

Synopsis
There is a strong genetic component to human obesity. Most genes that influence an individual’s predisposition to gain weight are not yet known. However, a glimpse into the long-term regulation of body weight has come from studying extreme human obesity caused by single gene defects. These monogenic obesity disorders have confirmed that the hypothalamic leptin-melanocortin system is critical for energy balance in humans because disruption of these pathways causes the most severe obesity phenotypes. Approximately 20 different genes and at least 3 different mechanisms implicated in monogenic causes of obesity have been identified, however, they account for less than 5% of all severe obesity. This suggests that the genetic basis for human obesity is likely to be extremely heterogeneous, with contribution from numerous different genes acting by various, yet undiscovered, molecular mechanisms.
Keywords: obesity, genetics, human, hypothalamus, energy intake
Introduction
The prevalence of common obesity has dramatically increased over the last 30 years.1 This rise is largely attributed to the increased caloric richness of our diet and the decreased physical activity in our lives. However, obesity is a multi-factorial disease that is influenced by genetic as well as environmental factors. There is an inherited component to body weight that accounts for 40-70% of an individual’s predisposition to obesity.2 Therefore, weight gain in an individual is due to dietary and lifestyle choices on a background of genetic susceptibility. Most genes that contribute to this predisposition are still not known but some insight about the hereditary nature of body weight has come from the discovery and characterization of single gene defects that cause severe human obesity.
Monogenic obesity is defined as obesity resulting from a mutation or deficiency of a single gene. See Table 1 for useful definitions. The monogenic forms of obesity known thus far can be divided into three broad categories. 1. Obesity caused by mutations in genes that have a physiologic role in the hypothalamic Leptin-Melanocortin system of energy balance. Specifically, obesity due to leptin, leptin receptor, melanocortin-4 receptor (MC4R), proopiomelanocortin (POMC), and prohormone convertase 1/3 (PC1/3) mutations will be addressed. These disorders are well-characterized, and extensively reviewed by others3–5, and therefore, will be summarized here only to highlight key clinical points. 2. Obesity resulting from mutations in genes that are necessary for the development of the hypothalamus. There are three such genes, SIM1, BDNF and NTRK2, with important roles during hypothalamic development, that when mutated lead to severe obesity. These conditions lend more support to the concept that the hypothalamus is critical for energy homeostasis, but the exact mechanisms by which these gene defects lead to obesity are not yet understood. 3. Obesity presenting as part of a complex syndrome caused by mutations in genes whose functional relationship to obesity is also not clear. In this review, we will focus on three such syndromes, Bardet-Biedl syndrome, Alstrom syndrome, and Carpenter syndrome, whose etiology has recently been ascribed to the dysfunction of the primary cilium. Consideration of these syndromes emphasizes the on-going discovery of new molecular mechanisms underlying the pathogenesis of obesity.
Table 1.
Useful Definitions
| Monogenic obesity | Obesity explained by mutation in a single gene |
| Early-onset obesity | Not clearly defined in the literature. Generally considered as abnormal weight gain occurring in children less than 10 years of age. In this review, referred to as the onset of rapid weight gain before the age of 2 years. |
| Common obesity | Obesity that is most frequently encountered in the general population and is not associated with any developmental syndromes. |
| The World Health Organization (WHO) BMI Classification in Adults | |
| Overweight | BMI between 25 and 29.99 kg/m2 |
| Obese | BMI greater than 30 |
| Obese class I | BMI between 30 and 34.99 |
| Obese class II (preferable to the term severe obesity) | BMI between 35 and 39.99 |
| Obese class III (preferable to the term morbid obesity) | BMI greater than 40 |
| The United States Center of Disease Control (CDC) BMI Classification in Children 2 to 20 years old | |
| At-risk for overweight | BMI greater than or equal to the 85th percentile on BMI-for-age curves |
| Overweight | BMI greater than or equal to the 95th percentile on BMI-for-age curves |
| The term obese is not defined in children. However, for the purposes of discussion and recruitment for research studies, a child with a BMI greater than the 97th percentile on BMI-for-age chart can be considered obese, and one with a BMI greater than the 99th percentile can be considered severely obese. | |
| For most recent US CDC BMI percentile curves go to http://www.cdc.gov/growthcharts | |
I. Obesity due to gene mutations that affect the Leptin-Melanocortin system
Naturally occurring mutations in mice that cause severe obesity led to the discovery and understanding of a neuronal system that regulates long-term energy homeostasis in mammals.6 Thereafter, the occurrence in humans of severe obesity-causing mutations affecting the same pathways as in mice has validated that this system of energy balance is conserved across species, and is in fact crucial to the maintenance of body weight in humans. Referred to as the Leptin-Melanocortin system, this specific network of neurons, centered in the hypothalamus, integrates information about peripheral energy stores relayed primarily by the hormone leptin. The effective output is a change in food intake behavior and basal energy expenditure. In Figure 1 we briefly summarize the current understanding of this system, as it pertains to our discussion of monogenic obesity. A detailed description of all molecules and pathways implicated in energy balance is beyond the scope of this review7–10.
Figure 1. Leptin-Melanocortin system of energy balance.
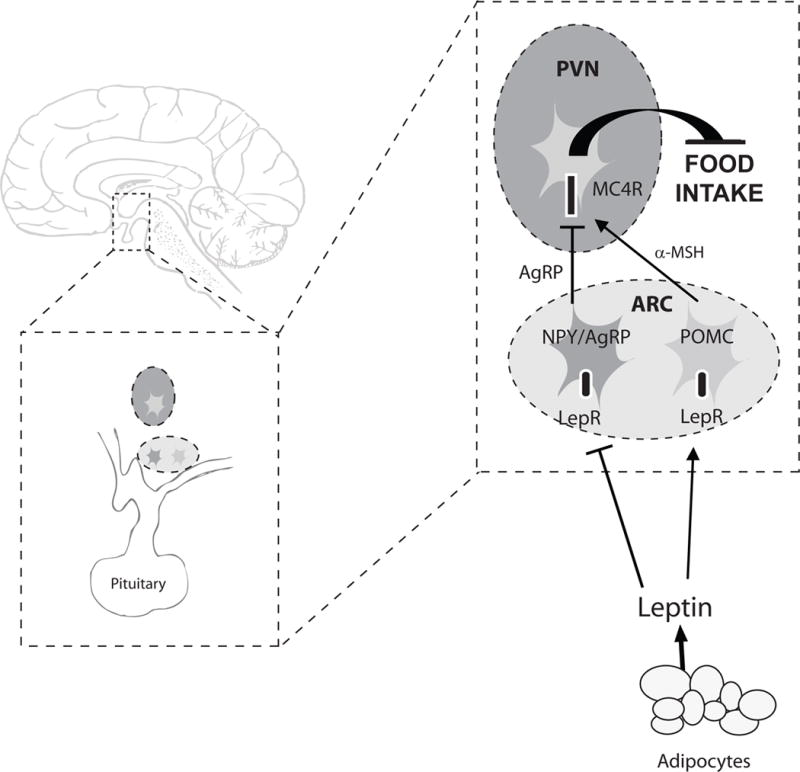
PVN paraventricular nucleus of the hypothalamus. ARC arcuate nucleus of the hypothalamus. LepR leptin receptor. NPY neuropeptide Y. AgRP agouti-related peptide. POMC proopiomelanocortin. α-MSH α-melanocyte stimulating hormone. MC4R melanocortin 4 receptor.
Information about the body’s energy stores is conveyed to the brain by hormones such as leptin. Leptin is secreted by adipocytes in proportion to the body’s fat mass. Leptin binds to its receptors on two populations of neurons in the arcuate nucleus of the hypothalamus: the orexigenic AgRP/NPY-expressing neurons, and the anorexigenic POMC-expressing neurons. These two groups of neurons have projections to the paraventricular nucleus of the hypothalamus, as well as to other regions of the brain. The PVN has a dense neuronal population that expresses MC4R. When leptin binds its receptors on POMC neurons, α-MSH, a cleavage product of the POMC transcript, is released. Activation of MC4R in the PVN by α-MSH relays a satiety signal and causes a decrease in food intake. AgRP is an antagonist of MC4R, and competes with α-MSH to bind MC4R. Binding of AgRP to MC4R leads to increased food intake. Leptin activates POMC neurons and inhibits AgRP neurons. Therefore, by activating its receptors on these two neuronal populations, leptin acts in a concerted way to increase MC4R activation by α-MSH and decrease its antagonism by AgRP, to cause a decrease in food intake.
Mutations in genes with critical roles in the Leptin-Melanocortin system cause early-onset, severe obesity. Autosomal recessive mutations in leptin, leptin receptor, POMC and PC1/3, and autosomal dominant MC4R mutations have been described.
Leptin
Severe early-onset obesity, extremely low serum leptin levels, and successful treatment with exogenous leptin distinguish congenital leptin deficiency from all other monogenic causes of obesity. This is a rare autosomal recessive disorder resulting from homozygous mutations in the leptin gene. There are only twelve reported individuals in the world with congenital leptin deficiency, all homozygous for one of two known mutations.11 Two cousins from a consanguineous Pakistani family, homozygous for a frameshift mutation (ΔG133) that leads to a truncated, unsecreted leptin molecule were first reported in 1997.12 Since then, three Turkish patients homozygous for a missense mutation (R105Y),13 and six patients from four unrelated Pakistani families with the ΔG133 mutation have been described.14,15
All reported patients share the clinical phenotype of severe obesity, hyperphagia, and serum leptin levels that are disproportionately low for their degree of fat mass. These patients are of normal birth weight, but their dramatic weight gain begins in the first three months of life and continues such that they weigh more than 20 kg by one year of age, and more than 50 kg by five years of age.12 Leptin deficient patients also have impressive adiposity with greater than 50 percent body fat where normal children have body fat in the 15 to 25 percent range.12
Aside from their striking weight phenotype, these patients come to clinical attention for their lack of pubertal development.13–15 The absent or delayed puberty results from hypogonadotropic hypogonadism and highlights leptin’s importance for the onset of puberty.16 Abnormal T cell number and function, which may present as frequent respiratory infections,17 is also present in congenital leptin deficiency and explained by leptin’s role in proliferation of CD4+ T cells and release of cytokines from T-helper-1 cells. Additionally, leptin regulates prohormone convertase 1/3 (PC1/3), which is necessary for the synthesis of TRH and GHRH. This role of leptin may explain the thyroid and growth hormone dysfunction reported in some of these patients.18 Since congenital leptin deficiency is so rare, extensive laboratory evaluation to uncover the hormonal and immunologic deficiencies associated with it can only be recommended if the degree of obesity and clinical presentation raise a strong suspicion for this disorder.
Daily subcutaneous administration of recombinant human leptin to children with congenital leptin deficiency results in dramatic weight loss and reduction in fat mass, as well as resumed pubertal progression, and improved thyroid and immune function.14,15 This is an exceptionally rare, but remarkable example of effective treatment of obesity arising from an understanding of its physiological basis. Furthermore, there is no similar benefit of treatment with supraphysiologic doses of leptin to obese non-leptin deficient patients.19 Thus congenital leptin deficiency is unique in that measurement of a serum leptin level that is extremely low compared to the patient’s fat mass can make the diagnosis, and daily administration of leptin can successfully treat it.
It is worth noting, however, that direct sequencing of the leptin gene is still the mainstay of diagnosis because a mutation could arise that does not affect synthesis or secretion of leptin, but impairs receptor binding or other downstream function. If such a mutation were to occur, the patient would have the clinical phenotype, but a serum leptin level that, like in other forms of obesity, is proportional to the patient’s fat mass rather than diagnostically low.
Leptin Receptor
The first report of leptin receptor deficiency was that of a homozygous mutation in three sisters from a consanguineous Algerian family.20 Much like the patients with congenital leptin deficiency, these three patients with no functional leptin receptor had severe, early-onset obesity. They had normal birth weight, rapid weight gain starting before six months of age, and weights greater than 15 kg at one year of age. When evaluated during adolescence, their BMIs were 50 to 70 kg/m2, their body fat was greater than 65%, and they lacked pubertal changes due to hypogonadotropic hypogonadism. Impaired growth hormone and thyrotropin secretion were subtle findings only evident with dynamic testing in these patients.20
Recently, eight more individuals with homozygous or compound heterozygous leptin receptor mutations were identified in a highly consanguineous cohort of severely obese and hyperphagic patients. Functional studies of these mutant receptors showed complete or partial loss of receptor signaling in response to leptin.11 The clinical features of severe obesity, hypogonadotropic hypogonadism, and impaired immune function were consistent with previous reports by Clement et al, and as expected, leptin levels were elevated proportional to fat mass.
Leptin and leptin receptor deficiencies are extraordinarily uncommon. The obesity resulting from these conditions is incomparable in severity, and associated with hypogonadism. If encountered in the clinical setting, a serum leptin level can help differentiate between the two conditions. While in leptin deficiency the leptin levels are typically low, in leptin receptor deficiency serum leptin reflects BMI and fat mass, like in common obesity, and all other forms of monogenic obesity. The fact that serum leptin levels in leptin receptor deficiency are not any higher than would be predicted by the degree of obesity emphasizes an important point. It shows that leptin synthesis occurs normally independent of a functional leptin receptor. Therefore, there is no feedback from leptin receptor signaling on the secretion of leptin from adipocytes.21
Proopiomelanocortin (POMC)
Proopiomelanocortin (POMC) is the precursor to five biologically active proteins made in the anterior pituitary and/or the hypothalamus and skin. POMC has a prominent role in the leptin-melanocortin system in that POMC-expressing neurons are targets of leptin signaling, and α-MSH is the POMC cleavage product that activates MC4R. Prohormone convertases 1/3 and 2 are necessary for the proteolytic cleavage of POMC to each of the active peptides. See Figures 1 and 2.
Figure 2. Processing of POMC.
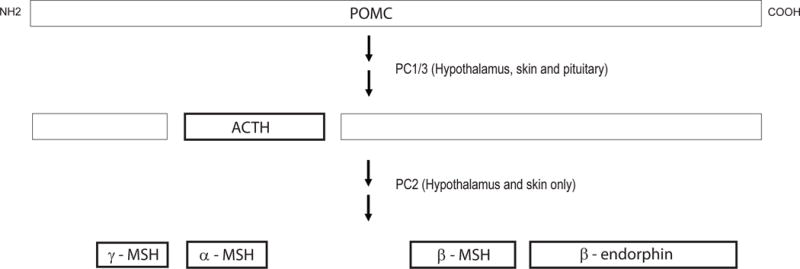
POMC proopiomelanocortin. ACTH adrenocorticotropic hormone. PC1/3 prohormone convertase 1/3. PC2 prohormone convertase 2. α-, β- and γ-MSH α-, β- and γ-melanocyte stimulating hormone.
POMC is processed by PC1/3 and PC2 into 5 biologically active proteins. In the corticotropes of the anterior pituitary, PC1/3 is expressed, but PC2 is not. Therefore, ACTH is the only biologically active POMC-derived peptide synthesized in the anterior pituitary. Both PC1/3 and PC2 are expressed in the melanotropes of the hypothalamus and skin. Thus, POMC is sequentially processed into α-, β- and γ-MSH and β-endorphin in these tissues. The phenotype of POMC deficiency is explained by the tissue-specific lack of these cleavage products.
The unique feature of complete POMC deficiency is that patients present in the newborn period with adrenal insufficiency. Their profound hypocortisolism is due to lack of the POMC substrate for ACTH synthesis in the anterior pituitary. Similar to patients with panhypopituitarism, on-going glucocorticoid replacement is required to prevent adrenal crises in these patients. The second salient feature of complete POMC deficiency that subsequently presents is the hyperphagia and severe obesity resulting from lack of MC4R activation by α-MSH. These patients have normal birth weight, onset of rapid weight gain before six months of age, and weights exceeding 15 kg by one year, and 25 kg by three years.
The first two patients with complete POMC deficiency were described in 1998. One patient was compound heterozygous for two nonsense mutations, and the other was homozygous for a base pair substitution that disrupted translation of the entire POMC protein.22 Three more patients with homozygous or compound heterozygous POMC mutations causing congenital POMC deficiency were described in 2003.23 And finally, the sixth case was reported in a Turkish patient with a homozygous frameshift loss of function mutation with severe obesity and ACTH deficiency, but dark hair.24 Red hair, due to lack of α-MSH activating MC1R in melanocytes, was initially reported as part of the clinical spectrum of congenital POMC deficiency. However, this finding is not essential for the diagnosis since the Turkish patient, and possibly his deceased, similarly affected brother, with complete POMC deficiency had dark hair.24
Prohormone convertase 1/3 (PC1/3)
Prohormone convertases (PCs) are a family of serine endoproteases that cleave inactive hormone precursors into biologically active secreted peptides. Of the seven members in this family of proteins, only PC1/3 and PC2 are selectively expressed in neuroendocrine tissues and are involved in the regulated secretory pathway of hormone biosynthesis.25 Substrates for PC1/3 and PC2 include proTRH, proinsulin, proglucagon, proGHRH, POMC, proproNPY, and proCART.18 Thus PC1/3 and PC2 are required for proper synthesis of many peptides involved in energy homeostasis. Moreover, the catalytic activities of PC1/3 and PC2 are tissue-specific, as seen in the example of POMC being processed differentially to ACTH in the pituitary and α-, β-, and γ-MSH in the hypothalamus. (Figure 2) Another example of the tissue-specificity of PC1/3 and PC2 is the cleavage of proglucagon to glucagon in the pancreatic α-cell, and to GLP-2 in the intestinal L cell.
Three cases of PC1/3 mutations that cause severe obesity have been reported.26–29 All three patients had hyperphagia and early-onset obesity thought to result from improper processing of POMC to α-MSH in hypothalamic neurons. Two of the patients had reported weights of more than 35 kg at 3 yrs of age. These patients also had mild hypocortisolism due to partial ACTH deficiency that was not as severe as in the patients with complete POMC deficiency. In addition, all three patients had malabsorption due to small bowel dysfunction, though with considerably variable severity. Improper processing of proglucagon in the intestinal cells to GLP-2, which has trophic effects on small-bowel epithelium, may contribute to poor integrity of the small bowel mucosa in these patients. Abnormalities of glucose homeostasis, namely postprandial hyperglycemia and subsequent reactive hypoglycemia, were noted in two of the three patients. This effect reflects abnormal processing of proinsulin to insulin in pancreatic β-cells.26,27 Other findings such as hypogonadotropic hypogonadism in one patient, and central hypothyroidism in another may be attributed to impaired proTRH and proGHRH processing by PC1/3.
It is important to note that these patients came to clinical attention due to reactive hypoglycemia in one case, and intractable neonatal diarrhea in the other two, rather than due to severe obesity. There are only three known cases of PC1/3 deficiency, the variability in their clinical phenotype is considerable, and obesity is not the distinguishing feature. Therefore, at this time, heterozygous PC1/3 mutations are extremely rare in the differential diagnosis of monogenic obesity. A better understanding of the various roles of PC1/3 in different tissues would improve our ability to clinically detect subtle deficiencies in its function. Currently, measurement of proinsulin and insulin levels after a glucose load to show a high proinsulin to insulin ratio is the only distinguishing lab evaluation to pursue if PC1/3 deficiency is suspected.
Melanocortin-4 Receptor (MC4R)
In 1998 two groups simultaneously reported the first two cases of severe obesity and hyperphagia due to MC4R mutations.30,31 Since then, MC4R has emerged as the most specialized and most crucial molecule for body weight regulation in the leptin-melanocortin system. First, MC4R mutations are inherited in an autosomal dominant fashion, with marked obesity resulting from only one affected allele. Second, aside from severe obesity and hyperphagia, there is no other physical, hormonal or developmental consequence of MC4R deficiency, making the function of this receptor very specific for energy balance. And third, mutations in MC4R are by far the most common cause of monogenic obesity known to date. Compared to the autosomal recessive mutations in leptin, leptin receptor, POMC and PC1/3 that together total only 32 reported cases of severe obesity in the world, the global prevalence of MC4R mutations is approximately 2.5% in severely obese individuals.32–35 Lubrano-Berthelier et al recently confirmed this prevalence of heterozygous, obesity-causing MC4R mutations to be 2.6% (2.83% in children with early-onset obesity and 2.35% in adults with later-onset obesity) in a large cohort of obese patients and non-obese controls.36
MC4R mutations segregate with obesity in the families of the probands, and are dominantly inherited with variable penetrance and expressivity. Therefore, the obesity phenotype of heterozygous MC4R mutation carriers can range from severely obese to lean. Functional studies of obesity-associated MC4R mutations show that more severely impaired receptor function in vitro correlates with earlier age of onset of obesity and higher BMI in the patient. The in vitro studies also show that each mutation impairs receptor function differently by affecting membrane expression, response to agonist and constitutive activity to a variable degree.36 The obesity phenotype of MC4R mutations is therefore determined not only by variable penetrance and expressivity, but also by allelic heterogeneity that contributes to different pathogenic mechanisms. Although no effective therapy for obesity due to MC4R mutations currently exists, there is hope that with on-going research, a better understanding of the mechanisms by which MC4R mutations cause obesity will lead to successful treatment options.
Heterozygous carriers of leptin, leptin receptor, and POMC mutations
The majority of obesity-causing MC4R mutations are heterozygous and dominantly inherited. Fewer than 10 cases of homozygous or compound heterozygous MC4R mutations have been reported.33, 37, 38 These individuals, lacking both alleles of MC4R, are significantly more obese than the heterozygotes, and are comparable to the patients with leptin, leptin receptor and POMC deficiency.
There is some evidence that an intermediate weight phenotype may exist for heterozygous carriers of leptin, leptin receptor and POMC mutations, implicating these genes in the susceptibility to common obesity. Farooqi et al. evaluated 13 heterozygous carriers of the leptin ΔG133 mutation, and found that serum leptin levels were significantly lower, while BMI and body fat mass were significantly higher in the heterozygotes than in controls.39 However, inter-individual variability in leptin measurements, and the small sample-size make interpretation of these results difficult. Heterozygous carriers of leptin receptor mutations were not severely obese, but had increased fat mass to the same extent as heterozygote leptin mutation carriers.11
Significantly higher BMIs were reported in the heterozygous relatives of a POMC deficient patient,24 and screening cohorts of severely obese patients has revealed heterozygous mutations in POMC that occur in the obese, but not in the controls.40,41, 42 These heterozygous POMC mutations segregate with obesity in the probands’ families and cause hyperphagia and obesity without any other clinical manifestations such as adrenal insufficiency. Thus, like heterozygous MC4R mutations, heterozygous POMC mutations may be a more common cause of monogenic obesity. The heterozygous carriers of PC1/3 mutations do not have an obvious phenotype. This is not surprising given the overlap of substrate specificity and functional redundancy between PC1/3 and PC2.
II. Obesity due to gene mutations that affect neurodevelopment
In this section we will discuss three genes, SIM1, BDNF and NTRK2, whose importance in hypothalamic development has been shown in mouse models. Recently, mutations in these genes have also been implicated in the development of obesity in both mice and humans. The mechanisms by which these genes regulate body weight are not known. It is possible that either abnormal development of the hypothalamus, or postnatal impairment of the function of these genes, or both are responsible for the obesity phenotype.
SIM1
In 2000, Holder et al described a girl with early-onset, severe obesity, hyperphagia, increased linear growth, and normal energy expenditure.43 Her rapid weight gain began at 3 months of age, such that she was almost 20 kg at 2 years, and more than 40 kg by 5 years. Her obesity was not associated with any developmental abnormalities, syndromic features or endocrine dysfunction. This patient had a de novo translocation that disrupted one of her SIM1 alleles on chromosome 6q.43
Mice missing one copy of Sim1 have the same phenotype as the patient, early-onset obesity with hyperphagia, normal energy expenditure, and increased linear growth, and also have a decrease in the total number of PVN (paraventricular nucleus) neurons.44,45 Since the PVN is the location of MC4R-expressing neurons that are critical for energy balance, it is hypothesized that abnormal development of the PVN causes obesity in Sim1 heterozygous mice and in the SIM1 haploinsufficient patient.
More recently there is evidence that SIM1 may have an ongoing, post-developmental role in energy balance, and specifically, that it may function downstream of MC4R to control food intake.46,47,48 However, the molecular pathways downstream of MC4R that regulate food intake are far from understood, and further studies are necessary to determine the exact role of SIM1 in these pathways.
Additional evidence for the role of SIM1 in the development of obesity comes from patients who are obese due to interstitial deletions of chromosome 6q that involve the SIM1 locus (6q16.2),49,50,51 and from significant linkage of childhood obesity related traits to the chromosomal region (6q22.31-q23.2) that contains SIM1.52
Therefore, haploinsufficiency of SIM1 has been shown to relate to severe, early-onset obesity in one patient with the translocation, and implicated in the cause of obesity in patients with interstitial deletions of chromosome 6 that include the SIM1 locus. Finally, rare point mutations in SIM1 are also shown to be significantly associated with obesity in a large screen of obese patients and matched controls.53 The extent of this association between rare SIM1 mutations and obesity was comparable only to that between MC4R mutations and obesity in this study. In vitro studies of these SIM1 mutations are needed to determine the functional significance and confirm the role in the development of obesity of these rare mutations.
BDNF and TRKB
BDNF (brain-derived neurotrophic factor) and its receptor TRKB (tropomyosin-related kinase B) regulate proliferation, survival and differentiation of neurons during development, and neuronal plasticity in the adult nervous system.54,55,56 Specific to energy balance, BDNF and TRKB modulate the development and postnatal plasticity of hypothalamic neurons, but both have also been shown to be important for memory, behavior and cognitive development.30,55,56 Partial deficiency of Bdnf and Trkb in mouse models causes hyperphagia and obesity.57–59 BDNF decreases food intake in mice,60,58,61 likely by acting downstream of MC4R.57
Last year the first human case of severe obesity due to haploinsufficiency of BDNF was reported.62 The patient was an 8 year-old girl who presented with hyperphagia and obesity. Her weight exceeded 20 kg at 2 years of age. She also had impaired cognition, memory and nociception, and hyperactivity. The patient had a de novo paracentric inversion on chromosome 11 that included the BDNF locus. Although it is possible that the inversion disrupts other unknown genes contributing to the patient’s phenotype, the marked similarity of this patient’s presentation to that of a patient with a mutation in NTRK, the gene that encodes TRKB, (see below) supports that her phenotype results from haploinsufficiency of BDNF.62 As in SIM1 haploinsufficiency, this patient’s obesity may result from a lack of BDNF during hypothalamic development, or from its impaired postnatal role in MC4R signaling and control of food intake.
One human case of a heterozygous de novo mutation in NTRK2 has been reported.63 The 8-year-old boy presented with hyperphagia and early-onset obesity of a similar magnitude to the BDNF haploinsufficent patient. He also had developmental delays, stereotyped behaviors, and impairment in memory, learning and nociception. Functional studies of his missense mutation showed significantly decreased BDNF-induced receptor autophosphorylation and activation of downstream signaling molecules.63, 64 The authors also found reduced neurite outgrowth and cell survival in response to BDNF in cells transfected with the mutant receptor,64 suggesting that post-developmental neuronal plasticity is also affected by NTRK2 mutations.
Screening a cohort of individuals with severe, early-onset obesity and developmental delay, revealed 3 other rare mutations in NTRK2 (I98V, P660L and T821A) that were not present in controls, but in vitro studies of these mutations did not show a significant difference in receptor function compared to wild-type.64
Although the exact role of these genes, SIM1, BDNF, and NTRK2, in the development of obesity has not been clearly delineated, their involvement in hypothalamic development, and their postnatal function possibly downstream of MC4R is suggested by evidence from mouse models.
III. Obesity associated with a pleiotropic developmental syndrome
There are a number of pleiotropic syndromes with obesity as a predominant phenotype in association with findings such as mental retardation, congenital organ defects, limb or facial dysmorphisms, and endocrine dysfunction. Prader-Willi Syndrome is the most common of such syndromes, characterized by neonatal hypotonia and failure to thrive, and subsequent obesity due to intense hyperphagia along with developmental delay, mental retardation, hypogonadism and small hands and feet. The genetic basis of these syndromes is complex. Although the genes or chromosomal regions implicated in the etiology of many of these syndromes are known, their relationship to the development of obesity is not clear. Many monogenic obesity-associated syndromes have been reviewed elsewhere,5, 65, 66 and therefore, will not be addressed in this review. We will focus on three syndromes with multiple phenotypic similarities in addition to obesity whose pathogenesis has recently been linked to the dysfunction of the primary cilium.
The primary cilium is an organelle extending from almost all eukaryotic cells. Its architecture differs from that of the more common motile cilium in that its axoneme is made up of 9 microtubule doublets only (9 + 0) without the additional central doublet present in motile cilia (9 + 2).67 Primary cilia are attached to the cell at the basal body, and are important for chemo- and mechano-sensation of the environment and transduction of intracellular signaling in the cell. Many important signaling pathways such as hedgehog signaling and the Wnt pathway localize to the primary cilium.68 Since protein synthesis does not occur in cilia, a mechanism called intraflagellar transport (IFT) is required to carry proteins necessary for ciliary maintenance and function into and out of the cilia.67
Bardet-Biedl Syndrome
Bardet-Biedl syndrome (BBS) is characterized by clinical findings of retinal degeneration, post-axial polydactyly, obesity, and structural or functional defects of the kidney. Other associated findings include anosmia, mental retardation, hepatic fibrosis, male hypogonadism or undescended testes, female urogenital tract abnormalities, type 2 diabetes mellitus, hypertension, cardiac abnormalities, Hirschsprung disease, situs inversus, and predisposition to malignancies.69, 70,71 Obesity in patients with BBS ranges from mild to severe, and is reversible with caloric restriction and exercise. Rapid weight gain in the first year of life is associated with hyperphagia. No difference in resting metabolic rate has been observed between BBS patients and matched obese controls, but lower level of spontaneous physical activity in BBS patients has been reported.72
BBS is rare and genetically heterogeneous. Mutations in 12 genes, BBS1-12, have been identified that contribute to the development of the phenotype.73 The functions of these genes are not well delineated, but are somehow linked to the primary cilium.74
Nachury et al. recently showed that 7 of the 12 BBS-causing genes encode highly conserved proteins that are necessary for primary cilia function. These proteins form a complex, called the BBSome, and associate with another factor, Rab8GTP, to facilitate transport of proteins to the primary cilium.73
Despite exciting advances in the understanding of BBS pathogenesis and its relationship to ciliary function, the etiology of obesity associated with this syndrome is still largely unclear. Dysfunction of cilia in specific neurons, could explain obesity due to hyperphagia and impaired satiety, since Davenport et al showed that deletion of cilia on neurons throughout the CNS and specifically from POMC-expressing neurons causes obesity in mice.75 This is the first evidence that even a novel mechanism of pathogenesis such as primary ciliary dysfunction, may relate to the hypothalamic regulation of food intake in causing obesity. But this evidence is preliminary, and further research is needed. That hypothalamic POMC neurons are critical for signaling satiety is well established, but how disruption of ciliary function in these neurons affects their role in energy balance is still not.
Alstrom Syndrome
Alstrom sydrome is another rare syndrome that shares many of the pleiotropic clinical findings of BBS, namely, retinal degeneration, early-onset obesity, type 2 diabetes mellitus and perceptive hearing loss. It is an autosomal recessive disorder caused by mutations in the ALMS1 gene. It is also associated with cardiomyopathy, liver and kidney dysfunction and delayed puberty. The pathogenesis of Alstrom syndrome has also been linked to dysfunction of the primary cilium, in that the ALMS1 protein localizes to the centrosome and ciliary basal body, and likely has a role in the formation or maintenance of primary cilia. Li et al show that ALMS1 has an important role in cilia formation in kidney cells.76 Human mutations in ALMS1 know to cause Alstrom syndrome, result in truncated ALMS1 proteins. These truncated proteins are able to support normal cilia formation, but may cause a subtler and as yet undetermined alteration in ciliary function that leads to the development of the Alstrom phenotype. Residual function of mutant ALMS1 in Alstrom syndrome explains lack of a more severe developmental phenotype.
Carpenter Syndrome
Carpenter syndrome is a pleiotropic disorder with the following features: craniosynostosis affecting primarily metopic and sagittal sutures, polydactyly, soft-tissue syndactyly, and obesity. Other associated findings include brachydactyly, molar agenesis, genu valgum, hypogenitalism, congenital heart defects, umbilical hernia, and learning disability. The disorder has an autosomal recessive inheritance, and was recently described to be due to a homozygous nonsense mutation L145X in RAB23 in 5 affected individuals from 3 families.77 Evaluation of additional patients with Carpenter syndrome identified four other mutations in RAB23. Similar nonsense mutations in mouse Rab23 gene leads to a far more severe phenotype of neural tube defect leading to exencephaly and embryonic lethality.
Rab23 is from Rab family of small GTPases that regulate intracellular trafficking of membrane-associated proteins. Rab23 negatively regulates the Sonic hedgehog signaling pathway.78,79,80,81 Yoshimura et al. recently showed that Rab23 is one of 3 Rab GTPases (Rab8a, Rab 17 and Rab 23) involved in the formation of the primary cilium.82
The phenotype of Carpenter syndrome shares findings of limb deformities (polysyndactyly and brachydactyly) with other syndromes that result from impaired hedgehog signaling. However, findings of craniosynostosis and obesity have not been previously associated with the hedgehog pathway. Given the evidence that obesity in BBS and Alstrom syndrome is associated with dysfunction of the primary cilium, and given that HH signaling occurs on the primary cilium in many cell types, it is possible to implicate ciliary dysfunction that disrupts hedgehog signaling in the pathogenesis of Carpenter syndrome.
Summary of lessons learned
The lessons gleaned from the study of extreme human obesity are as follows. 1. The long-term regulation of body weight in humans is centered in the hypothalamus. Within the hypothalamus, the leptin-melanocortin system is critical for energy balance, since disruption of these pathways that sense peripheral energy stores and signal satiety leads to the most severe forms of human obesity. Furthermore, MC4R is the most specialized molecule for body weight maintenance within this system because MC4R deficiency has no other clinical phenotype. 2. The monogenic causes of obesity identified thus far account for less than 5% of severe obesity, and are in themselves very heterogeneous. BBS for example can result from alterations in at least 12 different genes, and obesity due to MC4R mutations can result from different mechanisms that affect receptor function. Furthermore, novel mechanisms are emerging as important for pathogenicity of obesity such as abnormal hypothalamic development, alterations in neuronal plasticity and dysfunction of the primary cilium. Therefore, the currently characterized monogenic forms of obesity can be viewed as the “tip of the iceberg” giving us clues that the pathogenic mechanisms underlying common obesity is equally heterogeneous. 3. Treatment of congenital leptin deficiency with leptin, is a rare but powerful example of successful therapy arising from an understanding of the molecular pathogenesis. Thus further research to understand the pathogenic mechanisms underlying obesity is requisite for the development of similarly rational and effective treatments. However, this research is slow and challenging because of the great genetic heterogeneity of the disorder. 4. The unavailability of specific therapies to treat the various genetic causes of obesity highlights a dichotomy in the approach to an obese patient: although there is currently no direct benefit to the patient in knowing the genetic basis of his disease, it is important from a research perspective to further explore the genetic cause of this phenotype. By elucidating the molecular mechanisms underlying obesity alone can we rationally approach and effectively treat this devastating condition.
Acknowledgments
We thank Jimmy Chen for his contribution to the artwork.
This work was supported by the National Institutes of Health RO1 DK60540 and DK068152 Awards, and American Heart Association Established Investigator Award (to CV) and National Institutes of Health (T32) Award (to SR).
References
- 1.Ogden CL, Flegal KM, Carroll MD, Johnson CL. Prevalence and trends in overweight among US children and adolescents, 1999-2000. Jama. 2002 Oct 9;288(14):1728–1732. doi: 10.1001/jama.288.14.1728. [DOI] [PubMed] [Google Scholar]
- 2.Barsh GS, Farooqi IS, O’Rahilly S. Genetics of body-weight regulation. Nature. 2000 Apr 6;404(6778):644–651. doi: 10.1038/35007519. [DOI] [PubMed] [Google Scholar]
- 3.Farooqi S, O’Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006 Dec;27(7):710–718. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 4.Bell CG, Walley AJ, Froguel P. The genetics of human obesity. Nat Rev Genet. 2005 Mar;6(3):221–234. doi: 10.1038/nrg1556. [DOI] [PubMed] [Google Scholar]
- 5.Farooqi IS. Genetic and hereditary aspects of childhood obesity. Best Pract Res Clin Endocrinol Metab. 2005 Sep;19(3):359–374. doi: 10.1016/j.beem.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Ellacott KL, Cone RD. The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond B Biol Sci. 2006 Jul 29;361(1471):1265–1274. doi: 10.1098/rstb.2006.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005 May;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 8.Butler AA. The melanocortin system and energy balance. Peptides. 2006 Feb;27(2):281–290. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niswender KD, Baskin DG, Schwartz MW. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab. 2004 Oct;15(8):362–369. doi: 10.1016/j.tem.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Coll AP, Farooqi IS, O’Rahilly S. The hormonal control of food intake. Cell. 2007 Apr 20;129(2):251–262. doi: 10.1016/j.cell.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farooqi IS, Wangensteen T, Collins S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007 Jan 18;356(3):237–247. doi: 10.1056/NEJMoa063988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997 Jun 26;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 13.Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998 Mar;18(3):213–215. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 14.Gibson WT, Farooqi IS, Moreau M, et al. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J Clin Endocrinol Metab. 2004 Oct;89(10):4821–4826. doi: 10.1210/jc.2004-0376. [DOI] [PubMed] [Google Scholar]
- 15.Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002 Oct;110(8):1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998 Oct 22;395(6704):763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 17.Farooqi IS, O’Rahilly S. Monogenic human obesity syndromes. Recent Prog Horm Res. 2004;59:409–424. doi: 10.1210/rp.59.1.409. [DOI] [PubMed] [Google Scholar]
- 18.Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin. Endocrinology. 2007 Sep;148(9):4191–4200. doi: 10.1210/en.2007-0173. [DOI] [PubMed] [Google Scholar]
- 19.Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Jama. 1999 Oct 27;282(16):1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 20.Clement K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998 Mar 26;392(6674):398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 21.Lahlou N, Clement K, Carel JC, et al. Soluble leptin receptor in serum of subjects with complete resistance to leptin: relation to fat mass. Diabetes. 2000 Aug;49(8):1347–1352. doi: 10.2337/diabetes.49.8.1347. [DOI] [PubMed] [Google Scholar]
- 22.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998 Jun;19(2):155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 23.Krude H, Biebermann H, Schnabel D, et al. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4-10. J Clin Endocrinol Metab. 2003 Oct;88(10):4633–4640. doi: 10.1210/jc.2003-030502. [DOI] [PubMed] [Google Scholar]
- 24.Farooqi IS, Drop S, Clements A, et al. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006 Sep;55(9):2549–2553. doi: 10.2337/db06-0214. [DOI] [PubMed] [Google Scholar]
- 25.Rouille Y, Duguay SJ, Lund K, et al. Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases. Front Neuroendocrinol. 1995 Oct;16(4):322–361. doi: 10.1006/frne.1995.1012. [DOI] [PubMed] [Google Scholar]
- 26.O’Rahilly S, Gray H, Humphreys PJ, et al. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995 Nov 23;333(21):1386–1390. doi: 10.1056/NEJM199511233332104. [DOI] [PubMed] [Google Scholar]
- 27.Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997 Jul;16(3):303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 28.Jackson RS, Creemers JW, Farooqi IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003 Nov;112(10):1550–1560. doi: 10.1172/JCI18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farooqi IS, Volders K, Stanhope R, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 2007 Sep;92(9):3369–3373. doi: 10.1210/jc.2007-0687. [DOI] [PubMed] [Google Scholar]
- 30.Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998 Oct;20(2):113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 31.Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998 Oct;20(2):111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 32.Hainerova I, Larsen LH, Holst B, et al. Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis. J Clin Endocrinol Metab. 2007 Sep;92(9):3689–3696. doi: 10.1210/jc.2007-0352. [DOI] [PubMed] [Google Scholar]
- 33.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003 Mar 20;348(12):1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 34.Zakel UA, Wudy SA, Heinzel-Gutenbrunner M, et al. Prevalence of melanocortin 4 receptor (MC4R) mutations and polymorphismsin consecutively ascertained obese children and adolescents from a pediatric health care utilization population. Klin Padiatr. 2005 Jul-Aug;217(4):244–249. doi: 10.1055/s-2005-836589. [DOI] [PubMed] [Google Scholar]
- 35.Wang CL, Liang L, Wang HJ, Fu JF, Hebebrand J, Hinney A. Several mutations in the melanocortin 4 receptor gene are associated with obesity in Chinese children and adolescents. J Endocrinol Invest. 2006 Nov;29(10):894–898. doi: 10.1007/BF03349193. [DOI] [PubMed] [Google Scholar]
- 36.Lubrano-Berthelier C, Dubern B, Lacorte JM, et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab. 2006 May;91(5):1811–1818. doi: 10.1210/jc.2005-1411. [DOI] [PubMed] [Google Scholar]
- 37.Lubrano-Berthelier C, Le Stunff C, Bougneres P, Vaisse C. A homozygous null mutation delineates the role of the melanocortin-4 receptor in humans. J Clin Endocrinol Metab. 2004 May;89(5):2028–2032. doi: 10.1210/jc.2003-031993. [DOI] [PubMed] [Google Scholar]
- 38.Dubern B, Bisbis S, Talbaoui H, et al. Homozygous null mutation of the melanocortin-4 receptor and severe early-onset obesity. J Pediatr. 2007 Jun;150(6):613–617. 617 e611. doi: 10.1016/j.jpeds.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 39.Farooqi IS, Keogh JM, Kamath S, et al. Partial leptin deficiency and human adiposity. Nature. 2001 Nov 1;414(6859):34–35. doi: 10.1038/35102112. [DOI] [PubMed] [Google Scholar]
- 40.Challis BG, Pritchard LE, Creemers JW, et al. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum Mol Genet. 2002 Aug 15;11(17):1997–2004. doi: 10.1093/hmg/11.17.1997. [DOI] [PubMed] [Google Scholar]
- 41.Biebermann H, Castaneda TR, van Landeghem F, et al. A role for beta-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab. 2006 Feb;3(2):141–146. doi: 10.1016/j.cmet.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Lee YS, Challis BG, Thompson DA, et al. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab. 2006 Feb;3(2):135–140. doi: 10.1016/j.cmet.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000 Jan 1;9(1):101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 44.Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998 Oct 15;12(20):3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michaud JL, Boucher F, Melnyk A, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001 Jul 1;10(14):1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 46.Kublaoui BM, Holder JL, Jr, Tolson KP, Gemelli T, Zinn AR. SIM1 overexpression partially rescues agouti yellow and diet-induced obesity by normalizing food intake. Endocrinology. 2006 Oct;147(10):4542–4549. doi: 10.1210/en.2006-0453. [DOI] [PubMed] [Google Scholar]
- 47.Kublaoui BM, Holder JL, Jr, Gemelli T, Zinn AR. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol. 2006 Oct;20(10):2483–2492. doi: 10.1210/me.2005-0483. [DOI] [PubMed] [Google Scholar]
- 48.Holder JL, Jr, Zhang L, Kublaoui BM, et al. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab. 2004 Jul;287(1):E105–113. doi: 10.1152/ajpendo.00446.2003. [DOI] [PubMed] [Google Scholar]
- 49.Villa A, Urioste M, Bofarull JM, Martinez-Frias ML. De novo interstitial deletion q16.2q21 on chromosome 6. Am J Med Genet. 1995 Jan 30;55(3):379–383. doi: 10.1002/ajmg.1320550326. [DOI] [PubMed] [Google Scholar]
- 50.Gilhuis HJ, van Ravenswaaij CM, Hamel BJ, Gabreels FJ. Interstitial 6q deletion with a Prader-Willi-like phenotype: a new case and review of the literature. Eur J Paediatr Neurol. 2000;4(1):39–43. doi: 10.1053/ejpn.1999.0259. [DOI] [PubMed] [Google Scholar]
- 51.Faivre L, Cormier-Daire V, Lapierre JM, et al. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J Med Genet. 2002 Aug;39(8):594–596. doi: 10.1136/jmg.39.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyre D, Lecoeur C, Delplanque J, et al. A genome-wide scan for childhood obesity-associated traits in French families shows significant linkage on chromosome 6q22.31-q23.2. Diabetes. 2004 Mar;53(3):803–811. doi: 10.2337/diabetes.53.3.803. [DOI] [PubMed] [Google Scholar]
- 53.Ahituv N, Kavaslar N, Schackwitz W, et al. Medical sequencing at the extremes of human body mass. Am J Hum Genet. 2007 Apr;80(4):779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tapia-Arancibia L, Rage F, Givalois L, Arancibia S. Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol. 2004 Jul;25(2):77–107. doi: 10.1016/j.yfrne.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 57.Xu B, Goulding EH, Zang K, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003 Jul;6(7):736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. Embo J. 2000 Mar 15;19(6):1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lyons WE, Mamounas LA, Ricaurte GA, et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999 Dec 21;96(26):15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pelleymounter MA, Cullen MJ, Wellman CL. Characteristics of BDNF-induced weight loss. Exp Neurol. 1995 Feb;131(2):229–238. doi: 10.1016/0014-4886(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 61.Rios M, Fan G, Fekete C, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001 Oct;15(10):1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 62.Gray J, Yeo GS, Cox JJ, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006 Dec;55(12):3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeo GS, Connie Hung CC, Rochford J, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004 Nov;7(11):1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 64.Gray J, Yeo G, Hung C, et al. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int J Obes (Lond) 2007 Feb;31(2):359–364. doi: 10.1038/sj.ijo.0803390. [DOI] [PubMed] [Google Scholar]
- 65.Mutch DM, Clement K. Unraveling the genetics of human obesity. PLoS Genet. 2006 Dec 29;2(12):e188. doi: 10.1371/journal.pgen.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Rahilly S, Farooqi IS, Yeo GS, Challis BG. Minireview: human obesity-lessons from monogenic disorders. Endocrinology. 2003 Sep;144(9):3757–3764. doi: 10.1210/en.2003-0373. [DOI] [PubMed] [Google Scholar]
- 67.Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol. 2005 Dec;289(6):F1159–1169. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- 68.Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006 Aug 4;313(5787):629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 69.Kulaga HM, Leitch CC, Eichers ER, et al. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet. 2004 Sep;36(9):994–998. doi: 10.1038/ng1418. [DOI] [PubMed] [Google Scholar]
- 70.Lorda-Sanchez I, Ayuso C, Ibanez A. Situs inversus and hirschsprung disease: two uncommon manifestations in Bardet-Biedl syndrome. Am J Med Genet. 2000 Jan 3;90(1):80–81. doi: 10.1002/(sici)1096-8628(20000103)90:1<80::aid-ajmg14>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 71.Tobin JL, Beales PL. Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007 Jul;22(7):926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grace C, Beales P, Summerbell C, et al. Energy metabolism in Bardet-Biedl syndrome. Int J Obes Relat Metab Disord. 2003 Nov;27(11):1319–1324. doi: 10.1038/sj.ijo.0802420. [DOI] [PubMed] [Google Scholar]
- 73.Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007 Jun 15;129(6):1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 74.Ansley SJ, Badano JL, Blacque OE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003 Oct 9;425(6958):628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 75.Davenport JR, Watts AJ, Roper VC, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007 Sep 18;17(18):1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li G, Vega R, Nelms K, et al. A role for Alstrom syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007 Jan 5;3(1):e8. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jenkins D, Seelow D, Jehee FS, et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007 Jun;80(6):1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eggenschwiler JT, Espinoza E, Anderson KV. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001 Jul 12;412(6843):194–198. doi: 10.1038/35084089. [DOI] [PubMed] [Google Scholar]
- 79.Evans TM, Ferguson C, Wainwright BJ, Parton RG, Wicking C. Rab23, a negative regulator of hedgehog signaling, localizes to the plasma membrane and the endocytic pathway. Traffic. 2003 Dec;4(12):869–884. doi: 10.1046/j.1600-0854.2003.00141.x. [DOI] [PubMed] [Google Scholar]
- 80.Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev Biol. 2006 Feb 1;290(1):1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 81.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003 Nov 6;426(6962):83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 82.Yoshimura S, Egerer J, Fuchs E, Haas AK, Barr FA. Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol. 2007 Jul 30;178(3):363–369. doi: 10.1083/jcb.200703047. [DOI] [PMC free article] [PubMed] [Google Scholar]