

Abstract
Recent studies showed that human papillomavirus (HPV) integration contributes to the genomic instability seen in HPV-associated head and neck squamous cell carcinoma (HPV-HNSCC). However, the epigenetic alterations induced after HPV integration remains unclear. To identify the molecular details of HPV16 DNA integration and the ensuing patterns of methylation in HNSCC, we performed next-generation sequencing using a target-enrichment method for the effective identification of HPV16 integration breakpoints as well as the characterization of genomic sequences adjacent to HPV16 integration breakpoints with three HPV16-related HNSCC cell lines. The DNA methylation levels of the integrated HPV16 genome and that of the adjacent human genome were also analyzed by bisulfite pyrosequencing. We found various integration loci, including novel integration sites. Integration loci were located predominantly in the intergenic region, with a significant enrichment of the microhomologous sequences between the human and HPV16 genomes at the integration breakpoints. Furthermore, various levels of methylation within both the human genome and the integrated HPV genome at the integration breakpoints in each integrant were observed. Allele-specific methylation analysis suggested that the HPV16 integrants remained hypomethylated when the flanking host genome was hypomethylated. After integration into highly methylated human genome regions, however, the HPV16 DNA became methylated. In conclusion, we found novel integration sites and methylation patterns in HPV-HNSCC using our unique method. These findings may provide insights into understanding of viral integration mechanism and virus-associated carcinogenesis of HPV-HNSCC.
Keywords: human papillomavirus (HPV), DNA integration, epigenome, head and neck squamous cell carcinoma (HNSCC)
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, accounting for 3 – 5% of all cancers1. Recently, the incidence of oropharyngeal cancer (OPC) has been reported to be increasing markedly. This is mainly because the number of human papillomavirus (HPV)-related oropharyngeal cancers (HPV-OPC) has risen globally2, while the incidence of HNSCC associated with the use of tobacco and alcohol use has remained constant or decreased.
The tumor biology and clinical characteristics of HPV-OPC differ from those of HPV-negative HNSCCs, which are mainly alcohol- and smoking-related HNSCCs3. Most patients with HPV-OPC present with advanced nodal stage, and tend to be younger than patients with non-HPV associated tumors. Furthermore, HPV-OPC is associated with better response rates to treatment, and overall better prognosis when compared to non-HPV HNSCC. Therefore, the presence of HPV is now one of the most important prognostic factors in OPC patients [2].
While there are 15 high-risk subtypes of HPV with carcinogenic potential in humans, the vast majority of HPV-OPC is associated with HPV164, 5. High-risk HPV infection of mucosal epithelia leads to the expression of viral E6 and E7 oncoproteins inducing the inactivation of both the p53 and the retinoblastoma (Rb) tumor suppressive proteins, which contribute to the malignant formation of HPV-related cancers. In addition, immune escape, genomic instability, HPV DNA integration, and epigenetic alterations are also important for HPV-related cancer progression6. In fact, high-risk HPV integration is now known to be associated with the progression from low- to high-grade cervical intraepithelial neoplasia7. Many epigenetic alterations have also been reported during the process of cervical carcinogenesis in both the HPV and human genomes8. The mechanisms of these factors affecting HPV-induced cervical carcinogenesis have been thus well studied.
In HNSCC, a recent study using deep sequencing of HPV-HNSCC cell lines and HNSCC samples concluded that HPV16 integration itself directly promotes genomic instability and is important for carcinogenesis9. Park et al. have shown that hypomethylation status within the viral genome, especially in the long control region (LCR) including E2 binding sites (E2BSs), was related to E6 and E7 expression in HPV-related HNSCC10. On the other hand, several articles have also reported that hypermethylation status in the ESBSs might cause deregulated E6 and E7 expression11, 12. Thus, the roles of altered methylation status of HPV during HPV-HNSCC development remain unclear. Therefore, the HPV methylation status, including the association of epigenetic alterations in HPV integrants and the flanking host genomes, requires further characterization to allow us a better understanding of the detailed mechanisms of the viral integration and carcinogenesis of HPV-HNSCC.
In the present study, we identified the molecular details of HPV16 DNA integration and the ensuing patterns of methylation in HPV-HNSCC with a previously reported [11] next-generation sequencing (NGS)-based method for methylation analysis of integrated viral genomes (G-NaVI) using 3 HPV-related HNSCC cell lines to reveal the association of epigenetic alterations in HPV integrants and the flanking host genomes.
Materials and Methods
Cell lines
The UPCI:SCC090, UPCI:SCC152, and UPCI:SCC154 cell lines were kindly provided by Dr. Susanne M. Gollin, University of Pittsburgh. The clinicopathological data for the patients and tumors from which the cell lines were derived are shown in Table 113. Cell lines were maintained in appropriate media containing 10% fetal bovine serum in plastic culture plates. Adherent monolayer cultures were maintained on plastic and incubated at 37°C in 5% CO2 and 95% air. The integrity of each maintained cell line was clearly established by comparing the results from short tandem repeat profiling14 with that reported for the original stock15. The cultures were free of Mycoplasma species and were maintained for no longer than 12 weeks after recovery from frozen stocks.
Table 1.
Primary site and source of tumors used to derive the 3 HPV-HNSCC cell lines.
| Cell line | UPCI:SCC090 | UPCI:SCC152 | UPCI:SCC154 |
|---|---|---|---|
| Sex | Male | Male | Male |
| Age at surgery | 46 | 47 | 54 |
| Smoking | Yes | Yes | Yes |
| Alcohol | Yes | Yes | Yes |
| Primary tumor site | base of tongue | hypopharynx | tongue |
| TNM classification | T2N0 | recurrence | T4N2 |
| Degree of differentiation | Poorly to moderately | Moderately | NA |
Analysis of HPV16 integration site sequences using next-generation sequencing (NGS)
Genomic DNA was extracted from cultured cells using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The concentration and quantity of extracted DNA were measured using a NanoDrop spectrophotometer (Nano Drop Technologies, Wilmington, DE). A total of 427 custom-made RNA probes covering the entire HPV16 DNA sequences were designed using the Agilent SureSelect Target Enrichment System, a highly efficient hybrid selection technique for optimizing NGS. After capture and amplification, the templates were sequenced using the GS FLX Titanium system (Roche, Basel, Switzerland). Reads that aligned perfectly with the human or HPV16 genome were removed. Reads that aligned partially with the human genome and HPV16 genome were reserved. PCR and Sanger sequencing were used to verify the selected HPV integration breakpoints. PCR primers were designed to amplify the integrated HPV and the flanking human genome. As a template, 1 μL of genomic DNA solution was used in the subsequent PCR. Touchdown PCR was used for most of the assays. All PCR assays included a denaturation step at 95°C for 30 seconds, followed by an annealing step at various temperatures for 30 seconds, and an extension step at 72°C for 30 seconds. PCR products were analyzed using electrophoresis through 2% agarose gels.
Genomic elements around the integration positions
Genomic elements around the integration sites were analyzed using the RepeatMasker from the UCSC genome browser (https://genome.ucsc.edu/index.html). Each integration site was compared with previously reported integration and fragile sites16, 17. HPV16 breakpoints were mapped to the HPV16 genome (KO2718), and the distribution of the integration sites was analyzed for comparison with the expected HPV16 integration sites.
Microhomology (MH) calculation at the HPV16 genome integration sites
Bilateral sequences from the human and HPV16 genomes were extracted and the paired sequences were compared base by base to count the same sequences. The contiguous identical bases that could not be extended were defined as MH units and the MH lengths at the integration sites were also calculated.
DNA methylation analysis of the integrated HPV genome as well as the adjacent human genome
DNA methylation was analyzed using bisulfite pyrosequencing. Bisulfite PCR was performed according to the manufacturer’s protocol. One microliter of bisulfite-treated DNA, prepared using an EpiTect Bisulfite Kit (Qiagen, Valencia, CA), was used as a template. The primers used for amplifying the CpG sequences in the given sequence are described in Supplementary Table 1. After PCR, the biotinylated strand was captured on streptavidin-coated beads (Amersham Bioscience, Uppsala, Sweden) and incubated with sequencing primers (Supplementary Table 1). The pyrosequencing reactions were performed using the PyroMark Q24 Advanced (Qiagen) with the 3 HPV-HNSCC cell lines. Primers for the methylation analysis of integration sites in the UPCI:SCC090 line are shown in Supplementary Table 1.
DNA fragments, including 150 bp of the integrated HPV DNA and 150 bp of the human genome around the boundary, were then analyzed for average methylation to examine the correlation between the methylation pattern of the integrated HPV DNA and that of the human genome.
Lastly, allele-specific DNA methylation of both the integrated HPV genome and adjacent human genome was analyzed as described previously18. For this analysis, the pyrosequencing reactions were performed using the PyroMark Q24 Advanced (Qiagen).
LINE1 methylation analysis
To quantify the relatively high LINE1 methylation levels, pyrosequencing technology was again used as described previously19. Briefly, PCR and the subsequent pyrosequencing for LINE1 were performed using the PyroMark kit (Qiagen). This assay amplifies a region of the LINE1 elements that includes 3 CpG sites. PCR was conducted as follows: 45 cycles at 95°C for 20 seconds, 50°C for 20 seconds, and 72°C for 20 seconds, followed by 72°C for 5 min. The biotinylated PCR product was purified and converted to single strands to serve as a template for the pyrosequencing reaction using the Pyrosequencing Vacuum Prep Tool (Qiagen). The pyrosequencing reactions were performed using the PyroMark Q24 (Qiagen). The percentage of Cs relative to the total sum of the Cs and Ts at each CpG site was calculated. The average of the percentages of Cs at the 3 CpG sites was used to represent the overall LINE1 methylation level in each cell line.
Statistical analysis
The Spearman correlation coefficient was used to assess correlations between the average methylation of the HPV DNA and that of the human genome. Randomly selected breakpoints across the human genome were used to compare the observed and expected integration results. The chi-squared test was used for statistical analyses to compare variables between two groups. Statistical analyses were performed using GraphPad Prism Version 6.05 (GraphPad Software, La Jolla, CA) and IBM SPSS Statistics for Windows Version 22.0 (IBM Corp., Armonk, NY). A P-value of less than 0.05 was considered statistically significant.
Results
NGS analysis of HPV16 DNA integration site sequences
To detect the HPV16 integration sites effectively, we designed a target-enrichment technique using NGS analysis (Supplemental Fig. 1) with a total of 427 custom RNA probes to specifically target the HPV genome. The average read length was 414.18 bp, and the average read quality was 31.30 with >99.9% accuracy. The genome wide map of HPV16 integration sites is shown in (Fig. 1). The HPV integrants were mapped to chromosomes 2q23, 3p12, 6p21, and 9q22 in UPCI:SCC090, chromosomes 2q23, 3p12, 9q22, and 9q31 in UPCI:SCC152, and chromosome 21q21 in UPCI:SCC154. Integration sites were mainly located in the intergenic regions, and no integration sites were observed in the promoter or exon region (Fig. 1). The UPCI:SCC090 and UPCI:SCC152 cell lines, derived from a recurrent tumor from the same patient, showed almost identical integration sites, although a lack of integration in chromosome 6 was observed in UPCI:SCC152. Our mapping of the HPV16 integration sites provided greater detail than a previous report using FISH and APOT- and DIPS- PCR analysis in UPCI:SCC090 and UPCI:SCC152 cells20. Furthermore, our results for the UPCI:SCC090 cell line was consistent with those of a previous report based on the whole genome sequence (WGS)9. In addition, we found some new HPV integration sites in the present study.
Figure 1.
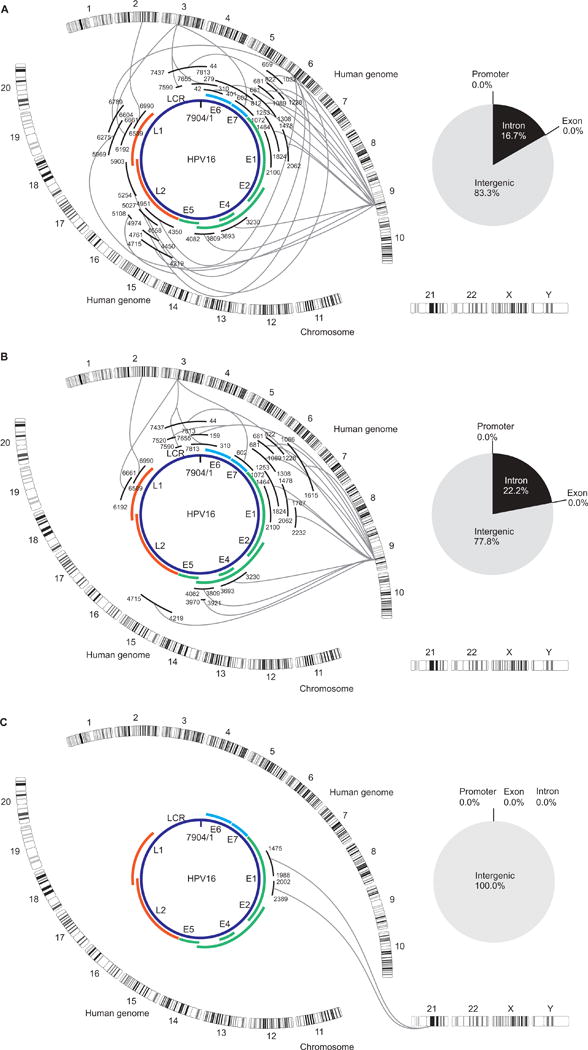
Distribution of integration breakpoints in the human chromosomes and HPV16 genome represented by Circos plots of the UCSC:SCC090 genome, the UCSC:SCC152 genome, and the UCSC:SCC154 genome
Characterization of genomic sequences adjacent to HPV16 integration breakpoints
To validate whether the HPV integrants in HNSCC target common fragile sites (CFSs) as reported previously in cervical cancers21, we compared the observed integration breakpoints in 3 cell lines with previously reported fragile sites16. We confirmed that these integration breakpoints are not only at or near CFSs but also at rare fragile sites (RFSs) in the UPCI:SCC090 and UPCI:SCC152 cell lines. Integration breakpoints in the UPCI:SCC154 cell line were detected at chromosome 21q21. This site was neither a CFS nor a RFS, however, it had previously been reported as one of the integration breakpoints in cervical cancer17. In addition, repetitive sequences at the integration sites were analyzed using RepeatMasker (http://www.repeatmasker.org/). Our results showed that 26/42 of the integration sites were distributed in repeating elements throughout the human genome. The number of integrations in long interspersed nuclear element (LINE) repeats (19/42) was found to be significantly larger than that of the expected occurrences by chi-squared test (Fig. 2a, P=0.006).
Figure 2. Analysis of the HPV16 integration sites.
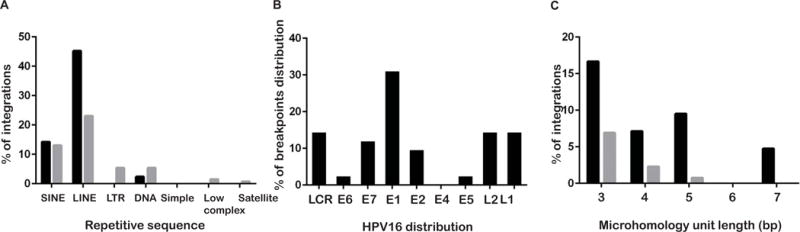
A. Repetitive elements analysis at integration breakpoints in the human genome, compared with expected the results. B. The percentage of breakpoints distribution in the HPV16 genome. C. Comparison between observed and expected integrations harboring MHs of different sizes at the integration sites.
HPV16 integration sites were distributed across almost the whole genome except for E4. Breakpoints were prone to occur in E1 13/42 (Fig. 2B), which is consistent with previous reports22, 23.
Distinct categories of viral-host sequence at the HPV16 integration breakpoints
Furthermore, viral-host junctional sequences were analyzed to reveal the existence of three distinct categories as reported previously24. In 4/42 direct ligations, the first category was defined as a seamless transition from one sequence to the next with a clearly defined breakpoint, as shown in Fig. 3A. The second category (n=15) was defined as the presence of short inserted sequences at the breakpoint that match neither reference sequence (Fig. 3B). The final category, which includes the majority of tumors (n=23), characterized by breakpoint microhomology (overlapping) was defined as the presence of several base of pairs of sequence homology at the breakpoint junction that could be assigned to either genome (Fig. 3C). In our samples, the breakpoint microhomology ranged from 1 to 7 bp in length. Furthermore, there was a significant enrichment of MHs when the MH length was 7bp between the human genome and the HPV genome at or near integration breakpoints compared to the expected integration (P=0.002). When MH lengths were 3, 4, or 5bp in length, the observed integrations tended to have more MHs than did the expected integrations; however, the differences were not significant (P=0.054, 0.17, and 0.21, respectively. Fig. 2C).
Figure 3. Categories of viral-host breakpoints.
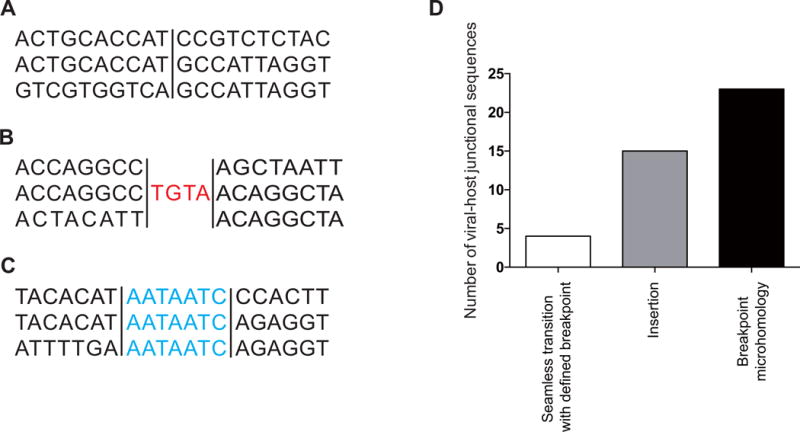
(Red) Nucleotides that align to neither reference gene (insertions); (blue) nucleotides that align to both reference genome (microhomology). A. Seamless transition with defined breakpoint. B. Insertion. C. Breakpoint microhomology. D. The number of viral-host junctional sequences.
Correlation between the methylation pattern of the integrated HPV16 DNA and the human genome DNA
To reveal the association between the epigenetic alterations in HPV integrants and the flanking host genomes, we first evaluated DNA methylation of the human genome at the HPV16 integration breakpoints by bisulfite pyrosequencing. We detected varying levels of methylation of the human genome at the HPV16 integration breakpoints in the genome of UPCI:SCC090, UPCI:SCC152, and UPCI:SCC154 cells as shown in Fig. 4B and Supplemental Fig. 2. However, no association was observed between the presence of integration occurrence and methylation level of the human genome.
Figure 4. Allele-specific methylation analysis of 3 cell lines.
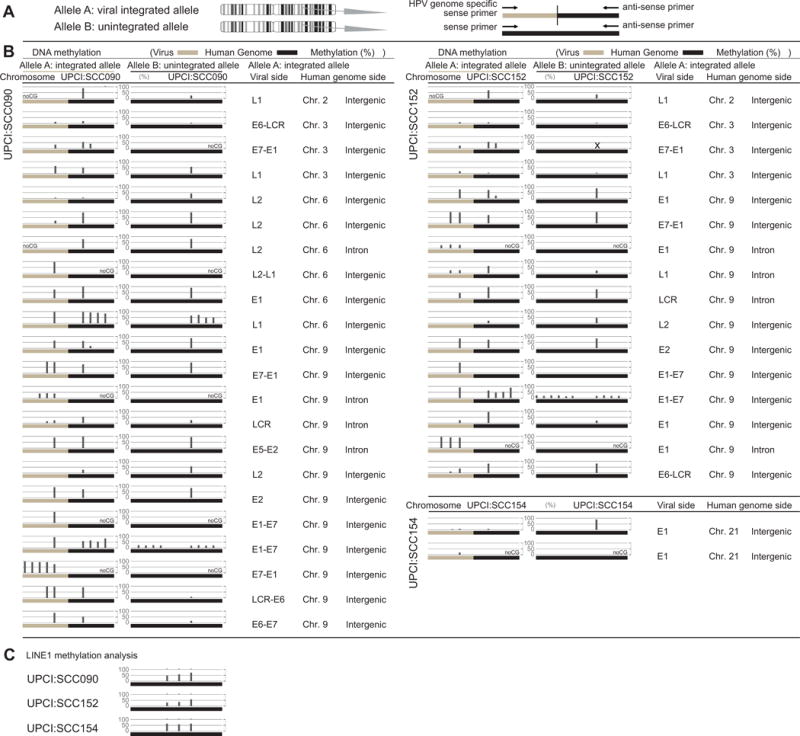
A. A schema of the allele-specific methylation analysis. B. Methylation levels of the HPV16 and human genomes for the integrated and unintegrated alleles. Detailed results of the HPV16 integrants (LCR, E6, E7, E1, E2, E4, E5, L2, L1) and flanking host genomes (chromosome and location of the genome) are shown. DNA methylation of the integrated HPV16 genomes as well as the flanking human genome was examined by allele-specific DNA methylation analysis using bisulfite pyrosequencing. C. The results of LINE1 methylation analysis.
We then evaluated DNA methylation of the integrated HPV16 genome by bisulfite pyrosequencing. We also found varying levels of methylation of the HPV16 sequences integrated into the genome of UPCI:SCC090, UPCI:SCC152, and UPCI:SCC154 cells. In addition, each integrant with the same viral gene showed differing methylation levels (Fig. 4B and Supplemental Fig. 2).
To evaluate if the levels of methylation in the HPV16 DNA were associated with that of human genome, DNA fragments including the 150 bp of the HPV16 DNA and 150 bp of the human genome around the boundary were analyzed for average methylation. A statistically significant correlation was observed between the average methylation of the HPV16 and that of the human genome in all 3 cell lines (Fig. 5A and 5B).
Figure 5. Correlation analysis between the methylation pattern of the integrated HPV16 DNA and that of the human genome.
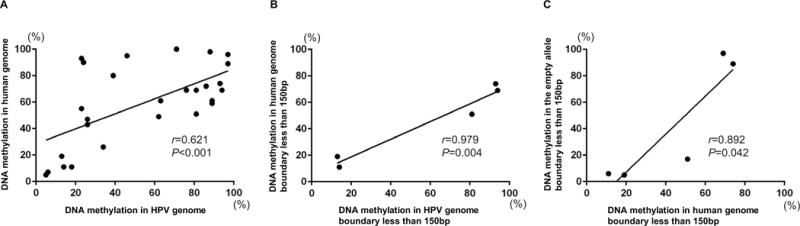
DNA fragments, including 150bp of the HPV16 DNA and 150 bp of the human genome around the boundary, were analyzed for average methylation. A. Correlation for total integration sites between the methylation levels of the HPV16 DNA and that of the human genome in the 3 cell lines. B. Correlation between 150bp of the HPV16 DNA and 150 bp of the human genome around the boundary analyzed for average methylation. C. Correlation between 150bp of the human genome and 150 bp of the empty allele around the boundary analyzed for average methylation. Correlations were analyzed with Pearson’s correlation coefficients, and P values were computed for the 2-tailed test.
These results suggested that the DNA methylation status of the integrated HPV16 genome was affected by the methylation status of the human genome flanking integration breakpoints. As our previous report also showed that methylation of the integrated HBV DNA is related to the methylation status of the flanking human genome25, we therefore hypothesized that DNA methylation in the integrated HPV16 genome is related to the methylation status of the integration sites within the host genome.
Allele-specific DNA methylation analysis of the HPV16 genome
To confirm our hypothesis, we further characterized the methylation status of the HPV genome and human genome by allele-specific DNA methylation analysis (Fig. 4A). We confirmed that the HPV genome was often highly methylated when integrated into highly methylated sites in the host genome, while the HPV16 genome remained largely unmethylated when integrated into unmethylated regions (Fig. 5C). Our results thus suggested that the HPV16 integrants became hypomethylated when the flanking host genomes were hypomethylated.
LINE1 methylation analysis
Despite the fact that integration mainly occurs in intergenic regions, which are usually considered to be highly methylated, we found varying levels of methylation in the human genome at the HPV16 integration breakpoints. Therefore, we planned to evaluate the global methylation levels of these cell lines. As LINE1 methylation level has been reported to be a good indicator of the global DNA methylation level26–28, we performed LINE1 methylation analysis in these cell lines. We found hypomethylation status of LINE1 in all of 3 cell lines as shown in Fig. 4C. These results indicate that global hypomethylation occurred in all 3 cell lines, consistent with the varying levels of methylation in intergenic regions observed in this study.
Discussion
In the present study, we revealed the detailed association between the epigenetic alterations in HPV integrants and the flanking host genomes in 3 HPV-related HNSCC cell lines using our novel approach with an NGS-based method for the structural methylation analysis of integrated viral genomes. Our NGS-based method using unique probes enabled us to identify some previously unreported integrants and that the HPV genome is often highly methylated when integrated into highly methylated sites in the host genome, while HPV16 integrants remained hypomethylated when the flanking host genome was hypomethylated.
The methylation of viral DNA integrated into the human genome has been studied over the past decade29 and in general it has been found that the local pattern of DNA methylation gradually spreads to involve the integrated viral genome30. The present study showed a statistically significant correlation between the average methylation level of the HPV16 DNA and that of the flanking host genome in 3 HPV-HNSCC cell lines. This correlation may be explained by our previous observation, indicating that the HBV genome often became significantly methylated when integrated into highly methylated host sites25. To the best of our knowledge, this is the first report to show a relationship between DNA methylation in the integrated HPV16 genome and the methylation status of the flanking host genome at the integration breakpoints in HPV-HNSCC.
The methylation of viral DNA in the virus-integrated cells has the potential to alter the expression levels of the viral genome31. A previous report showed a causal relationship between the demethylation of gene bodies and altered expression of the associated genes in cancer32. Our current and previous results thus suggested that the HPV integrants might be inactivated by methylation when integrated into a highly methylated human genome, while the HPV integrants could remain unmethylated after integrated into an unmethylated host genome, leading to tumorigenesis. However, there is still possibility that some alterations of methylation status of the HPV genome are occurred before the HPV integration into the human genome, as several publications have reported the hypothesis indicating that differential methylation of the E2BSs is related to the activation of both viral oncogene expression and the viral genome remains in the episomal state12, 33. In this case, the methylation status of the human genome might be affected by the methylation status of the HPV genome, as there were a couple of areas showing that the methylation status of human genome was not correlated with the methylation status of the HPV genome in our study. The biological impact of methylation on viral function needs to be further addressed. Since HPV integrations were mainly observed in non-coding regions in the present study, the association of epigenetic alteration with non-coding RNAs, including miRNA and long non-coding RNA, might be related to these epigenetic alterations.
In addition, the hypomethylation of LINE1 was also observed in all 3 cell lines, indicating that global hypomethylation could occur in HPV-HNSCC. Global hypomethylation of DNA sequences is often observed, not only during the early stage of tumorigenesis or in abnormal non-neoplastic tissues, but also on tumor progression34. As the degree of DNA hypomethylation increases with the grade of cervical neoplasia35, the DNA hypomethylation status might serve as oropharyngeal precancerous marker, since HPV-induced precancerous lesions, such as cervical intraepithelial neoplasias, have yet to be noted.
Recent genome-wide profiling of HPV integration in cervical cancer has shown that breakpoints could occur in any region of the viral genome, in contrast to the previously commonly held idea that integrated HPV16 would retain intact oncogene E6 and E7 with the long control region (LCR)23. Our results in HPV-HNSCC cells were consistent with these previous results. In the present study, the UPCI:SCC152 cell line, derived from a recurrent tumor in the patient from whom the UPCI:SCC090 cell line was derived, showed almost identical integration sites to UPCI:SCC090, which is consistent with a previous report that these cell lines have clonal expansion20. Interestingly, a lack of integration in chromosome 6 was observed only in the UPCI:SCC152 line. Further analysis would be required if this integration event itself caused genomic instability or the HPV integration tended to occur in the instable human genome regions.
There are several mechanisms for DNA repair, including non-homologous end joining (NHEJ), non-allelic homologous recombination (NAHR), fork stalling and template switching (FoSTeS)36, and microhomology-mediated break-induced replication (MMBIR)37. A recent report indicated that these mixed DNA-repair pathways participate in viral integration at genomic instability-related genomic elements23. The characteristics of the boundary including insertion and microhomology in the present study were consistent with this idea. In addition, a previous report showed that microhomology occurred not only at the breakpoint but also near the breakpoint, which was called flanking microhomology24. This phenomenon was also reported in association with HPV genome integration in cervical carcinoma23. This mechanism was also observed in HBV integration23, indicating that flanking microhomology might be a common mechanism for dsDNA virus integration. As several definitions exist for breakpoint and flanking microhomology23, 24, we only analyzed breakpoint microhomology in this study. Recently, Akagi et al. proposed a “looping model” to explain HPV integrant-mediated DNA replication and recombination leading to disruption of the expression and structure of neighboring genes9. Although our methodology was not able to examine the presence of this phenomenon, analysis around the boundary is thus important to understand molecular mechanism of HPV integration.
One of the main limitations in the present study was that only 3 cell lines were analyzed. A larger cohort, including clinical samples, should be analyzed to confirm our findings and to fully understand the pattern of methylation in the carcinogenesis of HPV-HNSCC. Other limitations might exist regarding the method itself using bisulfite pyrosequencing analysis with limited sequencing length from the boundary in this study. Particularly, the association between the methylation levels and transcription levels of the integrants in HPV-HNSCC cells could not be clarified in this study, since multiple HPV16 integration sites were present in the analyzed samples. We therefore have performed Amplification of Papillomavirus Oncogene Transcripts PCR (APOT-PCR) assay as described before38 to detect both integrated and episomal state of HPV. This PCR-based assay showed that all of 3 cell lines had the integration pattern and two of 3 cell lines had the episomal pattern (data not shown). Thus, the observed association between the methylation of the HPV integrants and the flanking host genome probably needs to be clarified as sequencing technology advances in the future. Lastly, while our results for the UPCI:SCC090 cell line were consistent with those of a previous report using WGS9 with some additional integration sites identified, there were also several integration sites identified by Akagi et al.9 which we could not observe in the present study. These small discrepancies could also be clarified with further advances in RNA sequencing technology. Despite these limitations, our results suggested that our G-NAVI method was a robust tool for the analysis of not only genomic sequences but also epigenetic alterations around integration breakpoints.
In conclusion, we here presented the molecular details of HPV16 DNA integration and various patterns of methylation in HPV-HNSCC using G-NaVI with 3 HPV-related HNSCC cell lines to reveal the association between epigenetic alterations in HPV integrants and the flanking host genomes. These results provided insights into the understanding of HPV integration in HNSCC with regard to HPV-related tumor progression, and also suggested that epigenetic alterations might provide candidate biochemical markers for establishing the precancerous stage in head and neck neoplasms. Together with previous reports on the whole genome and deep sequencing analysis9, 22, the detailed analysis performed in this study can help us understand the mechanisms for the viral integration and carcinogenesis of HPV-HNSCC.
Supplementary Material
Acknowledgments
We thank Mari Mitsuka (Department of Biology and Function in head and Neck, Yokohama City University Graduate School of Medicine, Yokohama, Japan) for her excellent technical assistance, and Dr. Jun Nakabayashi (Advanced medical research center bioinformatics laboratory, Yokohama City University Graduate School of Medicine, Yokohama, Japan) for his professional advice with regard to the statistical analysis.
Grant: P30CA016672.
Grant support
This work was supported by a Grant-in-Aid for Scientific Research 16K11240 (PI: DS), and 16K11242 (PI: HT).
Footnotes
Accession numbers
All raw sequence data for this study have been submitted to the DNA Data Bank of Japan (DDBJ; http://biosciencedbc.jp/en/) under the accession number DRA004152.
Disclosure of Potential Conflicts of Interest
The authors have no competing interests to disclose.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11:781–9. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 5.Ndiaye C, Mena M, Alemany L, Arbyn M, Castellsague X, Laporte L, Bosch FX, de Sanjose S, Trottier H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: a systematic review and meta-analysis. Lancet Oncol. 2014;15:1319–31. doi: 10.1016/S1470-2045(14)70471-1. [DOI] [PubMed] [Google Scholar]
- 6.Sano D, Oridate N. The molecular mechanism of human papillomavirus-induced carcinogenesis in head and neck squamous cell carcinoma. Int J Clin Oncol. 2016 doi: 10.1007/s10147-016-1005-x. [DOI] [PubMed] [Google Scholar]
- 7.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol. 2006;44:1755–62. doi: 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duenas-Gonzalez A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005;4:38. doi: 10.1186/1476-4598-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24:185–99. doi: 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park IS, Chang X, Loyo M, Wu G, Chuang A, Kim MS, Chae YK, Lyford-Pike S, Westra WH, Saunders JR, Sidransky D, Pai SI. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev Res (Phila) 2011;4:207–17. doi: 10.1158/1940-6207.CAPR-10-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vinokurova S, von Knebel Doeberitz M. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PloS one. 2011;6:e24451. doi: 10.1371/journal.pone.0024451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reuschenbach M, Huebbers CU, Prigge ES, Bermejo JL, Kalteis MS, Preuss SF, Seuthe IM, Kolligs J, Speel EJ, Olthof N, Kremer B, Wagner S, et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer. 2015;121:1966–76. doi: 10.1002/cncr.29315. [DOI] [PubMed] [Google Scholar]
- 13.White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, Gollin SM. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2007;43:701–12. doi: 10.1016/j.oraloncology.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, Kelland LR, Harrison M, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A. 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, Sturgis EM, Ow TJ, Lotan R, Carey TE, Sacks PG, Grandis JR, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res. 2011;17:7248–64. doi: 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukusa T, Fryns JP. Human chromosome fragility. Biochim Biophys Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Wentzensen N, Ridder R, Klaes R, Vinokurova S, Schaefer U, Doeberitz M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene. 2002;21:419–26. doi: 10.1038/sj.onc.1205104. [DOI] [PubMed] [Google Scholar]
- 18.Yamada Y, Ito T. Highly efficient PCR assay to discriminate allelic DNA methylation status using whole genome amplification. BMC Res Notes. 2011;4:179. doi: 10.1186/1756-0500-4-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, Nobuoka T, Nishida T, Miyazaki Y, Takamaru H, Yamamoto E, Yamamoto H, et al. A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin Cancer Res. 2010;16:5114–23. doi: 10.1158/1078-0432.CCR-10-0581. [DOI] [PubMed] [Google Scholar]
- 20.Olthof NC, Huebbers CU, Kolligs J, Henfling M, Ramaekers FC, Cornet I, van Lent-Albrechts JA, Stegmann AP, Silling S, Wieland U, Carey TE, Walline HM, et al. Viral load, gene expression and mapping of viral integration sites in HPV16-associated HNSCC cell lines. Int J Cancer. 2015;136:E207–18. doi: 10.1002/ijc.29112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22:1225–37. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 22.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova EV, Wilkerson MD, Protopopov A, Yang L, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111:15544–9. doi: 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47:158–63. doi: 10.1038/ng.3178. [DOI] [PubMed] [Google Scholar]
- 24.Lawson AR, Hindley GF, Forshew T, Tatevossian RG, Jamie GA, Kelly GP, Neale GA, Ma J, Jones TA, Ellison DW, Sheer D. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011;21:505–14. doi: 10.1101/gr.115782.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe Y, Yamamoto H, Oikawa R, Toyota M, Yamamoto M, Kokudo N, Tanaka S, Arii S, Yotsuyanagi H, Koike K, Itoh F. DNA methylation at hepatitis B viral integrants is associated with methylation at flanking human genomic sequences. Genome Res. 2015;25:328–37. doi: 10.1101/gr.175240.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33:6823–36. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, Luna R, Rashid A, Shen L, Estecio MR, Kantarjian HM, Garcia-Manero G, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66:5495–503. doi: 10.1158/0008-5472.CAN-05-2385. [DOI] [PubMed] [Google Scholar]
- 29.Doerfler W, Remus R, Muller K, Heller H, Hohlweg U, Schubbert R. The fate of foreign DNA in mammalian cells and organisms. Dev Biol (Basel) 2001;106:89–97. discussion 143–60. [PubMed] [Google Scholar]
- 30.Orend G, Kuhlmann I, Doerfler W. Spreading of DNA methylation across integrated foreign (adenovirus type 12) genomes in mammalian cells. J Virol. 1991;65:4301–8. doi: 10.1128/jvi.65.8.4301-4308.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, Pino I, Javierre B, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009;19:438–51. doi: 10.1101/gr.083550.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaiwongkot A, Vinokurova S, Pientong C, Ekalaksananan T, Kongyingyoes B, Kleebkaow P, Chumworathayi B, Patarapadungkit N, Reuschenbach M, von Knebel Doeberitz M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. International journal of cancer Journal international du cancer. 2013;132:2087–94. doi: 10.1002/ijc.27906. [DOI] [PubMed] [Google Scholar]
- 34.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–59. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, Selhub J, Mason JB. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer. 1994;74:893–9. doi: 10.1002/1097-0142(19940801)74:3<893::aid-cncr2820740316>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–47. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 37.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, Schneider A, Lotz B, Melsheimer P, von Knebel Doeberitz M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer research. 1999;59:6132–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.