

Abstract
Standard antibiotic-based strategies for the treatment of Clostridium difficile infections disrupt indigenous microbiota and commonly fail to eradicate bacterial spores, two key factors that allow recurrence of infection. As an alternative approach to controlling C. difficile infection, a series of bile acid derivatives have been prepared that inhibit taurocholate-induced spore germination. These analogues have been evaluated in a highly virulent NAP1 strain using optical density and phase-contrast microscopy assays. Heterocycle substitutions at C24 were well-tolerated and several tetrazole-containing derivatives were highly potent inhibitors in both assays, with complete inhibition of spore germination observed at 10–25 μM. To limit intestinal absorption, C7-sulfated analogues designed to avoid active and passive transport pathways were prepared. One of these derivatives, compound 21b, was found to be a potent inhibitor of C. difficile spore germination and poorly permeable in a Caco-2 model of intestinal epithelial absorption, suggesting that it is likely to be gut-restricted.
Graphical Abstract
INTRODUCTION
Clostridium difficile is an anaerobic, spore-forming, Gram-positive bacterium that causes a potentially fatal infection of the colon. C. difficile infection (CDI) is the most common hospital-acquired infection, and in recent years CDI has also become increasingly acquired in the community.1 The infection affects approximately 500000 patients annually and leads to death in 30000 patients per year in the U.S. alone.2–5 CDI typically occurs after the disruption of gut microbiota following a course of antibiotics and subsequent ingestion of C. difficile spores from the surrounding environment.6 The current standard of care also relies on antibiotics. However, this strategy can perpetuate recurrence of CDI because it fails to eliminate C. difficile spores and results in further suppression of indigenous microbiota. After the initial infection and antibiotic treatment, relapse can occur in 20–40% of patients, with a higher risk of recurrence associated with each episode of infection.7 A large fraction of patients ultimately develops an indefinite syndrome of recurrent CDI (R-CDI), which becomes refractory to eradication with antibiotics alone.8
To prevent R-CDI, there has been a shift in the design of C. difficile therapies toward developing antibiotics that are more selective for this bacterium and minimize the suppression of gut microbiota.9 In addition to the recently approved drug fidaxomicin, there are several new compounds being evaluated as narrow-spectrum antibiotics in clinical trials for CDI and prevention of R-CDI, including CRS3123 (phase I), SMT19969 (phase II), LFF571 (phase II), and ramoplanin (phase III, Figure 1).9 There are also existing drugs that are being repurposed for CDI in phase III trials (CB-183,315 and nitazoxanide, Figure 1).9 Outside of antibacterial therapeutics, recent evidence suggested that targeting the C. difficile toxins (TdcA and TdcB) through monoclonal antibody treatment reduces the recurrence of CDI.10
Figure 1.

Structures of narrow spectrum antibiotics currently in clinical trials for the treatment of C. difficile infection.
Recently, fecal microbiota transplantation (FMT) has emerged as a highly effective treatment in breaking the cycle of CDI recurrence. This procedure involves administration of fecal microbiota from healthy donors into the colon of patients.8 It results in prompt and sustained normalization of fecal microbial community structure in the recipients.11 A number of mechanistic investigations of FMT in treatment of R-CDI have converged on the critical role of secondary bile acid metabolism, which is normally mediated by gut microbiota, in inhibiting C. difficile spore germination and expansion in the colon.8,11–13 Spores of C. difficile germinate in the distal gut when they sense the appropriate host environment (signaled in large part by bile acids) and favorable nutrient conditions (signaled by glycine).14–17 Bile acids in the cholic acid (CA) family typically promote germination of C. difficile spores, while those in the chenodeoxycholic acid (CDCA) family generally inhibit C. difficile spore germination (Figure 2).13,17,18 Taurocholic acid (TCA), a potent progerminant, is routinely included in isolation media for C. difficile.19–21
Figure 2.

Structures of endogenous bile acids cholic acid (CA), taurocholic acid (TCA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA, 1a), and lithocholic acid (LCA).
Under normal physiologic conditions, colonic microbiota mediate 7α-dehydroxylation of primary bile acids, yielding deoxycholic acid (DCA) and lithocholic acid (LCA) from CA and CDCA, respectively.22,23 This metabolism is disrupted by the administration of antibiotics. The fecal bile acid composition in patients with R-CDI syndrome promotes C. difficile spore germination and vegetative growth when evaluated using in vitro assays.13 FMT helps restore normal bile acid concentrations, leading to compositions that are inhibitory to spore germination and vegetative growth.13
However, some patients are not good candidates for FMT because they continue to require frequent antibiotics for non-C. difficile indications. Common examples in our clinic include patients with osteomyelitis, recurrent urinary tract infections, and recurrent sinusitis. A potentially complementary approach to preventing CDI without further disrupting the indigenous microbiota is to directly inhibit the germination of C. difficile spores.
In 2010, Sorg and Sonenshein reported that several commercially available synthetic analogues of CDCA were more potent than the natural bile acid at inhibiting TCA-promoted spore germination in the UK1 epidemic strain.24 The most potent compound they reported was the C24 methyl ester derivative of CDCA (2a), with a Ki of 44 μM. Although this work provides one of the first examples of bile acid analogues successfully inhibiting C. difficile spore germination in vitro, the in vivo use of 2a is potentially problematic because of the susceptibility of methyl esters to chemical or enzymatic hydrolysis.25 In addition, bile acids are efficiently absorbed from the intestinal lumen by active and passive absorption mechanisms,26,27 and it is likely that enterohepatic recycling would prevent 2a from reaching the concentration in the colon necessary to inhibit C. difficile spore germination. More recently, Sorg and co-workers have focused on investigating the mechanism by which bile acids inhibit or promote C. difficile spore germination. In their 2013 paper, these researchers hypothesized that the germination-specific protease, CspC, served as a bile acid receptor for C. difficile and found that mutations in this receptor could alter germinant specificity.14 In their originally proposed model, CspC then activates CspB protease, which in turn triggers the degradation of the spore cortex by the spore cortex lytic enzyme SleC.28 In 2016, this model was revised based on data showing a potential inverse correlation between germination rate and CspC abundance. The researchers now hypothesize that CspC serves to both activate CspB and inhibit SleC but that the ability of CspC to inhibit SleC may vary depending on the presence of an additional protein GerS.28
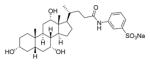
Other examples of bile acid-based inhibitors of spore germination include compounds discovered by Abel-Santos and co-workers. Their lead compound (3) was a potent inhibitor of spore germination in ATCC strain 630, with an IC50 of 58 ± 35 μM.29 These researchers also reported that compound 3 (Figure 3) eradicated CDI symptoms in a murine infection model of the same strain.30
Figure 3.
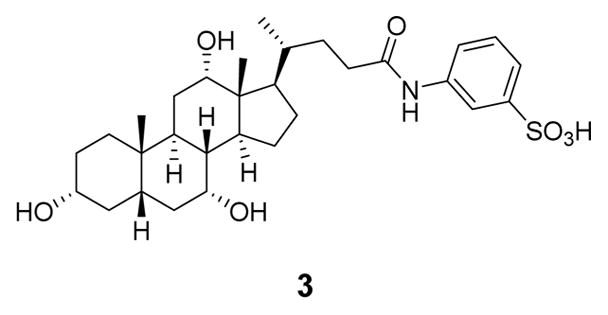
Likely structure of 3, the most potent inhibitor of strain ATCC 630 described by Abel-Santos and co-workers.29,31
The endogenous bile acid UDCA (1b, Figure 4) is another known inhibitor of spore germination and was active at ~500 μM across several strains of C. difficile, including a highly virulent NAP1 strain.24,32 Unfortunately, UDCA is unlikely to be a useful therapeutic agent in most patients because it is effectively reabsorbed into the enterohepatic circulation from the ileum, thus preventing the necessary intracolonic concentration needed to suppress C. difficile from being reached. However, we recently reported successful treatment of a patient with C. difficile ileal pouchitis with oral dosing of UDCA.32 We were able to achieve therapeutic levels of UDCA in the patient’s ileal pouch because she had undergone a colectomy, which disrupts enterohepatic recycling. This result provides further evidence that bile acids can be used to treat R-CDI if adequate concentrations can be achieved clinically.
Figure 4.
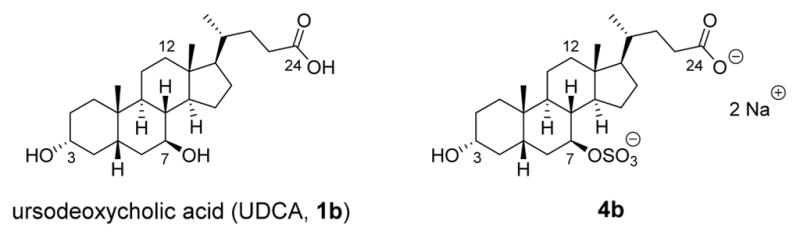
Structures of naturally occurring bile acids 1b and 4b.
One way to avoid bile acid reuptake into the enterohepatic circulation is with the addition of a sulfate group to the C7-position of UDCA (compound 4b in Figure 4). In 1995, Rodrigues and co-workers reported that 4b was gut restricted and when administered orally was found to be eliminated intact in the feces of rats.33 This observation agrees with the later findings of Kolhatkar and Polli, who observed that C7-substituted bile acids typically do not undergo active transport by the apical sodium-dependent bile acid transporter (ASBT), the primary transporter of bile acids from the gut.34 Additionally, the negatively charged sulfate group impedes passive absorption of the compound. This limited absorption is desirable for treating an infection in the lower intestine, as it provides a way to effectively increase the concentration of the therapeutic in the target area while minimizing unwanted systemic effects.35 An additional advantage is that bile acids sulfated at C7 are resistant to desulfation and conjugation with taurine or glycine and thus are typically excreted unchanged in fecal matter.36
With these concepts in mind, we evaluated the ability of 4b to prevent C. difficile spore germination. Unfortunately, while we determined that 4b inhibits NAP1 spore germination, it is significantly less potent than UDCA (3000 μM, Supporting Information, Figure S1). Thus, any bile acid derivative containing a C7-sulfate group to restrict intestinal absorption is likely to need additional structural modifications to increase potency in order to counteract the detrimental effect on potency that comes from incorporating this moiety.
As part of our continued efforts to develop improved methods of treating CDI, we set out to develop novel, gut-restricted, bile acid analogues capable of inhibiting C. difficile spore germination. This approach builds on the natural mechanisms that control the lifecycle of C. difficile and has the advantage of not placing selective pressure on dividing cells, which may limit the potential of developing resistance.6 Herein, we report the synthesis of modified bile acid derivatives of CDCA and UDCA. These compounds have been evaluated in optical density and microscope-based spore germination assays, and we have identified several analogues that are highly potent inhibitors of C. difficile spore germination.
RESULTS AND DISCUSSION
Chemistry
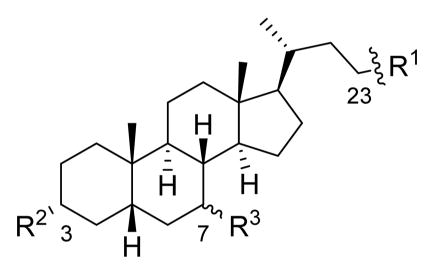
CDCA (1a) and UDCA (1b) are epimeric endogenous bile acids, with the C7-hydroxyl group of 1a in the α position and the C7-hydroxyl group of 1b in the β position. The syntheses of analogues based on the 1b scaffold are shown in the synthetic schemes below. Analogues of 1a were prepared in the same manner, except for compound 11a (see Scheme S1 in the Supporting Information). Initially, carboxylic acid and ester-containing derivatives were prepared to explore the SAR of modifying the C3- and C7-hydroxyl groups and for comparison with the inhibitory compounds described by Sorg and Sonenshein.24 A series of analogues containing bioisosteric replacements37 for carboxylic acids and esters, including amides and various heterocycles, were subsequently synthesized in order to obtain compounds that are less likely to be metabolized in the environment of the intestinal tract.
Synthesis of Esters and Carboxylic Acid Analogues
Treatment of 1b with p-toluenesulfonic acid in methanol afforded the methyl ester 2b (Scheme 1).38 Surprisingly, methylation of the C3 and/or C7 alcohols of 2b with sodium hydride and methyl iodide in THF generated the carboxylic acid analogues 5b and 6b instead of the expected esters 7b and 8b. The methyl ester analogues 7b and 8b were instead obtained by methylation with methyl triflate and 2,6-di-tert-butylpyridine.39 A three-step sequence of methylation, silyl deprotection, and esterification provided 11b from compound 9b, which was prepared as previously described.40 Addition of MeMgBr to the methyl ester of commercially available C7-ketone 12 produced the tertiary alcohols 13 and 14, with high stereoselectivity that matched the observed diastereomer reported by Une and co-workers.41
Scheme 1. Synthesis of Methylated Esters and Carboxylic Acidsa.

aReagents and conditions: (a) pTSA·H2O, MeOH; (b) NaH, MeI, THF; (c) MeOTf, 2,6-di-tert-butylpyridine, DCM; (d) TBAF, THF; (e) MeMgBr, Et2O.
Synthesis of Amide Analogues
Primary amide 16b was prepared by acylation of the C3 and C7 alcohols of 1b, followed by conversion to the acid chloride and amidation with ammonia gas (Scheme 2).42 Amides 17b and 18b were synthesized through HATU-mediated coupling of 1b with n-butylamine or pyrrolidine, respectively. Similarly, HATU coupling of 1a with 2-amino-2-methyl-propanol afforded 22a (the corresponding UDCA analogue 22b was not prepared). Compound 18b was methylated with sodium hydride and methyl iodide to afford bis-methylated amide 19b and monomethylated amide 20b. A sulfated derivative was synthesized by reaction of 20b with chlorosulfonic acid in pyridine and was isolated as a triethylammonium salt through purification via reverse-phase column chromatography. The triethylammonium salt was then converted to the sodium salt (21b) by ion-exchange chromatography.43
Scheme 2. Synthesis of Amide Analoguesa.

aReagents and conditions: (a) Ac2O, cat. DMAP, pyridine; (b) SOCl2, benzene, 80 °C, then NH3; (c) HATU, TEA, nbutylamine, DCM; (d) HATU, TEA, pyrrolidine, DCM; (e) NaH, MeI, THF; (f) ClSO3H, pyridine, 50 °C, ion exchange using Na+-Dowex resin; (g) HATU, TEA, 2-amino-2-methyl-propanol, DCM.
Synthesis of Oxadiazole Analogues
The 1,2,4-oxadiazole analogue was synthesized in a two-step procedure via a HATU-mediated amide coupling of 1b to (Z)-1-(hydroxyamino)prop-1-en-2-amine, followed by microwave-assisted dehydrative cyclization to afford 24b (Scheme 3).44 The isomeric 1,3,4-oxadiazole was synthesized by conversion of carboxylic acid 15b to hydrazine 25b, followed by dehydrative cyclization using tosyl chloride and TEA and removal of the acetate protecting groups under basic conditions to obtain 27b (Scheme 4).45
Scheme 3. Synthesis of 1,2,4-Oxadiazolea.

aReagents and conditions: (a) HATU, DIPEA, (Z)-1-(hydroxyamino)prop-1-en-2-amine, DCM; (b) toluene, THF, 160 °C, 45 min.
Scheme 4. Synthesis of 1,3,4-Oxadiazolea.

aReagents and conditions: (a) HATU, TEA, acetohydrazide, DCM; (b) TsCl, TEA, DCM; (b) NaOH, MeOH, 75 °C.
Synthesis of the Oxazoline Analogue
To prepare the oxazoline analogue of 1b, the C3 and C7 alcohols were first protected as formate esters (28b, Scheme 5). The carboxylic acid was converted to the corresponding acid chloride and reaction with 2-amino-2-methyl-propanol afforded the desired amide 29b. Thionyl chloride-mediated dehydrative cyclization of the amide afforded the oxazoline 30b, and the hydroxyl groups were deprotected under basic conditions to provide oxazoline analogue 31b.
Scheme 5. Synthesis of C24-Oxazoline 31ba.

aReagents and conditions: (a) cat. HClO4, HCO2H, 50 °C; (b) SOCl2, benzene, 80 °C, then 2-amino-2-methyl-propanol, DCM; (c) SOCl2, DCM; (d) NaOH, MeOH, 75 °C.
Synthesis of Tetrazole Analogues
Carboxylic acid 28b was converted to the primary amide 32b via treatment with thionyl chloride and then ammonia gas (Scheme 6). The amide was dehydrated with TFAA in pyridine to afford nitrile 33b. The formate esters were hydrolyzed with sodium methoxide in refluxing methanol and the deprotected nitrile 34b was heated with triethylammonium chloride and sodium azide to form the tetrazole,46 which was converted into the sodium salt 35b by ion-exchange chromatography.43
Scheme 6. Synthesis of Tetrazole Analoguea.

aReagents and conditions: (a) SOCl2, benzene, 80 °C, then NH3; (b) TFAA, pyridine, THF; (c) NaOMe, MeOH, 75 °C; (d) Et3N·HCl, NaN3, 1:1 toluene:DMF, 160 °C, ion-exchange using Na+-Dowex resin.
To synthesize the 3-methoxy-7-sulfate tetrazole analogue, the C3-hydroxyl of 34b was methylated with sodium hydride and methyl iodide to afford 36b (Scheme 7). The nitrile was heated with sodium azide and zinc(II) bromide to afford 37b as a sodium salt after ion-exchange chromatography.43,47 Tetrazole 37b was heated with chlorosulfonic acid in pyridine to incorporate a sulfate group, followed by ion-exchange chromatography using Dowex-50WX2 resin to obtain 38b as the disodium salt.
Scheme 7. Synthesis of 3-Methoxy-7-sulfate Tetrazole Analoguesa.

aReagents and conditions: (a) NaH, MeI, THF; (b) ZnBr2, NaN3, H2O/2-propanol, MW 170 °C, ion exchange using Na+-Dowex resin; (c) ClSO3H, pyridine, 50 °C, ion exchange using Na+-Dowex resin.
Evaluation of Biological Activity of Compounds
Some of the key steps in spore germination are depolymerization of peptidoglycan in the spore cortex followed by hydration of the spore core, which can be observed as a phase change from bright to dark by phase-contrast microscopy. This change also can be observed by measuring the decrease in optical density of the spore suspension at 600 nm (OD600) over time. An optical density assay was used as an initial screen to identify the most potent bile acid analogues. These compounds were then further evaluated using a phase-contrast microscopy assay to verify the initial findings.
Spores from a NAP1 strain of C. difficile were isolated from patient samples by previously reported methods.13 In the optical density assay, spores were incubated for 20 min on brain–heart infusion medium supplemented with 0.5% yeast extract and 0.1% L-cysteine (BHIS) along with 2000 μM TCA, a known germinant, under anaerobic conditions in the presence of the bile acid analogues. As a control for this experiment, spores were incubated with 2000 μM TCA for 20 min in the absence of test compounds, which resulted in a maximum change in OD600 to approximately 60% of the initial value. Compounds showing significant potency at inhibiting germination in the optical density were retested, with similar results obtained between runs.
In the phase-contrast spore count assay, NAP1 spores were plated with either DMSO as a control or the test compounds at 10 μM. The number of spores and germinated cells were counted at t0. The spores were then exposed to 2000 μM TCA for 20 min, and the number of spores and germinated cells were counted at t20 for both the DMSO control and the test compound plates. Each experiment was performed in triplicate and the percentage of germinated cells at t0 and t20 determined. In the DMSO control samples, the percent of germinated spores increased from approximately 20% to 75% of the total number of cells after 20 min.
Results of Optical Density Assay
Spores were incubated in BHIS supplemented with 2000 μM of the germinant TCA and with varying concentrations of the bile acid analogues. Compounds were divided into three categories based on the lowest concentration at which complete inhibition of spore germination was observed (defined as a relative OD600 reading after 20 min greater than or equal to 0.95). Good inhibitors were effective at 50 μM or less, moderate inhibitors were effective at 100–500 μM, and poor inhibitors were effective at concentrations ≥1000 μM. Examples of compounds that were good or moderate inhibitors are shown in Figures 5 and 6, respectively.
Figure 5.
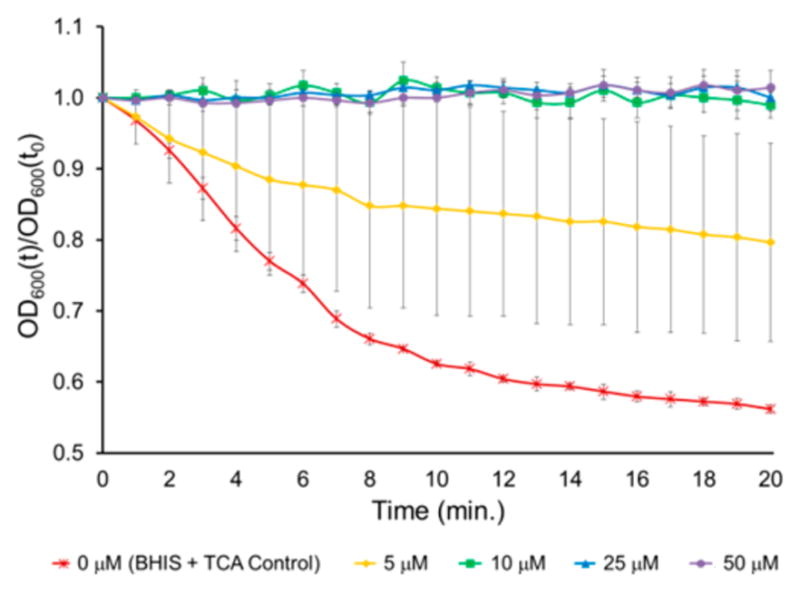
Example of a compound that was a potent inhibitor of C. difficile spore germination. The relative OD600 of spores in BHIS after 20 min exposure to 2000 μM TCA and 0, 5, 10, 25, or 50 μM 35a. OD600(t)/OD600(t0) = OD600 normalized to the initial OD600 (relative OD600). Data represent mean ± SEM.
Figure 6.
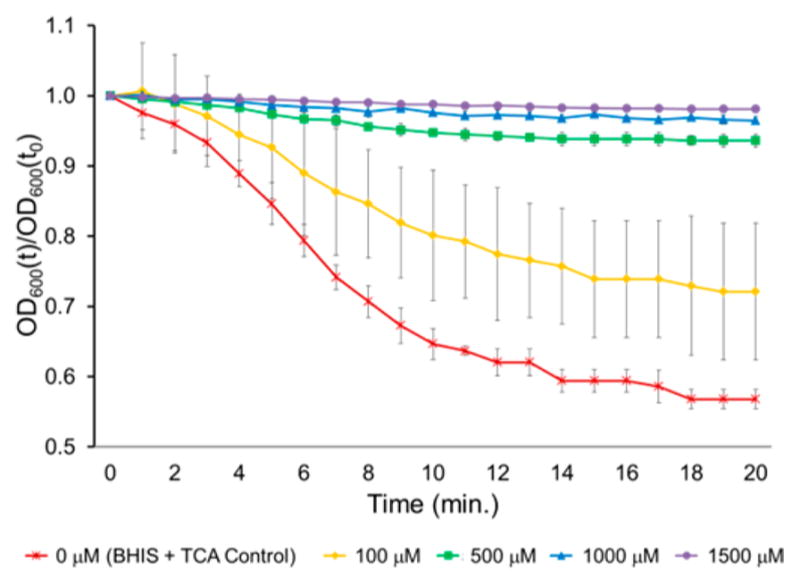
Example of a compound that was a moderate inhibitor of C. difficile spore germination. The relative OD600 of spores in BHIS after 20 min exposure to 2000 μM TCA and 0, 100, 500, 1000, or 1500 μM 19b. OD600(t)/OD600(t0) = OD600 normalized to the initial OD600 (relative OD600). Data represent mean ± SEM.
Modifications to the CDCA (1a) and UDCA (1b) scaffolds were made at the C24, C3, and C7 positions to determine how these changes affected the potency of the analogues at inhibiting TCA-promoted germination of C. difficile spores. Table 1 compares the lowest concentration of synthetic bile acids needed to obtain complete inhibition of C. difficile spore germination in the optical density assay (OD600 after 20 min. ≥ 0.95). Graphs of optical density vs time for all compounds tested can be found in the Supporting Information. Generally, most of the tested bile acid analogues demonstrated concentration-dependent inhibition of spore germination in the NAP1 strain examined, with higher concentrations of test compound leading to greater inhibition. However, a small number of compounds showed abnormal responses, where lower concentrations appeared to be more effective at preventing germination than higher concentrations.48 It is possible that these abnormal responses may be attributable to bile acid aggregation, micelle formation at the elevated concentrations,49,50 or were artifacts due to compound insolubility.51,52 Compounds showing significant potency in the optical density assay were subsequently tested in a phase contrast microscope assay to visually confirm that they actually inhibited C. difficile spore germination (see Microscope Spore Count Assay section below).
Table 1.
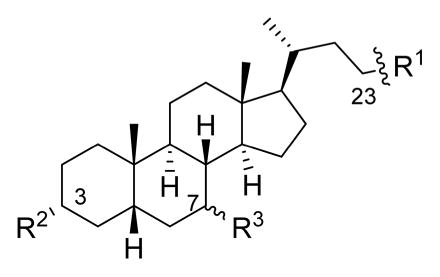
Lowest Concentration of Bile Acid Analogues That Showed Complete Inhibition of NAP1 C. difficile Spore Germination in the Presence of 2000 μM TCA in Optical Density Assay
| ||||
|---|---|---|---|---|
|
| ||||
| Compound number | R1 | R2 | R3 | Concentration Required for Complete Inhibition (μM)a |
| 1a |
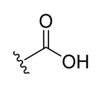
|
-OH | -αOH | 500 |
| 1b | -OH | -βOH | 500b | |
| 4bd | -OH | -βOSO3Na | 3000 | |
| 5a | -OMe | -αOH | >100d | |
| 5b | -OMe | -βOH | >500d | |
| 6b | -OMe | -βOMe | >1000 | |
| 10b | -OH | -βOMe | >500d | |
|
| ||||
| 2a |
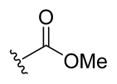
|
-OH | -αOH | 500 |
| 2b | -OH | -βOH | 50 | |
| 7a | -OMe | -αOMe | 500 | |
| 7b | -OMe | -βOMe | 500 | |
| 8a | -OMe | -αOH | 500 | |
| 8b | -OMe | -βOH | 500 | |
| 11a | -OH | -αOMe | 500 | |
| 11b | -OH | -βOMe | 500 | |
|
| ||||
| 16a |
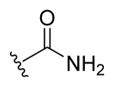
|
-OAc | -αOAc | 50 |
| 16b | -OAc | -βOAc | 50d | |
|
| ||||
| 17a |
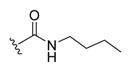
|
-OH | -αOH | 500 |
| 17b | -OH | -βOH | 500 | |
|
| ||||
| 18a |
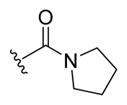
|
-OH | -αOH | 50 |
| 18b | -OH | -βOH | 100 | |
| 19a | -OMe | -αOMe | 500 | |
| 19b | -OMe | -βOMe | 1000 | |
| 20a | -OMe | -αOH | 100d | |
| 20b | -OMe | -βOH | 25 | |
| 21b | -OMe | -βOSO3Na | 50 | |
|
| ||||
| 22a |
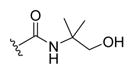
|
-OH | -αOH | 500 |
|
| ||||
| 24a |
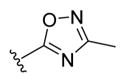
|
-OH | -αOH | 50 |
| 24b | -OH | -βOH | 50d | |
|
| ||||
| 27a |
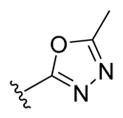
|
-OH | -αOH | 50 |
| 27b | -OH | -βOH | 50 | |
|
| ||||
| 31b |
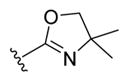
|
-OH | -βOH | 50 |
|
| ||||
| 35a |
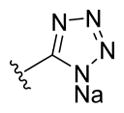
|
-OH | -αOH | 10 |
| 35b | -OH | -βOH | 25 | |
| 37a | -OMe | -αOH | 5 | |
| 37b | -OMe | -βOH | 25 | |
| 38a | -OMe | -αOSO3Na | 25 | |
| 38b | -OMe | -βOSO3Na | 10 | |
|
| ||||
| 3 |
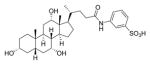
|
>1000 | ||
| 13 |
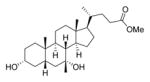
|
50 | ||
| 14 |
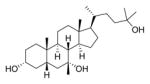
|
>1000 | ||
| 39 |
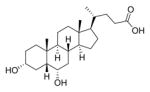
|
500d | ||
| 40 |
|
500 | ||
Complete inhibition = relative OD600 greater than 0.95 in optical density assay after exposure of spores to BHIS and 2000 μM TCA for 20 min with varying concentrations of bile acid analogues.
Value previously reported in Weingarden et al. (ref 32).
Tested as the sodium carboxylate.
Shows an abnormal concentration–response in the presence of TCA.
C24 Substitutions
Most synthetic ester derivatives (7a, 7b, 8a, 8b, 11a, 11b, and 40) had the same inhibitory activity as the most potent compound identified by Sorg and co-workers (2a, 500 μM in the optical density assay).24 Two exceptions are 2b and 13, which more potently inhibited spore germination (50 μM). Replacing the methyl ester of 13 with the tertiary alcohol in 14 resulted in a large decrease in potency (50 μM compared to >1000 μM, respectively).
Acids 1a and 1b both inhibited TCA-promoted spore germination of the NAP1 strain used in our assay at 500 μM, consistent with previously reported data for these endogenous bile acids.18,21,24 Carboxylic acid analogues 5a, 5b, 10b, and 39 showed abnormal responses where greater germination appeared to be observed at higher concentrations. However, further evaluation of one of these compounds (39) in the optical density assay in the absence of spores also showed a change in optical density with time, suggesting what initially appeared to be germination was likely actually an artifact due to compound insolubility. As many better alternatives were identified, these carboxylic acid analogues were not further investigated.
In contrast, the pyrrolidine amide derivatives 18a, 18b, 20b, and 21b were among the most potent inhibitors synthesized, with complete inhibition of spore germination observed at concentrations ≤100 μM in the presence of 2000 μM TCA. Secondary amides 17a, 17b, and 22a were moderate inhibitors of the NAP1 strain (500 μM). Both primary amide 16b and pyrrolidine amide 20a had abnormal concentration responses and were not investigated further. To confirm that the activity of our analogues was not limited to just the NAP1 strain, compound 20b was also tested against Nap2 and Nap10 strains of C. difficile isolated from patients and found to completely inhibit spore germination at 100 μM. The sulfonated amide 3, reported by Abel-Santos and co-workers to be active against strain ATCC 630,29 was also prepared but was inactive at concentrations up to 1000 μM in our assays (optical density and spore count data for 3 can be found in the Supporting Information, spectral data matched those previously reported for the compound50).
Heterocyclic substitutions were well-tolerated at the C24 position. The oxazoline analogue 31b and the oxadiazole analogues 24a, 24b, 27a, and 27b inhibit NAP1 spore germination at 50 μM, although with oxadiazole 24b incomplete inhibition of germination seemed to occur at higher concentrations. However, the tetrazole analogue series (35a, 35b, 37a, 37b, 38a, and 38b) contained the most potent analogues, which inhibit spore germination at concentrations as low as 5 μM (37a) and did not promote germination under the assay conditions.
C3 and C7 Substitutions
In the UDCA methyl ester series, methoxy substitutions at C3, C7, or C3/C7 decreased the potency of the compounds relative to 2b (50 μM). Conversely, trends in the CDCA methyl ester series showed that the inclusion of a methoxy group at C3, C7, or C3/C7 had little to no effect on potency.
In the UDCA carboxylic acid series, the 3,7-dimethoxy compound 6b did not inhibit spore germination at the highest concentration tested (1000 μM), whereas 1b inhibited germination of the NAP1 strain at 500 μM. These results, and the weak inhibition demonstrated by 4b, suggested that the C7-hydroxyl group may be important to inhibit spore germination in the UDCA carboxylic acid scaffold. The limited number of CDCA carboxylic acid analogues synthesized, and the abnormal concentration response exhibited by analogue 5a makes it difficult to reach any similar conclusions about the effect of methylation in this series.53
In the UDCA pyrrolidine amide series, 3,7-dimethoxy pyrrolidine amide 19b was a less potent inhibitor (500 μM) than the monosubstituted C3 methoxy compound 20b (25 μM) or diol 18b (100 μM). Trends in the CDCA pyrrolidine amide series suggest that inclusion of a C7-methoxy substituent in 19a (500 μM) decreases potency relative to the C7-hydroxyl group in 18a or 20a (50 and 100 μM, respectively).
Methylation of the C3-hydroxyl group in the tetrazole series did not seem to significantly affect the potency of the compounds for either 37a or 37b (5 and 25 μM, respectively) compared to 35a or 35b.
Effect of C7-Sulfation
As discussed above, the C7-sulfated analogue of UDCA (4b) is gut restricted but is a weaker germination inhibitor than UDCA (1b). With this in mind, we prepared C7-sulfated derivatives of some of our best inhibitors to determine whether potentially gut-restricted compounds that maintained good potency could be identified. Our results showed that this is indeed the case, as the pyrrolidine amide 20b and its C7-sulfated analogue 21b both showed similar potency in the optical density assay (25 and 50 μM, respectively). For the tetrazole series, a similar drop in potency was observed for the C7-sulfate 38a (25 μM) compared to 37a (5 μM). However, the UDCA tetrazole series showed a similar potency for compounds 37b (25 μM) and 38b (10 μM). These results confirm that by optimizing the rest of the scaffold, C7-sulfated analogues that are more potent inhibitors of spore germination than 4b could be identified.
Determination of Inhibitor Constants
The optical density assay can be used to determine an inhibitor constant (Ki) when it is run with varying germinant concentrations while holding the inhibitor concentration constant (see Figures S56–S60 in the Supporting Information for a detailed description).24 Selected compounds were evaluated at either 0.05 or 0.2 mM in this assay with varying concentrations of TCA and each experiment was repeated three times (Table 2). The results of this assay were similar to those obtained from the optical density assay using varying concentrations of inhibitor and a constant germinant concentration (Table 1), with tetrazole derivatives 37a and 37b showing greater potency than amides 20b and 21b, which in turn were more potent than ester derivative 2a. The results also confirmed that potent C7-sulfated derivatives could be identified.
Table 2.
Inhibitor Constants for Selected Compounds
| ||||
|---|---|---|---|---|
|
| ||||
| Number | R1 | R2 | R3 | Ki (μM)a |
|
| ||||
| 2ab | -CO2Me | -OH | -αOH | 118.5 ± 22.3 |
|
| ||||
| 20bb |
|
-OMe | -βOH | 49.2 ± 4.8 |
| 21bb | -OMe | -βOSO3Na | 20.0 ± 3.1 | |
|
| ||||
| 37ab |
|
-OMe | -αOH | 17.9 ± 4.6 |
| 38bc | -OMe | -βOSO3Na | 5.6 ± 3.3 | |
Data are averages of three experiments ± SEM.
Evaluated at 0.2 mM.
Evaluated at 0.05 mM.
Microscope Spore Count Assay
While the optical density assay allowed a large number of compounds to be evaluated at multiple concentrations in a reasonable amount of time, several compounds behaved unusually in this assay, most likely due to solubility issues. Therefore, a phase-contrast assay was used to directly confirm that the most potent analogues identified in Table 1 were actually inhibiting C. difficile spore germination. In this assay, spores appeared phase bright under the phase-contrast microscope, while germinated and vegetative cells appeared phase dark (Figure 7). The percentage of germinated cells ranged from 15 to 25% in both the BHIS + TCA control plates and BHIS + TCA + test compound plates at time = 0 min. In a typical assay, an additional 54–63% of spores germinated after 20 min in the absence of any inhibitor (see Supporting Information for assay results for all compounds evaluated).
Figure 7.

Cell count using phase-contrast microscopy to determine percentage of spore germination over 20 min. (A) Under the conditions of the control experiment (BHIS, 2000 μM TCA, and DMSO), the number of germinated spores rose from 20% to 71% over 20 min. (B) In contrast, far fewer spores germinated under the same conditions in the presence of 10 μM of 37a (from 20% to 25%). Inset: Phase-contrast microscope image of germinated cells (phase dark) and spores (phase bright).
In Table 3, the percent of total spores germinated at t0 was compared to the percent of total cells germinated at t20 when spores were exposed to 2000 μM TCA and bile acid analogues (10 μM). Each experiment was repeated three times. Substantial germination of spores was observed for 2a,24 3,29 and 6b at 10 μM (73%, 78%, and 75% germinated cells relative to control, respectively), which was consistent with the data from the optical density assay and suggested that these compounds are poor inhibitors of C. difficile spore germination in this NAP1 strain. The pyrrolidine amides 20b and 21b had similar strong inhibitory effects on spore germination at this concentration (15% and 9% germinated cells, respectively). At a higher concentration (50 μM), compound 21b showed complete inhibition of spore germination (see Table S1 in the Supporting Information). Tetrazoles 35a (12% germinated cells) and 35b (19% germinated cells) were similar in potency to the pyrrolidine amides. Methylation of the C3-hydroxyl group had little effect on the inhibitory activity of the compounds: compound 37b was slightly less potent (26% germinated cells) compared to the C3-hydroxyl compound 35b (19% germinated cells), while 37a showed a slight increase in potency (8% germinated cells) compared to 35a (12% germinated cells). Unlike the similar activities observed for 20b and 21b, sulfation of the C7-hydroxyl group in the tetrazole series had a larger effect on potency, as compounds 38b and 38a were much less inhibitory at 10 μM than the other tetrazole analogues (42% and 51% germinated cells, respectively). The similar inhibition of spore germination for 20b and its C7-sulfated derivative 21b was unlike what was observed with 1b and its C7-sulfated derivative 4b, and 21b may be a promising lead for further development due to its potency.
Table 3.
Percent Germination of NAP1 Spores in the Presence of 2000 μM TCA and 10 μM Bile Acid Analogues after 20 min
|
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Number | R1 | R2 | R3 | Percent Spore Germinationa
|
||
| t0 | t20 | Relative to controlb | ||||
|
| ||||||
| 2a | -CO2Me | -OH | -αOH | 25 ± 6 | 71 ± 1 | 73 ± 1 |
|
| ||||||
| 6b | -CO2H | -OMe | -βOMe | 25 ± 4 | 71 ± 2 | 75 ± 2 |
|
| ||||||
| 20b |
|
-OMe | -βOH | 18 ± 3 | 33 ± 6 | 15 ± 6 |
| 21b | -OMe | -βOSO3Na | 18 ± 2 | 30 ± 6 | 9 ± 5 | |
|
| ||||||
| 35a |
|
-OH | -αOH | 15 ± 6 | 22 ± 6 | 12 ± 6 |
| 35b | -OH | -βOH | 17 ± 4 | 29 ± 4 | 19 ± 4 | |
| 37a | -OMe | -αOH | 16 ± 4 | 21 ±4 | 21 ± 4 | |
| 37b | -OMe | -βOH | 16 ± 4 | 31 ± 4 | 26± 4 | |
| 38a | -OMe | -αOSO3Na | 20 ± 11 | 48 ± 11 | 51 ± 11 | |
| 38b | -OMe | -βOSO3Na | 21 ± 3 | 44 ± 3 | 42 ± 3 | |
|
| ||||||
| 3 |
|
25 ± 2 | 74 ± 2 | 78 ± 2 | ||
Total germinated cells and nongerminated spores were counted at the initial time and after 20 min and reported as a percentage of the total cell count. BHIS + TCA control had 18–25% germination at t0. Experiments were performed in triplicate (N = 3). Data represent mean ± standard deviation.
Percent germination (t20 − t0) with test compound/percent germination (t20 − t0) with control.
Cell Permeability
UDCA (1b) is a moderate inhibitor of C. difficile spore germination. However, it is both passively and actively absorbed from the intestine, resulting in much lower concentrations of 1b in the colon than in the upper digestive tract.54,55 As discussed in the Introduction, we hypothesized that we could greatly reduce both the active and passive absorption of our bile acid analogues by incorporating a C7-sulfate group. To test this theory, compound 20b and its sulfated derivative 21b were selected for comparison in an in vitro Caco-2 model of intestinal epithelial permeability (Table 4). At 10 μM, 20b was moderately permeable (Papp = 6.1 × 10−6 cm/s) in the apical to basolateral (A–B) direction and had low permeability (Papp = 0.5 × 10−6 cm/s) in the B–A direction, resulting in a transport ratio of approximately 12. This result suggests that 20b may be actively transported, which is consistent with an earlier report from Hidalgo and Borchardt in which they concluded that the high transport ratio of TCA (greater than 10) was attributed to the expression of bile acid carriers on the Caco-2 monolayers.56 In contrast, the sulfated compound 21b has much lower permeability (Papp = 0.1 × 10−6 cm/s) in the A–B direction, suggesting that this sulfated derivative is unlikely to be extensively absorbed from the digestive tract. Additionally, the permeability data suggested that if 21b is absorbed, it may be transported back into the gut as it is moderately permeable (Papp of 2.1 × 10−6 cm/s) in the B–A direction. Tetrazole CDCA derivatives 37a and 38a behaved similarly in the Caco-2 model (Table 4), with C7-hydroxy compound 37a (Papp of 10.7 × 10−6 cm/s), demonstrating over 25 times greater permeability in the A–B direction than its C7-sulfated analogue 38a (Papp of 0.4 × 10−6 cm/s).
Table 4.
In Vitro Absorption of Bile Acids at 10 μM in a Caco-2 Cell Monolayer Bidirectional Permeability Assay
| compd | mean permeability A–B (10−6 cm/s) | mean permeability B–A (10−6 cm/s) |
|---|---|---|
| 20b | 6.1 | 0.5 |
| 21b | 0.1 | 2.1 |
| 37a | 10.7 | 1.9 |
| 38a | 0.4 | 0.4 |
CONCLUSIONS
We have identified several bile acid analogues that are potent inhibitors of C. difficile spore germination in a highly virulent NAP1 strain. Some of the most potent compounds include C24 amide or tetrazole derivatives of UDCA. Substitutions at C3 were well tolerated, while C7 substitutions generally decreased the inhibitory activity of the compounds. To limit intestinal absorption, C7-sulfated analogues designed to avoid both active and passive transport pathways were prepared. One of these derivatives, compound 21b, was found to be both a potent inhibitor of C. difficile spore germination and poorly permeable in a Caco-2 model of intestinal epithelial absorption, suggesting that it is likely to be gut-restricted. These compounds work by a different mechanism of action than existing medications such as vancomycin or fidaxomicin, which do not inhibit spore germination.57 In future studies, we will evaluate the efficacy of our best compounds at preventing R-CDI in an animal model and investigate their effects on spore formation, outgrowth of vegetative cells, and toxin formation. We will also examine the effects of our bile acid derivatives on the indigenous gut microbiota. Our ultimate goal is to identify and clinically develop new bile acid inhibitors capable of preventing recurrent CDI in vulnerable patients.
EXPERIMENTAL SECTION
Chemistry
NMR spectra were recorded using a Bruker 400 spectrometer. 1H NMR data are reported as follows: chemical shift in parts per million downfield of tetramethylsilane (TMS) or relative to the residual solvent peak, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, quint = quintet and m = multiplet), coupling constant (Hz), and integrated value. Unless otherwise specified, all materials, reagents, and solvents were obtained from commercial suppliers and were used without further purification. The progress of a synthetic procedure was monitored, where possible, by thin layer chromatography (TLC), and the compounds of interest were visualized using PMA/Ce(SO4)2 stain. TLC was conducted on silica gel 250 μm, F254 plates. Flash column chromatography was performed using Teledyne-Isco Combiflash Rf+PurIon with Redisep Rf silica gel columns. The purity of the compounds evaluated in the phase-contrast spore count assay was determined to be >95% by LC-MS/UV/ELSD analysis.
Methyl (R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate (2b)
To a solution of UDCA (1.00 g, 2.55 mmol) in MeOH (26 mL) was added pTSA·H2O (0.048 g, 0.26 mmol) and the reaction stirred at room temperature for 24 h. The reaction was quenched by the addition of saturated aqueous NaHCO3 (5 mL), and the solvent was removed by rotary evaporation. The residue was partitioned between saturated aqueous NaHCO3 and EtOAc, and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (3 × 10 mL) and water (5 mL), dried over MgSO4, filtered, and concentrated to obtain 2b (0.933 g, 90% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.60 (tt, J = 10.4, 4.6 Hz, 2H), 2.37 (ddd, J = 15.3, 10.1, 5.0 Hz, 1H), 2.23 (ddd, J = 15.6, 9.5, 6.5 Hz, 1H), 2.01 (dt, J = 12.5, 3.2 Hz, 1H), 1.95–1.88 (m, 1H), 1.86–1.75 (m, 4H), 1.72–1.65 (m, 2H), 1.65–1.57 (m, 2H), 1.57–1.50 (m, 2H), 1.51–1.40 (m, 6H), 1.40–1.21 (m, 5H), 1.20–0.98 (m, 3H), 0.96 (s, 3H), 0.94 (d, J = 6.4 Hz, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.7, 71.4, 71.3, 55.7, 54.9, 51.5, 43.8, 43.8, 42.4, 40.1, 39.2, 37.3, 36.8, 35.3, 34.9, 34.1, 31.1, 31.0, 30.3, 28.6, 26.9, 23.4, 21.2, 18.4, 12.1. HRMS (ESI): m/z calcd C25H42NO4Na (M + Na+) 429.2981, found 429.2965. Compound 2a was prepared by the same method. Data for methyl (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate (2a): 1H NMR (400 MHz, CDCl3) δ 3.85 (q, J = 3.0 Hz, 1H), 3.67 (s, 3H), 3.46 (tt, J = 11.1, 4.4 Hz, 1H), 2.36 (ddd, J = 15.3, 10.1, 5.1 Hz, 1H), 2.29–2.14 (m, 3H), 2.03–1.05 (m, 22H), 1.00 (dd, J = 14.3, 3.3 Hz, 1H), 0.95–0.91 (m, 1H), 0.93 (d, J = 6.4 Hz, 3H), 0.91 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.7, 72.0, 68.5, 55.8, 51.5, 50.5, 42.7, 41.5, 39.9, 39.6, 39.4, 35.4, 35.3, 35.0, 34.6, 32.8, 31.0, 31.0, 30.7, 28.1, 23.7, 22.8, 20.6, 18.3, 11.8. HRMS (ESI): m/z calcd C25H42NO4Na (M + Na+) 429.2981, found 429.2961.
(R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-7-Hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic Acid (5b) and (R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dimethoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanoic Acid (6b)
To a solution of 2b (0.660 g, 1.62 mmol) in THF (8.1 mL) was added NaH (60% dispersion in mineral oil, 0.143 g, 3.57 mmol) and the reaction stirred at room temperature for 15 min. MeI (0.213 mL, 3.41 mmol) was added dropwise, and the mixture stirred for 3 h. The reaction was quenched by the addition of saturated aqueous NH4Cl (5 mL), and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (25–67% EtOAc in hexanes) to obtain 6b (0.018 g, 3% yield), followed by the elution of 5b (0.224 g, 34% yield). Data for 6b: 1H NMR (400 MHz, CDCl3) δ 3.35 (s, 3H), 3.23 (s, 3H), 3.13 (qd, J = 10.2, 9.5, 4.1 Hz, 1H), 2.99 (dq, J = 9.0, 5.0 Hz, 1H), 2.39 (ddd, J = 15.3, 10.2, 4.9 Hz, 1H), 2.24 (ddd, J = 15.5, 9.5, 6.3 Hz, 1H), 1.96 (dt, J = 12.7, 3.3 Hz, 1H), 1.89–0.95 (m, 22H), 0.93 (s, 3H), 0.92 (d, J = 5.7 Hz, 3H), 0.84 (ddd, J = 13.1, 9.5, 7.1 Hz, 1H), 0.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 179.7, 80.6, 80.3, 56.3, 55.8, 55.6, 55.4, 43.8, 42.4, 41.7, 40.4, 39.5, 35.5, 35.1, 34.6, 33.9, 32.3, 31.2, 31.1, 28.8, 26.8, 26.6, 23.6, 21.5, 18.6, 12.4. HRMS (ESI): m/z calcd C26H44NaO4 (M + Na+) 443.3137, found 443.3111. Data for 5b: 1H NMR (400 MHz, CDCl3) δ 3.60 (ddd, J = 11.5, 8.6, 5.1 Hz, 1H), 3.35 (s, 3H), 3.13 (tt, J = 10.4, 4.8 Hz, 1H), 2.40 (ddd, J = 15.3, 10.1, 5.0 Hz, 1H), 2.25 (ddd, J = 15.8, 9.5, 6.5 Hz, 1H), 2.05–1.03 (m, 23H), 1.00 (dd, J = 14.5, 3.0 Hz, 1H), 0.95 (s, 3H), 0.93 (d, J = 7.12 Hz, 3H), 0.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 179.4, 80.2, 71.5, 55.8, 55.7, 55.1, 43.9, 43.9, 42.5, 40.3, 39.2, 37.0, 35.4, 35.0, 34.5, 33.8, 31.1, 31.0, 28.8, 27.0, 26.7, 23.6, 21.3, 18.5, 12.3. HRMS (ESI): m/z calcd C25H42NO4Na (M + Na+) 429.2981, found 429.2960. Compound 6a was obtained in trace amounts and was not evaluated in the spore germination assays. Compound 5a was prepared in the same manner as 5b. Data for (R)-4-((3R,5R,7R,8R,9S,10S,13R,14S,17R)-7-hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanoic acid (5a): 1H NMR (400 MHz, CDCl3) δ 3.84 (d, J = 3.2 Hz, 1H), 3.34 (s, 3H), 3.02 (td, J = 10.9, 5.4 Hz, 1H), 2.40 (ddd, J = 15.5, 10.2, 5.1 Hz, 1H), 2.25 (ddd, J = 15.8, 9.7, 6.4 Hz, 1H), 2.18–1.02 (m, 23H), 0.99 (d, J = 3.3 Hz, 1H), 0.94 (d, J = 6.4 Hz, 3H), 0.90 (s, 3H), 0.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.5, 80.8, 68.7, 56.0, 55.7, 50.6, 42.9, 41.6, 39.9, 39.7, 36.0, 35.6, 35.6, 35.5, 34.9, 33.0, 31.0, 30.9, 28.4, 27.2, 23.9, 23.1, 20.8, 18.5, 12.0. HRMS (ESI): m/z calcd C25H42NO4Na (M + Na+) 429.2981, found 429.2998.
Methyl (R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dimethoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl) pentanoate (7b) and Methyl (R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-7-Hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-pentanoate (8b)
To a solution of 2b (0.260 g, 0.639 mmol) and 2,6-di-tert-butylpyridine (0.29 mL, 1.3 mmol) in DCM (6.4 mL) was added methyl triflate (approximately 0.1 mL). The reaction stirred at room temperature for 24 h and was quenched by the addition of 1 M aqueous HCl (5 mL). The aqueous layer was extracted with DCM (3 × 10 mL), and the combined organic layers were washed with saturated aqueous NaHCO3 (2 × 10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–50% EtOAc in DCM) to obtain 7b (0.147 g, 53% yield) as a clear colorless oil, followed by the elution of 8b (0.054 g, 21% yield) as clear, colorless oil that solidified over time to a white amorphous solid. Data for 7b: 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.36 (s, 3H), 3.24 (s, 3H), 3.13 (tt, J = 10.2, 4.4 Hz, 1H), 2.99 (ddd, J = 11.3, 8.8, 5.0 Hz, 1H), 2.36 (ddd, J = 15.2, 10.2, 4.9 Hz, 1H), 2.22 (ddd, J = 15.6, 9.6, 6.4 Hz, 1H), 1.98 (dt, J = 12.8, 3.3 Hz, 1H), 1.90–1.71 (m, 6H), 1.71–1.61 (m, 2H), 1.53–1.37 (m, 7H), 1.37–1.30 (m, 1H), 1.30–1.09 (m, 5H), 1.09–0.97 (m, 2H), 0.94 (s, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 80.6, 80.2, 56.4, 55.8, 55.7, 55.4, 51.7, 43.8, 42.4, 41.7, 40.5, 39.5, 35.6, 35.1, 34.6, 34.0, 32.3, 31.3, 31.3, 28.8, 26.9, 26.6, 23.6, 21.5, 18.6, 12.4. HRMS (ESI): m/z calcd C27H46NaO4 (M + Na+) 457.3294, found 457.3278. Data for 8b: 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 3H), 3.58 (s, 1H), 3.34 (s, 3H), 3.11 (tt, J = 10.6, 4.5 Hz, 1H), 2.36 (ddd, J = 15.3, 10.2, 5.0 Hz, 1H), 2.22 (ddd, J = 15.6, 9.5, 6.5 Hz, 1H), 2.02–0.96 (m, 24H), 0.94 (s, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.67 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 3.87 (d, J = 6.9 Hz, 1H), 3.57 (s, 3H), 3.31–3.22 (m, 1H), 3.20 (s, 3H), 3.05 (tt, J = 9.7, 5.3 Hz, 1H), 2.32 (ddd, J = 15.2, 9.7, 5.2 Hz, 1H), 2.20 (ddd, J = 15.7, 9.3, 6.8 Hz, 1H), 1.91 (dd, J = 11.4, 3.6 Hz, 1H), 1.88–1.79 (m, 1H), 1.78–1.59 (m, 6H), 1.47–1.28 (m, 7H), 1.27–1.09 (m, 6H), 1.06–0.88 (m, 3H), 0.88 (s, 3H), 0.86 (d, J = 6.8 Hz, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.9, 80.2, 71.6, 55.9, 55.8, 55.1, 51.7, 44.0, 44.0, 42.6, 40.3, 39.3, 37.2, 35.5, 35.1, 34.6, 33.9, 31.3, 31.2, 28.8, 27.1, 26.9, 23.6, 21.4, 18.6, 12.3. HRMS (ESI): m/z calcd C26H44NO4Na (M + Na+) 443.3137, found 443.3146. Compounds 7a and 8a were prepared by the same method. Data for methyl (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dimethoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate (7a): 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 3H), 3.33 (s, 3H), 3.23 (s, 3H), 3.17 (q, J = 2.9 Hz, 1H), 3.01 (tt, J = 11.2, 4.3 Hz, 1H), 2.35 (ddd, J = 15.2, 10.2, 5.0 Hz, 1H), 2.29–2.08 (m, 2H), 1.97–1.66 (m, 8H), 1.62 (ddd, J = 15.0, 5.4, 3.2 Hz, 1H), 1.57–1.39 (m, 5H), 1.37–1.10 (m, 8H), 1.08–0.99 (m, 1H), 0.92 (d, J = 6.4 Hz, 3H), 0.90 (s, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 80.8, 80.8, 68.7, 55.9, 55.7, 51.7, 50.6, 42.9, 41.6, 39.8, 39.7, 36.1, 35.6, 35.6, 35.5, 34.9, 33.0, 31.2, 31.2, 28.4, 27.2, 23.9, 23.1, 20.8, 18.5, 12.0. HRMS (ESI): m/z calcd C27H46NaO4 (M + Na+) 457.3294, found 457.3290. Data for methyl (R)-4-((3R,5R,7R,8R,9S,10S,13R,14S,17R)-7-hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanoate (8a): 1H NMR (400 MHz, CDCl3) δ 3.84 (q, J = 3.0 Hz, 1H), 3.67 (s, 3H), 3.35 (s, 3H), 3.02 (tt, J = 11.1, 4.3 Hz, 1H), 2.36 (ddd, J = 15.3, 10.1, 5.2 Hz, 1H), 2.23 (ddd, J = 15.6, 9.6, 6.6 Hz, 1H), 2.12 (td, J = 13.2, 11.2 Hz, 1H), 2.05–1.70 (m, 8H), 1.64 (dddd, J = 12.5, 9.9, 7.1, 2.9 Hz, 1H), 1.53–1.05 (m, 13H), 0.97 (dd, J = 15.2, 4.3 Hz, 1H), 0.93 (d, J = 6.5 Hz, 3H), 0.91 (s, 3H), 0.66 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 4.10 (d, J = 3.4 Hz, 1H), 3.62 (q, J = 3.1 Hz, 1H), 3.57 (s, 3H), 3.20 (s, 3H), 2.92 (tt, J = 11.1, 4.3 Hz, 1H), 2.32 (ddd, J = 15.2, 9.6, 5.3 Hz, 1H), 2.26–2.09 (m, 2H), 1.94–1.86 (m, 1H), 1.86–1.57 (m, 9H), 1.46–1.31 (m, 5H), 1.29–1.15 (m, 4H), 1.15–1.03 (m, 3H), 0.99 (dq, J = 11.8, 5.7 Hz, 1H), 0.88 (d, J = 6.4 Hz, 3H), 0.85 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 80.8, 68.7, 55.9, 55.7, 51.7, 50.6, 42.9, 41.6, 39.8, 39.7, 36.1, 35.6, 35.6, 35.5, 34.9, 33.0, 31.2, 31.2, 28.4, 27.2, 23.9, 23.1, 20.8, 18.5, 12.0. HRMS (ESI): m/z calcd C26H44NO4Na (M + Na+) 443.3137, found 443.3121.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3-Hydroxy-7-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic Acid (10b)
To a solution of 9b (0.300 g, 0.483 mmol) and 2,6-di-tert-butylpyridine (0.22 mL, 0.97 mmol) in DCM (5 mL) was methyl triflate (0.06 mL, 0.5 mmol). The reaction stirred at room temperature for 24 h and was quenched by the addition of 1 M aqueous HCl (5 mL). The aqueous layer was extracted with DCM (3 × 10 mL), and the combined organic layers were washed with saturated aqueous NaHCO3 (2 × 10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–50% EtOAc in DCM) to obtain tert-butyldimethylsilyl (R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-3-((tert-butyldimethylsilyl)oxy)-7-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate (0.207 g, 68% yield) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 3.54 (tt, J = 10.2, 4.7 Hz, 1H), 3.24 (s, 3H), 3.01 (ddd, J = 11.0, 9.1, 5.0 Hz, 1H), 2.36 (ddd, J = 15.2, 9.9, 5.1 Hz, 1H), 2.22 (ddd, J = 15.6, 9.4, 6.6 Hz, 1H), 1.97 (dt, J = 12.5, 3.1 Hz, 1H), 1.90–0.97 (m, 23H), 0.94 (s, 9H), 0.93 (d, J = 6.4 Hz, 3H), 0.92 (s, 3H), 0.90 (s, 9H), 0.66 (s, 3H), 0.27 (s, 3H), 0.27 (s, 3H), 0.07 (s, 3H), 0.07 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.9, 80.5, 77.4, 72.6, 56.2, 55.6, 55.4, 43.8, 42.5, 41.7, 40.4, 39.4, 38.0, 35.4, 35.3, 34.2, 33.3, 32.3, 31.4, 31.0, 28.7, 26.5, 26.1, 26.1, 25.7, 25.7, 23.5, 21.5, 18.6, 18.5, 17.8, 12.3, −4.4, −4.5, −4.6, −4.6. To tert-butyldimethylsilyl (R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-3-((tert-butyldimethylsilyl)oxy)-7-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanoate was added TBAF (0.58 mL, 0.58 mmol) in THF (2.7 mL) and the reaction mixture stirred for 3 h. Additional TBAF (0.58 mL, 0.58 mmol) was added, and the reaction mixture stirred at room temperature for 20 h. The solvent was evaporated, and the crude material was purified by flash column chromatography on silica gel (0–10% MeOH in DCM) to obtain 10b (0.094 g, 84% yield). 1H NMR (400 MHz, CDCl3) δ 3.61 (tt, J = 10.5, 4.7 Hz, 1H), 3.25 (s, 3H), 3.01 (td, J = 12.9, 11.0, 4.9 Hz, 2H), 2.41 (ddd, J = 15.2, 10.2, 4.9 Hz, 1H), 2.27 (ddd, J = 15.6, 9.5, 6.3 Hz, 1H), 2.02–1.94 (m, 1H), 1.89–1.01 (m, 22H), 0.95 (d, J = 6.4 Hz, 3H), 0.94 (s, 3H), 0.67 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 4.45 (d, J = 4.5 Hz, 1H), 3.22 (d, J = 79.8 Hz, 6H), 2.94 (td, J = 10.3, 5.0 Hz, 2H), 2.23 (ddd, J = 15.2, 9.7, 5.3 Hz, 1H), 2.15–2.04 (m, 1H), 1.93 (dd, J = 11.6, 3.7 Hz, 1H), 1.82–1.64 (m, 3H), 1.64–1.52 (m, 2H), 1.52–1.45 (m, 2H), 1.36 (dddd, J = 26.7, 20.5, 12.5, 8.3 Hz, 8H), 1.26–1.09 (m, 5H), 0.88 (d, J = 6.7 Hz, 3H), 0.86 (s, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.7, 80.6, 71.8, 56.4, 55.7, 55.4, 43.9, 42.4, 41.7, 40.5, 39.6, 37.5, 35.5, 35.1, 34.3, 32.2, 31.1, 30.6, 26.6, 25.5, 23.6, 20.3, 18.6, 13.8, 12.4. HRMS (ESI): m/z calcd C25H42NO4Na (M + Na+) 429.2981, found 429.2965. Compound 10a was not synthesized (see Scheme S2 for the synthetic route to 11a in the Supporting Information).
Methyl (R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3-Hydroxy-7-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanoate (11b)
To a solution of 10b (50 mg, 0.12 mmol) in MeOH (2.6 mL) was added pTSA·H2O (0.050 g, 0.26 mmol) and the reaction stirred at room temperature for 24 h. The solvent was removed by rotary evaporation. The crude material was purified by flash column chromatography on silica gel (33% EtOAc in DCM as eluent) to obtain 11b (0.031 g, 60% yield) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.59 (tt, J = 10.5, 4.7 Hz, 1H), 3.24 (s, 3H), 2.99 (ddd, J = 11.3, 9.0, 5.2 Hz, 1H), 2.36 (ddd, J = 15.2, 10.3, 4.9 Hz, 1H), 2.22 (ddd, J = 15.5, 9.6, 6.4 Hz, 1H), 1.98 (dt, J = 12.5, 3.3 Hz, 1H), 1.90–0.97 (m, 23H), 0.93 (s, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 80.5, 71.7, 56.4, 55.6, 55.4, 51.7, 43.8, 42.4, 41.7, 40.5, 39.6, 37.5, 35.5, 35.1, 34.3, 32.2, 31.3, 31.3, 30.6, 28.7, 26.6, 23.5, 21.5, 18.6, 12.4. HRMS (ESI): m/z calcd C26H44NaO4 (M + Na+) 443.3137, found 443.3119. Compound 11a was prepared by a different method than 11b (see Scheme S2 in the Supporting Information).
Methyl (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-7,10,13-trimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate (13) and (3R,5S,7R,8R,9S,10S,13R,14S,17R)-17-((R)-5-Hydroxy-5-methylhexan-2-yl)-7,10,13-trimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol (14)
3-α-7-oxo CDCA (0.800 g, 2.05 mmol) was dissolved in MeOH (20 mL), and pTSA·H2O (0.051 g, 0.27 mmol) was added. The reaction mixture stirred at room temperature for 12 h, and the solvent was removed under reduced pressure. The crude material was purified by flash column chromatography on silica gel (0–40% EtOAc in DCM) to obtain the methyl ester (0.671 g, 81% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.61 (dq, J = 10.7, 5.1 Hz, 1H), 2.86 (dd, J = 12.5, 6.0 Hz, 1H), 2.37 (qd, J = 10.7, 9.9, 6.2 Hz, 2H), 2.29–2.12 (m, 2H), 2.04–1.22 (m, 16H), 1.20 (s, 3H), 1.19–1.06 (m, 3H), 0.98 (dd, J = 12.3, 6.3 Hz, 1H), 0.93 (d, J = 6.3 Hz, 3H), 0.94–0.93 (m, 1H), 0.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 212.1, 174.9, 71.2, 55.0, 51.7, 49.7, 49.1, 46.3, 45.6, 42.9, 42.9, 39.2, 37.6, 35.4, 35.4, 34.4, 31.3, 31.2, 30.1, 28.5, 25.0, 23.3, 21.9, 18.6, 12.3. To a solution of the methyl ester (0.671 g, 1.66 mmol) in Et2O (17 mL) at room temperature was added MeMgBr (1.6 mL, 4.9 mmol) dropwise. The reaction was stirred for 2 h and was quenched by the addition of 1 M aqueous HCl (10 mL). The aqueous layer was extracted with Et2O (3 × 10 mL), and the combined organic layers were dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–100% EtOAc in DCM) to obtain recovered starting material (160 mg, 24% recovery), followed by the elution of 13 (0.107 g, 15% yield) and 14 (0.130 g, 19% yield) as white foams. Data for 13: 1H NMR (400 MHz, DMSO-d6) δ 4.30 (s, 1H), 3.57 (s, 3H), 3.36 (d, J = 18.1 Hz, 1H), 3.21 (tt, J = 10.3, 4.2 Hz, 1H), 2.32 (ddd, J = 15.2, 9.7, 5.2 Hz, 1H), 2.26–2.12 (m, 2H), 1.97–0.93 (m, 25H), 0.89 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 2.8 Hz, 1H), 0.80 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.7, 70.9, 70.9, 70.2, 54.4, 51.2, 50.8, 44.2, 43.3, 43.1, 41.9, 38.2, 35.6, 35.3, 34.8, 34.3, 33.1, 30.6, 30.4, 30.4, 28.0, 27.3, 22.8, 20.8, 18.3, 12.0. HRMS (ESI): m/z calcd C26H44NO4Na (M + Na+) 443.3137, found 443.3116. Data for 14: 1H NMR (400 MHz, DMSO-d6) δ 4.31 (d, J = 4.7 Hz, 1H), 4.01 (s, 1H), 3.65 (s, 1H), 3.32 (s, 3H), 3.20 (tt, J = 9.5, 4.7 Hz, 1H), 2.18 (q, J = 12.7 Hz, 1H), 1.96–1.06 (m, 23H), 1.05 (s, 3H), 1.04 (s, 3H), 1.04–1.03 (m, 1H), 0.89 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 2.3 Hz, 1H), 0.80 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 70.9, 70.2, 68.8, 64.9, 54.5, 50.8, 44.2, 43.2, 43.1, 41.9, 38.2, 35.6, 35.5, 35.3, 34.3, 33.2, 30.4, 29.8, 29.5, 29.1, 28.1, 27.3, 22.8, 20.8, 18.9, 15.2, 12.1. HRMS (ESI): m/z calcd C27H48NaO3 (M + Na+) 443.3501, found 443.3483.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Diacetoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-pentanoic Acid (15b)
UDCA (1.00 g, 2.55 mmol) was dissolved in pyridine (3 mL, 37 mmol) and Ac2O (2 mL, 21 mmol). The reaction stirred at room temperature 12 h and was diluted with Et2O (100 mL). The organic layer was washed with 1 M aqueous HCl (3 × 20 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–100% EtOAc in DCM as eluent) to obtain 15b (1.19 g, 98% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 4.65 (td, J = 11.0, 4.9 Hz, 1H), 4.55 (tt, J = 11.0, 4.9 Hz, 1H), 2.22 (ddd, J = 15.1, 9.6, 5.3 Hz, 1H), 2.09 (ddd, J = 15.7, 9.3, 7.1 Hz, 1H), 1.98 (s, 3H), 1.93 (s, 3H), 1.82–0.99 (m, 24H), 0.92 (s, 3H), 0.88 (d, J = 6.4 Hz, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.8, 169.8, 169.8, 73.0, 72.8, 64.9, 54.4, 54.3, 43.1, 41.2, 38.6, 34.7, 33.9, 33.5, 32.6, 32.5, 30.7, 30.7, 28.0, 26.0, 25.3, 22.8, 21.5, 21.0, 20.8, 18.2, 15.2, 11.8. TLC-MS (ESI): m/z calcd C28H44NaO6 (M + Na+) 499.3, found 499.9. Compound 15a was prepared by the same method. Data for (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-diacetoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid (15a): 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 4.76 (t, J = 3.0 Hz, 1H), 4.47 (tt, J = 11.0, 4.3 Hz, 1H), 2.23 (ddd, J = 15.0, 9.6, 5.3 Hz, 1H), 2.10 (ddd, J = 15.8, 9.1, 6.9 Hz, 1H), 1.98 (s, 3H), 1.97 (s, 3H), 1.95–1.90 (m, 3H), 1.83–0.95 (m, 21H), 0.90 (s, 3H), 0.88 (d, J = 6.7 Hz, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.8, 169.8, 169.7, 73.4, 70.5, 64.9, 55.3, 50.0, 42.2, 37.1, 34.8, 34.7, 34.4, 34.3, 34.3, 33.6, 30.8, 30.7, 30.6, 27.5, 26.4, 23.0, 22.3, 21.3, 21.1, 20.2, 18.1, 11.5. TLC-MS (ESI): m/z calcd C28H43O6 (M − H−) 475.6, found 475.9.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-5-Amino-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthrene-3,7-diyl Diacetate (16b)
Compound 15b (0.840 g, 1.76 mmol) was dissolved in benzene (8.8 mL), and SOCl2 (0.52 mL, 7.1 mmol) was added dropwise. The reaction was heated to reflux for 4 h and cooled to room temperature. The solvent was removed under reduced pressure, and excess SOCl2 was removed by coevaporation with benzene (3 × 5 mL). The residue was dissolved in ammonia solution (0.5 M in THF, 15 mL) and stirred for 12 h. The solvent was removed under reduced pressure, and the crude material was purified by flash column chromatography on silica gel (15–100% EtOAc in DCM as eluent) to obtain 16b (0.581 g, 69% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 7.20 (s, 1H), 6.63 (s, 1H), 4.66 (td, J = 10.7, 4.8 Hz, 1H), 4.55 (tt, J = 10.7, 4.8 Hz, 1H), 2.05 (ddd, J = 14.6, 9.9, 5.3 Hz, 1H), 1.98 (s, 3H), 1.93 (s, 3H), 1.95–1.89 (m, 1H), 1.86–0.96 (m, 26H), 0.92 (s, 3H), 0.88 (d, J = 6.5 Hz, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.6, 169.8, 169.8, 73.0, 72.8, 54.9, 54.5, 54.4, 43.1, 41.2, 38.6, 34.8, 33.9, 33.5, 32.7, 32.5, 32.0, 31.4, 28.0, 26.0, 25.3, 22.8, 21.5, 21.0, 20.8, 18.4, 11.8, 0.1. TLC-MS (ESI): m/z calcd C28H47NO6 (M + H2O+) 493.7, found 493.1. Compound 16a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-17-((R)-5-amino-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl diacetate (16a): 1H NMR (400 MHz, DMSO-d6) δ 7.21 (s, 1H), 6.63 (s, 1H), 4.76 (d, J = 3.2 Hz, 1H), 4.47 (dt, J = 11.5, 6.7 Hz, 1H), 2.12–1.87 (m, 4H), 1.98 (s, 3H), 1.97 (s, 3H), 1.87–0.96 (m, 21H), 0.91 (s, 3H), 0.89–0.87 (m, 1H), 0.88 (d, J = 6.6 Hz, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 176.0, 170.8, 170.6, 74.3, 71.4, 55.9, 42.8, 41.1, 39.6, 38.0, 35.5, 35.0, 34.9, 34.8, 34.2, 32.9, 31.7, 31.4, 28.2, 26.9, 23.7, 22.8, 21.7, 21.7, 21.6, 20.8, 18.5, 11.8. TLC-MS (ESI): m/z calcd C28H43O6 (M − H−) 475.6, found 475.9.
(R)-N-Butyl-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanamide (17b)
To a solution of UDCA (0.393 g, 1.00 mmol) in THF (10 mL) was added carbonyldiimidazole (0.243 g, 1.50 mmol) and the reaction stirred for 1 h at room temperature. To the solution was added butan-1-amine (0.198 mL, 2.00 mmol) and the reaction stirred for 18 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl (10 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aqueous NH4Cl (3 × 5 mL) and water (5 mL), and then dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (2–10% MeOH in EtOAc as eluent) to obtain 17b (0.162 g, 36% yield) as an off-white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 5.45 (s, 1H), 3.59 (tq, J = 10.0, 4.5, 4.1 Hz, 2H), 3.25 (td, J = 7.1, 5.7 Hz, 2H), 2.23 (ddd, J = 15.0, 10.4, 4.8 Hz, 1H), 2.12–0.97 (m, 28H), 0.96–0.90 (m, 9H), 0.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 173.8, 72.0, 68.4, 56.1, 50.5, 42.8, 41.7, 39.8, 39.5, 39.3, 35.6, 35.5, 35.2, 34.8, 33.7, 32.9, 32.1, 31.8, 30.7, 29.8, 28.4, 23.8, 22.9, 20.7, 20.2, 18.5, 13.9, 11.9. HRMS (ESI): m/z calcd C28H49NO3Na (M + Na+) 470.3610, found 470.3615. Compound 17a was prepared by the same method. Data for (R)-N-butyl-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-pentanamide (17a): 1H NMR (400 MHz, CDCl3) δ 5.57 (t, J = 5.8 Hz, 1H), 3.83 (q, J = 2.9 Hz, 1H), 3.44 (tt, J = 11.0, 4.3 Hz, 1H), 3.23 (q, J = 6.7 Hz, 2H), 2.30–1.03 (m, 27H), 1.03–0.83 (m, 11H), 0.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 173.8, 72.0, 68.4, 56.1, 50.5, 42.8, 41.7, 39.9, 39.8, 39.5, 39.3, 35.6, 35.5, 35.2, 34.8, 33.7, 32.9, 32.1, 31.8, 30.7, 28.4, 23.8, 22.9, 20.7, 20.2, 18.5, 13.9, 11.9. HRMS (ESI): m/z calcd C28H49NO3Na (M + Na+) 470.3610, found 470.3614.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin-1-yl)pentan-1-one (18b)
To a solution of UDCA (0.500 g, 1.27 mmol), TEA (0.53 mL, 3.8 mmol), and pyrrolidine (0.21 mL, 2.6 mmol) in DCM (13 mL) was added HATU (0.533 g, 1.40 mmol). The reaction mixture was stirred at room temperature for 16 h. The reaction was quenched by the addition of saturated aqueous NaHCO3 (6 mL), and the aqueous layer was extracted with DCM (3 × 5 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (2–5% MeOH in EtOAc as eluent) to obtain 18b (0.495 g, 87% yield) as an off-white foam. 1H NMR (400 MHz, CDCl3) δ 3.48 (dp, J = 10.2, 4.2 Hz, 2H), 3.14 (q, J = 6.7 Hz, 2H), 2.12 (ddd, J = 14.9, 10.5, 4.9 Hz, 1H), 2.00–0.86 (m, 26H), 0.86–0.77 (m, 11H), 0.57 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.2, 76.7, 71.5, 71.4, 55.7, 55.0, 46.6, 45.6, 43.8, 43.8, 42.4, 40.1, 39.2, 37.3, 36.8, 35.5, 34.9, 34.1, 31.7, 31.0, 30.3, 28.6, 26.9, 26.2, 24.4, 23.4, 21.2, 18.6. HRMS (ESI): m/z calcd C28H47NNaO3 (M + Na+) 468.3454, found 468.3479. Compound 18a was prepared by the same method. Data for (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin-1-yl)pentan-1-one (18a): 1H NMR (400 MHz, CDCl3) δ 3.85 (q, J = 3.0 Hz, 1H), 3.44 (dt, J = 13.8, 6.7 Hz, 6H), 2.30 (ddt, J = 16.7, 10.9, 5.5 Hz, 1H), 2.21–1.06 (m, 27H), 1.00 (dd, J = 14.2, 3.3 Hz, 1H), 0.95 (d, J = 6.4 Hz, 3H), 0.91 (s, 3H), 0.67 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.2, 72.0, 68.5, 55.8, 50.5, 46.6, 45.6, 42.7, 41.5, 39.9, 39.7, 39.4, 35.5, 35.3, 35.0, 34.6, 32.8, 31.6, 30.9, 30.7, 28.2, 26.2, 24.4, 23.7, 22.8, 20.6, 18.5, 11.8. HRMS (ESI): m/z calcd C28H47NO3Na (M + Na+) 468.3454, found 468.3454.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dimethoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin - 1 - yl) pentan-1-one (19b) and (R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-7-Hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin-1-yl)pentan-1-one (20b)
To a solution of 18b (0.480 g, 1.08 mmol) in THF (10 mL) was added NaH (60% dispersion in mineral oil, 0.065 g, 1.6 mmol). The reaction stirred at room temperature for 15 min, and MeI (0.08 mL, 1.3 mmol) was added in one portion. The reaction stirred overnight 13 h and quenched with saturated aqueous NH4Cl (8 mL). The organic and aqueous layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (1/3/0.1 DCM/EtOAc/MeOH) to obtain 19b (0.041 g, 8% yield) as white amorphous solid, followed by the elution of 20b (0.327 g, 66% yield) as a white amorphous solid. Data for 19b: 1H NMR (400 MHz, DMSO-d6) δ 3.38 (t, J = 6.8 Hz, 2H), 3.24 (t, J = 6.9 Hz, 2H), 3.22 (s, 3H), 3.17 (d, J = 5.3 Hz, 2H), 3.13 (s, 3H), 3.07 (td, J = 10.6, 5.0 Hz, 1H), 3.00–2.89 (m, 1H), 2.23 (ddd, J = 15.5, 10.4, 5.1 Hz, 1H), 2.09 (ddd, J = 15.5, 10.1, 5.8 Hz, 1H), 1.97–1.01 (m, 24H), 1.00–0.91 (m, 2H), 0.89 (d, J = 6.6 Hz, 3H), 0.87 (s, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.7, 79.4, 79.3, 55.5, 54.9, 54.8, 54.7, 54.7, 48.6, 45.9, 45.2, 43.1, 41.5, 40.9, 35.0, 34.4, 33.9, 33.2, 31.7, 30.8, 30.6, 28.1, 26.4, 26.1, 25.7, 24.0, 23.2, 20.9, 18.5, 12.0. TLC-MS (ESI): m/z calcd C30H52NO4 (M + OH−) 490.4, found 490.2. Data for 20b: 1H NMR (400 MHz, CDCl3) δ 3.59 (q, J = 7.0, 4.6 Hz, 1H), 3.44 (dt, J = 16.0, 6.8 Hz, 4H), 3.35 (s, 3H), 3.12 (dp, J = 10.6, 4.5 Hz, 1H), 2.31 (ddd, J = 15.4, 10.7, 5.0 Hz, 1H), 2.16 (ddd, J = 15.2, 10.3, 5.7 Hz, 1H), 2.04–1.04 (m, 27H), 1.01 (dd, J = 14.2, 3.0 Hz, 1H), 0.95 (s, 3H), 0.94 (d, J = 6.4 Hz, 3H), 0.68 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 3.86 (d, J = 6.8 Hz, 1H), 3.38 (t, J = 6.7 Hz, 2H), 3.24 (t, J = 6.9 Hz, 3H), 3.20 (s, 3H), 3.04 (td, J = 10.4, 4.6 Hz, 1H), 2.23 (ddd, J = 15.5, 10.5, 5.2 Hz, 1H), 2.09 (ddd, J = 15.5, 9.9, 5.9 Hz, 1H), 1.98–0.98 (m, 27H), 0.94 (dd, J = 14.3, 2.7 Hz, 1H), 0.89 (d, J = 6.3 Hz, 3H), 0.88 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.4, 80.2, 71.6, 55.9, 55.8, 55.2, 46.8, 45.8, 44.0, 42.6, 40.3, 39.3, 37.1, 35.7, 35.1, 34.6, 34.6, 33.9, 31.9, 31.2, 28.8, 27.1, 26.9, 26.4, 24.6, 23.6, 21.4, 18.8, 12.4. HRMS (ESI): m/z calcd C29H49NO3Na (M + Na+) 482.3610, found 482.3612. Compounds 19a and 20a were prepared by the same method. Data for (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dimethoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin-1-yl)pentan-1-one (19a): 1H NMR (400 MHz, CDCl3) δ 3.45 (dt, J = 16.5, 6.8 Hz, 4H), 3.34 (s, 3H), 3.24 (s, 3H), 3.18 (q, J = 2.8 Hz, 1H), 3.01 (tt, J = 11.1, 4.2 Hz, 1H), 2.31 (ddd, J = 15.5, 10.9, 5.0 Hz, 1H), 2.24–2.08 (m, 2H), 2.01–1.67 (m, 12H), 1.63 (ddd, J = 15.0, 5.5, 3.2 Hz, 1H), 1.59–1.41 (m, 5H), 1.42–1.15 (m, 8H), 1.05 (tt, J = 10.3, 5.7 Hz, 1H), 0.94 (d, J = 6.6 Hz, 3H), 0.91 (s, 3H), 0.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.5, 80.9, 80.9, 56.1, 55.9, 55.6, 50.4, 46.8, 45.8, 42.7, 42.2, 39.8, 39.7, 35.7, 35.6, 35.5, 34.9, 33.9, 31.7, 31.1, 28.4, 28.1, 26.9, 26.4, 24.6, 23.9, 23.2, 21.1, 18.7, 11.9. HRMS (ESI): m/z calcd C30H51NNaO3 (M + Na+) 496.3767, found 496.3769. Data for (R)-4-((3R,5R,7R,8R,9S,10S,13R,14S,17R)-7-hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1-(pyrrolidin-1-yl)pentan-1-one (20a): 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 1H), 3.44 (dt, J = 16.8, 7.0 Hz, 4H), 3.35 (d, J = 1.0 Hz, 3H), 3.02 (td, J = 11.0, 5.4 Hz, 1H), 2.31 (ddd, J = 15.5, 10.8, 5.1 Hz, 1H), 2.24–2.09 (m, 2H), 2.03–1.03 (m, 26H), 0.99–0.95 (m, 1H), 0.95 (d, J = 6.5 Hz, 3H), 0.91 (s, 3H), 0.67 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.4, 80.8, 68.7, 56.0, 55.7, 50.7, 46.8, 45.8, 42.9, 41.6, 39.9, 39.7, 36.1, 35.7, 35.6, 35.5, 34.9, 33.0, 31.7, 31.1, 28.4, 27.2, 26.4, 24.6, 24.0, 23.1, 20.8, 18.7, 12.0. HRMS (ESI): m/z calcd C29H49NO3Na (M + Na+) 482.3610, found 482.3624.
Sodium (3R,5S,7S,8R,9S,10S,13R,14S,17R)-3-Methoxy-10,13-dimethyl-17-((R)-5-oxo-5-(pyrrolidin-1-yl) pentan-2-yl)-hexadecahydro-1H-cyclopenta[a]phenanthren-7-yl Sulfate (21b)
To a solution of 20b (0.050 g, 0.11 mmol) in pyridine (0.54 mL) was added dropwise chlorosulfonic acid (0.05 mL, 0.7 mmol). The reaction mixture was heated to 50 °C for 30 min, then cooled to room temperature. The reaction was terminated by the addition of water (1 mL), and the solvents were removed by rotary evaporation. The crude material was dissolved in DMSO (0.5 mL) and 1 M triethylammonium acetate buffer (0.1 mL) and purified by flash column chromatography (5–100% 20 mM triethylammonium acetate buffer in acetonitrile in water as eluent, C18 column) to yield a white solid after lyophilization. To prepare the sodium salt of 21b, a 1 cm wide column was filled with 12 cm of Dowex-50WX2 (50–100 mesh, strongly acidic) ion-exchange resin. The column was prepared by sequentially washing with 1:1 acetonitrile/water, ~1 M aqueous NaHCO3 (caution: gas evolution), water, and finally 1:1 acetonitrile/water. The reaction product was dissolved in 1:1 acetonitrile/water and loaded onto the column, which was eluted with 1:1 acetonitrile/water. The fractions containing the product were lyophilized to furnish 21b as an off-white solid (0.054 g, 88% yield). 1H NMR (400 MHz, D2O) δ 4.32 (app q, J = 9.9, 8.5 Hz, 1H), 3.51 (t, J = 6.9 Hz, 2H), 3.40 (d, J = 7.0 Hz, 2H), 3.36 (s, 3H), 3.31 (s, 1H), 2.37 (d, J = 10.4 Hz, 1H), 2.32–2.16 (m, 1H), 2.14–1.05 (m, 28H), 1.01 (s, 3H), 1.00 (d, J = 6.6 Hz, 6H), 0.72 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 3.93 (s, 1H), 3.38 (t, J = 6.7 Hz, 2H), 3.25 (t, J = 6.8 Hz, 2H), 3.21 (s, 3H), 3.05 (s, 1H), 2.29–2.00 (m, 4H), 1.99–1.80 (m, 3H), 1.80–1.53 (m, 8H), 1.37 (d, J = 7.8 Hz, 6H), 1.29–0.97 (m, 8H), 0.94 (d, J = 14.8 Hz, 1H), 0.89 (d, J = 6.4 Hz, 3H), 0.89 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, D2O) δ 174.5, 81.0, 80.0, 54.8, 54.5, 54.3, 47.5, 46.1, 43.4, 41.7, 41.1, 39.8, 39.2, 35.2, 34.4, 33.8, 33.5, 32.8, 31.1, 30.9, 28.1, 26.0, 25.8, 25.5, 24.1, 23.1, 21.4, 18.7, 11.9. TLC-MS (ESI): m/z calcd C29H47NO6S (M − H)− 537.3, found 537.2.
(R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-N-(1-hydroxy-2-methylpropan-2-yl)pentanamide (22a)
To a suspension of CDCA (0.589 g, 1.50 mmol) in DCM (15 mL) was added TEA (0.63 mL, 4.5 mmol), 2-amino-2-methylpropan-1-ol (0.147 g, 1.65 mmol), and HATU (0.627 g, 1.65 mmol). The reaction was stirred at room temperature for 2 h and was quenched by the addition of water (10 mL). The aqueous and organic layers were separated, and the organic layer was washed with water (2 × 10 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (10–100% EtOAc in DCM as eluent) to obtain 22a (0.375 g, 54% yield) as an off-white foam. 1H NMR (400 MHz, DMSO-d6) δ 7.22 (s, 1H), 4.88 (t, J = 5.9 Hz, 1H), 4.30 (d, J = 4.7 Hz, 1H), 4.10 (d, J = 3.4 Hz, 1H), 3.62 (s, 1H), 3.42–3.33 (m, 2H), 3.17 (dd, J = 10.5, 5.0 Hz, 1H), 2.32–1.15 (m, 22H), 1.15 (s, 6H), 1.13–0.93 (m, 3H), 0.88–0.86 (m, 1H), 0.87 (d, J = 6.6 Hz, 3H), 0.83 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 72.0, 70.9, 68.5, 56.2, 55.9, 50.5, 42.8, 41.6, 39.9, 39.8, 39.5, 35.5, 35.4, 35.2, 34.7, 34.1, 32.9, 31.9, 30.7, 28.4, 24.8, 24.8, 23.8, 22.9, 20.7, 18.6, 11.9. TLC-MS (ESI): m/z calcd C28H43O6 (M + OH + 2H2O+) 516.4, found 516.0.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-N-((Z)-1-(hydroxyamino)prop-1-en-2-yl)pentanamide (23b)
To a suspension of UDCA (1.00 g, 2.55 mmol) in DCM (8.5 mL) was added (Z)-N′-hydroxyacetimidamide (0.189 g, 2.55 mmol), HATU (1.07 g, 2.80 mmol), and diisopropylethylamine (1.3 mL, 7.6 mmol). The reaction stirred at room temperature for 24 h and was quenched with water (5 mL). The organic phase was washed with water (2 × 10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (1–10% MeOH in EtOAc as eluent) to obtain 23b (0.639 g, 56% yield) as a white amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 6.29 (s, 2H), 4.43 (d, J = 4.6 Hz, 1H), 3.87 (d, J = 6.8 Hz, 1H), 3.31–3.22 (m, 2H), 2.37 (ddd, J = 15.2, 9.8, 5.2 Hz, 1H), 2.24 (ddd, J = 15.7, 9.3, 6.7 Hz, 2H), 1.94 (d, J = 11.2 Hz, 1H), 1.90–1.73 (m, 2H), 1.72 (s, 3H), 1.70–1.58 (m, 4H), 1.56–0.93 (m, 16H), 0.90 (d, J = 6.6 Hz, 3H), 0.87 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.1, 155.7, 69.7, 69.5, 64.9, 55.9, 54.6, 43.1, 43.0, 42.2, 38.7, 37.7, 37.3, 34.9, 34.8, 33.8, 30.7, 30.2, 29.3, 28.1, 26.7, 23.3, 20.8, 18.4, 16.3, 12.0. TLC-MS (ESI): m/z calcd C26H44N2O4 (M−) 448.3, found 448.2. Compound 23a was prepared by the same method. Data for (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-N-((Z)-1-(hydroxyamino)prop-1-en-2-yl)pentanamide (23a): 1H NMR (400 MHz, DMSO-d6) δ 4.43 (d, J = 4.6 Hz, 1H), 3.87 (d, J = 6.8 Hz, 1H), 3.30–3.21 (m, 2H), 2.90 (ddd, J = 15.1, 10.0, 4.4 Hz, 1H), 2.84–2.70 (m, 1H), 2.29 (s, 3H), 1.97–1.00 (m, 25H), 0.94 (d, J = 5.9 Hz, 3H), 0.89–0.87 (m, 1H), 0.84 (s, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 179.8, 166.6, 69.6, 69.3, 64.8, 55.7, 54.3, 43.0, 42.9, 42.1, 38.8, 38.6, 37.6, 37.2, 34.7, 33.6, 32.2, 30.1, 28.0, 26.6, 23.2, 22.5, 20.7, 18.1, 11.9, 11.0. TLC-MS (ESI): m/z calcd C26H43N2O4 (M − H−) 447.3, found 447.4.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-10,13-Dimethyl-17-((R)-4-(3-methyl-1,2,4-oxadiazol-5-yl)butan-2-yl)hexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol (24b)
Compound 23b (0.300 g, 0.669 mmol) was dissolved in toluene (2.2 mL) and THF (2.5 mL). The solution was heated in the MW at 160 °C for 45 min. The solvent was removed under reduced pressure, and the crude residue was purified by flash column chromatography on silica gel (0–10% MeOH in EtOAc as eluent) to obtain 24b (0.155 g, 54% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 4.43 (d, J = 4.6 Hz, 1H), 3.87 (d, J = 6.8 Hz, 1H), 3.31–3.23 (m, 2H), 2.90 (ddd, J = 15.1, 10.0, 4.4 Hz, 1H), 2.86–2.72 (m, 2H), 2.29 (s, 3H), 2.01–1.59 (m, 7H), 1.57–1.00 (m, 16H), 0.94 (d, J = 5.9 Hz, 3H), 0.87 (s, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 180.0, 166.8, 69.8, 69.5, 55.9, 54.5, 43.2, 43.1, 42.2, 38.8, 38.8, 37.8, 37.3, 34.9, 33.8, 33.8, 32.4, 30.3, 28.2, 26.8, 23.4, 22.7, 20.9, 18.3, 12.1, 11.2. HRMS (ESI): m/z calcd C26H42N2NaO3 (M + Na+) 453.3093, found 453.3074. Compound 24a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-10,13-dimethyl-17-((R)-4-(3-methyl-1,2,4-oxadiazol-5-yl)butan-2-yl)hexadecahydro-1H-cyclopenta-[a]phenanthrene-3,7-diol (24a): 1H NMR (400 MHz, DMSO-d6) δ 4.30 (d, J = 4.7 Hz, 1H), 4.11 (d, J = 3.4 Hz, 1H), 3.62 (t, J = 3.2 Hz, 1H), 3.25–3.10 (m, 1H), 2.90 (ddd, J = 14.8, 10.1, 4.5 Hz, 1H), 2.84–2.73 (m, 1H), 2.29 (s, 3H), 2.19 (q, J = 13.0 Hz, 1H), 1.97–1.61 (m, 7H), 1.40 (tdd, J = 21.4, 13.9, 10.1 Hz, 8H), 1.29–1.06 (m, 7H), 1.00 (td, J = 11.8, 6.1 Hz, 1H), 0.94 (d, J = 5.6 Hz, 3H), 0.84 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 180.3, 167.1, 72.0, 68.5, 55.7, 50.5, 42.8, 41.6, 39.9, 39.7, 39.5, 35.5, 35.4, 35.1, 34.8, 32.9, 32.8, 30.8, 28.3, 23.8, 23.5, 22.9, 20.7, 18.4, 11.9, 11.7. HRMS (ESI): m/z calcd C26H42N2NaO3 (M + Na+) 453.3093, found 453.3075.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-5-(2-Acetylhydrazinyl)-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta-[a]phenanthrene-3,7-diyl Diacetate (25b)
To a solution of 15b (1.19 g, 2.50 mmol) in DCM (25 mL) was added HATU (1.23 g, 3.25 mmol), DIPEA (1.2 mL, 6.7 mmol), and acetohydrazide (0.222 g, 3.00 mmol). The reaction stirred at room temperature for 18 h and was quenched by the addition of saturated aqueous NaHCO3 (20 mL). The aqueous layer was extracted with DCM (3 × 20 mL), and the combined organic layers were washed with saturated aqueous NaHCO3 (3 × 20 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (1–10% MeOH in DCM as eluent) to obtain 25b (0.633 g, 48% yield) as a yellow foam. 1H NMR (400 MHz, DMSO-d6) δ 9.66 (d, J = 2.0 Hz, 1H), 9.62 (d, J = 2.0 Hz, 1H), 4.66 (td, J = 10.7, 4.9 Hz, 1H), 4.55 (tt, J = 10.6, 4.9 Hz, 1H), 2.12 (ddd, J = 14.3, 9.5, 5.1 Hz, 1H), 2.07–1.99 (m, 1H), 1.98 (s, 3H), 1.93 (s, 3H), 1.83 (s, 3H), 1.80–0.96 (m, 24H), 0.93 (s, 3H), 0.89 (d, J = 6.4 Hz, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.3, 169.8, 169.8, 167.9, 73.0, 72.8, 54.9, 54.5, 54.4, 43.1, 41.3, 39.2, 38.6, 34.7, 33.9, 33.5, 32.7, 32.5, 31.3, 30.1, 28.0, 26.0, 25.3, 22.8, 21.5, 21.0, 20.8, 20.5, 18.4, 11.8. TLC-MS (ESI): m/z calcd C30H48N2O6 (M−) 532.4, found 532.5. Compound 25a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-17-((R)-5-(2-acetylhydrazinyl)-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthrene-3,7-diyl diacetate (25a): 1H NMR (400 MHz, DMSO-d6) δ 9.67 (d, J = 2.0 Hz, 1H), 9.63 (d, J = 2.0 Hz, 1H), 4.76 (q, J = 2.9 Hz, 1H), 4.47 (tt, J = 11.2, 4.4 Hz, 1H), 2.13 (ddd, J = 14.5, 9.6, 5.2 Hz, 1H), 2.08–1.88 (m, 10H), 1.82 (s, 3H), 1.81–0.97 (m, 20H), 0.90 (s, 3H), 0.89 (d, J = 7.6 Hz, 3H), 0.88–0.84 (m, 1H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.3, 169.8, 169.7, 167.9, 73.4, 70.5, 55.3, 50.0, 42.2, 40.2, 39.0, 37.1, 34.8, 34.4, 34.3, 34.3, 33.6, 31.2, 30.8, 30.0, 27.6, 26.4, 23.0, 22.3, 21.3, 21.1, 20.5, 20.2, 18.2, 11.6. TLC-MS (ESI): m/z calcd C30H48N2O6 (M + H+) 533.4, found 533.3.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-10,13-Dimethyl-17-((R)-4-(5-methyl-1,3,4-oxadiazol-2-yl)butan-2-yl)hexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl Diacetate (26b)
To a solution of 25b (0.623 g, 1.17 mmol) and triethylamine (0.41 mL, 2.9 mmol) in DCM (12 mL) was added tosyl chloride (0.223 g, 1.17 mmol) and the reaction stirred at room temperature for 24 h. The solvent was removed under reduced pressure, and the crude material was purified by flash column chromatography on silica gel (1–5% MeOH in DCM as eluent) to obtain 26b (0.491 g, 0.954 mmol, 82% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 4.66 (td, J = 10.7, 4.9 Hz, 1H), 4.55 (dq, J = 10.4, 5.2, 4.8 Hz, 1H), 2.81 (ddd, J = 14.2, 9.7, 4.2 Hz, 1H), 2.70 (dt, J = 15.5, 7.7 Hz, 1H), 2.44 (s, 3H), 1.97 (s, 3H), 1.93 (s, 3H), 1.84–0.99 (m, 23H), 0.94 (d, J = 5.9 Hz, 3H), 0.92 (s, 3H), 0.88–0.76 (m, 1H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.8, 169.7, 166.7, 163.3, 72.9, 72.8, 54.3, 54.2, 43.1, 41.8, 41.2, 39.2, 38.6, 34.6, 33.9, 33.5, 32.6, 32.5, 32.1, 27.9, 26.0, 25.2, 22.8, 21.4, 21.0, 20.9, 18.1, 14.1, 11.8, 10.4. TLC-MS (ESI): m/z calcd C30H47N2O5 (M + H+) 515.3, found 515.7. Compound 26a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-10,13-dimethyl-17-((R)-4-(5-methyl-1,3,4-oxadiazol-2-yl)butan-2-yl)hexadecahydro-1H-cyclopenta-[a]phenanthrene-3,7-diyl diacetate (26a): 1H NMR (400 MHz, DMSO-d6) δ 4.76 (t, J = 3.1 Hz, 1H), 4.47 (tt, J = 10.9, 4.6 Hz, 1H), 2.82 (ddd, J = 14.6, 9.7, 4.4 Hz, 1H), 2.70 (dt, J = 15.5, 7.6 Hz, 1H), 2.44 (s, 3H), 2.07–1.87 (m, 2H), 1.98 (s, 3H), 1.97 (s, 3H), 1.86–0.98 (m, 22H), 0.94 (d, J = 6.0 Hz, 3H), 0.91 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.8, 169.7, 166.8, 163.3, 73.4, 70.5, 55.0, 50.0, 42.2, 41.8, 39.0, 37.1, 34.7, 34.4, 34.3, 34.3, 33.6, 32.0, 30.8, 27.5, 26.4, 23.0, 22.3, 21.4, 21.3, 21.1, 20.2, 18.0, 11.5, 10.4. TLC-MS (ESI): m/z calcd C30H47N2O5 (M + OH−) 531.3, found 531.3.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-10,13-Dimethyl-17-((R)-4-(5-methyl-1,3,4-oxadiazol-2-yl)butan-2-yl)hexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol (27b)
To a solution of 26b (0.491 g, 0.954 mmol) in MeOH (5 mL) was added NaOH (0.095 g, 2.4 mmol), and the reaction was heated to reflux for 12 h. The solution was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The aqueous layer was extracted with EtOAc (2 × 10 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel to obtain 27b (0.250 g, 61% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 4.43 (d, J = 4.5 Hz, 1H), 3.87 (d, J = 6.8 Hz, 1H), 3.32–3.22 (m, 2H), 2.82 (ddd, J = 14.4, 10.0, 4.3 Hz, 1H), 2.70 (ddd, J = 15.5, 9.1, 6.6 Hz, 1H), 2.44 (s, 3H), 2.01–1.00 (m, 23H), 0.94 (d, J = 5.7 Hz, 3H), 0.93–0.91 (m, 1H), 0.87 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 163.3, 69.7, 69.4, 55.8, 54.9, 54.4, 43.1, 43.0, 42.2, 38.7, 37.7, 37.3, 34.8, 34.8, 33.7, 32.2, 30.2, 28.1, 26.7, 23.3, 21.5, 20.8, 18.2, 12.0, 10.4. TLC-MS (ESI): m/z calcd C26H43N2O3 (M + H−) 431.3, found 431.6. Compound 27a was prepared by the same method. Data for (3R,5S,7S,8R,9S,10S,13R,14S,17R)-10,13-dimethyl-17-((R)-4-(5-methyl-1,3,4-oxadiazol-2-yl)butan-2-yl)hexadecahydro-1H-cyclopenta[a]-phenanthrene-3,7-diol (27a): 1H NMR (400 MHz, DMSO-d6) δ 4.30 (d, J = 4.6 Hz, 1H), 4.11 (d, J = 3.4 Hz, 1H), 3.74–3.52 (m, 1H), 3.18 (qt, J = 9.5, 4.4 Hz, 1H), 2.82 (ddd, J = 14.4, 9.8, 4.4 Hz, 1H), 2.70 (ddd, J = 15.6, 9.2, 6.7 Hz, 1H), 2.44 (s, 3H), 2.19 (app q, J = 12.8 Hz, 1H), 1.96–1.05 (m, 21H), 1.00 (td, J = 12.2, 11.7, 6.2 Hz, 1H), 0.94 (d, J = 6.0 Hz, 3H), 0.88 (dd, J = 14.4, 3.2 Hz, 1H), 0.83 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 163.3, 70.3, 66.1, 59.7, 55.3, 50.0, 42.0, 41.4, 39.6, 35.3, 34.9, 34.8, 34.7, 32.3, 32.1, 30.6, 27.7, 23.1, 22.7, 21.5, 20.8, 20.2, 18.1, 11.6, 10.4. TLC-MS (ESI): m/z calcd C26H43N2O3 (M + H−) 431.3, found 431.2.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-5-((1-Hydroxy-2-methylpropan-2-yl)amino)-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl Diformate (29b)
UDCA (1.00 g, 2.55 mmol) was dissolved in formic acid (20 mL), and perchloric acid (0.05 mL, 0.7 mmol) was added. The solution was heated to 50 °C for 12 h, then cooled to 40 °C. Acetic anhydride (10 mL) was added over 10 min, and the solution was cooled to room temperature. The reaction was quenched with water (100 mL), and the aqueous layer was extracted with Et2O (3 × 100 mL). The combined organic layers were washed with water (10 × 50 mL), dried over Na2SO4, filtered, and concentrated. Crude (R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-bis(formyloxy)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid (28b) was used for the next step without further purification. Thionyl chloride (1.71 mL, 23.4 mmol) was added dropwise to a solution of the crude material in benzene (12 mL), and the reaction was heated to reflux for 4 h. The reaction was cooled to room temperature, and the solvent was evaporated. The residual thionyl chloride was removed by coevaporation with benzene (2 × 6 mL), and the crude material was dissolved in DCM (5 mL). A solution of 2-amino-2-methylpropan-1-ol (0.498 g, 5.59 mmol) in DCM (1 mL) was added dropwise at 0 °C. After 1.5 h, the reaction was filtered and the solids were rinsed with DCM. The filtrate was evaporated to dryness, and the crude material was purified by flash column chromatography on silica gel (0–40% EtOAc in DCM as eluent) to obtain 29b (0.824 g, 62% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 8.16 (s, 1H), 4.87 (t, J = 5.9 Hz, 1H), 4.77 (td, J = 10.8, 4.9 Hz, 1H), 4.66 (dq, J = 10.6, 5.3, 4.7 Hz, 1H), 3.35 (d, J = 5.3 Hz, 2H), 2.07 (ddd, J = 14.5, 9.8, 5.2 Hz, 1H), 1.98–1.87 (m, 2H), 1.84–1.49 (m, 12H), 1.46–1.16 (m, 8H), 1.15 (s, 6H), 1.13–0.96 (m, 4H), 0.94 (s, 3H), 0.88 (d, J = 6.5 Hz, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.7, 152.1, 151.7, 63.1, 63.0, 57.6, 54.9, 44.5, 44.3, 44.1, 33.1, 31.2, 24.7, 23.8, 23.5, 23.0, 22.8, 22.5, 21.6, 17.9, 16.1, 15.6, 13.7, 13.7, 12.8, 10.8, 8.5, 5.2, 1.8, −9.9. TLC-MS (ESI): m/z calcd C30H48NO3 (M − H+) 518.4, found 518.9.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-4-(4,4-Dimethyl-4,5-dihydrooxazol-2-yl)butan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl Diformate (30b)
To a solution of 29b (0.824 g, 1.59 mmol) in THF (4 mL) was added dropwise SOCl2 (0.64 mL, 8.7 mmol) at 0 °C. After stirring for 18 h overnight, the reaction was quenched with saturated aqueous NaHCO3 (10 mL). The aqueous layer was extracted with Et2O (5 × 10 mL). and the combined organic layers were dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–30% EtOAc in DCM as eluent) to obtain 30b (0.505 g, 64% yield) as an off-white foam. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.98 (s, 1H), 4.90 (td, J = 10.8, 5.2 Hz, 1H), 4.81 (dq, J = 10.2, 5.4 Hz, 1H), 2.37–1.02 (m, 34H), 0.99 (s, 3H), 0.93 (d, J = 6.4 Hz, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.8, 161.2, 160.8, 79.1, 73.8, 73.6, 71.1, 56.4, 55.5, 55.2, 43.9, 43.8, 42.2, 40.1, 40.0, 39.6, 35.5, 34.6, 34.4, 34.2, 33.1, 33.0, 32.0, 28.6, 26.6, 26.0, 23.4, 21.4, 18.7, 12.3. TLC-MS (ESI): m/z calcd C32H51N2O5 (M + H + CH3CN+) 543.4, found 543.1.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-4-(4,4-Dimethyl-4,5-dihydrooxazol-2-yl)butan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol (31b)
NaOH (0.078 g, 1.9 mmol) was added to a solution of 30b (0.390 g, 0.777 mmol) in MeOH (4 mL), and the reaction was heated to reflux for 2 h. The reaction was cooled to room temperature and diluted with EtOAc (25 mL). The organic layer was washed with water (3 × 10 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (0–100% EtOAc in DCM) to obtain 31b (0.254 g, 73% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 4.43 (dd, J = 4.7, 2.7 Hz, 1H), 3.86 (d, J = 6.8 Hz, 1H), 3.83 (d, J = 1.0 Hz, 2H), 3.32–3.21 (m, 2H), 2.18 (ddd, J = 14.8, 9.6, 5.1 Hz, 1H), 2.11–1.99 (m, 1H), 1.98–1.60 (m, 6H), 1.57–0.98 (m, 24H), 0.89 (d, J = 6.5 Hz, 3H), 0.87 (s, 3H), 0.61 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.7, 77.9, 69.7, 69.4, 66.7, 64.9, 55.8, 54.5, 43.1, 43.0, 42.2, 38.7, 37.7, 37.3, 34.8, 34.7, 33.7, 31.8, 30.2, 28.3, 28.2, 28.2, 26.7, 24.3, 23.3, 20.8, 18.4, 12.0. HRMS (ESI): m/z calcd C28H48NO3 (M + H+) 446.3634, found 446.3608.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-5-Amino-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthrene-3,7-diyl Diformate (32b)
To a solution of 28b (3.30 g, 7.36 mmol) in benzene (38 mL) was added SOCl2 (2.8 mL, 38 mmol), followed by 3 drops of DMF. The reaction was heated to 80 °C for 4 h, then cooled to room temperature. The excess SOCl2 was removed by coevaporation with benzene (3 × 5 mL). The residue was dissolved in benzene (20 mL), and ammonia gas was bubbled through the solution for 20 min. The solvent was removed under reduced pressure, and the residue was partitioned between water (50 mL) and EtOAc (50 mL). The aqueous phase was extracted with EtOAc (2 × 20 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (5–100% EtOAc in hexanes) to obtain 32b (1.72 g, 52% yield) as a white foam. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 8.16 (s, 1H), 7.37–7.10 (m, 1H), 6.63 (s, 1H), 4.77 (td, J = 10.7, 4.9 Hz, 1H), 4.68 (dq, J = 11.0, 5.9, 5.3 Hz, 1H), 2.04 (dt, J = 10.0, 5.0 Hz, 1H), 1.99–0.96 (m, 25H), 0.94 (s, 3H), 0.88 (d, J = 6.4 Hz, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.6, 162.1, 161.7, 73.1, 73.0, 54.5, 54.3, 43.1, 41.2, 39.2, 38.5, 34.8, 33.8, 33.5, 32.8, 32.5, 32.0, 31.4, 30.7, 27.9, 26.1, 25.6, 22.8, 20.8, 18.4, 11.8. TLC-MS (ESI): m/z calcd C26H41NNaO5 (M + Na+) 470.3, found 470.9. Compound 32a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-17-((R)-5-amino-5-oxopentan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl diformate (32a): 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.17 (s, 1H), 7.20 (s, 1H), 6.63 (s, 1H), 4.88 (d, J = 3.2 Hz, 1H), 4.59 (tt, J = 10.9, 4.5 Hz, 1H), 2.11–0.99 (m, 26H), 0.92 (s, 3H), 0.88 (d, J = 6.4 Hz, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.7, 161.9, 161.7, 73.3, 70.8, 55.3, 49.9, 42.2, 42.2, 39.0, 37.0, 34.8, 34.3, 34.2, 33.5, 31.9, 31.3, 31.1, 27.5, 26.5, 22.9, 22.3, 20.8, 20.2, 18.3, 11.6. TLC-MS (ESI): m/z calcd C26H41NNaO5 (M + Na+) 470.3, found 470.8.
(3R,5S,7S,8R,9S,10S,13R,14S,17R)-17-((R)-4-Cyanobutan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl Diformate (33b)
To a solution of 32b (0.780 g, 1.74 mmol) in THF (17 mL) was added pyridine (0.28 mL, 3.4 mmol) and TFAA (0.50 mL, 3.5 mmol). The reaction was stirred overnight at room temperature, and the solvent was removed under reduced pressure. The residue was partitioned between 1 M aqueous HCl (50 mL) and EtOAc (50 mL). The aqueous layer was extracted with EtOAc (2 × 50 mL), and the combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated. The crude material (33b) was used for the next reaction without further purification. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 8.16 (s, 1H), 4.77 (td, J = 10.8, 4.9 Hz, 1H), 4.67 (tt, J = 10.7, 4.8 Hz, 1H), 2.60–2.33 (m, 2H), 1.98–0.98 (m, 24H), 0.94 (s, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.65 (s, 3H). 1H NMR (400 MHz, CD3OD) δ 3.57–3.40 (m, 2H), 2.56–2.31 (m, 2H), 2.09–1.01 (m, 24H), 0.99 (d, J = 6.5 Hz, 3H), 0.97 (s, 3H), 0.74 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.1, 160.7, 120.2, 73.6, 73.5, 55.4, 54.9, 43.8, 42.1, 39.9, 39.9, 39.5, 35.1, 34.5, 34.1, 33.0, 32.9, 31.6, 28.5, 26.5, 25.9, 23.3, 21.3, 18.1, 14.4, 12.2. HRMS (ESI): m/z calcd C26H39NNaO4 (M + Na+) 452.2777, found 452.2747. Compound 33a was prepared by the same method. Data for (3R,5S,7R,8R,9S,10S,13R,14S,17R)-17-((R)-4-cyanobutan-2-yl)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,7-diyl diformate (33a): 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.17 (s, 1H), 4.89 (q, J = 3.0 Hz, 1H), 4.59 (tt, J = 11.0, 4.5 Hz, 1H), 2.52 (m, 2H), 2.41 (dt, J = 16.8, 8.1 Hz, 1H), 2.14–0.98 (m, 22H), 0.92 (s, 3H), 0.91 (d, J = 6.7 Hz, 3H), 0.92–0.87 (m, 1H), 0.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 160.8, 160.8, 120.2, 74.1, 71.4, 55.6, 50.2, 42.9, 41.0, 39.5, 37.9, 35.2, 34.9, 34.9, 34.7, 34.0, 31.5, 31.5, 28.1, 26.8, 23.5, 22.7, 20.7, 18.0, 14.3, 11.8. TLC-MS (ESI): m/z calcd C26H39NNaO4 (M + Na+) 452.3, found 452.0.
(R)-4-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-Dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-pentanenitrile (34b)
To a suspension of 33b (0.749 g, 1.74 mmol) in MeOH (8.72 mL) was added NaOH (0.174 g, 4.36 mmol), and the reaction was heated to reflux for 12 h. The reaction was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was partitioned between 1 M aqueous HCl (15 mL) and EtOAc (30 mL). The aqueous layer was extracted with EtOAc (2 × 30 mL), and the combined organic layers were washed with water (10 mL) and brine (10 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (50–100% EtOAc in hexanes as eluent) to obtain 34b as a white amorphous solid (0.445 g, 68% yield over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ 4.43 (d, J = 4.5 Hz, 1H), 3.88 (d, J = 7.0 Hz, 1H), 3.33–3.19 (m, 2H), 2.62–2.30 (m, 2H), 2.00–0.96 (m, 24H), 0.91 (d, J = 6.5 Hz, 3H), 0.87 (s, 3H), 0.63 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 121.1, 69.7, 69.4, 55.8, 54.3, 43.2, 43.0, 42.2, 38.7, 37.7, 37.3, 34.8, 34.8, 33.8, 33.8, 31.0, 30.2, 28.1, 26.7, 23.3, 20.8, 17.8, 13.4, 12.0. TLC-MS (ESI): m/z calcd C24H39NO2 (M+) 373.3, found 373.5. Compound 34a was prepared by the same method. Data for (R)-4-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanenitrile (34a): 1H NMR (400 MHz, DMSO-d6) δ 4.31 (d, J = 4.7 Hz, 1H), 4.12 (d, J = 3.4 Hz, 1H), 3.63 (q, J = 3.0 Hz, 1H), 3.19 (ddp, J = 15.5, 9.5, 4.7, 4.3 Hz, 1H), 2.60–2.31 (m, 2H), 2.19 (q, J = 12.8 Hz, 1H), 1.94–0.95 (m, 22H), 0.91 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 3.1 Hz, 1H), 0.84 (s, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 120.3, 71.8, 68.3, 55.6, 42.7, 41.5, 40.9, 39.7, 39.7, 39.4, 35.4, 35.2, 35.1, 34.8, 32.8, 31.5, 30.6, 28.2, 23.6, 22.8, 20.6, 17.9, 14.3, 11.8. TLC-MS (ESI): m/z calcd C24H41NO3 (M + H2O+) 391.3, found 391.6.
Sodium 5-((R)-3-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)butyl)tetrazol-1-ide (35b)
To a suspension of 34b (0.200 g, 0.535 mmol) in toluene (1.1 mL) in a MW vial was added NaN3 (0.104 g, 1.61 mmol) and triethylamine hydrochloride (0.258 g, 1.87 mmol). The reaction was heated via MW at 130 °C for 3 h, then cooled to room temperature. DMF (1 mL) was added, and the reaction was heated via MW at 160 °C for 4 h, then cooled to room temperature. The reaction was quenched with water (1 mL), and 1 M aqueous HCl (2 mL) was added. The precipitate was collected via vacuum filtration and rinsed with water (3 mL), then dried on the frit. To prepare the sodium salt of 35b, a 1 cm wide column was filled with 12 cm of Dowex-50WX2 (50–100 mesh, strongly acidic) ion-exchange resin. The column was prepared by sequentially washing with 1:1 acetonitrile/water, ~1 M aqueous NaHCO3 (caution: gas evolution), water, and finally 1:1 acetonitrile/water. The reaction product was dissolved in 1:1 acetonitrile/water and loaded onto the column, which was eluted with 1:1 acetonitrile/water. The fractions containing the product were lyophilized to furnish 35b as a white solid (98 mg, 42% yield). 1H NMR (400 MHz, CD3OD) δ 3.48 (t, J = 6.9 Hz, 2H), 2.88 (ddd, J = 14.5, 10.7, 4.2 Hz, 1H), 2.72 (dt, J = 14.8, 8.4 Hz, 1H), 2.05 (d, J = 12.4 Hz, 1H), 1.99–1.74 (m, 4H), 1.70–1.09 (m, 18H), 1.06–1.02 (m, 1H), 1.04 (d, J = 5.9 Hz, 3H), 0.96 (s, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 163.8, 72.2, 72.0, 57.5, 56.6, 44.8, 44.5, 44.1, 41.6, 40.7, 38.6, 38.0, 36.8, 36.7, 36.1, 35.2, 31.1, 29.7, 28.0, 23.9, 22.9, 22.4, 19.2, 12.6. LC/MS (ESI): m/z calcd C24H39N4O2 (M+) 415.3, found 415.6. Compound 35a was prepared by the same method. Data for sodium 5-((R)-3-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)butyl)tetrazol-1-ide (35a): 1H NMR (400 MHz, CD3OD) δ 3.79 (d, J = 2.8 Hz, 1H), 3.38 (td, J = 11.0, 5.5 Hz, 1H), 2.99 (ddd, J = 15.0, 10.4, 4.3 Hz, 1H), 2.93–2.78 (m, 1H), 2.27 (q, J = 12.9 Hz, 1H), 2.06–1.16 (m, 21H), 1.16–1.07 (m, 1H), 1.05 (d, J = 5.9 Hz, 3H), 1.00 (dd, J = 14.2, 3.3 Hz, 1H), 0.93 (s, 3H), 0.68 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 159.2, 72.8, 69.0, 57.1, 51.5, 43.7, 43.2, 41.0, 40.7, 40.5, 36.8, 36.5, 36.2, 35.9, 35.4, 34.0, 31.3, 29.2, 24.6, 23.4, 21.8, 21.3, 18.8, 12.1. LC/MS (ESI): m/z calcd C24H39N4O2 (M+) 415.3, found 415.4.
(R)-4-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-7-Hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanenitrile (36b)
To a suspension of NaH (60% dispersion in mineral oil, 0.072 g, 1.8 mmol) in THF (3 mL) was added a solution of 34b (0.270 g, 0.723 mmol) in THF (3 mL). After 1 h at room temperature, MeI (0.050 mL, 0.79 mmol) was added as a solution in THF (3 mL). After 12 h at room temperature, additional MeI (0.050 mL, 0.79 mmol) was added and the reaction stirred an additional 24 h. The reaction was quenched by the addition of 1 M aqueous HCl (5 mL), and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aqueous sodium thiosulfate (10 mL), saturated aqueous NaHCO3 (10 mL), water (10 mL), and brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (5–100% EtOAc in hexanes as eluent) to obtain 36b (0.200 g, 71% yield) as a white amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 3.88 (d, J = 6.7 Hz, 1H), 3.32–3.23 (m, 1H), 3.20 (s, 3H), 3.05 (dd, J = 10.6, 5.1 Hz, 1H), 2.56–2.52 (m, 2H), 2.41 (dt, J = 16.8, 8.1 Hz, 1H), 1.99–0.94 (m, 22H), 0.91–0.89 (m, 1H), 0.90 (d, J = 6.9 Hz, 3H), 0.89 (s, 3H), 0.63 (s, 3H). 1H NMR (400 MHz, CD2Cl2) δ 3.52 (ddd, J = 11.5, 9.0, 5.1 Hz, 1H), 3.28 (s, 3H), 3.07 (dp, J = 10.2, 4.7 Hz, 1H), 2.36 (ddd, J = 17.0, 8.8, 5.2 Hz, 1H), 2.25 (dt, J = 16.7, 8.2 Hz, 1H), 2.07–0.97 (m, 24H), 0.92 (d, J = 7.4 Hz, 3H), 0.91 (s, 3H), 0.68 (s, 3H). 13C NMR (100 MHz, CD2Cl2) δ 120.7, 80.5, 71.5, 56.1, 55.7, 55.1, 44.2, 44.1, 42.9, 40.5, 39.4, 37.7, 35.6, 35.2, 34.7, 34.2, 32.0, 28.9, 27.3, 27.1, 23.6, 21.5, 18.2, 14.6, 12.3. Compound 36a was prepared by the same method. Data for (R)-4-((3R,5R,7R,8R,9S,10S,13R,14S,17R)-7-hydroxy-3-methoxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)pentanenitrile (36a): 1H NMR (400 MHz, CDCl3) δ 3.84 (d, J = 3.4 Hz, 1H), 3.34 (s, 3H), 3.02 (tt, J = 11.1, 4.3 Hz, 1H), 2.37 (ddd, J = 16.9, 8.5, 5.1 Hz, 1H), 2.27 (dt, J = 16.8, 8.2 Hz, 1H), 2.17–1.05 (m, 23H), 1.00–0.93 (m, 1H), 0.97 (d, J = 6.6 Hz, 3H), 0.91 (s, 3H), 0.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 120.2, 80.5, 68.4, 55.5, 55.5, 50.4, 42.8, 41.4, 39.6, 39.4, 35.9, 35.4, 35.3, 35.2, 34.8, 32.8, 31.5, 28.2, 27.0, 23.7, 22.8, 20.5, 17.9, 14.3, 11.8. TLC-MS (ESI): m/z calcd C25H42NO2 (M + H+) 388.3, found 388.7.
Sodium 5-((R)-3-((3R,5R,7S,8R,9S,10S,13R,14S,17R)-7-Hydroxy-3-methoxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]-phenanthren-17-yl)butyl)tetrazol-1-ide (37b)
To a suspension of 36b (0.150 g, 0.387 mmol) in iPrOH (2 mL) was added zinc bromide (0.096 g, 0.43 mmol) and sodium azide (0.028 g, 0.43 mmol) in water (2 mL). The reaction was heated via MW at 160 °C for 9 h. Starting material remained, and additional zinc bromide (0.096 g, 0.43 mmol) and sodium azide (0.028 g, 0.43 mmol) were added and the reaction was heated for 5 h. The reaction was poured into 1 M aqueous HCl (10 mL), and the aqueous layer was extracted with EtOAc (4 × 10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The crude material was purified by flash column chromatography on silica gel (2–10% MeOH in DCM as eluent) to obtain the tetrazole (0.086 g, 52% yield) as a white solid. To prepare the sodium salt of 37b, a 1 cm wide column was filled with 12 cm of Dowex-50WX2 (50–100 mesh, strongly acidic) ion-exchange resin. The column was prepared by sequentially washing with 1:1 acetonitrile/water, ~1 M aqueous NaHCO3 (caution: gas evolution), water, and finally 1:1 acetonitrile/water. The reaction product was dissolved in 1:1 acetonitrile/water and loaded onto the column, which was eluted with 1:1 acetonitrile/water. The fractions containing the product were lyophilized to furnish 37b as a white solid. 1H NMR (400 MHz, CD3OD) δ 3.50–3.39 (m, 1H), 3.33 (s, 3H), 3.16 (td, J = 10.7, 4.8 Hz, 1H), 3.02–2.92 (m, 1H), 2.81 (ddd, J = 15.0, 9.4, 6.6 Hz, 1H), 2.74–2.62 (m, 1H), 2.04 (dt, J = 12.4, 3.1 Hz, 2H), 1.98–1.09 (m, 20H), 1.04 (d, J = 6.0 Hz, 3H), 1.00 (dd, J = 10.8, 3.4 Hz, 1H), 0.97 (s, 3H), 0.70 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 160.6, 81.7, 71.9, 57.4, 56.4, 55.9, 44.8, 44.5, 43.9, 41.5, 40.7, 38.6, 36.7, 35.9, 35.8, 35.4, 34.7, 29.6, 27.9, 27.7, 23.9, 22.4, 21.8, 19.0, 12.6. TLC-MS (ESI): m/z calcd C25H41N4O2 (M+) 429.3, found 429.7. Compound 37a was prepared by the same method. Data for sodium 5-((R)-3-((3R,5R,7R,8R,9S,10S,13R,14S,17R)-7-hydroxy-3-methoxy-10,13-dimethyl-hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)butyl)-tetrazol-1-ide (37a): 1H NMR (400 MHz, CD3OD) δ 3.80 (s, 1H), 3.33 (s, 3H), 3.06 (dq, J = 10.7, 5.4, 4.1 Hz, 1H), 3.00–2.88 (m, 1H), 2.88–2.75 (m, 1H), 2.28–1.16 (m, 22H), 1.14–1.08 (m, 1H), 1.05 (d, J = 5.8 Hz, 3H), 1.03–0.96 (m, 1H), 0.94 (s, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 156.0, 82.4, 69.0, 57.3, 55.6, 51.5, 43.7, 43.1, 41.0, 40.8, 37.0, 36.8, 36.5, 36.3, 36.1, 35.8, 34.0, 29.3, 28.0, 24.6, 23.4, 22.3, 21.8, 18.9, 12.1. HRMS (ESI): m/z calcd C25H42N4NaO2 (M + Na+) 453.3206, found 453.3164.
Sodium 5-((R)-3-((3R,5S,7S,8R,9S,10S,13R,14S,17R)-3-Methoxy-10,13-dimethyl-7-(sulfonato-oxy)hexadecahydro-1H-cyclopenta-[a]phenanthren-17-yl)butyl)tetrazol-1-ide (38b)
To a solution of 37b (0.060 g, 0.14 mmol) in pyridine (0.7 mL) was added dropwise chlorosulfonic acid (0.06 mL, 0.9 mmol). The reaction was heated to 50 °C for 30 min and was cooled to room temperature. The reaction was quenched by the addition of 1 mL of water, and the solvents were removed by rotary evaporation. The crude material was dissolved in DMSO (0.5 mL) and 1 M triethylammonium acetate buffer (0.1 mL) and purified by flash column chromatography (5–100% 20 mM triethylammonium acetate buffer in acetonitrile in water as eluent, C18 column) to yield a white solid after lyophilization. To prepare the sodium salt of 38b, a 1 cm wide column was filled with 12 cm of Dowex-50WX2 (50–100 mesh, strongly acidic) ion-exchange resin. The column was prepared by sequentially washing with 1:1 acetonitrile/water, ~1 M aqueous NaHCO3 (caution: gas evolution), water, and finally 1:1 acetonitrile/water. The reaction product was dissolved in 1:1 acetonitrile/water and loaded onto the column, which was eluted with 1:1 acetonitrile/water. The fractions containing the product were lyophilized to furnish 38b as an off-white solid (0.025 g, 32% yield) as the disodium salt. 1H NMR (400 MHz, CD3OD) δ 4.28 (td, J = 10.8, 5.0 Hz, 1H), 3.33 (s, 3H), 3.16 (dt, J = 10.8, 5.8 Hz, 1H), 2.93 (ddd, J = 14.7, 10.7, 4.4 Hz, 1H), 2.86–2.63 (m, 1H), 2.28–1.10 (m, 21H), 1.06 (s, 1H), 1.04 (d, J = 5.8 Hz, 3H), 0.98 (s, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 161.4, 81.6, 80.8, 56.5, 56.4, 55.8, 44.9, 43.7, 42.7, 41.2, 40.7, 36.8, 36.0, 35.8, 35.3, 35.0, 34.4, 29.7, 27.8, 27.1, 23.9, 22.4, 22.2, 19.1, 12.5. TLC-MS (ESI): m/z calcd C25H41N4O5S (M+) 509.3, found 509.5. Compound 38a was prepared by the same method. Data for sodium 5-((R)-3-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3-methoxy-10,13-dimethyl-7-(sulfonatooxy)hexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-butyl)tetrazol-1-ide (38a): 1H NMR (400 MHz, DMSO-d6) δ 4.11 (d, J = 3.1 Hz, 1H), 3.19 (s, 3H), 2.98–2.82 (m, 2H), 2.76 (dt, J = 15.1, 8.0 Hz, 1H), 2.21–1.02 (m, 23H), 0.95 (d, J = 5.7 Hz, 3H), 0.90–0.79 (m, 1H), 0.85 (s, 3H), 0.58 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 158.8, 82.4, 78.2, 56.9, 55.6, 51.2, 43.8, 43.0, 40.8, 40.7, 36.7, 36.4, 36.2, 36.0, 35.2, 34.7, 31.8, 29.2, 28.3, 24.4, 23.3, 21.8, 21.1, 18.8, 12.2. HRMS (ESI): m/z calcd C25H41N4Na2O5S (M + H+) 555.2593, found 555.2586.
Biology
Isolation and characterization of C. difficile isolates was performed as previously reported.13
C. difficile Spore Germination Assays. Optical Density Assay
BHIS media was spiked with TCA to a final concentration of 2000 μM to test compounds for inhibition of germination. Either 130 μL of BHIS media (negative control) or the BHIS with 2000 μM TCA was added to separate wells of a 96-well plate in triplicate. The test compound (10 μL) in solvent (typically DMSO or DMSO/ethanol) was added to both the BHIS and BHIS with TCA in triplicate. In addition, BHIS with 2000 μM TCA (positive control) was amended, in triplicate, with solvent (DMSO or DMSO/ethanol). BHIS with DMSO or DMSO/ethanol and filter sterilized water were also run in duplicate as a negative control. The 96-well plates containing the solutions were reduced in an anaerobic chamber for 2–3 h. Spores, at a ~0.5 McFarland standard concentration, were heated to 65 °C for 30 min before being inoculated into the test media, with or without spiked bile acid analogues solutions, within an anaerobic bag under nitrogen gas. The OD600 was measured once every minute for 20 min with an EL808 microplate reader (Biotek Instruments, Inc., Winooski, VT) and normalized using the OD600 obtained at time zero [relative OD600 = OD600(t)/OD600(t0)].
Microscope Spore Count Assay
Media and spore preparation for phase contrast assay was performed in the same method as outlined in the optical density assay above. t0 samples were immediately placed on ice, while the t20 samples were incubated at room temperature for 20 min. Following the 20 min incubation, the t20 samples were also placed on ice to prevent further germination. Then 10 μL of each of the triplicate samples were removed and counted on a hemocytometer under a Plan 40/0.65 Ph2 lens for counts of spores and germinated cells. Experiments were performed in triplicate and average percentage of germination was determined.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by Faculty Research Development Grant 14.30 from the University of Minnesota Academic Health Center (A.K., P.I.D., M.J.S.) and by NIH grant 1R21-AI114722-01 (M.J.S. and A.K.). Caco-2 assays were performed by Eurofins Panlabs.
ABBREVIATIONS USED
- A-B
apical to basolateral
- app
apparent
- ASBT
apical sodium-dependent bile acid transporter
- BHIS
brain–heart infusion supplemented
- C. difficile
Clostridium difficile
- CDAD
C. difficile-associated disease
- CDI
C. difficile infection
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMA
dimethylacetamide
- DMAP
dimethylaminopyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- ELSD
evaporative light scattering detection
- EtOAc
ethyl acetate
- Et2O
diethyl ether
- FMT
fecal microbiota transplantation
- HATU
1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexa-fluorophosphate
- HRMS
high resolution mass spectrometry
- LC-MS
liquid chromatography–mass spectrometry
- MeOTf
methyl trifluoromethanesulfonate
- MW
microwave
- NAP1
North American pulsotype 1
- NMR
nuclear magnetic resonance
- Papp
apparent permeability
- pTSA
para-toluene-sulfonic acid
- R-CDI
recurrent C. difficile infection
- SAR
structure–activity relationship
- SEM
standard error of the mean
- TBAF
tetrabutylammonium fluoride
- TBS
tertbutyldimethylsilyl-
- TCA
taurocholic acid, taurocholate
- TEA
triethylamine
- TFAA
trifluoromethylacetic anhydride
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- TMS
trimethylsilane
- UDCA
ursodeoxycholic acid
- UV
ultraviolet
Footnotes
Author Contributions
The laboratories of Khoruts, Sadowsky, and Dosa contributed equally to this work.
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.7b00295.
Inhibitory activity of 7-UDCA in a NAP1 strain of C. difficile, synthetic scheme for 11a, and graphs of relative OD600 vs time and phase-contrast microscopy data for all compounds tested (PDF)
Molecular formula strings (CSV)
References
- 1.Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, St Sauver JL, Harmsen WS, Zinsmeister AR. The Epidemiology of Community-Acquired Clostridium difficile Infection: A Population-Based Study. Am J Gastroenterol. 2012;107:89–95. doi: 10.1038/ajg.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antibiotic Resistance Threats in the United States, 2013. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; Atlanta, GA: 2013. pp. 1–114. [Google Scholar]
- 3.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. Burden of Clostridium difficile Infection in the United States. N Engl J Med. 2015;372:825–833. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly CP, LaMont JT. Clostridium difficile - More Difficult Than Ever. N Engl J Med. 2008;359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 5.Black SR, Weaver KN, Jones RC, Ritger KA, Petrella LA, Sambol SP, Vernon M, Burton S, Garcia-Houchins SW, Weber SG, Lavin MA, Gerding D, Johnson S, Gerber IS. Clostridium difficile Outbreak Strain BI is Highly Endemic in Chicago Area Hospitals. Infect Control Hosp Epidemiol. 2011;32:897–902. doi: 10.1086/661283. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez Z, Abel-Santos E. Potential Use of Inhibitors of Bacteria Spore Germination in the Prophylatic Treatment of Anthrax and Clostridium difficile-associated disease. Expert Rev Anti-Infect Ther. 2007;5:783–792. doi: 10.1586/14787210.5.5.783. [DOI] [PubMed] [Google Scholar]
- 7.Kelly CR, de Leon L, Jasutkar N. Fecal Microbiota Transplantation for Relapsing Clostridium difficile Infection in 26 Patients: Methodology and Results. J Clin Gastroenterol. 2012;46:145–149. doi: 10.1097/MCG.0b013e318234570b. [DOI] [PubMed] [Google Scholar]
- 8.Borody TJ, Khoruts A. Fecal Microbiota Transplantation and Emerging Applications. Nat Rev Gastroenterol Hepatol. 2011;9:88–96. doi: 10.1038/nrgastro.2011.244. [DOI] [PubMed] [Google Scholar]
- 9.Jarrad AM, Karoli T, Blaskovich MAT, Lyras D, Cooper MA. Clostridium difficile Drug Pipeline: Challenges in Discovery and Development of New Agents. J Med Chem. 2015;58:5164–5185. doi: 10.1021/jm5016846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kociolek LK, Gerding DN. Breakthroughts in the Treatment and Prevention of Clostridium difficile Infection. Nat Rev Gastroenterol Hepatol. 2016;13:150–160. doi: 10.1038/nrgastro.2015.220. [DOI] [PubMed] [Google Scholar]
- 11.Khoruts A, Sadowsky MJ. Understanding the Mechanisms of Faecal Microbiota Transplantation. Nat Rev Gastroenterol Hepatol. 2016;13:508–516. doi: 10.1038/nrgastro.2016.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weingarden AR, Chen C, Bobr A, Yao D, Lu Y, Nelson VM, Sadowsky MJ, Khoruts A. Microbiota Transplantation Restores Normal Fecal Bile Acid Composition in Recurrent Clostridium difficile Infection. Am J Physiol Gastrointest Liver Physiol. 2014;306:G310–G319. doi: 10.1152/ajpgi.00282.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weingarden AR, Dosa PI, DeWinter E, Steer CJ, Shaughnessy MK, Johnson JR, Khoruts A, Sadowsky MJ. Changes in Colonic Bile Acid Composition Following Fecal Microbiota Transplantation Are Sufficient to Control Clostridium difficile Germination and Growth. PLoS One. 2016;11:e0147210. doi: 10.1371/journal.pone.0147210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis MB, Allen CA, Shrestha R, Sorg JA. Bile Acid Recognition by the Clostridium difficile Germinant Receptor, CspC, is Important for Establishing Infection. PLoS Pathog. 2013;9:e1003356. doi: 10.1371/journal.ppat.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kevorkian Y, Shirley DJ, Shen A. Regulation of Clostridium difficile Spore Germination by the CspA Pseudoprotease Domain. Biochimie. 2016;122:243–254. doi: 10.1016/j.biochi.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams CM, Eckenroth BE, Putnam EE, Doublié S, Shen A. Structural and Functional Analysis of the CspB Protease Required for Clostridium difficile Spore Germination. PLoS Pathog. 2013;9:e1003165. doi: 10.1371/journal.ppat.1003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorg JA, Sonenshein AL. Bile Salts and Glycine as Cogerminants for Clostridium difficile Spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heeg D, Burns DA, Cartman ST, Minton NP. Spores of Clostridium difficile Clinical Isolates Display a Diverse Germination Response to Bile Salts. PLoS One. 2012;7:e32381. doi: 10.1371/journal.pone.0032381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arroyo LG, Rousseau J, Willey BM, Low DE, Staempfli H, McGeer A, Weese JS. Use of a Selective Enrichment Broth to Recover Clostridium difficile From Stool Swabs Stored Under Different Conditions. J Clin Microbiol. 2005;43:5341–5343. doi: 10.1128/JCM.43.10.5341-5343.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burns DA, Heap JT, Minton NP. SleC is Essential for Germination of Clostridium difficile Spores in Nutrient-Rich Medium Supplemented with the Bile Salt Taurocholate. J Bacteriol. 2010;192:657–664. doi: 10.1128/JB.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez N, Liggins M, Abel-Santos E. Kinetic Evidence for the Presence of Putative Germination Receptors in Clostridium difficile spores. J Bacteriol. 2010;192:4215–4222. doi: 10.1128/JB.00488-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridlon JM, Kang DJ, Hylemon PB. Bile Salt Transformations by Human Intestinal Bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Masuda N, Oda H. 7alpha-Dehydroxylation of Bile Acids by Resting Cells of an Unidentified, Gram-Positive, Nonsporeforming Anaerobic Bacterium. Appl Environ Microbiol. 1983;45:456–462. doi: 10.1128/aem.45.2.456-462.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorg JA, Sonenshein AL. Inhibiting the Initiation of Clostridium difficile Spore Germination Using Analogs of Chenodeoxycholic Acid, a Bile Acid. J Bacteriol. 2010;192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: Design and Clinical Applications. Nat Rev Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 26.Alrefai WA, Gill RK. Bile Acid Transporters: Structure, Function, Regulation, and Pathophysiological Implications. Pharm Res. 2007;24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 27.Meier PJ. Molecular Mechanisms of Hepatic Bile Salt Transport from Sinusoidal Blood into Bile. Am J Physiol. 1995;269:G801–G812. doi: 10.1152/ajpgi.1995.269.6.G801. [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharjee D, Francis MB, Ding X, McAllister KN, Shrestha R, Sorg JA. Reexamining the Germination Phenotypes of Several Clostridium difficile Strains Suggests Another Role for the CspC Germinant Receptor. J Bacteriol. 2016;198:777–786. doi: 10.1128/JB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howerton A, Ramirez N, Abel-Santos E. Mapping Interactions Between Germinants and Clostridium difficile Spores. J Bacteriol. 2011;193:274–282. doi: 10.1128/JB.00980-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howerton A, Patra M, Abel-Santos E. A New Strategy for the Prevention of Clostridium difficile Infection. J Infect Dis. 2013;207:1498–1504. doi: 10.1093/infdis/jit068. [DOI] [PubMed] [Google Scholar]
- 31.Although the hydroxyl groups in compound 3 are described as all α in the text, the C12 hydroxyl group is drawn as β for all structures in the publication, including cholic acid, which is known to contain all α hydroxyl groups. Additionally, compound 3 is drawn with all α hydroxyl groups in ref 50.
- 32.Weingarden AR, Chen C, Zhang N, Graiziger CT, Dosa PI, Steer CJ, Shaughnessy MK, Johnson JR, Sadowsky MJ, Khoruts A. Ursodeoxycholic Acid Inhibits Clostridium difficile Spore Germination and Vegetative Growth, and Prevents the Recurrence of Ileal Pouchitis Associated With the Infection. J Clin Gastroenterol. 2016;50:624–630. doi: 10.1097/MCG.0000000000000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodrigues CM, Kren BT, Steer CJ, Setchell KD. The Site-Specific Delivery of Ursodeoxycholic Acid to the Rat Colon by Sulfate Conjugation. Gastroenterology. 1995;109:1835–1844. doi: 10.1016/0016-5085(95)90750-5. [DOI] [PubMed] [Google Scholar]
- 34.Kolhatkar V, Polli JE. Structural Requirements of Bile Acid Transporters: C-3 and C-7 Modifications of Steroidal Hydroxy Groups. Eur J Pharm Sci. 2012;46:86–89. doi: 10.1016/j.ejps.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filipski KJ, Varma MV, El-Kattan AF, Ambler CM, Ruggeri RB, Goosen TC, Cameron KO. Intestinal Targeting of Drugs: Rational Design Approaches and Challenges. Curr Top Med Chem. 2013;13:776–802. doi: 10.2174/1568026611313070002. [DOI] [PubMed] [Google Scholar]
- 36.Alnouti Y. Bile Acid Sulfation: A Pathway of Bile Acid Elimination and Detoxification. Toxicol Sci. 2009;108:225–246. doi: 10.1093/toxsci/kfn268. [DOI] [PubMed] [Google Scholar]
- 37.Meanwell NA. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 38.Pellicciari R, Gioiello A, Sabbatini P, Venturoni F, Nuti R, Colliva C, Rizzo G, Adorini L, Pruzanski M, Roda A, Macchiarulo A. Avicholic Acid: A Lead Compound from Birds on the Route to Potent TGR5 Modulators. ACS Med Chem Lett. 2012;3:273–277. doi: 10.1021/ml200256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicolaou KC, Lim Yh, Piper JL, Papageorgiou CD. Total syntheses of 2,2′-epi-Cytoskyrin A, Rugulosin, and the Alleged Structure of Rugulin. J Am Chem Soc. 2007;129:4001–4013. doi: 10.1021/ja0685708. [DOI] [PubMed] [Google Scholar]
- 40.Setchell KDR. Sulfate Conjugates of Ursodeoxycholic Acid, and Their Beneficial Use in Inflammatory Disorders and Other Applications. 6,251,884. US Patent. 2001 Jun 26;
- 41.Une M, Yamanaga K, Mosbach E, Kuroki S, Hoshita T. Synthesis of Bile Acid Analogs: 7-Alkylated Chenodeoxycholic Acids. Steroids. 1989;53:97–105. doi: 10.1016/0039-128x(89)90148-7. [DOI] [PubMed] [Google Scholar]
- 42.Karabulut HRF, Rashdan SA, Dias JR. Notable Chenodeoxycholic Acid Oligomers - Synthesis, Characterization, and 7alpha-OR Steric Hindrance Evaluation. Tetrahedron. 2007;63:5030–5035. [Google Scholar]
- 43.Dosa PI, Ward T, Castro RE, Rodrigues CMP, Steer CJ. Synthesis and Evaluation of Water-Soluble Prodrugs of Ursodeoxycholic Acid (UDCA), an Anti-Apoptotic Bile Acid. Chem Med Chem. 2013;8:1002–1011. doi: 10.1002/cmdc.201300059. [DOI] [PubMed] [Google Scholar]
- 44.Porcheddu A, Cadoni R, de Luca L. A Fast and Efficient One-Pot Microwave Assisted Synthesis of Variously Di-Substituted 1,2,4-Oxadiazoles. Org Biomol Chem. 2011;9:7539–7546. doi: 10.1039/c1ob06055d. [DOI] [PubMed] [Google Scholar]
- 45.Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan ZQ, Boger DL. Intramolecular Diels-Alder/1,3-Dipolar Cycloaddition Cascade of 1,3,4-Oxadiazoles. J Am Chem Soc. 2006;128:10589–10595. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bukiya AN, Patil SA, Li W, Miller DD, Dopico AM. Calcium- and Voltage-Gated Potassium (BK) Channel Activators in the 5-Beta-Cholanic Acid-3-Alpha-Ol Analogue Series with Modifictions in the Lateral Chain. ChemMedChem. 2012;7:1784–1792. doi: 10.1002/cmdc.201200290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demko ZP, Sharpless KB. Preparation of 5-Substituted 1H-Tetrazoles from Nitriles in Water. J Org Chem. 2001;66:7945–7950. doi: 10.1021/jo010635w. [DOI] [PubMed] [Google Scholar]
- 48.When the optical density assay was run in the absence of TCA, a small number of compounds appeared to cause germination at high concentrations (1500–2000 μM). However, further experiments showed that these compounds cause a change in optical density even in the absence of spores, suggesting that the initial results were likely due to problems with solubility.
- 49.Sharma R, Majer F, Peta VK, Wang J, Keaveney R, Kelleher D, Long A, Gilmer JF. Bile Acid Toxicity Structure-Activity Relationships: Correlations Between Cell Viability and Lipophilicity in a Panel of New and Known Bile Acids Using an Oesophageal Cell Line (HET-1A) Bioorg Med Chem. 2010;18:6886–6895. doi: 10.1016/j.bmc.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 50.Howerton AJ. PhD Dissertation. University of Nevada; Las Vegas: 2012. Anti-Germinants as a New Strategy to Prevent Clostridium difficile Infections. [Google Scholar]
- 51.Hofmann AF, Mysels KJ. Bile Acid Solubility and Precipitation In Vitro and In Vivo: The Role of Conjugation, pH, and Ca2+ Ions. J Lipid Res. 1992;33:617–626. [PubMed] [Google Scholar]
- 52.Some compounds that had abnormal responses at high concentrations in the presence of TCA were retested with a shorter reducing time, which resulted in a normal concentration response and suggests that insolubility of the analogues may contribute to the appearance of spore germination at high concentrations.
- 53.Sorg and Sonenshein concluded that the C7-α hydroxyl group of CDCA is not required for germination or inhibition of UK1 spores.
- 54.Martinez-Augustin O, Sanchez de Medina F. Intestinal Bile Acid Physiology and Pathophysiology. World J Gastroenterol. 2008;14:5630–5640. doi: 10.3748/wjg.14.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Northfield TC, McColl I. Postprandial Concentrations of Free and Conjugated Bile Acids Down the Length of the Normal Human Small Intestine. Gut. 1973;14:513–518. doi: 10.1136/gut.14.7.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hidalgo IJ, Borchardt RT. Transport of Bile Acids in a Human Intestinal Epithelial Cell Line, Caco-2. Biochim Biophys Acta, Gen Subj. 1990;1035:97–103. doi: 10.1016/0304-4165(90)90179-z. [DOI] [PubMed] [Google Scholar]
- 57.Allen CA, Babakhani F, Sears P, Nguyen L, Sorg JA. Both Fidaxomicin and Vancomycin Inhibit Outgrowth of Clostridium difficile Spores. Antimicrob Agents Chemother. 2013;57:664–667. doi: 10.1128/AAC.01611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.