

Abstract
Campylobacter jejuni infections are a leading cause bacterial food-borne diarrheal illness worldwide, and Campylobacter infections in children are associated with stunted growth and therefore long-term deficits into adulthood. Despite this global impact on health and human capital, how zoonotic C. jejuni responds to the human host remains unclear. Unlike other intestinal pathogens, C. jejuni does not harbor pathogen-defining toxins that explicitly contribute to disease in humans. This makes understanding Campylobacter pathogenesis challenging and supports a broad examination of bacterial factors that contribute to C. jejuni infection. Here we use a controlled human infection model to characterize C. jejuni transcriptional and genetic adaptations in vivo, along with a non-human primate infection model to validate our approach. We found variation in 11 genes is associated with either acute or persistent human infections and include products involved in host cell invasion, bile sensing, and flagella modification, plus additional potential therapeutic targets. Particularly, a functional version of the cell invasion protein A (cipA) gene product is strongly associated with persistently infecting bacteria and we went on to identify its biochemical role in flagella modification. These data characterize the adaptive C. jejuni response to primate infections and suggest therapy design should consider the intrinsic differences between acute and persistently infecting bacteria. Additionally, RNA-sequencing revealed conserved responses during natural host commensalism and human infections. 39 genes were differentially regulated in vivo across hosts, lifestyles, and C. jejuni strains. This conserved in vivo response highlights important C. jejuni survival mechanisms such as iron acquisition and evasion of the host mucosal immune response. These advances highlight pathogen adaptability across host species and demonstrate the utility of multidisciplinary collaborations in future clinical trials to study pathogens in vivo.
Introduction
Studying bacterial evolution during infection exposes how pathogens adapt and survive in the host. High-resolution whole genome sequencing enables accurate tracing of pathogen transmission between patients, identification of antibiotic resistance loci, and understanding of selective pressures in vivo1–6. These developments have led to the identification of pathogen genetic variants that predict the success of treatments and guide therapy design. Such studies reveal the extent of bacterial adaptability in vivo while regarding the genetic fitness compromises that would only arise in the complex host environment.
Some bacteria adapt to new hosts and environments using a common population-level adaptation method known as phase variability7. Phase variation is driven by relatively high frequency genome mutations in homopolynucleotide tracts which result in a functionally “on” or “off” phase of affected gene(s)7. Subpopulations of on/off gene variants are intrinsically more fit for a new environment and therefore increase in frequency in a population as they outcompete less fit variants. This adaptive process genetically regulates virulence factors and is important for pathogen colonization and immune evasion8–10. To fully understand phase variable populations, whole genome sequencing can be used to identify the frequency of every “on” or “off” gene variant at the population level and then identify influential variants that are selected for during adaption to new host environments11,12.
Campylobacter jejuni is a phase variable Gram-negative intestinal pathogen that causes bloody diarrhea, fever, and abdominal pain in humans. Notable secondary sequelae include Guillain-Barré syndrome, a potentially fatal paralytic autoimmune disorder. C. jejuni is a prevalent commensal bacterium in the intestinal tracts of chickens and other livestock, and consumption of contaminated animal products makes C. jejuni a leading cause of food-borne bacterial diarrhea worldwide. In low-resource areas, asymptomatic and sometimes persistent Campylobacter infections are common in children younger than 1 year old and correlate with stunted growth and therefore life-long physical and cognitive deficits13. In line with our understanding of in vivo pathogen evolution, small animal models of C. jejuni infection show host-passaged isolates are more fit in the host than the initial inoculum, and this genetic advantage remains even after multiple passages in vitro14. Here we use samples from a controlled human infection model to characterize the C. jejuni response to the human host. High-resolution genomic sequencing was used to track pathogen adaptation and phase variation in vivo, from inoculum through acute and persistent disease. In the challenge, a single liquid inoculum of C. jejuni strain CG8421 was used to simultaneously infect a group of human volunteers, making this study a highly-controlled pathogen adaptation experiment in humans. The challenge was designed to evaluate the prophylactic efficacy of rifaximin on C. jejuni infection in humans and is described elsewhere (ClinicalTrials.gov Identifier NCT02280044)15. Briefly, rifaximin treatment had no effect on the primary clinical outcome of campylobacteriosis. To be released from the trial, volunteers were treated with azithromycin and ciprofloxacin and produced feces negative for C. jejuni. However, five volunteers experienced recrudescent16 infections. The clinical trial outcomes enabled us to compare pathogen gene variants that were selected for during both acute and recrudescent16 infections, as well as in volunteers who experienced severe disease or received prophylactic antibiotic treatment. We found variants of genes involved in host cell invasion, bile sensing, and flagella modification are selected for in recurrent human infections. To validate our approach, we performed a similar analysis in a symptomatic non-human primate infection model. Beyond defining genetic adaptation in primate hosts, we also used RNA-sequencing to determine the C. jejuni transcriptome directly in human infection samples.
Campylobacter Gene Expression in Human Infection Samples
Responding to the host environment via transcriptional changes is essential for C. jejuni colonization and infection17,18. To identify the transcriptional adaptations that occur in a human host we determined the C. jejuni transcriptome directly in infected human feces of three volunteers. Compared to in vitro microaerobic growth on blood agar, 264 genes were differentially regulated in vivo by at least 3 fold (false-discovery rate adjusted p-value <0.05) (Fig. 1a, Sup. Table 1). A similar number of genes were differentially regulated compared to growth on Mueller-Hinton agar and in Mueller-Hinton broth (Fig. 1a). No matter which comparison is made, a conserved group of 110 genes were differentially regulated in vivo. These gene products include diverse characterized colonization factors, including Peb1A, PldA, CsrA, and the capsule polysaccharide transporter KpsM (Sup. Table 1)19–22. These data also reveal only between 10 and 15 percent of annotated open reading frames show significant differential regulation in infected human feces compared to growth in standard laboratory conditions.
Figure 1. The C. jejuni Transcriptome in Human Infection and Chick Commensalism.
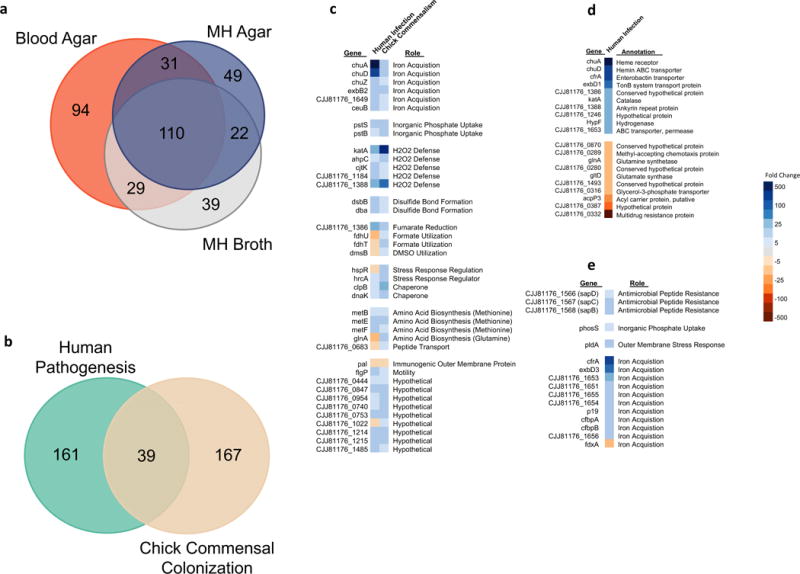
a, Differentially regulated genes in vivo. A Venn diagram showing the number of differentially regulated C. jejuni CG8421 genes (fold change >|3|, FDR p-value <0.05) in the infected feces of three volunteers compared to laboratory control growths which each represent three biological replicates. b, Human infection versus chicken commensalism transcriptomes. A Venn diagram showing the number of differentially regulated (fold change >|3|, FDR p-value <0.05) gene homologs in vivo between C. jejuni CG8421 infected human feces (3 samples) and a previously published RNA-seq transcriptome of C. jejuni 81-176 in the chicken cecum, which represents 3 pools of 5 infected chick ceca each. Both data sets use mid-log phase growth in Mueller-Hinton broth biological triplicates as the in vitro lab comparison. c, Conserved differential gene regulation across hosts, lifestyles, and strains. The 39 differentially regulated homologs from Fig. 1b conserved between human infection and chicken commensalism with transcriptional fold changes noted. d, The top most upregulated and downregulated CG8421 genes in human infection samples. e, Notable genes uniquely differentially regulated in human infection samples.
Although primates can experience disease symptoms during infection, C. jejuni has a commensal lifestyle in the natural bird host. To relate transcriptional adaptations that occur during infection and commensalism, we compared our data to a previously published RNA-sequencing data set of C. jejuni strain 81-176 in the chick cecum (Fig 1b, Sup. Table 2)23. We identified a core set of 39 conserved genes that are differentially regulated in vivo across hosts, colonization lifestyles, and strains. These gene products share strikingly similar roles, including iron acquisition, inorganic phosphate uptake, and protection from peroxide stress (Fig. 1c). Epithelial cell production of hydrogen peroxide plays an important role in mucosal immunity against C. jejuni. Hydrogen peroxide-mediated inactivation of the C. jejuni outer membrane tyrosine kinase Cjtk leads to a decrease in capsule production and therefore decreased virulence24. Here we see the importance and conserved nature of this response as cjtK along with two major hydrogen peroxide scavengers, katA and ahpC, are upregulated during both chicken commensalism and human infection. Finally, 7 of these 39 genes showed discrepant expression patterns. Notably, the fdhTU operon, which is important for formate dehydrogenase activity25, is oppositely expressed between the human and chick data.
The most upregulated genes in human infection samples represent two distinct iron acquisition pathways. The chuA and chuD hemin uptake genes, and cfrA, a ferric-enterobactin uptake gene, are all upregulated >100 fold in vivo (Fig. 1d). chuA is not required for chicken colonization26 and is modestly upregulated in the chick cecum (<10 fold increase in vivo), although it is highly upregulated in human infection samples (>1000 fold increase in vivo). There are 161 genes that are differentially regulated specifically in our human samples and not in the chick cecum (Fig. 1b) and includes the highly upregulated cfrA. C. jejuni does not produce siderophores, but CfrA enables import of a wide array of siderophores made by other organisms and, unlike chuA, is required for chicken colonization27. This suggests microbiome derived siderophores and cfrA play an important role in C. jejuni infection of humans. Interestingly, components of the Sap antimicrobial peptide resistance efflux pump, which is required for chicken colonization28, are also uniquely upregulated in human infection (Fig. 1e).
Genome Variant Selection During Human Infection
To identify genetic variants that are selected for during human infection we examined the genome of C. jejuni before and during acute and persistent human infections. We sequenced the genomes of 49 C. jejuni CG8421 infection isolate populations from 14 volunteers during acute and recrudescent infections and compared them to the inoculum genome (Fig. 2a, Sup. Fig. 1). These infection isolate populations represent bacterial colonies harvested directly from primary isolation plates and are pooled together per sample with no further outgrowth, and is consistent with previous work that validated C. jeuni isolate sequencing to study genetic variation in vivo7. We achieved exceptional coverage of the infection population genomes across all samples, with an average genome sequencing coverage >1000x and 97 percent of genes had at least 25x coverage at every nucleotide position (Sup. Fig. 2). Due to the complexity of phase variable populations, different subjective methods have been used to determine noteworthy changes in phase variant frequencies over time7,11,29. We first called any variant that occurred in at least 1 percent of any sample population, which resulted in over 600 variants identified across all samples (Sup. Table 3). To focus on variants that had both a large change in frequency during infection and represented a large portion of the population, we called variants in a sample if they occurred in at least 25 percent of the population. This enriched for variants that had, on average, a large statistically significant fold change in frequency between the inoculum and an infection isolate populations (~12 fold) while also representing a majority of the infection population sample (~60 percent) (Sup. Fig. 3).
Figure 2. C. jejuni Genomic Variants are Consistently Selected for During Human Infections.
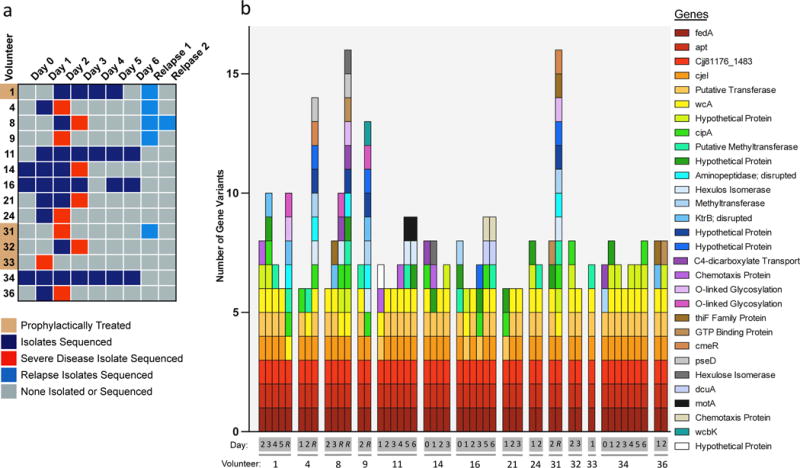
a, Infection populations sequenced. Whole genome sequencing was performed on 49 infection isolate populations, taken from 14 volunteers across 6 days of primary infection. 5 volunteers relapsed after the clinical trial, and those infection isolate populations were also sequenced. Some volunteers experienced severe disease on the noted days (red) and therefore received early antibiotic intervention. Some volunteers received exploratory prophylactic treatment with the antibiotic Rifaximin as noted. Sequencing details are noted in Sup. Fig. 2. b, Genomic variants detected per volunteer isolate population, noted by the genes the variants affect. The number of genomic variants detected per infection population is noted on the y-axis, and the x-axis denotes the day and volunteer the sample was taken from, with R denoting a relapse sample. The corresponding genes are listed by their gene name or annotation when considering homologs across CG8421, 11168, and 81176 C. jejuni strains.
This enrichment analysis resulted in 48 unique genomic variants with major changes in frequency in at least one sample, including small nucleotide polymorphisms (SNPs), phase variations (PV), and multinucleotide variants (MNV) (Sup. Table 4). 47 of the variants clearly affected 30 annotated coding sequences and/or promoters and are noted per isolate population in Figure 2b. Slip-strand mutations in poly G/C nucleotide tracts are the dominant phase variable mechanisms in C. jejuni7, and 19 of 28 tracts in the CG8421 genome varied in at least one infection isolate population (Sup. Fig. 4). The inoculum population had 12 genetic variants that differed from the reference genome, 4 of which remained intact in all infection isolate populations (Sup. Table 5).
Three genetic variants were selected for in all 49 infection isolate populations. One of these completely conserved variants is a PV that occurs in the in last 10 percent of the fedA gene open reading frame and in the -10 promoter region of an overlapping downstream gene of unknown function. fedA is putative hemerythrin that contributes to C. jejuni colonization in chicks and is coregulated with flagellar gene expression30. A second completely conserved variant is a SNP in the house keeping gene adenine phosphoribosyltransferase (apt) which salvages adenine to make adenosine monophosphate. The inoculum population contained an apt SNP that is selected for during osmotic stress29, however the wild type apt sequence was selected for in vivo. The final completely conserved variant is a 9 nucleotide in-frame deletion in the CG8421 homolog of the DNA response regulator CJJ81176_1483 that was selected against in vivo. Selection for the wild type version CJJ81176_1483 in vivo was confirmed by structural genomic variant analysis (Sup. Table 6). CJJ81176_1483 is part of a newly identified two-component system that regulates a gluconate dehydrogenase complex important for chick colonization31. These data represent 3 distinct genetic variations (PV, SNP, and MNV) in genes with diverse roles (chicken colonization, osmotic stress/house keeping, two-component system) and demonstrate the genetic adaptability of C. jejuni in vivo.
Gene Variants Associated with Treatment or Human Disease State
Rifaximin prophylactic treatment did not impact the rate of campylobacteriosis during the clinical trial. Nevertheless, we hypothesized the antibiotic treatment environment in the host may have selected for unique genomic variants in these volunteers. However, we found no variants were more likely to be called in these volunteers when compared to control volunteers (Sup. Table 7) using Fisher’s Exact Test. We concluded rifaximin did not exert a noticeable pressure on C. jejuni in vivo. We also hypothesized some variants may be more virulent, and therefore would be more likely to be found in volunteers who experienced severe disease symptoms. However, a Fisher Exact Test showed no variants were associated with isolates from volunteers experiencing severe disease compared to typical symptomatic disease isolate populations (Sup. Table 8). This indicates host factors, more so than genetic drift of more virulent genotypes, may influence the severity of disease symptoms.
Despite being treated with azithromycin and ciprofloxacin and repeatedly producing feces negative for C. jejuni isolates at the end of the trial, 5 volunteers returned to the clinic with at least one bout of recrudescent infection (Fig. 2a). This rate of recrudescence is higher than previous trials likely due to enhanced culture techniques, such as not refrigerating stool before culturing, that were employed specifically for increased vigilance of recrudescence15. Recrudescent infection isolates were confirmed to be the inoculum strain and, in all cases, were sensitive to azithromycin and ciprofloxacin. Remarkably, relapse infection isolate populations had twice as many genomic variants as primary infection isolates (Fig 3a). We hypothesized the increased variant count would correlate with increased time in vivo. To test this, we compared the number of variants called per sample on every day of infection. Interestingly, there was not an increase in the number of variants called over time during the primary infection (Fig. 3b) and a Fisher exact test showed no particular variants are associated with early or late primary infection periods (Sup. Table 9). These data suggest there is an immediate and consistent selection pressure during primary infection, and that either a secondary selection event or additional time within the host results in increased genetic variation of relapse isolates.
Figure 3. Particular Genome Variants are Associated with C. jejuni Recrudescent Infection Isolate Populations.
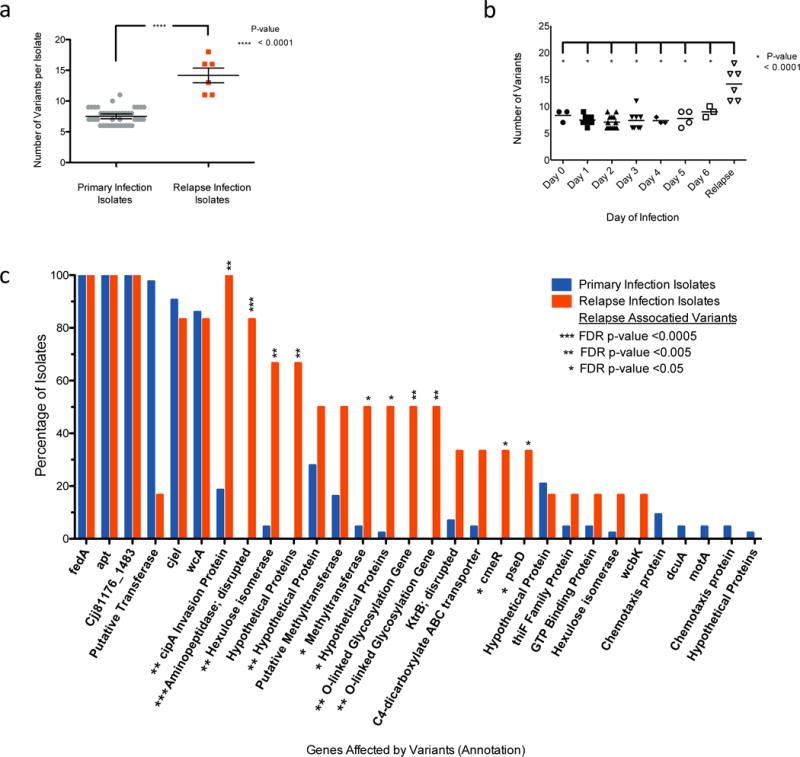
Primary infection isolates (days 0-6) represent 43 isolate samples while relapse infection isolate populations represent 6 samples. a, Bulk genome variation between primary and relapse infection isolates. On average, relapse infection isolates have twice as many genomic mutations compared to primary infection isolates (Unpaired two tailed T-test with bars representing the mean and standard error of the mean). b, Variant accumulation over time. Variants are selected for immediately in human infections (day 0 is the day of inoculation); however, they do not increase over time during the primary infection (One-way ANOVA, Tukey’s multiple comparison test). c, Genome variants that are associated with relapse infection. After the genome variants were determined for all samples (Fig 2b), a Fisher’s Exact test was used to determine if variation in particular genes is associated with relapse infection isolate populations. Variation of 10 genes was statistically associated with relapse infection populations and is noted with a False-Discovery Rate adjusted (FDR) one-sided p-value of < 0.05. The y-axis represents the percent of samples that had a variant in the gene noted on the x-axis. More detailed information is noted in Sup. Table 4.
There are a considerable number of genetic variants that are associated with relapse infection isolates as determined by Fisher Exact Test (false-discovery rate corrected p-value <0.05) (Fig. 3c, Sup. Table 10). Of the 10 gene variants associated with relapse infection only 3 are characterized, named genes. The most striking is a phase variation in cipA (cell invasion protein A), which turned the gene “on” in every relapse infection isolate population, but does not appear to be important for establishing a primary infection. cipA was identified in three independent studies to contribute to C. jejuni invasion of host cells in vitro32,33; however, this gene has not been shown to play a role in vivo until this work in a human challenge model. Mechanisms of C. jejuni cellular invasion are not well defined and no biochemical functions of CipA have been previously identified. Although, BLAST search revealed cipA harbors a domain of unknown function (DUF2972) that is also found genes annotated as glycosyl transferases. To determine the role of CipA during human infection we therefore considered possible glycosylation targets. We find the “on” variant of cipA that was selected for in vivo results in modification of the flagella (Sup. Figure 6a). This suggests cipA likely contributes to cellular invasion and persistence in humans via flagella modification.
Another notable gene associated with relapse infection is cmeR, a pleotropic bile-sensing transcriptional regulator that is required for chicken colonization34. CmeR functions as a dimer to bind DNA and repress gene expression until C. jejuni is exposed to bile in the intestinal environment35. CmeR repression is relieved upon bile acid binding, which induces expression of the CmeR regulon including the multi-drug efflux pump cmeABC36. Two unique SNPs that each resulted in truncation of the cmeR open reading frame were selected for independently in relapse infection isolates and are in non-phase variable loci (Sup. Table 4). Similar truncations have been shown to prevent dimerization of CmeR and therefore prevent CmeR repression regardless of environmental bile concentrations37. cmeR was intact in every primary infection isolate, and likewise, functional CmeR protein is important for colonization of chickens. However, the independent and functionally conserved relapse variants suggest a cmeR “off” mutation can be advantageous in persisting human infections. This may be due to a resulting constitutive expression of the CmeABC efflux pump that increases resistance to bile acids in the host38, although losing the ability to sense bile is an unexpected in vivo phenotype for an intestinal pathogen.
pseD is the final named gene variant that is associated with relapse infection. PseD attaches pseudaminic acid to flagellin which contributes to virulence-associated autoagglutination of the bacteria39. pseD was considered “on” in every primary infection sample but turned “off” in a one third of relapse infection samples. The remaining 7 relapse associated gene variants included hypothetical genes, a disrupted aminopeptidase gene, a methyltransferase gene, and two additional O-linked glycosylation locus genes. Interestingly, an annotated methyltransferase and multiple O-linked glycosylation locus genes were identified to be under positive selection in a study of Burkholderia infection of cystic fibrosis patients1, although these genes share low sequence homology with genes in CG84211.
One variant is statistically significantly associated with primary infection isolates and not relapse infection isolates (Sup. Fig. 5, Sup. Table 11). This is a phase variant in the promoter region of an uncharacterized putative transferase gene, CJ8421_RS06560 (homolog of Cj1321 in strain NCTC_11168), located in the flagellin glycosylation locus40 and was strongly selected against in primary infection isolates, but not in relapse infection isolates. In total, 7 of the 8 flagella modification genes identified in our analysis are associated with either primary or relapse infection isolates, indicating the significance and variability of flagellar modification during human infections (Sup. Fig 6). Finally, Figure 4a depicts a SNP-tree analysis to visually represent the differences in genomic variations amongst primary and relapse infection isolates. The closer two samples are on the tree, the more similar their genomic variant profiles are to one another. Primary infection isolates from the same volunteer are more similar to each other than they are to the isolates of other volunteers. However, the relapse infection isolates from different volunteers are more similar to each other than they are to the founding primary infection isolates from the same volunteer. Overall, the relapse-associated variants demonstrate the profound genetic differences that occur in persisting bacteria.
Figure 4. Validating C. jejuni Genomic Variant Selection in non-Human Primates.
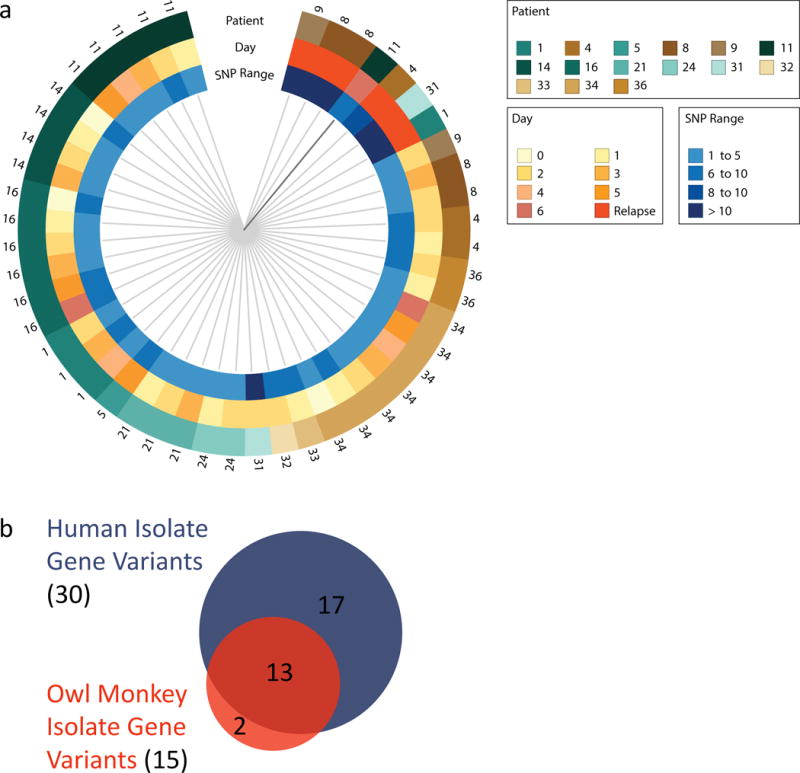
a, A SNP tree representing how similar human infection isolate populations are to one another. Each wedge represents one sequenced isolate population and the closer two samples are to each other the more similar their genomic variations. Volunteer identification numbers per sample are noted on the outside of the ring, and the day of infection and the number of SNPs detected are denoted by the inner rings. Note that primary infection isolates from the same volunteer are more similar to each other than they are to isolates from other volunteers. However, relapse infection isolates (bright orange) are more similar to each other despite the volunteer they come from. b, Conserved gene variant selection across primates. A second, different C. jejuni CG8421 inoculum was used to infect owl monkeys, and the genomes of 12 diarrheal primary infection isolate populations from 5 monkeys were sequenced (Sup. Tables 12 and 13). The Venn diagram shows the C. jejuni genome variants selected for in owl monkey infection closely resemble those of human infection (Sup. Table 13). This supports the human data and suggests non-human primates harbor a similar fitness selection environment found in humans. The smaller sample size and no relapse infections contributed to fewer total variants detected in the monkeys.
Genome Variant Selection in Aotus Primates
The variants selected for during human infection replicated strongly across volunteer isolate populations. This powerfully suggests a conserved selective pressure and adaptive response in vivo. However, over 30 coding sequences were implicated in adaptation, and understanding the fitness contribution of variant combinations within the population is challenging. To validate our data and respect the complexity of population-level adaptation, we performed a similar analysis in a symptomatic non-human primate infection model. We hypothesized adaptations similar to those in human infection would be selected for in a New World monkey, Aotus nancymaae. An independently prepared CG8421 liquid inoculum was used to infect Aotus nancymaae and we compared this inoculum genome to 12 diarrheal isolate populations taken over 8 days of infection from 5 monkeys (Sup. Table 12). Despite differences between the human and Aotus inoculums (Sup. Table 12), we found the genetic adaptations selected for in Aotus closely resemble those in humans (Fig 4b, Sup. Table 13). The 3 completely conserved variants that were selected for in human infection were already present as a majority in the Aotus inoculum, and congruently, were maintained during infection. Two genes were uniquely varied in Aotus infection, including a SNV in the promoter region of CJJ81176_1215, a gene encoding an uncharacterized, putative NLPA-family lipoprotein (Sup. Table 13). Interestingly, the second variant is a E70K amino acid substitution in chuA, which is also the most upregulated gene during human infection. Therefore, chuA can undergo both transcriptional and genetic adaptations during primate infection. Few of the genetically variable genes were also differentially regulated in vivo, yet this is consistent with analyses done in vitro41 and in vivo42 that show genes that contribute to fitness are rarely also differentially regulated.
Discussion
Understanding the bacterial pathogen response to the human host revealed genetic mechanisms of success during both establishment of infection and persistent human colonization. Notably, we determined the cipA gene had the strongest association with recrudescent, persistent infections, and we went on to identify its role in C. jejuni flagellar modification. 9 other variable genes are part of the conserved relapse infection genotype. We suspect this genetic background may enable bacteria to reside in a protected niche within the host during primary infection where they withstand being shed from the host, resist antibiotic treatment and the host immune response, and result in recrudescent infection. This genotype would then be the dominant recrudescent population, and a similar pattern was seen in this study. Future therapy design should consider the “on/off” status of a therapeutic target throughout primary and recrudescent infections, as well as appreciate target expression alterations due genetic variation of promoter regions or transcription factors such as CmeR.
Tracking pathogen evolution in vitro requires studying multiple cultures grown in parallel, and variants that arise consistently across replicates are considered adaptive. Studying pathogen evolution in vivo can reveal important virulence determinants, although it is difficult to study well-controlled human infections in parallel. Here we used a C. jejuni clinical trial to examine bacterial adaptation during human infection and found many genomic variants are well conserved across infected volunteers. We also found human infection exemplifies rapid bacterial adaptation, as variants are selected for in vivo on the day of inoculation. This study characterizes pathogen genetic and transcriptional adaptations across hosts and lifestyles, and similar collaborative approaches in future clinical studies may increase the possibilities for therapeutic design.
Materials and Methods
C. jejuni Infected Human Feces Sample Preparation for RNA-Sequencing
Campylobacter jejuni strain CG8421 infected diarrhea was weighed and added 1:1 in RNA Later reagent as quickly as possible after it was produced and frozen at −80°C. Samples came from volunteers who were not receiving antibiotics and therefore represent untreated infections. Preserved samples were thawed on ice and total RNA extracted via Trizol-chloroform phase separation. DNA was removed (Turbo DNA-free DNAse kit, Ambion), rRNA depleted (Ribo-Zero human and bacterial rRNA removal kits, Illumina), and libraries built for Illumina Sequencing (Ultra Directional RNA Library Prep Kit for Illumina, New England Biolabs).
In vitro C. jejuni Sample Preparation for RNA Sequencing
Campylobacter jejuni strain CG8421 was grown at 37°C in a microaerobic atmosphere (85% nitrogren, 10% carbon dioxide, 5% oxygen) in a Coy Laboratories atmosphere controlled chamber. For RNA-seq of samples grown on agar plates: Mueller-Hinton agar or 5 percent sheep blood agar plates were streaked in triplicate from a frozen stock of C. jejuni strain CG8421. >10 Colonies from these plates were re-streaked on fresh plates and mid-log phase bacteria were harvested and suspended in Trizol reagent for RNA extraction. For RNA-seq of samples grown in liquid broth: 5 mL aliquots of Mueller-Hinton broth were equilibrated in the microaerobic chamber for 24 hours before being inoculated and were grown with shaking. First, Mueller-Hinton agar plates were steaked in triplicate from a frozen stock of C. jejuni strain CG8421. Colonies from these plates were used to inoculate equilibrated broth and grown with shaking to stationary phase. These cultures were then used to inoculate freshly equilibrated Mueller-Hinton broth aliquots, which were grown with shaking until mid-log phase. Bacteria were then harvested via centrifugation and resuspended in Trizol reagent for immediate RNA extraction. Extracted RNA was DNAse treated (Turbo DNA Free DNAse Kit, Ambion), rRNA was removed (Ribo-Zero Bacterial rRNA Removal Kit, Illumina) and the samples were prepared for Illumina Hiseq using the TruSeq Stranded mRNA Library Kit (Illumina).
Illumina RNA Sequencing and Analysis
For RNA sequencing of samples from infected human feces, three RNA libraries built from the infected feces samples of different volunteers were sequenced using Illumina Hiseq Single-End 50bp reads. Approximately 13 billion reads were obtained in total across all three samples. Using CLC Genomics Workbench software, reads were trimmed to 21bp and aligned to the CG8421 published reference genome using 100 percent exact homology match mapping parameters. At least 3 million reads mapped to the reference genome per sample. For in vitro samples, RNA libraries were sequenced and aligned in an identical manner, with at least 15 million reads mapping to the reference genome per sample. To determine differential expression between samples, Reads Per Kilobase per Million reads (RPKM) values were determined for each gene, normalized by quintile, and then were used in a Baggerley’s test to determine the weighted proportions fold change between groups of biological replicates (all groups were in triplicate) with a false discovery rate corrected (FDR) p-value. Fold changes ≥ |3| with a FDR p-value of ≤ 0.05 were considered significant. Annotation of differentially regulated CG8421 genes was made by considering the annotations of homologous genes in strains 81-176 and 11168 as determined by NCBI’s online blastp suite. Additional functional annotations were made after considering published literature, using NCBI’s PubMed literature search online. Comparison of the CG8421 transcriptome in human feces to the C. jejuni 81-176 transcriptome in the chicken cecum was made by using the published list of expression fold changes per 81-176 gene in the chicken cecum as described in Sup. Table 2. Both datasets use mid-log phase microaerobic growth in Mueller-Hinton broth as the reference transcriptome. We compared the list of differentially regulated CG8421 genes in human feces (>|3| fold, <0.05 FDR p-value) to the list of differentially regulated 81-176 genes (>|3| fold, < 0.05 adjusted p-value) in the chicken cecum. Only genes with a homolog in both strains (as determined by similarity of predicted amino acid sequence) were included in the comparison.
Human Infection and Inoculum Preservation
Samples were obtained from a previously described15 controlled human infection model (ClinicalTrials.gov Identifier NCT02280044, Navy Medical Research Center IRB NMRC.2014.0013, and Western Institutional Review Board study number WIRB 1147996). Volunteers were healthy adults recruited from the mid-Atlantic area, signed informed consent prior to participating in the study, and allowed for all future use of the samples obtained during the study. Volunteers who were admitted to the inpatient facility at Johns Hopkins University Center for Immunization Research for the study had ages ranging from 18-50 years old (median 30), normal stool patterns, were not using antidiarrheal or antacid therapy, and had no history of clinically significant diseases. Subjects with a history of Campylobacter exposure, a personal and/or family history of Guillain-Barre syndrome and/or inflammatory arthritis, were positive for HLA-B27, or whose serum immunoglobulin A titer to CG8421 glycine extract was greater than 1:4000 were excluded from the study. Admitted volunteers were inoculated on Day 0 with a C. jejuni CG8421 liquid inoculum that was generated using Good Manufacturing Processing (GMP) and kept on ice. The inoculum is derived from Mueller-Hinton agar plate growths that are suspended in phosphate buffered saline, and therefore has complex standing genetic variation (detailed in Sup. Table 3) and is not derived from a single colony. This resuspension method was used for the Aotus nancumaae inoculum preparation as well. Immediately prior to inoculation, the PBS resuspension human inoculum (target concentration of 5×105 colony forming units per ml (CFU/mL) was diluted in sodium bicarbonate buffer (NaHCO3) to neutralize stomach acids and ingested. After the last patient was inoculated, a portion of the remaining inoculum was used to verify the infection dosage via growth and colony enumeration on Campylobacter CVA (cefoperazone, vancomycin, and amphotericin B) agar (dosage was 1.7×105 CFU) and another portion was frozen at −80°C to later be used for genomic sequencing.
Human Infection Isolate Collection/Preservation
Campylobacter isolation from infected feces was performed as previously described15. Briefly, infected feces were diluted and temporarily incubated in thioglycollate broth, plated on selective Campylobacter media, and incubated in a microaerobic atmosphere at 37C. Colonies on these primary isolation plates were screened by eye and all putative C. jejuni colonies were collected and pooled together in glycerol media per sample and frozen at −20°C. An effort was made to collect at least 10 colonies per sample, and samples with insufficient amounts of C. jejuni isolates were identified during analysis of the sample genomes (samples with less than 50x coverage of the genome) and removed from the study.
Genomic Sequencing
The frozen inoculum and isolate samples were thawed on ice, genomic DNA was extracted (Easy DNA Kit, Invitrogen) from each sample, followed by processing for Illumina NextSeq Single-Read sequencing. All genome analysis was preformed using CLC Genomics Workbench. Reads were aligned to the CG8421 reference genome for each isolate population. Only isolate population samples with at least 50x coverage of the CG8421 genome were included for genomic variant analysis. These samples averaged >1000x genome coverage (Sup. Fig. 2).
Genomic Variant Calling
Mapped reads were locally realigned and genomic variations compared to the reference genome were identified using a no ploidy assumption low frequency variant detection method. For the highest quality data we manually removed clear false positive variant calls, such as SNPs called in extensive pileups of single reads or in regions of unreliable coverage. By calling variants that occurred in at least 1 percent of any sample population, we identified over 600 genomic variants across all samples. This list of variants, including their frequency in the population, per sample, is provided in Sup. Table 1. To enrich this list for meaningful variants that may impact the human disease state, we aimed to identify genome variants that had major changes in frequency between the inoculum and isolate populations, such as a minority variant in the inoculum becoming a majority variant in an infection isolate population. By defining a variant as a mutation that occurred in at least 25 percent of a sample population, and identifying variants that differed between the inoculum and an isolate population, we achieved this goal (Sup. Fig 1). For example, using this approach, variants that were only called in an isolate population(s) on average had a 12-fold increase in frequency between the inoculum and the infection isolate populations (Sup. Fig. 3). These variants represented a minority of the inoculum population (~5 percent on average), but a majority in the isolate population (~60 percent on average across samples) (Sup. Fig. 3). These variants were used in the analyses throughout the study and are listed in Sup. Table 4.
Genomic Variant Analysis
Our variant calling procedure identified 48 genomic variants with major changes in frequency between the inoculum and least one isolate population. Based on variant location in the published annotated CG8421 genome, these variants likely affect ~30 annotated genes. In many cases, different variants across samples had an equivalent predicted affect on the same gene (such as an insertion or deletion induced frameshift at the same location in the same gene) and this is shown for each variant in Sup Table 1d. To incorporate these shared variant affects into our analysis, we manually refined the variant lists to reflect the genes affected by each variant (Sup Fig 1, Step 5), so that different variants with the same affects were considered as equivalent. This enabled us to determine if variation in a particular gene was associated with the human disease state (Sup Fig 1, Step 6) such as if a variation in a particular gene is associated with relapse isolate populations compared to primary infection isolate populations. To do this, we preformed Fisher’s exact test with a false discovery rate (FDR) corrected p-value in CLC Genomics Workbench to compare variants between sample groups. The Microbial Genomics module of CLC Genomics Workbench was used to generate the SNP-tree seen in Figure 4a using the work done by Kass et al43. Briefly, the tree visually represents how similar genomic variations are between samples. The closer two samples are to each other, the more similar the genomic variations. When examining genomic variants between samples, different variant alleles at the same genomic location are scored as different variants. Other statistical tests used to compare variant frequencies were performed in the statistics and graphing software Prism 6 and are noted in the appropriate figure legends.
Aotus nancymaae Infection, Isolate Collection, and Analysis
The Aotus nancymaae infection protocol was approved by the Naval Medical Research Unit-6 Institutional Animal Care and Use Committee (IACUC approval number NAMRU6-16-03) in compliance with all applicable Federal regulations governing the protection of animals in research. Aotus nancymaae monkeys were infected as described previously44,45 with a single C. jejuni CG8421 liquid inoculum that was harvested from Mueller-Hinton agar plates and resuspended in PBS similarly to harvesting bacteria for the human inoculum. Animals were treated with rantidine and CeraVacx to neutralize stomach acid and were anesthetized with ketamine intramuscularly. Animals were inoculated with 5 × 1011 bacteria in 5 ml of PBS orogastrically for infection and a portion of the inoculum was frozen at −80°C for preservation and genomic sequencing. C. jejuni infected diarrhea was plated on Campylobacter selective media as previously described44,45 and isolates were collected and pooled per sample, processed for sequencing, and data analyzed in a parallel manner as described for the human infection isolate population samples. After genomic sequencing, 12 diarrheal samples from 5 monkeys covering 8 days of infection had high enough C. jejuni isolate counts to be included in this study (as determined by CG8421 genome coverage), as indicated in Sup. Table 12, and were produced by male and female monkeys with a median age of 14 months as noted in Sup. Table 12. The Aotus inoculum was prepared independently of the human inoculum, and similarly, is not derived from a single colony. This accounts for differences in standing genetic variation between the two inoculums and is detailed in Sup. Table 12. Therefore, the Aotus infection isolate population genomes were compared to the Aotus inoculum during genetic analysis of the Aotus samples. The samples used in this study were selected from an Aotus nancymaae infection preformed as a randomized controlled immunization and C. jejuni CG8421 challenge model. Power calculations were used to determine the appropriate number of animals to be used in that preventative treatment investigation model. To be consistent with our human isolate data, we only used samples if: 1) the sample represented symptomatic disease (diarrhea), 2) the sample was produced by an animal that did not receive therapy before or at the time the sample was produced and 3) genomic sequencing yielded sufficient coverage of the C. jejuni CG8421 genome for genetic analysis. Therefore, it was necessary that choosing samples for use in our study be performed in a nonblinded manner.
Cj0617/618 Flagellin Mobility Shift Assay
The following genetic studies were performed on strain 81-176 because strain GC8421 is not amenable to genetic analyses. The homopolymeric tract in Cj0617/618 was repaired as previously described46. Briefly, the genes were PCR amplified and cloned behind a sigma28 promoter. The G tract was repaired such that the two genes were fused into a single open reading frame, and the codons were modified to introduce silent changes that removed the homopolymeric tract by Quick Change mutagenesis using primers pg.16.18 (5-CTCCATTTAAACTAATGAGAGGCGGCGGTATTAGAACGATTTTGTTTGG-3′) and pg.16.19 (5′-CCAAACAAAATCGTTCTAATACCGCCGCCTCTCATTAGTTTAAATGGAG-3′). The repaired gene and a kanamycin resistance cassette were introduced into the arylsulfatase gene of 81-176. Purified flagellins were separated using ampholytes ranging from 4-6 (Biolyte 4/6; BioRad) as previously described47. IEF protein markers were purchased from Serva.
Data Availability
All genomic and RNA sequencing data used in this study have been deposited in the NCBR sequence read archive under Bioproject PRJNA392448.
Supplementary Material
Acknowledgments
We thank Erica J. Rubin for support in developing nucleic acid extraction protocols; Howard Ochman and Mathew A. Leibold for feedback on this project; Simone M. Giovanetti for insightful discussion on figures. This work was supported by the National Institutes of Health (NIH) (AI064184 to M.S.T.), the Military Infectious Diseases Research Program (6000.RAD1.DA3.A0308), and the Navy Advanced Medical Development Program (NMRC enterprise Work Unit NumberA1406).
This work was funded by the Military Infectious Diseases Research Program, Navy Work Unit 6000.RAD1.DA3.A0308 the Naval Medical Research Center’s Advanced Medical Development Program (NMRC enterprise Work Unit Number A1406).
Footnotes
Financial interest: The authors declare no competing financial interests with this work.
Contributions:
A.A.C. conceived, designed, preformed, and interpreted genetic and transcriptomic analyses and wrote the paper. P.G. and M.S.T. established this collaborative project and oversaw this study. B.W.D. and M.S.T. oversaw computational analyses. A.A.C., M.S.T. and P.G. provided critical biological interpretations of the data. J. E. R, C. H., D. S., K. R. T., C. K. P., R. L. G., B. D., J. B., R.M.L., A.C.M., K. J., A. A., D. R. T. and M. S. R. conducted the clinical trial. F. M. P. and J.M.K. collected specimens for transcriptomics. C. P. E. performed experiments relevant to biological interpretation of the data. A.R. and A.J.M. performed the non-human primate experiments. B.W.D., P.G., and M.S.T supervised this work and edited the manuscript.
Disclaimer
The study protocol was approved by the Naval Medical Research Center and the Western Institutional Review Boards in compliance with all applicable local and Federal regulations governing the protection of human subjects.
The animal protocol was reviewed and approved by the Naval Medical Research Unit-6 Institutional Animal Care and Use Committee in compliance with all applicable Federal regulations governing the protection of animals in research.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. government.
PG, DRT, FMP, CKP, RLG, MSR and AJM are either employees of the U.S. government or military service members and this work was prepared as part of their official duties. Title 17 USC 105 provides that ‘Copyright protection under this title is not available for any work of the United States government.’ Title 17 USC 101 defines a U. S. government work as a work prepared by a military service member or employee of the U. S. government as part of that person’s official duties.
References
- 1.Lieberman TD, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, et al. Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci. 2011;108:7481–7486. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris SR, et al. Evolution of MRSA During Hospital Transmission and Intercontinental Spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennemann L, et al. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci U S A. 2011;108:5033–5038. doi: 10.1073/pnas.1018444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zdziarski J, et al. Host Imprints on Bacterial Genomes—Rapid, Divergent Evolution in Individual Patients. PLOS Pathog. 2010;6:e1001078. doi: 10.1371/journal.ppat.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copin R, et al. Within Host Evolution Selects for a Dominant Genotype of Mycobacterium tuberculosis while T Cells Increase Pathogen Genetic Diversity. PLOS Pathog. 2016;12:e1006111. doi: 10.1371/journal.ppat.1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayliss CD, et al. Phase variable genes of Campylobacter jejuni exhibit high mutation rates and specific mutational patterns but mutability is not the major determinant of population structure during host colonization. Nucleic Acids Res. 2012;40:5876–5889. doi: 10.1093/nar/gks246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergman M, Del Prete G, van Kooyk Y, Appelmelk B. Helicobacter pylori phase variation, immune modulation and gastric autoimmunity. Nat Rev Microbiol. 2006;4:151–159. doi: 10.1038/nrmicro1344. [DOI] [PubMed] [Google Scholar]
- 9.Srikhanta YN, Fox KL, Jennings MP. The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat Rev Microbiol. 2010;8:196–206. doi: 10.1038/nrmicro2283. [DOI] [PubMed] [Google Scholar]
- 10.Kim JS, et al. Passage of Campylobacter jejuni through the chicken reservoir or mice promotes phase variation in contingency genes Cj0045 and Cj0170 that strongly associates with colonization and disease in a mouse model. Microbiology. 2012;158:1304–1316. doi: 10.1099/mic.0.057158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jerome JP, et al. Standing Genetic Variation in Contingency Loci Drives the Rapid Adaptation of Campylobacter jejuni to a Novel Host. PLOS ONE. 2011;6:e16399. doi: 10.1371/journal.pone.0016399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohawk KL, Poly F, Sahl JW, Rasko DA, Guerry P. High Frequency, Spontaneous motA Mutations in Campylobacter jejuni Strain 81-176. PLOS ONE. 2014;9:e88043. doi: 10.1371/journal.pone.0088043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amour C, et al. Epidemiology and Impact of Campylobacter Infection in Children in 8 Low-Resource Settings: Results From the MAL-ED Study. Clin Infect Dis. 2016;63:1171–1179. doi: 10.1093/cid/ciw542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cawthraw SA, Wassenaar TM, Ayling R, Newell DG. Increased colonization potential of Campylobacter jejuni strain 81116 after passage through chickens and its implication on the rate of transmission within flocks. Epidemiol Infect. 1996;117:213–215. doi: 10.1017/s0950268800001333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rimmer JE, et al. Rifaximin Fails to Prevent Campylobacteriosis in the Human Challenge Model: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin Infect Dis. doi: 10.1093/cid/cix1014. [DOI] [PubMed] [Google Scholar]
- 16.Baqar S, et al. Recrudescent Campylobacter jejuni infection in an immunocompetent adult following experimental infection with a well-characterized organism. Clin Vaccine Immunol CVI. 2010;17:80–86. doi: 10.1128/CVI.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palyada K, Threadgill D, Stintzi A. Iron Acquisition and Regulation in Campylobacter jejuni. J Bacteriol. 2004;186:4714–4729. doi: 10.1128/JB.186.14.4714-4729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brás AM, Chatterjee S, Wren BW, Newell DG, Ketley JM. A Novel Campylobacter jejuniTwo-Component Regulatory System Important for Temperature-Dependent Growth and Colonization. J Bacteriol. 1999;181:3298–3302. doi: 10.1128/jb.181.10.3298-3302.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Rocio Leon-Kempis M, Guccione E, Mulholland F, Williamson MP, Kelly DJ. The Campylobacter jejuni PEB1a adhesin is an aspartate/glutamate-binding protein of an ABC transporter essential for microaerobic growth on dicarboxylic amino acids. Mol Microbiol. 2006;60:1262–1275. doi: 10.1111/j.1365-2958.2006.05168.x. [DOI] [PubMed] [Google Scholar]
- 20.Ziprin RL, et al. Role of Campylobacter jejuni potential virulence genes in cecal colonization. Avian Dis. 2001;45:549–557. [PubMed] [Google Scholar]
- 21.Jones MA, et al. Adaptation of Campylobacter jejuni NCTC11168 to High-Level Colonization of the Avian Gastrointestinal Tract. Infect Immun. 2004;72:3769–3776. doi: 10.1128/IAI.72.7.3769-3776.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fields JA, Thompson SA. Campylobacter jejuni CsrA Mediates Oxidative Stress Responses, Biofilm Formation, and Host Cell Invasion. J Bacteriol. 2008;190:3411–3416. doi: 10.1128/JB.01928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taveirne ME, Theriot CM, Livny J, DiRita VJ. The Complete Campylobacter jejuni Transcriptome during Colonization of a Natural Host Determined by RNAseq. PLOS ONE. 2013;8:e73586. doi: 10.1371/journal.pone.0073586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corcionivoschi N, et al. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe. 2012;12:47–59. doi: 10.1016/j.chom.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pryjma M, Apel D, Huynh S, Parker CT, Gaynor EC. FdhTU-Modulated Formate Dehydrogenase Expression and Electron Donor Availability Enhance Recovery of Campylobacter jejuni following Host Cell Infection. J Bacteriol. 2012;194:3803–3813. doi: 10.1128/JB.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson JG, Gaddy JA, DiRita VJ. The PAS Domain-Containing Protein HeuR Regulates Heme Uptake in Campylobacter jejuni. mBio. 2016;7:e01691–16. doi: 10.1128/mBio.01691-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naikare H, et al. Campylobacter jejuni ferric-enterobactin receptor CfrA is TonB3 dependent and mediates iron acquisition from structurally different catechol siderophores. Met Integr Biometal Sci. 2013;5:988–996. doi: 10.1039/c3mt20254b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoang KV, Wang Y, Lin J. Identification of genetic loci that contribute to Campylobacter resistance to fowlicidin-1, a chicken host defense peptide. Front Cell Infect Microbiol. 2012;2 doi: 10.3389/fcimb.2012.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cameron A, et al. High-Frequency Variation of Purine Biosynthesis Genes Is a Mechanism of Success in Campylobacter jejuni. mBio. 2015;6:e00612–15. doi: 10.1128/mBio.00612-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrero-Tobon AM, Hendrixson DR. Identification and analysis of flagellar coexpressed determinants (Feds) of Campylobacter jejuni involved in colonization. Mol Microbiol. 2012;84:352–369. doi: 10.1111/j.1365-2958.2012.08027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luethy PM, Huynh S, Parker CT, Hendrixson DR. Analysis of the Activity and Regulon of the Two-Component Regulatory System Composed by Cjj81176_1484 and Cjj81176_1483 of Campylobacter jejuni. J Bacteriol. 2015;197:1592–1605. doi: 10.1128/JB.02564-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Javed MA, et al. Transposon mutagenesis in a hyper-invasive clinical isolate of Campylobacter jejuni reveals a number of genes with potential roles in invasion. Microbiology. 2010;156:1134–1143. doi: 10.1099/mic.0.033399-0. [DOI] [PubMed] [Google Scholar]
- 33.Poli VFS, Thorsen L, Olesen I, Wik MT, Jespersen L. Differentiation of the virulence potential of Campylobacter jejuni strains by use of gene transcription analysis and a Caco-2 assay. Int J Food Microbiol. 2012;155:60–68. doi: 10.1016/j.ijfoodmicro.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 34.Guo B, et al. CmeR Functions as a Pleiotropic Regulator and Is Required for Optimal Colonization of Campylobacter jejuni In Vivo. J Bacteriol. 2008;190:1879–1890. doi: 10.1128/JB.01796-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei HT, et al. Crystal structures of CmeR-bile acid complexes from Campylobacter jejuni. Protein Sci Publ Protein Soc. 2011;20:712–723. doi: 10.1002/pro.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin J, Overbye Michel L, Zhang Q. CmeABC Functions as a Multidrug Efflux System in Campylobacter jejuni. Antimicrob Agents Chemother. 2002;46:2124–2131. doi: 10.1128/AAC.46.7.2124-2131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grinnage-Pulley T, Zhang Q. Genetic Basis and Functional Consequences of Differential Expression of the CmeABC Efflux Pump in Campylobacter jejuni Isolates. PLOS ONE. 2015;10:e0131534. doi: 10.1371/journal.pone.0131534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin J, Sahin O, Michel LO, Zhang Q. Critical Role of Multidrug Efflux Pump CmeABC in Bile Resistance and In Vivo Colonization of Campylobacter jejuni. Infect Immun. 2003;71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerry P, et al. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol. 2006;60:299–311. doi: 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howard SL, et al. Campylobacter jejuni glycosylation island important in cell charge, legionaminic acid biosynthesis, and colonization of chickens. Infect Immun. 2009;77:2544–2556. doi: 10.1128/IAI.01425-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray JL, Kwon T, Marcotte EM, Whiteley M. Intrinsic Antimicrobial Resistance Determinants in the Superbug Pseudomonas aeruginosa. mBio. 2015;6:e01603–1615. doi: 10.1128/mBio.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A. 2015;112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLOS ONE. 2014;9:e104984. doi: 10.1371/journal.pone.0104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monteiro MA, et al. Capsule Polysaccharide Conjugate Vaccine against Diarrheal Disease Caused by Campylobacter jejuni. Infect Immun. 2009;77:1128–1136. doi: 10.1128/IAI.01056-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones FR, et al. New World monkey Aotus nancymae as a model for Campylobacter jejuni infection and immunity. Infect Immun. 2006;74:790–793. doi: 10.1128/IAI.74.1.790-793.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pequegnat B, et al. Phase variable changes in the position of O-methyl phosphoramidate modifications on the polysaccharide capsule of Campylobacter jejuni modulate serum resistance. J Bacteriol. 2017 doi: 10.1128/JB.00027-17. JB.00027-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thibault P, et al. Identification of the Carbohydrate Moieties and Glycosylation Motifs in Campylobacter jejuni Flagellin. J Biol Chem. 2001;276:34862–34870. doi: 10.1074/jbc.M104529200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic and RNA sequencing data used in this study have been deposited in the NCBR sequence read archive under Bioproject PRJNA392448.