 0.55 | Int | PD2 |
| Rh41 | Alveolar RMS | M6620 | 7.6 | 0.094 | 1.1 | >4 | 0.88 | Low | PD1 |
| Rh41 | Alveolar RMS | Combo | 20.1 | <0.001 | 3 | >4 | 0.44 | Int | PD2 |
| Rh18 | Embryonal RMS | Cisplatin | 22.8 | 0.023 | 1.6 | >4 | 0.53 | Low | PD2 |
| Rh18 | Embryonal RMS | M6620 | 15.8 | 0.831 | 1.1 | >4 | 0.93 | Low | PD1 |
| Rh18 | Embryonal RMS | Combo | 20.6 | 0.03 | 1.5 | >4 | 0.66 | Low | PD1 |
| BT-50 | Medulloblastoma | Cisplatin | 38.1 | 0.754 | 0.8 | >4 | 0.86 | Low | PD1 |
| BT-50 | Medulloblastoma | M6620 | 40 | 0.368 | 0.9 | >4 | 0.94 | Low | PD1 |
| BT-50 | Medulloblastoma | Combo | 42 | 0.101 | 0.9 | >4 | 0.85 | Low | PD1 |
| GBM2 | Glioblastoma | Cisplatin | 17.4 | <0.001 | 1.7 | >4 | 0.46 | Low | PD2 |
| GBM2 | Glioblastoma | M6620 | 9.2 | 0.619 | 0.9 | >4 | 1.13 | Low | PD1 |
| GBM2 | Glioblastoma | Combo | 14.3 | 0.002 | 1.4 | >4 | 0.61 | Low | PD1 |
| D645 | Glioblastoma | Cisplatin | 15.8 | 0.006 | 1.2 | >4 | 0.75 | Low | PD1 |
| D645 | Glioblastoma | M6620 | 12.9 | 0.637 | 1 | >4 | 0.97 | Low | PD1 |
| D645 | Glioblastoma | Combo | 13.5 | 0.77 | 1 | >4 | 0.93 | Low | PD1 |
| D456 | Glioblastoma | Cisplatin | 12.5 | <0.001 | 1.4 | >4 | 0.75 | Low | PD1 |
| D456 | Glioblastoma | M6620 | 10.4 | 0.146 | 1.2 | >4 | 0.85 | Low | PD1 |
| D456 | Glioblastoma | Combo | 13 | 0.002 | 1.5 | >4 | 0.59 | Low | PD1 |
| D212 | Glioblastoma | Cisplatin | 21.6 | 0.005 | 1.4 | >4 | 0.74 | Low | PD1 |
| D212 | Glioblastoma | M6620 | 13.5 | 0.783 | 0.9 | >4 | 1.11 | Low | PD1 |
| D212 | Glioblastoma | Combo | 21.7 | 0.002 | 1.4 | >4 | 0.65 | Low | PD1 |
| NB-1691 | Neuroblastoma | Cisplatin | 10.2 | 0.002 | 1.5 | >4 | 0.8 | Low | PD1 |
| NB-1691 | Neuroblastoma | M6620 | 11.2 | 0.002 | 1.6 | >4 | 0.8 | Low | PD2 |
| NB-1691 | Neuroblastoma | Combo | 12.2 | 0.012 | 1.8 | >4 | 0.69 | Low | PD2 |
| NB-EBc1* | Neuroblastoma | Cisplatin | 7.3 | 0.095 | 1.1 | >4 | 0.81 | Low | PD1 |
| NB-EBc1* | Neuroblastoma | M6620 | 9.9 | 0.02 | 1.5 | >4 | 0.72 | Low | PD1 |
| NB-EBc1* | Neuroblastoma | Combo | 12.7 | <0.001 | 1.9 | >4 | 0.47 | Low | PD2 |
| NB-1643 | Neuroblastoma | Cisplatin | 9 | 0.065 | 1.1 | >4 | 0.82 | Low | PD1 |
| NB-1643 | Neuroblastoma | M6620 | 8.6 | 0.369 | 1.1 | >4 | 0.99 | Low | PD1 |
| NB-1643 | Neuroblastoma | Combo | 8.9 | 0.232 | 1.1 | >4 | 0.8 | Low | PD1 |
| OS-2 | Osteosarcoma | Cisplatin | 44.4 | 0.073 | 1.1 | >4 | 0.3 | Low | PD1 |
| OS-2 | Osteosarcoma | M6620 | 26 | 0.259 | 0.7 | >4 | 1.13 | Low | PD1 |
| OS-2 | Osteosarcoma | Combo | 52.4 | 0.018 | 1.3 | >4 | 0.4 | Low | PD1 |
| OS-17 | Osteosarcoma | Cisplatin | 17 | <0.001 | 1.5 | >4 | 0.67 | Low | PD1 |
| OS-17 | Osteosarcoma | M6620 | 17.6 | 0.009 | 1.5 | >4 | 0.72 | Low | PD2 |
| OS-17 | Osteosarcoma | Combo | 17.5 | <0.001 | 1.5 | >4 | 0.64 | Low | PD1 |
| OS-9* | Osteosarcoma | Cisplatin | 39.6 | 0.869 | 1 | >4 | 0.98 | Low | PD1 |
| OS-9* | Osteosarcoma | M6620 | 39.1 | 0.521 | 1 | >4 | 0.84 | Low | PD1 |
| OS-9* | Osteosarcoma | Combo | 69.5 | 0.001 | 1.7 | >4 | 0.54 | Low | PD2 |
Cisplatin as a single agent induced significant differences in EFS distribution compared to control in 18 of 24 (75%) of the solid tumor xenografts studied (Table 3). Cisplatin induced tumor growth inhibition meeting criteria for intermediate EFS T/C activity in 4 of 24 (17%) solid tumor xenografts studied. Three objective responses were observed among the 24 solid tumor xenografts.
The M6620 and cisplatin combination induced significant differences in EFS distribution compared to control in 21 of 24 (88%) of the solid tumor xenografts studied (Table 3). The combination induced tumor growth inhibition meeting criteria for intermediate EFS T/C activity in 5 of 24 (21%) solid tumor xenografts studied. Four objective responses were observed among the 24 solid tumor xenografts. The objective response results for solid tumor models are represented using ‘COMPARE’ format (Figure 1) and heatmep fomat (Supplemental Figure S2), with the former format based on the objective response scoring criteria centered around the midpoint score of 0 that represents stable disease (SD).
Figure 1.

COMPARE representation of tumor sensitivity based on the difference of individual tumor lines from the midpoint response (stable disease). Bars to the right of the median represent lines that are more sensitive, and to the left are tumor models that are less sensitive. Red bars indicate lines with a significant difference in EFS distribution between treatment and control groups, while blue bars indicate lines for which the EFS distributions were not significantly different.
Specific comparisons were made between the EFS distributions of the M6620 plus cisplatin combination and single agent cisplatin and single agent M6620, respectively (Table 4). Of special interest were models for which the combination was significantly superior to single agent cisplatin (p<0.05). Four of 24 models met this criterion, including BT-29 (rhabdoid tumor), KT-5 (Wilms tumor), NB-EBc1 (neuroblastoma), and OS-9 (osteosarcoma). Examples of tumor volume growth curves and Kaplan-Meier event-free survival for these solid tumor xenografts for which the EFS distribution for the M6620 plus cisplatin combination significantly exceeded that for single agent cisplatin are shown in Figure 2. The most substantial potentiation of activity was observed for KT-5, for which the addition of M6620 led to complete responses in many animals that were maintained for 4 or more weeks. The potentiation observed for BT-29 and NB-EBc1, although significant, was more modest.
Table 4.
Comparison of EFS distributions for the M6620 plus cisplatin combination to single agent M6620 and single agent cisplatin
| Xenograft | Cisplatin T-C (Days) | M6620 T-C (Days) | Combination T-C (Days) | P-value [combination vs. cisplatin] | P-value [combination vs. M6620] |
|---|---|---|---|---|---|
| BT-29 | 7.6 | 7.7 | 20.8 | 0.02 | 0.03 |
| KT-10 | 81.0 | −0.6 | 81.0 | 1 | <0.01 |
| KT-13 | 8.0 | −0.1 | 9.2 | 0.31 | <0.01 |
| SK-NEP-1 | 80.9 | 3.3 | 80.9 | 1 | <0.01 |
| KT-12 | 8.5 | 1.4 | 8.0 | 0.28 | <0.01 |
| KT-5 | 28.7 | 0.5 | 74.0 | <0.01 | <0.01 |
| Rh30R | 5.4 | −0.3 | 5.4 | 0.53 | 0.39 |
| TC-71 | 3.4 | −1.6 | 6.7 | 0.19 | <0.01 |
| EW5 | 24.4 | −17.2 | 7.1 | 0.48 | <0.01 |
| EW8 | −0.7 | −1.4 | −1.0 | 0.55 | 0.76 |
| Rh18 | 8.8 | 1.9 | 6.7 | 0.77 | <0.01 |
| Rh30 | 6.1 | −8.0 | 5.8 | 0.73 | <0.01 |
| Rh41 | 8.3 | 0.9 | 13.4 | 0.61 | <0.01 |
| BT-50 | −8.5 | −6.5 | −4.5 | 0.82 | 0.79 |
| GBM2 | 7.4 | −0.8 | 4.3 | 0.08 | <0.01 |
| D645 | 2.9 | −0.1 | 0.6 | 0.02 | 0.84 |
| D456 | 3.8 | 1.7 | 4.3 | 0.18 | <0.01 |
| D212 | 6.3 | −1.8 | 6.4 | 0.69 | <0.01 |
| NB-1691 | 3.3 | 4.4 | 5.3 | 0.65 | 0.13 |
| NB-EBc1 | 0.7 | 3.3 | 6.1 | <0.01 | 0.07 |
| NB-1643 | 0.9 | 0.6 | 0.9 | 0.84 | 0.57 |
| OS-2 | 4.6 | −13.8 | 12.6 | 0.61 | <0.01 |
| OS-17 | 5.7 | 6.2 | 6.1 | 0.23 | 0.75 |
| OS-9 | −0.4 | −0.9 | 29.6 | <0.01 | 0.22 |
Figure 2.
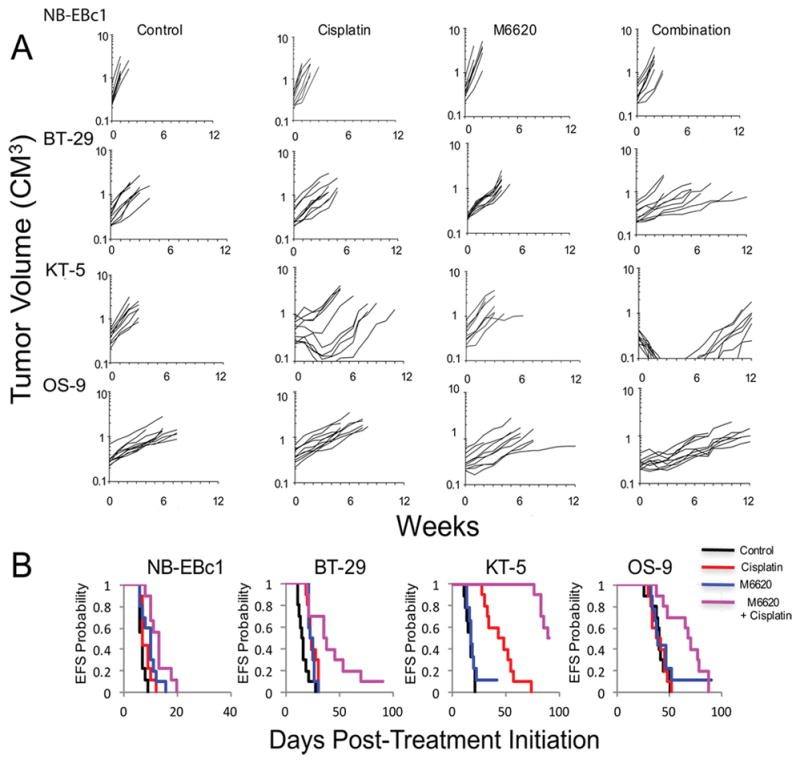
Tumor models where M6620 statistically significantly increased the efficacy of cisplatin; NB-EBc1 (neuroblastoma), BT-29(CNS rhabdoid), KT-5 (Wilms tumor), and OS-9 (osteosarcoma). Individual tumor volume graphs are shown for Control, cisplatin, M6620 and combination treatment groups. Kaplan Meier Event-Free survival plots are shown in the right panels. Control (black solid line), cisplatin orange broken line), M6620 (blue broken line) or combination (purple broken line) treated groups.
As ATM deficiency has been reported to predict for sensitivity to ATR inhibitors, the mutation status of ATM was examined. Exome sequencing identified predicted deleterious mutations in ATM in only the glioblastoma line GBM2 (p.G494C in exon 10) and the ALL cell line MOLT-4 (p.S2165F in exon 45) (sequencing data deposited at NCI TARGET Data Matrix). MOLT-4 showed a relatively low rIC50 and was the cell line that showed the greatest potentiation of cisplatin activity by M6620. The rIC50 for GBM2 was at the median for the entire panel, and M6620 did not potentiate either cisplatin or melphalan activity for this cell line.
DISCUSSION
Several studies have shown that ATR inhibition can potentiate cytotoxicity of chemotherapy and radiation therapy in vitro and in vivo [18–20] in preclinical models, although the single agent activity of ATR inhibitors such as M6620 is relatively modest. Here we examined the activity of M6620 as a single agent, and in combination with cisplatin or melphalan against panels of pediatric tumor cell lines in vitro and with cisplatin in solid tumor xenograft models.
In vitro, the median rIC50 value was 0.19 μM, and at higher concentrations clearly caused cytotoxicity with Relative I/O% values approaching −100% for all cell lines. The sensitivity of the pediatric cancer cell lines in the PPTP panel to single agent M6620 was greater than that reported for lung cancer cell lines (IC50 of approximately 1.5 μM) [18]. In combination with cisplatin, modulation by M6620 was relatively modest, being about 1.48-fold (range 0.85–14.31) over the entire tumor panel. In the study of Hall et al [18] similar potentiation was reported for some human lung tumor cell lines (0.9–4.2-fold), but there were two lines for which modulation was significantly greater (43- and 1112-fold). M6620 modulation of the activity of the bifunctional alkylating agent, melphalan, was also relatively modest (mean 1.98-fold, range 0.91–7.55-fold). There was no significant impact of TP53 status on the extent of modulation of cytotoxic activity by M6620 for either cisplatin or for melphalan. Furthermore, when comparing the potentiation of individual cell lines, there was little association between the fold potentiation induced by M6620 for these two chemotherapeutic agents. For example, Kasumi-1 was the only line common to the top quartile of lines showing potentiation of cisplatin and melphalan. The Pearson correlation coefficient (r) for the potentiation ratios for cisplatin and melphalan was −0.16.
As a single agent M6620 induced statistically significant differences in EFS distribution in a minority (21%) of solid tumor xenografts, but induced no objective responses. Among the tumor lines showing significant differences in EFS distribution (P<0.05, Supplemental Table S1), the magnitude of the treatment effect was small (EFS T/C values ranging from 1.3 to 1.5). Cisplatin as a single agent induced significant differences in EFS distribution compared to control in three-fourths of solid tumor xenografts, but induced only three objective responses. As reported previously the KT-10 Wilms tumor (PALB2 mutant) was highly sensitive to cisplatin [14]. Other cisplatin responsive tumors were Ewing sarcomas EW-5 and SK-NEP-1.
M6620 did not significantly increase toxicity of cisplatin, and the M6620-cisplatin combination induced significant differences in EFS distribution compared to control in the vast majority of lines tested (88%), with four objective responses. However there were only four models for which the antitumor response to cisplatin was significantly enhanced in combination with M6620. The greatest effect was observed for KT-5 Wilms tumor for which the combination induced CR with a marked increase in time to event compared to cisplatin (P<0.0001). Other statistically significant differences between cisplatin and the combination were observed for BT-29 CNS rhabdoid tumor, NB-EBc1 neuroblastoma and OS-9 osteosarcoma models (Supplemental Table S1). For BT-29 the response was changed from PD1 to PD2, whereas addition of M6620 to cisplatin did not change the overall group response for the other models, the time to event was extended over that for cisplatin alone treatment. Hall, et al., reported more consistent in vivo potentiation of cisplatin by M6620 using non-small cell lung cancer (NSCLC) xenograft lines that that observed for the PPTP pediatric xenograft lines [18]. Potential reasons include differences in the biology of NSCLC lines compared to our pediatric lines, as well as differences in dose and schedule for M6620 and cisplatin.
Results of phase 1 trials of M6620 as a single agent and in combination with either cisplatin or carboplatin have been reported. As a single agent, intravenous doses of up to 480 mg/m2 administered weekly were tolerated without dose-limiting toxicities observed [26]. In combination with carboplatin (AUC 5) administered on day 1, recommended phase 2 dose of M6620 is 90 mg/m2 administered intravenously on days 2 and 9 [26]. Dose-limiting toxicities at higher M6620 doses were primarily hematologic and included febrile neutropenia as well as carboplatin dose delays due to neutropenia and/or thrombocytopenia. The phase 1 evaluation of cisplatin and M6620 used the same administration schedule as that studied for carboplatin, and the recommended phase 2 doses are 75 mg/m2 for cisplatin and 140 mg/m2 for M6620 [27]. The maximum tolerated combination dose was not reached, because dose escalation was stopped as M6620 systemic exposure at 140 mg/m2 exceeded drug levels shown in preclinical models to produce target engagement and tumor regression in combination with cisplatin [27]. Objective responses were observed in 4 patients (among 28 patients enrolled), including 3 patients with platinum-resistant/refractory tumors.
ATM deficiency has been reported to predict for sensitivity to ATR inhibitors in preclinical testing [15,17]. As well, a patient with colorectal cancer whose tumor was ATM negative by immunohistochemical testing showed a complete response to single agent M6620 [26]. Among the PPTP lines tested, exome sequencing identified predicted deleterious mutations in ATM in only the glioblastoma line GBM2 (p.G494C in exon 10) and the ALL cell line MOLT-4 (p.S2165F in exon 45). MOLT-4 did show a relatively low IC50 and was the cell line that showed the greatest potentiation of cisplatin activity by M6620. The IC50 for GBM2 was at the median for the entire panel, and M6620 did not potentiate either cisplatin or melphalan in vitro, or cisplatin in vivo.
In summary, M6620 showed modest potentiation of cisplatin and melphalan activity for some PPTP cell lines. M6620 showed little single agent activity against pediatric solid tumor xenografts. The addition of M6620 to cisplatin significantly prolonged time to event for a minority of tested xenografts across several histologies.
Supplementary Material
Supplemental Figure S1. M6620 in vitro activity
Supplemental Figure S2. Heatmap representation of single drug and combination activity.
Supplemental Table S1. Efficacy of cisplatin, M6620 and cisplatin combined with M6620 against PPTP solid tumor Xenograft Models
Supplemental Response Definitions S1
Acknowledgments
This work was supported by NO1-CM-42216, CA21765, and CA108786 from the National Cancer Institute and used M6620 supplied the Vertex Pharmaceuticals (Europe) Inc. In addition to the authors this paper represents work contributed by the following: Sherry Ansher, Catherine Billups, Hulyun Wu, and Jian Zhang.
Abbreviations
- ATR
Ataxia telangiectasia and Rad3 related,
- ATM
Ataxia-telangiectasia mutated
- CHK1
Checkpoint kinase 1
- CHK2
Checkpoint kinase 2
- PALB2
Partner and localizer of BRCA2
- BRCA2
Breast cancer type 2 susceptibility protein
- RAD51
Radiation sensitive gene 51
- ERCC1
Excision Repair Cross-Complementing Rodent Repair Deficiency, Complementation Group 1
- PARP
Poly-ADP ribose polymerase
- HRR
Homologous recombination repair
- EFS
Event-Free Survival
- EFS T/C
Event-Free Survival Treated/Control
- DDR
DNA Damage Response
- PPTP
Pediatric Preclinical Testing Program
Footnotes
Abstract previously presented at EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium in November 2016
Conflict of interest statement: The authors consider that there are no actual or perceived conflicts of interest.
References
- 1.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopes M, Cotta-Ramusino C, Pellicioli A, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412(6846):557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 5.Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 6.Bartek J, Mistrik M, Bartkova J. Thresholds of replication stress signaling in cancer development and treatment. Nature structural & molecular biology. 2012;19(1):5–7. doi: 10.1038/nsmb.2220. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 8.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 9.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual review of biochemistry. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 10.Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(22):3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carser JE, Quinn JE, Michie CO, et al. BRCA1 is both a prognostic and predictive biomarker of response to chemotherapy in sporadic epithelial ovarian cancer. Gynecologic oncology. 2011;123(3):492–498. doi: 10.1016/j.ygyno.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Lesnock JL, Darcy KM, Tian C, et al. BRCA1 expression and improved survival in ovarian cancer patients treated with intraperitoneal cisplatin and paclitaxel: a Gynecologic Oncology Group Study. British journal of cancer. 2013;108(6):1231–1237. doi: 10.1038/bjc.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. The New England journal of medicine. 2006;355(10):983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 14.Smith MA, Hampton OA, Reynolds CP, et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer. 2015;62(1):91–98. doi: 10.1002/pbc.25201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 16.Menezes DL, Holt J, Tang Y, et al. A synthetic lethal screen reveals enhanced sensitivity to ATR inhibitor treatment in mantle cell lymphoma with ATM loss-of-function. Mol Cancer Res. 2015;13(1):120–129. doi: 10.1158/1541-7786.MCR-14-0240. [DOI] [PubMed] [Google Scholar]
- 17.Kwok M, Davies N, Agathanggelou A, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–595. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 18.Hall AB, Newsome D, Wang Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–5685. doi: 10.18632/oncotarget.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Josse R, Martin SE, Guha R, et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer research. 2014;74(23):6968–6979. doi: 10.1158/0008-5472.CAN-13-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fokas E, Prevo R, Pollard JR, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell death & disease. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown JS, O’Carrigan B, Jackson SP, et al. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer discovery. 2016 doi: 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang MH, Smith MA, Morton CL, et al. National Cancer Institute Pediatric Preclinical Testing Program: Model description for in vitro cytotoxicity testing. Pediatr Blood Cancer. 2011;56(2):239–249. doi: 10.1002/pbc.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houghton PJ, Morton CL, Gorlick R, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Molecular cancer therapeutics. 2010;9(1):101–112. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houghton PJ, Morton CL, Tucker C, et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr Blood Cancer. 2006 doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 25.Jin W, Liu H, Zhang Y, et al. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Human genetics. 2008;123(6):643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Carrigan B, de Miguel Luken MJ, Papadatos-Pastos D, et al. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J Clin Oncol. 2016;34 (suppl; abstr 2504) [Google Scholar]
- 27.Shapiro G, Wesolowski R, Middleton M, et al. Phase 1 trial of first-in-class ATR inhibitor VX-970 in combination with cisplatin (Cis) in patients (pts) with advanced solid tumors ( NCT02157792) Cancer research. 2016;76(14 Suppl) Abstract nr CT012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. M6620 in vitro activity
Supplemental Figure S2. Heatmap representation of single drug and combination activity.
Supplemental Table S1. Efficacy of cisplatin, M6620 and cisplatin combined with M6620 against PPTP solid tumor Xenograft Models
Supplemental Response Definitions S1