

Abstract
Multiple sclerosis (MS) involves damage to white matter microstructures. This damage has been related to grey matter function as measured by standard, physiologically‐nonspecific neuroimaging indices (i.e., blood‐oxygen‐level dependent signal [BOLD]). Here, we used calibrated functional magnetic resonance imaging and diffusion tensor imaging to examine the extent to which specific, evoked grey matter physiological processes were associated with white matter diffusion in MS. Evoked changes in BOLD, cerebral blood flow (CBF), and oxygen metabolism (CMRO2) were measured in visual cortex. Individual differences in the diffusion tensor measure, radial diffusivity, within occipital tracts were strongly associated with MS patients’ BOLD and CMRO2. However, these relationships were in opposite directions, complicating the interpretation of the relationship between BOLD and white matter microstructural damage in MS. CMRO2 was strongly associated with individual differences in patients’ fatigue and neurological disability, suggesting that alterations to evoked oxygen metabolic processes may be taken as a marker for primary symptoms of MS. This work demonstrates the first application of calibrated and diffusion imaging together and details the first application of calibrated functional MRI in a neurological population. Results lend support for neuroenergetic hypotheses of MS pathophysiology and provide an initial demonstration of the utility of evoked oxygen metabolism signals for neurology research. Hum Brain Mapp 38:5375–5390, 2017. © 2017 Wiley Periodicals, Inc.
Keywords: calibrated imaging, blood flow and metabolism, white matter, diffusion tensor imaging, multiple sclerosis
INTRODUCTION
Multiple sclerosis (MS) is a complex neurological disease characterized by micro‐ and macrostructural central nervous system damage. MS pathology is deleterious to neural tissue, wherein the most extensive damage is largely incurred by white matter microstructures [WMMS; i.e., astrocytes, myelin, oligodendrocytes; cf. Frohman et al., 2006; Lassman, 2014; Trapp and Nave, 2008]. Diffusion tensor imaging (DTI) studies reveal MS‐related increases in regional and whole‐brain isotropic diffusion of molecular water, indexing potential WMMS damage [see Ge et al., 2005]. A central question in MS research concerns how such structural alterations are related to brain function. Research suggests that such alterations to WMMS could potentially influence grey matter function [e.g., Alshowaeir et al., 2014; Au Duong et al., 2005; Hubbard et al., 2016a; see Paling et al., 2011; Trapp and Nave, 2008].
Grey matter function is commonly studied in MS using blood‐oxygen‐level dependent (BOLD) signal [e.g., Au Duong et al., 2005; Cader et al., 2006; Genova et al., 2009; Hubbard et al., 2016a,b; Janssen et al., 2013; Passamonti et al., 2009; White et al., 2009]. MS patients show relationships between their BOLD functional responses and white matter diffusion characteristics, demonstrating a link between grey matter function and WMMS in this group [Hubbard et al., 2016, 2014a; see also Alshowaeir et al., 2014; Au Duong et al., 2005]. Although the two are related, it is important to emphasize a distinction between white matter macrostructural (i.e., lesions) and microstructural (i.e., WMMS) alterations in MS because (1) MS‐related alterations to WMMS exist outside of apparent lesioned tissue [e.g., De Keyser et al., 1999; Klistorner et al., 2016; Sorbara et al., 2014] and (2) measures of WMMS account for variance in measures of MS patients' neural function separate from the degree of patients' macrostructural damage [e.g., Hannoun et al., 2012; Hubbard et al., 2016]. In one study, for instance, we found that grey matter BOLD response amplitudes were strongly related to individual differences in MS patients' white matter diffusion characteristics, however, no such relationship was found between grey matter BOLD and the extent of white matter macrostructural damage [i.e., lesion burden; Hubbard et al., 2016a].
BOLD signal changes are based upon multiple physiological processes. A precise understanding of what grey matter physiological processes are related to WMMS in MS cannot be deduced from BOLD signal alone. However, two hypotheses concerning pathophysiological alterations in MS could potentially explain BOLD‐WMMS relationships in this group. One hypothesis is that alterations to WMMS influence MS patients' hemodynamic responses—which are intimately linked to BOLD signal [see Kim and Ogawa, 2012]. MS‐related damage to WMMSs that couple neural and vascular responses could result in decreased cerebral blood flow (CBF) and decreased functional hyperemia [see Brosnan and Raine, 2013; De Keyser et al., 2008; D'haeseeler et al., 2011]. Although many studies have examined the relationship between cerebral blood flow and lesion burden, few studies have actually examined relationships between WMMS and CBF [Saindane et al., 2007]. Moreover, no study has examined the link between WMMS and evoked CBF in grey matter. Thus, the relationship between hemodynamics and WMMS in MS has not been thoroughly evaluated.
A second, but not necessarily mutually exclusive, hypothesis that might explain BOLD‐WMMS relationships involves WMMS damage and neurometabolism [see Campbell et al., 2014; Mahad et al., 2015; Paling et al., 2011]. Neurometabolism is also intimately linked to changes in BOLD signal [see Kim and Ogawa, 2012]. Neuroimaging research has produced extensive evidence of altered neurometabolism across MS subtypes [e.g., Cader et al., 2007; Ge et al., 2012; Hannoun et al., 2012; Kahn et al., 2017; Kindred et al., 2015; Sijens et al., 2005; Sun et al., 1998; see Stromillo et al., 2013 for preclinical MS]. Postmortem studies in advanced MS have also traced neurometabolic deficits to the level of the mitochondria and electron transport chain complexes in lesioned and nonlesioned neural tissue [Dutta et al., 2006; see also Singhal et al., 2015]. Relationships between MS patients' metabolism markers in white matter and measures of WMMS have also been demonstrated [e.g., Hannoun et al., 2012; Sijens et al., 2005]. For example, one magnetic resonance spectroscopy study of centrum semiovale white matter in MS patients showed that N‐acetylaspertate (NAA) and NAA:creatine ratios were strongly related to white matter diffusion in this region [Hannoun et al., 2012]. However, such studies have been limited to examination of metabolism in white matter, precluding a direct understanding of the relationship between WMMS and grey matter metabolism in MS.
Several studies have assessed MS alterations in white matter diffusion characteristics, metabolism, and blood‐flow signals. To date however, no research has evaluated combined relationships between WMMS and physiological processes in grey matter. Advances in functional neuroimaging now permit temporally‐adjacent acquisition of BOLD and blood flow signals which can be used along with biophysical modeling to non‐invasively acquire oxygen metabolic signals via BOLD‐signal decomposition [calibrated functional magnetic resonance imaging [cfMRI]; Davis et al., 1998; Hoge et al., 1999]. To be clear, many neurophysiological processes are linked to BOLD signal changes. For instance, changes to the electrochemical properties of populations of neurons (i.e., neural activity) are associated with BOLD signal changes [e.g., Logothetis et al., 2001]. However, with cfMRI, we can isolate two distinct, neurophysiological processes (i.e., CBF and oxygen metabolism) that largely form the basis for BOLD signal changes [see Kim and Ogawa, 2012]. In combination with DTI, these techniques could provide new insights about the grey matter physiological processes associated with WMMS damage and yield data for further hypotheses about MS pathophysiology.
Here, we directly investigated relationships between grey matter physiological processes and white matter diffusion in MS using cfMRI and DTI. With cfMRI we examined evoked BOLD, CBF, and oxygen metabolism within occipital cortex in MS patients during visual stimulation. The visual system is one of the most affected neural systems in MS and tends to show alterations in both structure and function even at early stages of the disease course [see Frohman et al., 2005, 2008; Grahm and Klistorner, 2017; Kolappan et al., 2009]. With DTI, we were able to examine how cfMRI‐based evoked physiological processes and BOLD signal were related to occipital‐tract radial diffusivity (RD)—an index of potential WMMS damage [e.g., Harsan et al., 2007; Song et al., 2002] of particular relevance to MS [e.g., Alshowaeir et al., 2014; Klistorner et al., 2016]. Further, we elucidated which evoked signals were associated with individual variability in primary MS symptomology (i.e., fatigue and neurological disability).
METHOD
Participants
Twelve MS patients were recruited from the University of Texas Southwestern Medical Center. These patients were required to be between the ages of 18 and 65, free of MR‐contraindicators and concurrent substance abuse, have normal or corrected‐to‐normal vision, and speak fluent English. Because study procedures included a gas‐inhalation challenge, patient selection was limited to nonsmokers. Patients did not have histories of respiratory or pulmonary problems, cerebral vascular issues, or cardiac disease. Patients were required to have a score greater than 21 on the Telephone Interview for Cognitive Status (TICS) [Brandt et al., 1988]. Patients were also required to be at least 1 month past their most recent exacerbation and their last corticosteroid treatment. Patients did not report a history of optic neuritis and their vision was normal or corrected‐to‐normal. Two patients declined to complete the gas challenge (total n = 10). Patients who completed the scanning protocol had an average of 9.90 years since their initial MS diagnosis (SEM = 1.61 years) and an average estimated 28.60 months since their last exacerbation (SEM = 11.32 months; see Table 1). Because we have shown relationships between MS patients DTI‐derived WMMS and visuocortical BOLD activity and we have not found these relationships for control participants [Hubbard et al., 2016a], we did not have evidence to warrant similar examinations in control participants. Thus, MS patients were the sole focus of these analyses.
Table 1.
Patient characteristics
| Case | Age/sex | Course | Disease duration | Last flare‐up | Neurological disability | MFIS total | TICS score | Lesion burden |
|---|---|---|---|---|---|---|---|---|
| 1 | 59/F | RR | 15 | 054 | 05 | 23 | 26 | 46,243.90 |
| 2 | 55/F | RR | 14 | 012 | 32 | 73 | 24 | 2,707.11 |
| 3 | 46/F | RR | 11 | 002 | 21 | 78 | 26 | 20,758.90 |
| 4 | 55/F | RR | 20 | 001 | 36 | 44 | 28 | 14,816.00 |
| 5 | 49/F | RR | 06 | 067 | 12 | 39 | 26 | 2,789.85 |
| 6 | 43/F | SP | 04 | 010 | 07 | 17 | 25 | 677.03 |
| 7 | 48/F | RR | 07 | 007 | 04 | 07 | 32 | 475.23 |
| 8 | 28/M | RR | 05 | 016 | 06 | 24 | 26 | 13,272.20 |
| 9 | 63/F | RR | 09 | 108 | 10 | 44 | 31 | 7,469.53 |
| 10 | 55/F | RR | 08 | 009 | 24 | 42 | 26 | 1,751.60 |
Disease duration in years. Last flare‐up (i.e., exacerbation) in months. Neurological disability based upon total self‐reported neurological disability [Verdier‐Taillefer et al., 1994].
Study Procedures
Study procedures were approved by the University of Texas Southwestern Medical Center Institutional Review Board. Patients meeting inclusion criteria were asked to refrain from caffeine use at least two hours before their scheduled appointment time [e.g., Perthen et al., 2008]. They were also asked not to consume alcohol on the same calendar day before their scheduled appointment. Patients gave informed consent before undergoing procedures and were compensated for their time. Patients underwent functional and structural neuroimaging on a Philips 3 T magnet (Philips Medical Systems, Best, The Netherlands) with an eight‐channel SENSE radiofrequency head coil. Foam padding was placed around the head to minimize motion during MRI scan acquisition. Patients completed standard neuropsychological and clinical measures, as well as measures of symptom severity (Modified Fatigue Impact Scale [MFIS; Fisk et al., 1994] and a neurological disability scale [Verdier‐Taillefer et al., 1994]). Fatigue and neurological disability were assessed as estimators of symptom severity because both fatigue and neurological disability are common features of MS and contribute significantly to MS patients' quality of life [Amato et al., 2001].
cfMRI Parameters and Theory
Dual‐echo pseudocontinuous arterial spin labeling (pCASL) and BOLD images (together referred to as dual‐echo images) were acquired near‐simultaneously using an interleaved echo scanning protocol [see Hoge et al., 1999; Lu and van Zijl, 2005]. Together, the perfusion (Echo 1) and BOLD‐weighted (Echo 2) images along with biophysical modeling procedures allowed for estimation of the cerebral metabolic rate of oxygen (CMRO2) associated with steady‐state stimulation [Davis et al., 1998; Hoge et al., 1999]. One task run of dual‐echo imaging data and one gas‐challenge run of dual‐echo imaging data were collected with the following parameters: Echo 1: labeling duration 1,650 ms, labeling RF flip angle 18°, labeling gap = 63.5 mm, 3.44 × 3.44 × 5 mm voxel, TR = 4,000 ms, TE = 14 ms, 1,525 ms postlabel delay, 0 mm slice gap. Echo 2: 90° flip angle, 3.44 × 3.44 × 5 mm voxel, TR = 4,000 ms, TE = 40 ms, 0 mm slice gap. Total scan time for the visual stimulation task = 600 s (72 dual‐echo dynamics). Total scan time for the gas challenge = 624 s (75 dual‐echo dynamics).
Estimation of CMRO2 was based upon the Davis model of BOLD signal change [Davis et al., 1998; Hoge et al., 1999]:
| (1) |
where Δx/x 0 denotes a change from baseline (where x = S, CBF, or CMRO2), α is an empirically‐derived constant linking cerebral blood flow and cerebral blood volume, and β is an empirically‐derived constant related to vascular exchange and susceptibility of deoxyhemoglobin at specific field strengths [e.g., Ances et al., 2011; Leontiev and Buxton, 2007]. M is a subject‐specific scaling factor dependent upon the washout of resting deoxyhemoglobin [see Buxton, 2010]. M was estimated in each participant, using the gas‐challenge procedure detailed below.
The measurement of BOLD, CBF, and M allows for the estimation of CMRO2. Here, ΔCMRO2 reflects the task‐related change in neurometabolism of oxygen from resting baseline:
| (2) |
where Δx/x 0 reflects percent change of signal during task compared to resting baseline.
cfMRI Task and Gas Challenge
Participants completed a visual stimulation task during dual‐echo task imaging. This task was chosen for two reasons. First, differences in the functional response to visual stimulation have been observed in MS visual cortex and these results have generalized to other regions [e.g., motor and prefrontal cortices; see Hubbard et al., 2016, 2014a,b]. Second, because this task involved minimal cognitive demand, individual differences in performance were not expected to be a factor.
Patients were trained on the task before entering the MR environment. During the task, patients focused on a fixation cross in the center of their visual field. Patients were required to respond via bilateral, thumb‐button press when a change in the luminance of the fixation cross occurred. This secondary‐task was used to control the center of the participants' visual field [Hutchison et al., 2013a,2013b; Pasley et al., 2007]. Luminance changes were jittered and occurred every 2, 3, 4, or 6 s. Visual stimulation occurred in a block format. There were six visual stimulation “on” blocks consisting of 60 s of continual annulus flickering in the participants' near‐foveal visual field. Annuli alternated at orthogonal orientations (0°–90°) to avoid neural adaptation [Pasley et al., 2007]. Alterations occurred at a constant frequency of 8 Hz because both electrochemical neural activity and BOLD signal peak at this frequency, yielding the greatest signal‐to‐noise estimates [e.g., Lin et al., 2008; Singh et al., 2003]. Visual stimulation “off” blocks were jittered at 32, 34, 36, 38, and 40 s intervals (see Fig. 1).
Figure 1.
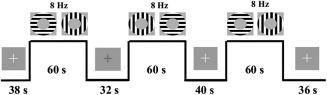
Example of three‐trial visual stimulation task. Participants viewed a fixation cross at the center of the screen. This cross changed color at jittered intervals throughout task. Rest periods were also jittered. Continuous stimulation blocks lasted 60 s with 0° to 90° flickering annuli (at 8 Hz).
Patients also completed a gas‐challenge in order to estimate M. In this challenge, participants breathed room air for 4 min (≈0.03% CO2: 21% O2: 78% N2) and iso‐oxic, CO2 solution for 6 min (5% CO2: 21% O2: 74% N2) during dual‐echo imaging. During this portion of the study, each participant was fitted with a two‐way, nonrebreathing valve/mouthpiece and a nose clip. Baseline end‐tidal CO2 (EtCO2), O2 saturation, breath rate, and heart rate measures were collected. After 4 min of room air breathing, a valve was opened to release the CO2 solution from a Douglas airbag which then flowed into the participants' breathing apparatus for 6 min [Hutchison et al., 2013a,2013b]. Hypercapnic challenge, via an inhaled 5% CO2 solution, increases global CBF, but exerts no, or minimal, depressant effects on oxygen metabolism [e.g., Peng et al., 2017; Xu et al., 2011; Zappe et al., 2008]. Hypercapnia‐induced increase in CBF, but null or decreasing resting response in oxygen metabolism washes out local baseline concentrations of deoxyhemoglobin, yielding a local maximum estimate of resting BOLD signal. Potential changes to oxygen metabolism due to hypercapnic challenge do not appreciably alter the estimation of M; relationships between hypercapnia‐derived M and M derived from non‐hypercapnic techniques show high correspondence [Yücel et al., 2014].
cfMRI Processing
Task and gas‐challenge Echo 1 and Echo 2 data were processed in Analysis of Functional Neuroimages (AFNI) [Cox, 1996] and FMRIB Software Library (FSL; FMRIB Analysis Group, http://www.fsl.fmrib.ox.ac.uk/fsl). Data were transformed into cardinal planes. Anomalous data points in each voxel time series were then attenuated using an interpolation method based upon the average signal. Data were volume registered to correct for motion to the fourth functional volume of each dataset's (task or gas challenge) Echo 2 sequence using a heptic polynomial interpolation method. CBF was estimated from Echo 1 images using the surround subtraction method [as described in Liu and Wong, 2005]. Dual‐echo BOLD data were also interpolated by pairwise averaging of temporally adjacent images.
For the visual stimulation task, Echo 2 data were linearly registered (12 degrees‐of‐freedom) to each participant's anatomical data using the align_epi_anat.py program. The transformation matrix from this registration was then applied to Echo 1 data. For gas‐challenge data, a binary mask was created for functional voxels in Echo 2 to aid in co‐registration. This mask was then registered to the respective participant's anatomical space using the align_epi_anat.py program. Gas‐challenge Echo 2 and Echo 1 data were also aligned to the mask which was registered in native anatomical space. After alignment, Echoes 1 and 2 data from both the visual task and gas challenge were visually inspected for registration errors. Echoes 1 and 2 data from the visual task and gas challenge were then spatially smoothed using a Gaussian kernel (FWHM = 8 mm) and high‐pass filtered (1/256 Hz). The 1/256 Hz cutoff produced BOLD model fit estimates and BOLD and CMRO2 percent signal changes estimates that were significantly greater than the more typically applied 1/100 Hz cutoff, indicating increased signal‐to‐noise using this filter criterion (see Supporting Information Fig. 1). No change was observed between these cutoffs on CBF.
Preprocessed data from echoes 1 and 2 in the visual stimulation task were analyzed via generalized linear modeling of task versus rest periods using a boxcar reference function. This modeling quantified task‐related CBF and BOLD changes from baseline. BOLD and CBF β‐values were scaled to each voxel's resting baseline signal and were multiplied by 100, yielding percent signal change estimates from baseline (ΔBOLD and ΔCBF). Data were averaged from a visual (functional) region of interest (ROI) comprised of overlapping ΔBOLD and ΔCBF suprathreshold signals within occipital lobe [see Structural and Functional ROI; Hutchison et al., 2013a,2013b].
For the gas challenge, resting baseline BOLD and CBF signals during room air breathing were averaged for each voxel time‐series (BOLD0 and CBF0). The first 2 min of hypercapnia BOLD and CBF time‐series were discarded to allow participants' blood flow to stabilize to the CO2 solution [e.g., Hutchison et al., 2013a,2013b]. The last 4 min of hypercapnia BOLD and CBF time‐series were averaged to yield BOLDhc and CBFhc, respectively. Average values were extracted from a functional region of interest (see Structural and Functional ROI) using overlapping BOLDhc and CBFhc suprathreshold signals within occipital lobe, and were used to calculate M, using the following equation:
| (3) |
where (x hc – x 0)/x 0 reflects percent change in signal from normocapnic to hypercapnic states, normalized by the signals during normocapnia and multiplied by 100. We assumed α = 0.38 [Grubb et al., 1974] and β = 1.3 [Lu and van Zijl, 2005]; these values were chosen because they have been shown to yield plausible estimates of visual cortex CMRO2 and n [Hutchison et al., 2013a]. There is some debate about the optimal specification of these parameters [e.g., Griffeth and Buxton, 2011]. However, the outcomes of associations between diffusion characteristics and CMRO2, as well as CMRO2 associations with MS symptom severity measures, were similar between our chosen α, β parameters and others used in extant literature [e.g., Chen and Pike, 2009; Davis et al., 1998; Griffeth and Buxton, 2011; see Supporting Information Fig. 2]. Once M was estimated, ΔCMRO2 was also estimated [see Eq. (2); see Fig. 2] within a functional region of interest (see Structural and Functional ROI; Fig. 3).
Figure 2.
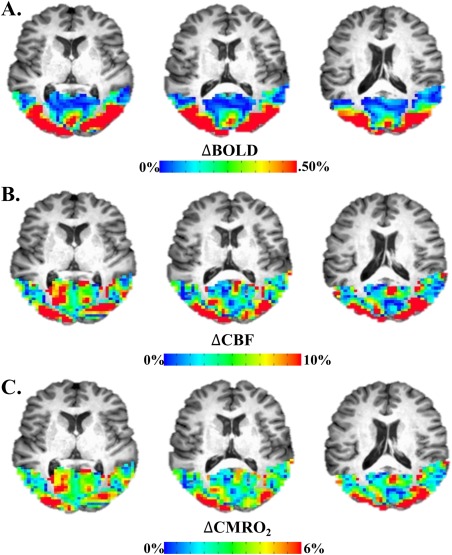
Example of visually‐evoked cfMRI signal in occipital cortex. (A) Evoked BOLD response to visual stimulation. (B) Evoked CBF response to visual stimulation. (C) Evoked CMRO2 response to visual stimulation derived from BOLD and CBF via the Davis model. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
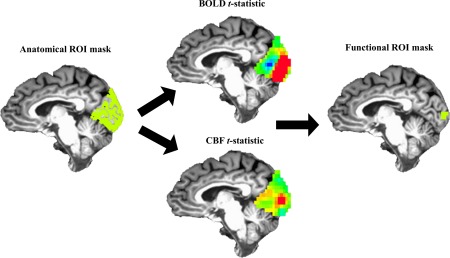
Graphical overview of masking procedure. Top 5%, overlapping BOLD and CBF signals within the anatomical ROI (green) were used to create the functional ROI mask (green). Average BOLD, CBF, CMRO2 estimates were taken from the functional ROI mask for each participant. [Color figure can be viewed at http://wileyonlinelibrary.com]
Structural and Functional ROIs
One T1‐weighted magnetization‐prepared rapid acquisition gradient‐echo (MPRAGE) image was acquired for each participant: 160 slices, TE = 3.7 ms, repetition time TR = 8.1 ms (fast field gradient‐echo), sagittal slice orientation, 1 × 1 × 1 mm3 voxel, 12° flip angle. The MPRAGE data were processed to create a native‐space, occipital ROI. The skull was removed using AFNI's 3dSkullStrip command, separating parenchyma and cerebral spinal fluid from the skull. An intensity‐based automated segmentation algorithm (FSL FAST) was used to delineate primarily white matter, grey matter, and cerebral spinal fluid voxels yielding a partial volume estimate of each tissue type, for each voxel. A grey matter mask was then created, retaining voxels with only a greater‐than‐or‐equal‐to grey‐matter partial volume estimate of 80%.
A structural ROI of occipital lobe was manually delineated on each participant's MPRAGE image. Manual delineation is generally the standard by which automatic labeling programs are compared [e.g., Desikan et al., 2006]. Further, previous work in our laboratories has shown that in order to get the most robust estimates of BOLD signal from older or patient populations, automated spatial normalization/parcellation programs (i.e., Freesurfer) require manual landmark demarcation and considerable quality assurance. In total, these processes can take up to four working days per subject with atypical neuroanatomy [Hutchison et al., 2014], whereas our manual delineation of occipital cortex approach took on average less than one hour per subject. The structural ROI was drawn using gyral and sulcal landmarks and encompassed most of occipital cortex including calcarine sulcus, cuneus, and occipital portions of lingual gyrus. Several anatomical landmarks were used in the demarcation of this ROI (parieto‐occipital sulcus, occipital pole, pre‐occipital notch). Within the anatomically defined occipital lobe, only voxels with partial volume estimates of grey matter (≥80%) were retained. Final masks were down‐sampled to the functional voxel size.
A visual‐task functional ROI was created within the structural ROI described above to estimate ΔBOLD, ΔCBF, and ΔCMRO2 for this task [see Hutchison et al., 2013a,2013b; see Fig. 1]. This procedure was done because, by eschewing noise from inactive voxels, functional ROIs have greater signal estimates compared to structural ROIs [Hutchison et al., 2014]. Voxels comprising each participants' functional ROI were the overlapping top 5% of BOLD and top 5% of CBF t‐values obtained from the generalized model, within the structural ROI. This was done to ensure that ΔBOLD and ΔCBF estimates were being derived from the same, task‐responsive voxels and that ΔCMRO2 was derived in voxels with both CBF and BOLD task‐related increases (see Fig. 3; see Supporting Information text). ΔCMRO2 was calculated voxel‐wise within this functional ROI using ΔBOLD, ΔCBF, M. Average positive ΔBOLD, ΔCBF, and ΔCMRO2 were extracted from the functional ROI (see Fig. 3A,B).
Because the gas challenge data differed for some participants in occipital coverage compared to the visual task data, M was estimated ex situ. To create a functional ROI for the gas challenge, ΔBOLDhc/BOLD0 and ΔCBFhc/CBF0 maps were thresholded and extracted from the structural ROI detailed above. The criteria for retention of a voxel within these maps required that the voxel was within the top 15% (top 20% for one participant) of ΔBOLDhc/BOLD0 and ΔCBFhc/CBF0 voxels in the structural ROI; and that these ΔBOLDhc/BOLD0 and ΔCBFhc/CBF0 voxels overlapped. This procedure ensured complementary maximum ΔBOLDhc/BOLD0 and ΔCBFhc/CBF0 signals in the retained voxels. Average ΔBOLDhc/BOLD0 and ΔCBFhc/CBF0 signals were extracted from this ROI and M was calculated [see Eq. (4)]. Importantly, this approach to calculating M did not bias our primary results (see Supporting Information Fig. 3).
T2 Images
A T2 fluid attenuated inversion recovery (FLAIR) scan was also acquired for each participant: 33 slices, TE = 125 ms, TR = 11,000 ms, no slice gap, transverse slice orientation, 0.45 × 0.45 × 5.00 mm3 voxel, 120° refocusing angle.
FLAIR images were used to estimate the extent of gross lesion burden for each participant. Lesion burden was calculated using a semi‐automated procedure [Hart et al., 2013; Hubbard et al., 2016a,b]. Here, hyperintense voxels were demarcated using in‐house MATLAB code based upon slice‐wise, signal intensity (i.e., voxels that were ≥1.25 SD over the slice mean intensity). Next, lesions were manually delineated from the hyperintense tissue by two trained researchers (L.H. and S.F.). Manual delineation was used to rule out false positives in the lesion classification due to fat signals, motion, ventricular edge effects, skull, or signal inhomogeneites [Hubbard et al., 2016b, p. 6; see also Hart et al., 2013]. Lesion burden was estimated by extracting the number of voxels that were demarcated by the automated and manual procedures. Inter‐rater agreement of lesion burden was calculated using a Dice ratio (κ) of the lesion burden estimates made by the two researchers on a sample of several subjects [Dice, 1945]. After the researchers were trained on lesion classification, inter‐rater agreement was found to be high, κ = 0.89; where κ > 0.70 is generally thought to reflect excellent inter‐rater agreement [Zhang et al., 2007].
Diffusion Images
DTI images were acquired using a single‐shot, echo‐planar imaging sequence with a Sensitivity Encoding parallel imaging scheme (SENSE, reduction factor = 2.3), 112 × 112 matrix, field of view = 224 × 224 mm2 (nominal resolution of 2 mm), 65 slices (0 mm gap), slice thickness = 2 mm, TR = 7.78 s, TE = 97 ms. The diffusion weighting was encoded along 30 independent orientations [Jones et al., 1999] and the b‐value was 1,000 s/mm2. Imaging time was 5 min and 15 s.
Automatic image registration [Woods et al., 1998] was performed on raw diffusion‐weighted images to correct distortion caused by eddy currents. Six elements of the 3 × 3 diffusion tensor were determined by multivariate least‐squares fitting. The tensor was diagonalized to obtain three eigenvalues ( ) and eigenvectors ( ). Standard tensor fitting was conducted with DTIStudio [Jiang et al., 2006] to generate the DTI‐derived diffusion characteristic, fractional anisotropy (FA), which was used in the registration/alignment/skeletonization process, and the diffusion characteristic of interest, RD. RD was chosen as the diffusion characteristic of interest because this measure is highly sensitive to WMMS changes [e.g., Harsan et al. 2007; Song et al., 2002] and is often operationalized as reflecting the degree of WMMS damage in MS. Further, RD has been demonstrated in MS to show considerable alterations regardless of macrostructural damage to tissue, whereas diffusion characteristics of other DTI measures (e.g., axial diffusivity) tend to fluctuate appreciably within patients across normal‐appearing and lesioned tissue types [Klistorner et al., 2016]. Also, RD has been shown in optic neuritis, a syndrome closely tied to MS, to be significantly related to occipital lobe electrical activity [Trip et al., 2006]. Thus, for these reasons and to limit comparisons and minimize potential Type‐I error, RD alone was evaluated here.
DTI measurements were obtained at the skeletons of the white matter using FSL [Smith et al., 2006] to alleviate partial volume effects with tract‐based spatial statistics. Participant FA maps were registered nonlinearly to the EVE single‐subject FA template [Huang et al., 2012a, 2012b; Ouyang et al., 2016] for better alignment with a digital white‐matter atlas [JHU ICBM‐DTI‐81; Mori et al., 2008]. Registered FA maps of all subjects were averaged to generate a mean FA map, from which an FA skeleton mask was created (see Fig. 4). Skeletonized FA images of all subjects were obtained by projecting the registered FA images onto the mean FA skeleton mask. Occipital tract‐specific RD was obtained by averaging RD in the skeletal voxels of tracts with connections to and from occipital lobe (occipital tracts; i.e., bilateral fronto‐occipital fasciculus, bilateral superior longitudinal fasciculus, bilateral superior fronto‐occipital fasciculus, bilateral posterior thalamic/optic radiations.
Figure 4.
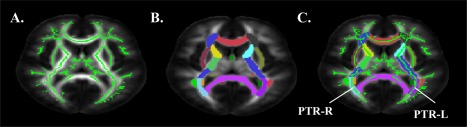
DTI occipital tract processing stream. (A) White matter skeleton. (B) JHU ICBM‐DTI‐81 atlas labels. (C) Diffusion characteristics within white matter skeleton extracted from occipital tracts (e.g., posterior thalamic radiations). [Color figure can be viewed at http://wileyonlinelibrary.com]
Statistical Analyses
Data were analyzed in JMP Pro version 12.2.0 (SAS Institute, Inc.) and R version 3.3.2 (R Core Development Team; http://r-project.org). Distributions were scrutinized for outliers (≥±2 SD mean) which were censored from these tests. Single‐sample t‐tests were used to test distribution differences against hypothesized means. Individual difference hypotheses were tested using ordinary least‐squares regression. Bootstrapped 95% confidence intervals (B = 1,000) and Pearson product‐moment correlations are also reported for significant individual differences effects in order to provide a thorough overview of the effect sizes for primary results. Multivariate ordinary‐least squares modeling was used to assess the effects of multiple independent variables; partial Pearson correlations were used to demonstrate significant effect sizes. The Monte Carlo method for assessing mediation [Preacher and Selig, 2012; code retrieved from Selig and Preacher, 2008] with 20,000 resampling iterations was used for assessing the significance of indirect effects (unstandardized ab—a measure indirect effects independent of sample size [Preacher and Kelley, 2011]) via 95% confidence intervals. This method was employed when both independent variables in the multivariate models were significant. Descriptive statistics are expressed as means ± 1 standard error.
RESULTS
Response to CO2 and Visual Stimulation Task Performance/Activity
We formally tested whether patients showed a significant change (>0%) in BOLD ([BOLDhc‐BOLD0]/BOLD0) and CBF response to the CO2 solution ([CBFhc‐CBF0]/CBF0). Patients showed a significant increase in BOLD (3.85% ± 0.47), t (9) = 8.12, P < 0.001, and CBF (167.48% ± 19.8), t (8) = 8.44, P < 0.001, in response to CO2 administration. Other physiological responses to CO2 solution are in Table 2. MS patients (92.75% ± 1.11) showed significantly greater than chance accuracy (>50%) on the visual stimulation secondary task, t (8) = 38.28, P < 0.001, with the high degree of accuracy verifying that foveae were appropriately positioned throughout the task. ΔBOLD, ΔCBF, and ΔCMRO2 responses within the visual cortex ROI were formally tested against a mean of 0 (no change from baseline; see Table 3).
Table 2.
Non‐neural physiological data
| Baseline | 5% CO2 | P | |
|---|---|---|---|
| Breath Rate | 11.20 (1.00) | 13.35 (1.28) | 0.124 |
| EtCO2 | 42.70 (1.81) | 48.95 (1.45) | <0.001 |
| Heart Rate | 66.90 (2.38) | 69.67 (2.38) | 0.156 |
| SpO2 | 98.10% (0.35%) | 97.58% (0.39%) | 0.443 |
Mean (SEM). Breath rate in breaths per minute. Heart rate in beats per minute. P values were based on paired‐samples t‐tests.
EtCO2, end‐tidal CO2 in mm Hg. SpO2, peripheral oxygen saturation in percent hemoglobin saturation.
Table 3.
Visual ROI response to visual stimulation task
| Mean | SEM | Range | P | |
|---|---|---|---|---|
| ΔBOLD | 1.12 | 0.08 | 0.77–1.36 | <0.001a |
| ΔCBF | 48.06 | 12.58 | 21.77–137.85 | <0.001a |
| ΔCMRO2 | 9.59 | 0.90 | 6.87–14.72 | <0.001a |
ΔBOLD, BOLD percent signal change response to visual stimulation.
P values were based on single‐sample t‐test against a mean of 0 (no change from baseline).
ΔCBF, cerebral blood flow percent signal change response to visual stimulation; ΔCMRO2, cerebral metabolic rate of oxygen percent signal change response to visual stimulation.
RD Associations with Grey Matter Functional Measures
Similar to our previous findings [Hubbard et al., 2014], occipital‐tract RD significantly predicted visual ROI ΔBOLD, β = −3,280.38, t (8) = −2.71, P = 0.027, r = −0.69 (95% CI: −0.96 to −0.05; see Fig. 5A). Occipital‐tract RD also significantly predicted visual ROI ΔCMRO2, β = 39,048.28, t (7) = 3.10, P = 0.017, r = 0.76 (95% CI: 0.41 to 0.94; see Fig. 5B). Occipital‐tract RD was not a significant predictor of MS patients' visual ROI ΔCBF, β = 363,579.67, t (7) = 1.56, P = 0.163.
Figure 5.
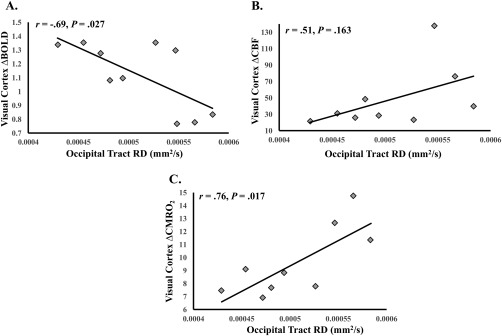
WMMS associations with visual cortex function. (A) Occipital tract RD and visual cortex ΔBOLD. (B) Occipital tract RD and visual cortex ΔCBF. (C) Occipital tract RD and visual cortex ΔCMRO2. Degrees of freedom differ due to outlier removal (see Statistical Analyses).
RD Associations With Grey Matter Functional Measures, Independence From Lesion Burden
We assessed whether the associations between occipital‐tract RD and visual ROI ΔBOLD and ΔCMRO2 remained significant when controlling for lesion burden. The association between occipital‐tract RD and visual ROI ΔBOLD remained significant when controlling for lesion burden, β = −4,396.60, t (6) = −4.18, P = 0.006, rXY | Z = −0.86. Lesion burden was also a significant predictor in this model, β = 0.00002, t (6) = 2.63, P = 0.039, rXY | Z = 0.73. Because both occipital‐tract RD and lesion burden were significant predictors of ΔBOLD, we formally assessed the significance of the independent, indirect (mediation magnitude) effect of lesion burden on the RD‐ΔBOLD relationship. The indirect effect was not significant, ab = 1,544.97 (95% CI: −174 to 3,268). The significant associations between occipital‐tract RD and visual ROI ΔCMRO2 remained when controlling for lesion burden, β = 42,384.83, t (6) = 2.62, P = 0.040, rXY | Z = 0.73. There was no significant association between lesion burden and visual ROI ΔCMRO2 in this model (P > 0.05).
Grey Matter Function Associations With MS Symptom Severity Factors
We sought to test whether the visual cortex functional measures could reflect a marker of individual variability in MS symptom severity (fatigue and neurological disability). Visual ROI ΔBOLD was not a significant predictor of MFIS scores, β = −24.36, t (8) = −0.76, P = 0.469, nor neurological disability scores, β = −26.50, t (8) = −1.87, P = 0.100. Visual ROI ΔCBF was a significant predictor of MS patients' MFIS scores, β = 0.50, t (7) = 3.62, P = 0.009, r = 0.81 (95% CI: 0.24 to 0.95). Visual ROI ΔCBF was not a significant predictor of neurological disability, β = 0.14, t (7) = 1.37, P = 0.212. Visual ROI ΔCMRO2 was a significant predictor of patients' MFIS scores, β = 7.01, t (7) = 3.59, P = 0.009, r = 0.81 (95% CI: 0.27 to 0.95), and neurological disability, β = 3.21, t (7) = 2.88, P = 0.024, r = 0.74 (95% CI: 0.15 to 0.97; see Fig. 6).
Figure 6.
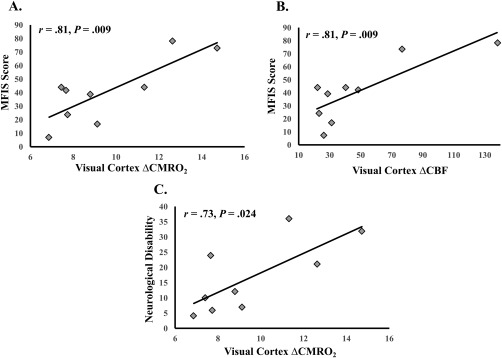
Significant visual cortex functional and MS symptom severity associations. (A) ΔCMRO2 and fatigue (MFIS scores). (B) ΔCBF and fatigue (MFIS scores). (C) ΔCMRO2 and neurological disability ratings. Degrees of freedom differ due to outlier removal (see Statistical Analyses).
Grey Matter ΔCMRO2 Associations With MS Symptom Severity Factors, Independence From Lesion Burden and RD
We sought to assess whether ΔCMRO2 accounted for unique variance in symptom severity factors, when controlling for occipital‐tract RD or lesion burden. ΔCMRO2 accounted for significant variance in MFIS scores when controlling for RD, β = 7.98, t (6) = 2.49, P = 0.047, rXY | Z = 0.78. There was no significant association between RD and MFIS in this model (P > 0.05). ΔCMRO2 also accounted for significant variance in MFIS scores when controlling for lesion burden, β = 6.32, t (6) = 3.03, P = 0.023, rXY | Z = 0.78. There was no significant association between lesion burden and MFIS in this model (P > 0.05). Visual ROI ΔCBF also remained a significant predictor of MFIS scores when controlling for occipital‐tract RD and lesion burden (Ps < 0.05). ΔCMRO2 did not account for significant variance in neurological disability when controlling for occipital‐tract RD (P > 0.05). There was also no significant association between occipital tract RD and neurological disability in this model (P > 0.05). ΔCMRO2 did, however, account for significant variance in neurological disability when controlling for lesion burden, β = 3.16, t (6) = 2.46, P = 0.049, rXY | Z = 0.71. There was no significant association between lesion burden and neurological disability in this model (P > 0.05).
DISCUSSION
Our primary aim was to investigate relationships between WMMS and grey matter function in MS. We tested associations between occipital‐tract WMMS damage, as indexed by RD, and visual cortex BOLD, blood flow, and oxygen metabolism during visual stimulation. Results demonstrated that, consistent with our previous work, increases in occipital‐tract RD were significantly associated with decreased BOLD response to visual stimulation. Moreover, increases in RD were associated with increases in CMRO2 responses to visual stimulation. These effects were independent of individual differences in brain‐wide macrostructural white matter damage (i.e., lesion burden). Increases in the CMRO2 response were also strongly related to increases in factors of MS symptom severity (i.e., fatigue and neurological disability). These relationships remained significant after controlling for lesion burden. Further, the relationship between fatigue and CMRO2 remained significant when accounting for WMMS damage. Taken together, these findings indicated that evoked grey matter oxygen metabolism in MS is (1) strongly linked to WMMS damage, and (2) can largely account for unique variance in MS symptom severity regardless of the extent of white matter micro‐ and macrostructural damage.
This study was the first to use cfMRI in a neurological population. Although the technique is not as easily employed as standard BOLD imaging, it does offer several advantages relative to measurement of BOLD signal exclusively. First, the blood flow and oxygen metabolism measures gathered from cfMRI reflect unambiguous physiological processes, whereas BOLD reflects a confluence of processes and as such, is physiologically nonspecific. This is particularly beneficial for study questions like ours, wherein a specific understanding of grey matter physiological (i.e., cerebral blood flow and oxygen metabolism) alterations is necessary to gain more nuanced insight into individual differences in brain function. Second, calibration‐derived CMRO2 is strongly coupled to electrical and chemical measures of neural activity [e.g., Herman et al., 2009, 2013; Hyder et al., 2001; Hyder, 2004; Lin et al., 2010; Smith et al., 2002]. For example, calibration‐derived CMRO2 has been shown to account for approximately 98% of the production of the primary neuroenergetic substrate, adenosine triphosphate (ATP), in visual cortex during visual stimulation [Lin et al., 2010]. Thus, the metabolic process measured by this signal is crucial for the substrate that drives neuronal work. Finally, CBF and oxygen metabolism measures used here are not dependent upon the hemodynamic assumptions of BOLD, making them optimal measures of brain function in populations with atypical hemodynamics, like MS [e.g., Hutchison et al., 2013a,2013b; see Iannetti and Wise, 2007]. Although calibration‐derived CMRO2 relies upon BOLD signal fluctuations, it is estimated using concurrent changes in CBF. Further, implementation of the BOLD scaling factor, M, within the Davis model can account for individual differences in baseline BOLD physiology at regional and voxel resolutions [e.g., Davis et al., 1998; Hoge et al., 1999; see Buxton, 2010]. Thus, the calibration approach provides data about individual differences in hemodynamics and models these data to derive CMRO2.
Using the cfMRI approach along with DTI permitted new observations regarding the relationships between grey matter functioning and WMMS in MS. We demonstrated that visual cortex BOLD and CMRO2 had large‐effect relationships with occipital‐tract RD [cf. Cohen, 1988]. Of note was the opposite directions of these relationships; RD and BOLD were negatively correlated, and RD and CMRO2 were positively correlated (a formal test indicated that these correlations were significantly different (P < 0.05; see Supporting Information Material). These findings complicate the interpretation of relationships between BOLD signal and white matter measures in MS. It is possible that the relationship observed between BOLD signal and WMMS damage is misrepresenting the relationship between WMMS damage and neurophysiological processes in these patients. The healthy system uses active hyperemic processes to ensure a surfeit of oxygen delivery for ongoing neurometabolic processes during periods of increased neuronal activity [e.g., Malonek and Grinvald, 1996; see Attwell et al., 2010; Hillman, 2014]. Even minor alterations in the coupling of increased blood flow/volume changes to neural activity (i.e., neurovascular coupling) are thought to result in dramatic changes to the magnitude of the BOLD signal [cf. Ances et al., 2008]. In MS, neurovascular coupling is suspected to be altered given known alterations to perfusion and neural‐vascular communicating structures [i.e., astrocytes; see D'haeseeler et al., 2011; De Keyser et al., 1999; Lassman, 2014]. Potential neurovascular uncoupling in MS could result in the magnitude of the BOLD response reflecting an attenuated or even inverted reflection of the actual, evoked neural or neurometabolic activity [Hubbard et al., 2016a; see Iannetti and Wise, 2007].
Precisely understanding the directionality of WMMS‐grey matter function relationships is important for understanding the pathophysiology of MS. For instance, energy failure hypotheses postulate links between increased white matter damage and increased energy demand in early MS, eventually leading to mitochondrial damage and neuronal death in later stages of the disease [Campbell et al., 2014; Mahad et al., 2015; Paling et al., 2011]. Specifically, Paling et al. (2011) posited that WMMS damage in MS leads to axon sodium channel upregulation and consequent increased energy burden. Because metabolic substrate and mitochondrial production are largely supported by the cell bodies [see MacAskill and Kittler, 2010], this increase in energetic burden for white matter related to its extent of damage, would weigh heavily upon the grey matter. Increased burden on grey matter, coupled with decreased transport of metabolic substrate [see Paling et al., 2011], probably results in decreases in basal levels of neurometabolism [for work on mitochondrial inhibition and neuroimaging see Kannurpati, 2017; Sanganahalli et al., 2013]. For example, negative relationships have been observed between basal NAA:creatine ratios and RD in MS white matter [Hannoun et al., 2012; see also Kahn et al., 2017]. Our results showing positive relationships between evoked CMRO2 and RD/symptom combined with the prospect of MS‐related decreases in basal neurometabolism suggest energy failure as a plausible explanation.
The metabolic process studied here was measured from relative changes scaled by each patient's baseline BOLD and CBF. Previous work using cfMRI has established that reduced baselines of BOLD and CBF demonstrate greater evoked changes in CMRO2 relative to higher baselines [Pasley et al., 2007; see also Hyder et al., 2002; Restom et al., 2007; Shulman et al., 2007]. Negative relationships between whole‐brain resting CMRO2 and neurological disability [Ge et al., 2012; see also Kindred et al., 2015], and basal NAA:creatine ratios and RD [Hannoun et al., 2012; see also Kahn et al., 2017] have also been observed in MS. These baseline associations demonstrate that decreased basal metabolism is associated with increased WMMS damage and more severe MS symptomology. Consistent with the basal neurometabolism associations, the baseline metabolism findings from the cfMRI literature [e.g., Pasley et al., 2007], and with the predictions of energy failure hypotheses, it possible that the more WMMS damaged and more symptomatic patients in this study also had reduced basal oxygen metabolism. This reduced metabolic baseline could result in increased metabolism relative to this baseline for the more WMMS damaged and more symptomatic patients compared to their less damaged and less symptomatic counterparts. Such a mechanism would account for the positive relationships between ΔCMRO2 and RD, and ΔCMRO2 and the symptom indices we observed.
Relative changes from different neurometabolic baselines provide one explanation for our results. Even with the assumption of homogeneous peak absolute oxygen metabolism (e.g., µmol/100g parenchyma), a reduced metabolic baseline for more WMMS damaged/symptomatic MS patients implies that these patients produced greater neurometabolic responses to stimulation in order to reach the same absolute metabolic peak as their less affected cohorts [Fig. 7; e.g., Hyder et al., 2002; see also Pasley et al., 2007; Restom et al., 2007; Shulman et al., 2007]. Foregoing the unlikely scenario of increased basal oxygen metabolism for more WMMS damaged and more symptomatic MS patients [cf. Ge et al., 2012; Hannoun et al., 2012; Kahn et al., 2017], conclusions related to either absolute or relative increases in oxygen metabolism suggest a similar interpretation: stimulation induced oxygen metabolism increased as of function of the extent of WMMS damage and symptomology. Of note is that our MS cohort was predominantly earlier stage MS patients—90% of patients had relapsing‐remitting diagnoses and were on average over 2 years removed from their last exacerbation; all were ambulatory with intact cognitive status (TICS > 21). It is likely that in cases such as acute exacerbations (including acute optic neuritis) or more advanced MS, extreme damage at either anterior or posterior portions of the afferent visual pathway would result in a potential decrease in absolute and relative visuocortical neurometabolic activity, due to cell death and/or a paucity of signal throughput [cf. Trip et al., 2006].
Figure 7.
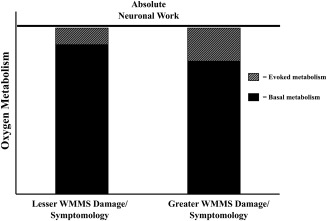
Interpretation of CMRO2 differences between MS patients with lesser WMMS damage/symptomology versus those with greater WMMS damage/symptomology. This shows differential basal and task metabolism but similar absolute neuronal work across MS participants [baseline + task; see Hyder et al., 2002; Pasley et al., 2007; Shulman et al., 2007].
Our findings provide new evidence of an evoked oxygen metabolism facet of fatigue and neurological disability in MS. The study of neural factors related to fatigue and neurological disability is of considerable importance as these maladies are key features of MS [e.g., Sandry et al., 2014; Sehle et al., 2014] and contribute substantially to these patients' quality of life [Amato et al., 2001]. Here, evoked CMRO2 responses accounted for 65% of the variance in MS patients' fatigue scores and 55% of the variance in their neurological disability scores (CBF also showed a large effect relationship with fatigue, but no significant relationship with neurological disability). These shared variances remained large when controlling for lesion burden. Further, the shared variance remained large for the fatigue‐CMRO2 relationship while also controlling for occipital‐tract RD. These results establish a relative independence, particularly in the case of fatigue, of the relationship between evoked grey matter metabolism and MS symptomology. These findings also suggest that evoked grey matter metabolic processes play a unique role, along side macro‐ and microstructural white matter damage, in the manifestation of individual differences in symptomology. This interpretation is also consistent with energy failure hypotheses, because these models posit that mitochondria and neuroenergetic dysfunction are integral in the pathogenic cascade resulting in MS‐related neurodegeneration, and thus, progressive symptomology [cf. Mahad et al., 2015].
The present results elucidated WMMS and evoked, grey matter physiological relationships in MS. Increased occipital‐tract RD was associated with increased visual cortex oxygen metabolism, establishing a link between increased WMMS damage and increased evoked neurometabolism. Evoked oxygen metabolism was also highly related to individual variability in MS fatigue and neurological disability, demonstrating that evoked neurometabolism could be an indicator of MS symptom severity. The associations between evoked oxygen metabolism, WMMS damage, and MS symptomology suggests this measure as a focus for elucidating pathophysiological mechanisms of MS and provides a neurophysiological target for future works to understand progression of MS symptomology.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank Hannah Grotzinger and Ashti M. Shah for contributions to manuscript preparation.
N.A.H. contributed to study design, data analysis, and wrote the manuscript. M.P.T., L.H., B.P.T., and J.L.H. contributed to study design, data collection and analyses, and manuscript writing. M.O., J.F.S., and H.H., contributed to data analysis. S.F. contributed to data collection and manuscript writing. S.L.D., J.S., D.C.K., H.L., J.H., T.F., E.F., D.T.O., and B.R. contributed to study design, conceptualization, and manuscript writing.
T.F. received speaker fees from Genzyme, Novartis, and Acorda. E.F. received speaker and consulting fees from Genzyme, Novartis, TEVA, and Acorda. D.T.O. received lecture fees from Acorda, Genzyme, and TEVA Neuroscience, consulting and advisory board fees from EMD Serono, Genentech, Genzyme, Novartis and TEVA Neuroscience, and research support from Biogen.
REFERENCES
- Alshowaeir D, Yiannikas C, Garrick R, Parratt J, Barnett MH, Graham SL, Klistorner A (2014): Latency of multifocal visual evoked potentials in nonoptic neuritis eyes of multiple sclerosis patients associated with optic radiation lesions. ARVO 55:3758–3764. [DOI] [PubMed] [Google Scholar]
- Amato MP, Ponziani G, Rossi F, Liedl CL, Stefanile C, Rossi L (2001): Quality of life in multiple sclerosis: The impact of depression, fatigue, and disability. Mult Scler 7:340–344. [DOI] [PubMed] [Google Scholar]
- Ances BM, Leontiev O, Perthen JE, Liang C, Lansing AE, Buxton RB (2008): Regional differences in the coupling of cerebral blood flow and oxygen metabolism changes in response to activation: Implications for BOLD‐fMRI. NeuroImage 39:1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA (2010): Glial and neuronal control of brain blood flow. Nature 468:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au Duong M‐V, Audoin B, Boulanouar K, Ranjeva J‐P (2005): Altered functional connectivity related to white matter changes inside the working memory network at the very early stage of MS. JCBFM 25:1245–1253. [DOI] [PubMed] [Google Scholar]
- Brandt J, Spencer M, Folstein M (1988): The telephone interview for cognitive status. Neuropsychiatry Neuropsychol Behav Neurol 1:111–117. [Google Scholar]
- Brosnan CF, Raine CS (2013): The astrocyte in multiple sclerosis revisited. Glia 61:453–465. [DOI] [PubMed] [Google Scholar]
- Buxton RB (2010): Interpreting oxygenation‐based neuroimaging signals: The importance and the challenge of understanding brain oxygen metabolism. Front Neuroenergetics 2:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cader S, Cifelli A, Abu‐Omar Y, Palace J, Matthews PM (2006): Reduced brain functional reserve and altered functional connectivity in patients with multiple sclerosis. Brain 29:527–537. [DOI] [PubMed] [Google Scholar]
- Cader S, Johansen‐Berg H, Wylezinska M, Palace J, Beherens TE, Smith S, Matthews PM (2007): Discordant white matter N‐acetylasparate and diffusion MRI measure suggest that chronic metabolic dysfunction contributes to axonal pathology in multiple sclerosis. NeuroImage 36:19–27. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Worrall JT, Mahad DJ (2014): The central role of mitochondrial in axonal degeneration in multiple sclerosis. Mult Scler 20:1806–1813. [DOI] [PubMed] [Google Scholar]
- Cohen J (1988): Statistical power analysis for the behavioral sciences, (2nd ed. Hillsdale, NJ: Lawrence Earlbaum Associates. [Google Scholar]
- Cox RW (1996): AFNI: Software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res 29:162–173. [DOI] [PubMed] [Google Scholar]
- D'haeseleer M, Cambron M, Vanopdenbosch L, De Keyser J (2011): Vascular aspects of multiple sclerosis. Lancet Neurol 10:657–666. [DOI] [PubMed] [Google Scholar]
- Davis TL, Kwong KK, Weisskoff RM, Rosen BR (1998): Calibrated functional MRI: Mapping the dynamics of oxidative metabolism. PNAS 95:1834–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keyser J, Steen C, Mostert JP, Koch MW (2008): Hypoperfusion of the cerebral white matter in multiple sclerosis: Possible mechanisms and pathophysiological significance. JCBFM 28:1645–1651. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Wilczak N, Leta R, Streetland C (1999): Astrocytes in multiple sclerosis lack beta‐2 adrenergic receptors. Neurology 53:1628–1633. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl BM, Quin BT, Dickerson BC, Blacker D, et al. (2006): NeuroImage 31:968–980. [DOI] [PubMed] [Google Scholar]
- Dice LR (1945): Measures of the amount of ecologic association between species. Ecology 26:297–302. [Google Scholar]
- Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD (2006): Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 59:478–489. [DOI] [PubMed] [Google Scholar]
- Fisk JD, Pontefract A, Ritvo PG, Archibald CJ, Murray TJ (1994): The impact of fatigue on patients with multiple sclerosis. Can J Neurol Sci 21:9–14. [PubMed] [Google Scholar]
- Frohman EM, Frohman TC, Zee DS, McColl R, Galetta S (2005): The neuro‐ophthalmology of multiple sclerosis. The Lancet Neurology 4:111–121. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS (2006): Multiple sclerosis: The plaque and its pathogenesis. NEJM 354:942–955. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Fujimoto JG, Frohman TC, Calabresi PA, Cutter G, Balcer LJ (2008): Optical coherence tomography: A window into the mechanisms of multiple sclerosis. Nature Clinical Practice Neurology 4:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Law M, Grossman RI (2005): Applications of diffusion tensor MR imaging in multiple sclerosis. Ann NY Acad Sci 1064:202–219. [DOI] [PubMed] [Google Scholar]
- Ge Y, Zhang Z, Lu H, Tang L, Jaggi H, Herbert J, Babb JS, Rusinek H, Grossman R (2012): Characterizing brain oxygen metabolism in patients with multiple sclerosis with T2‐relaxation‐under‐spin‐tagging MRI. JCBFM 32:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova HM, Hillary FG, Wylie G, Rypma B, Deluca J (2009): Examination of processing speed deficits in multiple sclerosis using functional magnetic resonance imaging. JINS 15:383–393. [DOI] [PubMed] [Google Scholar]
- Grahm SL, Klistorner A (2017): Afferent visual pathways in multiple sclerosis: a review. Clin Exp Ophthalmol 45:62–72. [DOI] [PubMed] [Google Scholar]
- Griffeth VEM, Buxton RB (2011): A theoretical framework for estimating cerebral oxygen metabolism changes using the calibrated‐BOLD method: Modeling the effects of blood volume distribution, hematocrit, oxygen extraction fraction, and tissue signal properties on the BOLD signal. NeuroImage 58:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb RL, Raichle ME, Eichling JO, Ter‐Pogossian MM (1974): The effects of changes in PaCO2 cerebral blood volume, blood flow, and vascular mean transit time. Stroke 5:630–639. [DOI] [PubMed] [Google Scholar]
- Hannoun S, Bagory M, Durand‐Dubief F, Ibarrola D, Comte JC, Confavreux C, Cotton F, Sappey‐Marinier D (2012): Correlation of diffusion and metabolic alterations in different clinical forms of multiple sclerosis. PLoS One 7:e32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart J Jr, Kraut MA, Womack KB, Strain J, Didehbani N, Bartz E, Conover H, Mansinghani S, Lu H, Cullum CM (2013): Neuroimaging of cognitive dysfunction and depression in aging retired national football league players. JAMA Neurol 70:326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Parizel N, Skoff RP, Ghandour MS (2007): Astrocytic hypertrophy in dysmyelination influences the diffusion anisotropy of white matter. J Neurosci Research 85:935–944. [DOI] [PubMed] [Google Scholar]
- Herman P, Sanganahalli BG, Blumenfeld H, Hyder F (2009): Cerebral oxygen demand for shot‐lived and steady‐state events. J Neurochem 109(Suppl 1):73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman P, Sanganahalli BG, Blumenfeld H, Rothman DL, Hyder F (2013): Quantitative basis for neuroimaging of cortical laminae with calibrated functional MRI. PNAS 110:15115–15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman EMC (2014): Coupling mechanism and significance of the BOLD signal: A status report. Annu Rev Neurosci 37:161–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge RD, Atikinson J, Gill B, Marrett S, Pike GB (1999): Investigation of BOLD signal dependence on cerebral blood flow and oxygen consumption: The deoxyhemoglobin dilution model. Magnet Reson Med 42:849–863. [DOI] [PubMed] [Google Scholar]
- Huang H, Guandapuneedi T, Rao U (2012a): White matter disruptions in adolescents exposed to childhood maltreatment and vulnerability to psychopathology. Neuropsychopharmacology 37:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Fan X, Weiner M, Martin‐Cook K, Xiao G, Davis J, Devous M, Rosenberg R (2012b): Distinctive disruption patterns of white matter tracts in Alzheimer's disease with full diffusion tensor characterization. Neurobiology of Aging 33:2029–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard NA, Hutchison JL, Turner MP, Sundaram S, Oasay L, Robinson D, Strain J, Weaver T, Davis SL, Remington GM, Huang H, Biswal BB, Hart J, Frohman TC, Frohman EM, Rypma B (2016): Asynchrony in executive networks predicts cognitive slowing in multiple sclerosis. Neuropsychology 30:75. [DOI] [PubMed] [Google Scholar]
- Hubbard NA, Turner M, Hutchison JL, Ouyang A, Strain J, Oasay L, Sundaram S, Davis S Remington G, Brignate R, Huang H, Hart J, Frohman T, Frohman E, Biswal BB, Rypma B (2016): Multiple sclerosis‐related white matter microstructural change alters the BOLD hemodynamic response. JCBFM 36:1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard NA, Turner MP, Robinson DM, Sundaram S, Oasay L, Hutchison JL, Ouyang A, Huang H, Rypma B (2014): Attenuated BOLD hemodynamic response predicted by degree of white matter insult, slows cognition in multiple sclerosis. Mult Scler J 20:267. [Google Scholar]
- Hutchison JL, Hubbard NA, Brigante RM, Turner M, Sandoval TI, Hillis GA, Weaver T, Rypma B (2014): The efficiency of fMRI region of interest analysis methods for detecting group differences. J Neurosci Methods 226:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison JL, Lu H, Rypma B (2013): Neural mechanisms of age‐related slowing: The ΔCBF/ΔCMRO2 ratio mediates age‐differences in BOLD signal and human performance. Cereb Cortex 23:2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison JL, Shokri‐Kojori E, Lu H, Rypma B (2013): A BOLD perspective on age‐related neurometabolic‐flow coupling and neural efficiency changes in human visual cortex. Front Psychol 4:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder F (2004): Neuroimaging with calibrated FMRI. Stroke 35(11 Suppl 1):2635–2641. [DOI] [PubMed] [Google Scholar]
- Hyder F, Kida I, Behar KL, Kennan RP, Maciejewski PK, Rothman DL (2001): Quantitative functional imaging of the brain: Towards mapping neuronal activity by BOLD fMRI. NMR Biomed 14:413–431. [DOI] [PubMed] [Google Scholar]
- Hyder F, Rothman DL, Shulman RG (2002): Total neuroenergetics support localized brain activity: Implications for the interpretation of fMRI. PNAS 99:10771–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetti GD, Wise RG (2007): BOLD functional MRI in disease and pharmacological studies: room for improvement? Magn Reson Imag 25:978–988. [DOI] [PubMed] [Google Scholar]
- Janssen AL, Boster A, Patterson BA, Abduljalil A, Prakash RS (2013): Resting‐state functional connectivity in multiple sclerosis: an examination of group differences and individual differences. Neuropsychologia 51:2918–2929. [DOI] [PubMed] [Google Scholar]
- Jiang H, van Zijl PC, Kim J, Pearlson GD, Mori S (2006): DtiStudio: resource program for diffusion tensor computation and fiber bundle tracking. Comput Methods Programs Biomed 81:106–116. [DOI] [PubMed] [Google Scholar]
- Jones DK, Horsfield MA, Simmons A (1999): Optimal strategies for measuring diffusion in anisotropic systems by magnetic resonance imaging. Magn Reson Med 42:515–525. [PubMed] [Google Scholar]
- Kahn O, Seraji‐Bozorgzad N, Bao F, Razmjou S, Caon C, Santiago C, Latif Z, Aronov R, Zak I, Ashtamker M, Kolodny S, Ford C, Sidi Y (2017): The relationship between brain MR spectroscopy and disability in multiple sclerosis: 20‐year data from the U.S. glatiramer acetate extension study. J Neuroimaging 27:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannurpati SS (2017): Mitochondrial calcium homeostasis: Implicaitons for neurovascular and neurometabolic coupling. JCBFM 37:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐G, Ogawa S (2012): Biophysical and physiological origins of blood oxygenation level‐dependent fMRI signals. JCBFM 32:1188–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindred JH, Tuulari JJ, Bucci M, Kalliokoski KK, Rudroff T (2015): Walking speed and brain glucose uptake are uncoupled in patients with multiple sclerosis. Front in Hum Neurosci 9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klistorner A, Wang C, Fofanova V, Barnett MH, Yiannikas C, Parratt J, You Y, Graham SL (2016): Diffusivity in multiple sclerosis lesions: At the cutting edge? NeuroImage: Clinical 12:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolappan M, Henderson APD, Jenkins TM, Wheeler‐Kingshott CA, Plant GT, Thompson AJ, Miller DH (2009): Assessing structure and function of the afferent visual pathway in multiple sclerosis and associated optic neuritis. J Neurol 256:305–319. [DOI] [PubMed] [Google Scholar]
- Lassman H (2014): Mechanisms of white matter damage in multiple sclerosis. Glia 62:1816–1830. [DOI] [PubMed] [Google Scholar]
- Leontiev O, Buxton RB (2007): Reproducibility of BOLD, perfusion, and CMRO2 measurements with calibrated‐BOLD fMRI. NeuroImage 35:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A‐L, Fox PT, Hardies J, Duong TQ, Gao JH (2010): Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. PNAS 107:8446–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Fox PT, Yang Y, Lu H, Tan LH, Gao JH (2008): Evaluation of MRI models in the measurement of CMRO2 and its relationship with CBF. Magn Reson Medicine 60:380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT, Wong EC (2005): A signal processing model for arterial spin labeling functional MRI. NeuroImage 24:207–215. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A (2001): Neurophysiological investigation of the basis of the fMRI signal. Nature 412:150–157. [DOI] [PubMed] [Google Scholar]
- Lu H, van Zijl P (2005): Experimental measurement of extravascular parenchymal BOLD effects and tissue oxygen extraction fractions using multi‐echo VASO fMRI at 1.5 and 3.0 T. Magnetic Reson Medicine 53:808–816. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT (2010): Control of mitochondrial transport and localization in neurons. Trends in Cell Biology 20:102–112. [DOI] [PubMed] [Google Scholar]
- Mahad DH, Trapp BD, Lassmann H (2015): Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193. [DOI] [PubMed] [Google Scholar]
- Malonek D, Grinvald A (1996): Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: Implications for functional brain mapping. Science 272:551–554. [DOI] [PubMed] [Google Scholar]
- Mori S, Oishi K, Jiang H, et al. (2008): Stereotaxic white matter atlas based on diffusion tensor imaging in an ICBM template. NeuroImage 40:570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M, Cheng H, Mishra V, Gong G, Mosconi M, Sweeney J, Peng Y, Huang H (2016): Atypical age‐dependent effects of autism on white matter microstructure in children of 2‐7 years. Human Brain Mapping 37:819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paling D, Golay X, Wheeler‐Kingshott C, Kapoor R, Miller D (2011): Energy failure in multiple sclerosis and its investigation using MR techniques. J Neurol 258:2113–2127. [DOI] [PubMed] [Google Scholar]
- Pasley BN, Inglis BA, Freeman RD (2007): Analysis of oxygen metabolism implies a neural origin for the negative BOLD response in human visual cortex. NeuroImage 36:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti L, Cerasa A, Liguori M, Gioia MC, Valentino P, Nisticò R, Quattrone A, Fera F (2009): Neurobiological mechanisms underlying emotional processing in relapsing‐remitting multiple sclerosis. Brain 132:3380–3391. [DOI] [PubMed] [Google Scholar]
- Peng S‐L, Ravi H, Sheng M, Thomas BP, Lu H (2017): Searching for a truly “iso‐metabolic” gas challenge in physiological MRI. JCBFM 37:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perthen JE, Lansing AE, Liau J, Liu TT, Buxton RB (2008): Caffeine‐induced uncoupling of cerebral blood flow and oxygen metabolism: a calibrated BOLD fMRI study. NeuroImage 40:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preacher KJ, Kelley K (2011): Effect size measures for mediation models: Quantitative strategies for communicating indirect effects. Psychol Methods 16:93–115. [DOI] [PubMed] [Google Scholar]
- Preacher KJ, Selig JP (2012): Advantages of Monte Carlo confidence intervals for indirect effects. Commun Methods Meas 6:77–98. [Google Scholar]
- Restom K, Bangen KJ, Bondi MW, Perthen JE, Liu TT (2007): Cerebral blood flow and BOLD responses to a memory encoding task: A comparison between healthy young and elderly adults. NeuroImage 37:430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanganahalli BG, Herman P, Hyder F, Kannurpatti SS (2013): Mitochondrial calcium uptake capacity modulates neocortical excitability. JCBFM 33:1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saindane AM, Law M, Ge Y, Johnson G, Babb JS, Grossman RI (2007): Correlation of diffusion tensor and dynamic perfusion MR imaging metrics in normal‐appearing corpus callosum: Support for primary hypoperfusion in multiple sclerosis. AJNR 28:767–772. [PMC free article] [PubMed] [Google Scholar]
- Sandry J, Genova H, Dobryakova E, DeLucca J, Wylie G (2014): Subjective cognitive fatigue in multiple sclerosis depends on task length. Front Neurol 5:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehle A, Vieten M, Mündermann A, Dettmers C (2014): Difference in motor fatigue between patients with stroke and patients with multiple sclerosis: a pilot study. Front Neurol 5:36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig JP, and Preacher KJ (2008): Monte Carlo method for assessing mediation: An interactive tool for creating confidence intervals for indirect effects [Computer software]. Available at: http://quantpsy.org/.
- Shulman RG, Rothman DL, Hyder F (2007): A BOLD search for baseline. NeuroImage 36:277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijens PE, Irwan R, Potze JH, Mostert JP, De Keyser J, Oudkerk M (2005): Analysis of the human brain in primary progressive multiple sclerosis with mapping of the spatial distributions using 1H MR spectroscopy and diffusion tensor imaging. Eur Radiol 15:1686–1693. [DOI] [PubMed] [Google Scholar]
- Singh M, Kim S, Kim T (2003): Correlation between BOLD‐fMRI and EEG signal changes in response to visual stimulus frequency in humans. Magn Reson Medicine 49:108–114. [DOI] [PubMed] [Google Scholar]
- Singhal NK, Li S, Arning E, Alkhayer K, Clements R, Dassanayake RS, Brasch NE, Freeman EJ, Bottiglieri T, McDonough J (2015): Changes in methionine metabolism and histone H3 trimethylation are linked to mitochondrial defects in multiple sclerosis. J Neurosci 35:15170–15186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Blumenfeld H, Behar KL, Rothman DL, Shulman RG, Hyder F (2002): Cerebral energetics and spiking frequency: The neurophysiological basis of fMRI. PNAS 99:10765–10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Johansen‐Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TE (2006): Tract‐based spatial statistics: Voxelwise analysis of multi‐subject diffusion data. NeuroImage 31:1487–1505. [DOI] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH (2002): Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. NeuroImage 17:1429–1436. [DOI] [PubMed] [Google Scholar]
- Sorbara CD, Wagner NE, Ladwig A, Nikic I, Merler D, Leele T, Marinkovic P, Naumann R, Godinho L, Bareyre FM, Bishop D, Misgeld T, Kerschensteiner M (2014): Pervasive axonal transport deficits in multiple sclerosis models. Neuron 84:1183–1190. [DOI] [PubMed] [Google Scholar]
- Stromillo ML, Giorgio A, Rossi F, Battaglini M, Hakiki B, Malentacchi G, Santangelo M, Gasperini C, Bartolozzi ML, Portaccio E, Amato MP, De Stefano N (2013): Brain metabolic changes suggestive of axonal damage in radiologically isolated syndrome. Neurology 80:2090–2094. [DOI] [PubMed] [Google Scholar]
- Sun X, Tanaka M, Kondo S (1998): Clinical significance of reduced cerebral metabolism in multiple sclerosis: A combined PET and MRI study. Ann Nuclear Med 12:89–94. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nave K (2008): Multiple sclerosis: An immune or neurodegenerative disorder? Ann Rev of Neurosci 31:247–269. [DOI] [PubMed] [Google Scholar]
- Trip AS, Wheeler‐Kingshott C, Jones SJ, Li WY, Barker GJ, Thompson AJ, Plant GT, Miller DH (2006): Optic nerve diffusion tensor imaging in optic neuritis. NeuroImage 30:498–505. [DOI] [PubMed] [Google Scholar]
- Verdier‐Taillefer M, Roullet E, Cesaro P, Alperovitch A (1994): Validation of Self‐Reported Neurological Disability in Multiple Sclerosis. Int J Epidemiol 23:148–154. [DOI] [PubMed] [Google Scholar]
- White AT, Lee JN, Light AR, Light KC (2009): Brain activation in multiple sclerosis: a BOLD fMRI study of the effects of fatiguing hand exercise. Multi Scler 15:580–586. [DOI] [PubMed] [Google Scholar]
- Woods RP, Grafton ST, Holmes CJ, Cherry SR, Mazziotta JC (1998): Automated image registration: I. General methods and intrasubject, intramodality validation. J Comput Assist Tomogr 22:139–152. [DOI] [PubMed] [Google Scholar]
- Xu F, Uh J, Brier MR, Hart J, Yezhuvath US, Gu H, Yang Y, Lu H (2011): The influence of carbon dioxide on brain activity and metabolism in conscious humans. JCBFM 31:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yücel MA, Evans KC, Selb J, Huppert TJ, Boas DA, Gagnon L (2014): Validation of the hypercapnic calibrated fMRI method using DOT‐fMRI fusion imaging. NeuroImage 102:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappe AC, Uludağ K, Oeltermann A, Ugurbil K, Logothetis NK (2008): The influence of moderate hypercapnia on neural activity in the anesthetized nonhuman primate. Cereb Cortex 18:2666–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Dean D, Liu JZ, Saghal V, Wang X, Yue GH (2007): Quantifying degeneration of white matter in normal aging using fractal dimension. Neurobiol Aging 28:1543–1555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information