

Abstract
A method of cysteine alkylation using cyclopropenyl ketones is described. Due to the significant release of cyclopropene strain energy, reactions of thiols with cyclopropenyl ketones are both fast and irreversible and give rise to stable conjugate addition adducts. The resulting cyclopropenyl ketones have a low molecular weight and allow for simple attachment of amides via N-hydroxysuccinimide (NHS)-esters. While cyclopropenyl ketones do display slow background reactivity toward water, labeling by thiols is much more rapid. The reaction of a cyclopropenyl ketone with glutathione (GSH) proceeds with a rate of 595 M−1s−1 in PBS at pH 7.4, which is considerably faster than α-halocarbonyl labeling reagents, and competitive with maleimide/thiol couplings. The method has been demonstrated in protein conjugation, and an arylthiolate conjugate was shown to be stable upon prolonged incubation in either GSH or human plasma. Finally, cyclopropenyl ketones were used to create PEG-based hydrogels that are stable to prolonged incubation in a reducing environment.
Introduction
An ideal bioconjugation should display high selectivity, efficiency and rapid kinetics at 25–37 °C in order to facilitate fast labeling under biologically relevant conditions. Among the varied methods of bioconjugation, the covalent modification of thiols remains an important technique for the selective functionalization of proteins and other biomolecules.1–5 There has been considerable effort to develop reagents capable of selective cysteine modification.6–9 Historically, the most broadly used reagents for the modification of cysteine residues have been maleimide6,10 and α-halocarbonyl6,11 reagents. α-Halocarbonyl reagents give rise to stable conjugates, but reaction rates are modest (< 1 M−1s−1 at pH 7.0),12 and therefore reactions are typically conducted at high concentrations (mM) and elevated pH levels (pH 8-9).13,14 The conjugate addition reactions of maleimides are highly selective for cysteine modification and proceed efficiently under physiological conditions.6,10 Biomolecular rate constants of maleimides with thiols range from 102 – 104 M−1s−1 dependent on pH and reagent structure.14–16 A limitation of maleimide-based protein conjugation is that the retro conjugate addition reaction can occur under physiological conditions.17–20 Thus, competing thiols can scavenge maleimides from the molecule of interest in a biological context. Indeed, the retro-thiol/maleimide conjugate addition has been pursued as the basis for biomaterial designs and drug release applications.18–20 However, in many instances a stable thioether linkage is needed such as for use as linkers in protein drug conjugates.21 It has been shown possible to improve the stability of maleimide thioethers by increasing their propensity to hydrolyze,15,22–24 as the ring opened variant does not participate in the retro conjugate addition chemistry Recently, exocyclic maleimides have also been shown to give stable thiol conjugate addition products.25,26
A number of alternate strategies have been introduced for selective cysteine functionalization.6–9 Heteroatomic sulfones have been introduced as reagents that can react with cysteines in aqueous buffer containing organic cosolvents.27 While the adducts were more stable than their maleimide counterparts under acidic, basic and reducing conditions, high concentrations are required to achieve complete reactivity (11 mM, 60 min) in 1:1 THF/PBS at 25°C). Arylpalladium reagents have been developed that label cysteines, with site specific protein labeling within 5–30 min using 10–60 μM of the organometallic reagent.28 Finn and coworkers have described the use of electron deficient oxanorbornadienes (ONDs) as thiol-selective alkylating agents with second order rate constants for glutathione alkylation ranging from 40 – 200 M−1s−1 at pH 7 (25 °C) 29–31 These reagents have been designed to undergo retro Diels-Alder reactions with tunable rates, providing a novel mechanism for temporally controlled payload release and attenuation of material properties. Cysteine phosphorylation through sequential treatment with Ellman’s reagent and nucleophilic phosphites has also been described.32
Described here is a new class of cysteine alkylating agents based on cyclopropenyl ketones. The use of cyclopropenes in addition reactions with heteroatomic nucleophiles has been developed by Rubin for applications in synthesis.33 While the use of cyclopropenes in bioorthogonal reactions with tetrazines and tetrazoles has been documented recently,34–40 the use of cyclopropenyl ketones has not been described. Based on our prior work where cyclopropenyl ketones were found to serve as very reactive and selective dienophiles,41,42 we hypothesized that the significant release of cyclopropene olefinic strain43,44 upon conjugate addition should render the reaction both fast and irreversible (Scheme 1). Additionally, cyclopropenyl ketones described here are small (MW 168) and allow for simple attachment of pendant groups via amide bond construction based on N-hydroxysuccinimide (NHS)-ester conjugation.
Scheme 1.

(A) Bioconjugation reactions involving thiols with maleimides are fast but can be limited by reversibility. (B) Strain-driven reactions of cyclopropenylketones with thiols are both fast and irreversible.
Results and discussion
Cyclopropenyl ketones were prepared using the cyclopropene carboxylate dianion method previously developed by our group.41 The conjugatable NHS ester 1, available in 4 steps, could be conjugated to amines as illustrated in Scheme 2. Oxidation with the Dess-Martin periodinane provided the enone 2, which participated in rapid conjugation with thiols as exemplified by the reaction with 4-mercaptophenylacetic acid to give conjugate 3 as a mixture of diastereomers.
Scheme 2.

Conjugation/oxidation gives rise to functionalized cyclopropenyl ketones that undergo rapid conjugation with thiols.
In order to demonstrate the specificity of cyclopropenyl ketones toward the cysteine residues of a protein, we examined the reaction of enone 4 with the redox active protein Thioredoxin (Trx),45 which contains a single disulfide bond (Scheme 3). Trx ionizes readily by ESI-MS with a molecular weight of 11658 as deconvoluted using MagTran software (Figure S1). Trx (10 μM) in pH 6 acetate buffer was reduced by tris(hydroxypropyl)phosphine (50 μM) and subsequently treated with 4 (500 μM) and aliquots analyzed by ESI-MS after 10 minutes. Labeling of reduced Trx was observed to give an adduct 5 with m/z 11994, corresponding to the addition of two cyclopropenyl ketones 4 to the Trx core (Scheme 3a and Figure S1). In an otherwise identical experiment where the reduction step with THP was omitted, no labeling of the protein was observed (Scheme 3b and Figure S1). These observations support that the labeling of Trx takes place selectively at the cysteine residues and that lysines do not react with 4 under these experimental conditions.
Scheme 3.

Thioredoxin (Trx) is an 11.7 kDa protein with a single disulfide. (A) The reduced form of the Trx is readily alkylated by enone 4, whereas (B) the oxidized form of Trx is unreactive, providing evidence that the labeling of the protein is selective for cysteine functionality. (C) The reaction of 4 (250 μM) with variable concentrations of glutathione (2.5, 5.0 and 10 mM) was monitored by the disappearance of UV absorption at 252 nm by stopped flow kinetics. A bimolecular rate constant of 595 M−1s−1 was observed.
We evaluated the stability of 2 by 1H NMR of a 20 mM solution of 2 in 9:1 D2O/CD3OD (Figure S2). The study indicated that aqueous solutions of 2 can be handled, but that water and/or methanol slowly adds to cyclopropenyl ketone 2. The half-life for the disappearance of 2 at room temperature was approximately 6 hours. Thus, while the cyclopropenyl ketones described here can be handled in aqueous solution for moderate periods, they should not be stored in aqueous environment for prolonged periods. In practice, we find it most practical to store compounds as their allylic alcohol precursors and to generate the cyclopropenyl ketones as needed using Dess-Martin reagent. Cyclopropenyl allylic alcohols such as 1 show improved stability, but should be stored in cold solution as the neat compounds show variable amounts of decomposition products when stored overnight on the bench.
Experiments were conducted in order to measure the second order rate constant for the conjugation of a thiol to cyclopropenyl ketone 4, which has an absorption maximum in the UV at 252 nm. The reaction of 4 (250 μM) with glutathione (GSH) was monitored under pseudo first order conditions (2.5 mM, 5.0 mM or 10 mM in GSH). Stopped flow kinetics experiments were performed with monitoring at 252 nm at room temperature in pH 7.4 in 50 mM phosphate buffer with 1 mM EDTA and 0.5 % MeOH. The second order rate constant was calculated to be 595 ± 30 M−1s−1 (Figure S3). Thus, 4 is considerably more reactive than α-halocarbonyl labeling reagents,12 and the rate of 4 with GSH is within the realm of maleimide/thiol couplings.14–16 In a competition experiment using equimolar amounts (500 μM) of 4 and N-ethylmaleimide and a limiting amount of glutathione (50 μM), ESI-MS showed adducts of both N-ethylmaleimide and 4 in an uncorrected 4:1 ratio, respectively (Figure S4). This result provides additional qualitative support that the cyclopropenyl ketone reagent approaches the reactivity of the maleimides as alkylating agents.
As arylthiolate conjugates are especially susceptible to retro-Michael addition,18 we chose to demonstrate the robustness of cyclopropenyl ketone-arylthiolate conjugate 3 (Scheme 4). We first studied the stability of 3 in the presence of glutathione. If the elimination of α-(4-mercaptophenyl)acetic acid (MPA) from 3 to give 2 were to take place in the presence of excess glutathione, one or more of the compounds 6–8 would be expected to form. In a control experiment where 2 (0.2 mM) was combined with a 10:1 mixture of glutathione:MPA (2 mM and 0.2 mM, respectively), both 3 and 8 were observed by ESI-MS (Scheme 4A and Figure S5). Compound 3 (0.2 mM) did not undergo retroaddition when incubated with GSH (2 mM) at 37 °C in PBS (pH 7.4). After 55 hours, compound 3 remained fully intact and compounds 6–8 were not detected (Scheme 4A and Figure S6). The high stability of 3 can be contrasted to the N-ethyl maleimide adduct of MPA, which is known to undergo retro-Michael addition.19,20,46
Scheme 4.
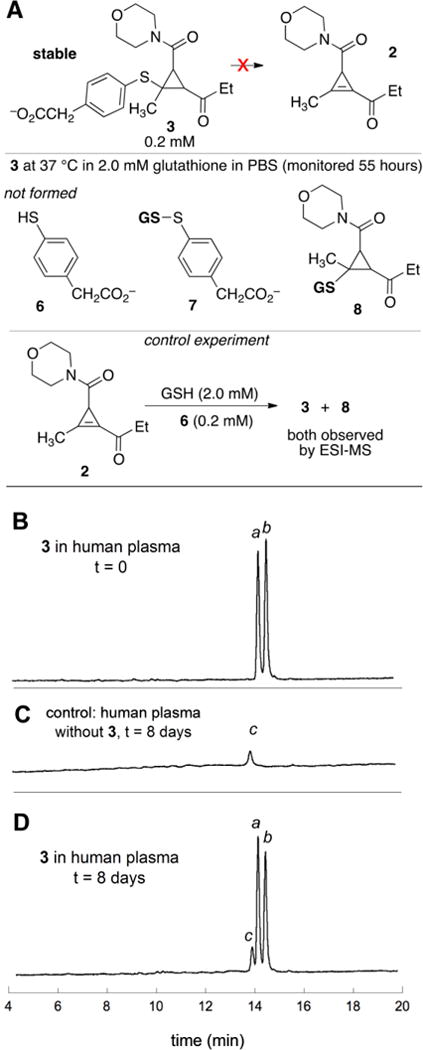
(A) Conjugate 3 was shown to be stable upon incubation in the presence of excess glutathione in PBS buffer. Adducts 6–8 were not observed (Figure S7) (B–D) Demonstration of stability of 3 in human plasma. HPLC with detection at 254 nm was used to monitor the stability of 3 (0.2 mM) incubated in human plasma at 37 °C over a period of 8 days. After 8 days there was not a significant change in the intensity of the peaks labeled a and b, which are due to isomers of 3. The ratio of peaks a:b was 47:53 at t=0, and 49:51 at t = 8 days. The peak labeled c is due to background from the plasma.
Stability toward retroaddition was also evaluated in human plasma (Scheme 4B-D). Thus, a solution of 3 (200 μM) in human plasma was incubated at 37 °C over a period of 8 days. Aliquots were removed and subjected to centrifugal filtration to allow for large protein removal, and the filtrate obtained assayed by HPLC. Immediately upon mixing, an aliquot was taken and HPLC analysis showed peaks due to diastereomers of 3 (labeled a and b, Scheme 4B). After 8 days, the peaks due to the 3 were remained, in addition to small peak (labeled c) assignable to background reactivity in plasma (Scheme 4C). By HPLC integration, 95% of compound 3 was unchanged after incubation in human plasma at 37 °C for 8 days (Scheme 4D).
Hydrogel-based materials have found wide-ranging applications as three-dimensional (3D) scaffolds for cell culture and tissue engineering, and thiol-based conjugations have become essential tools for the crosslinking reactions used to form many hydrogel materials.47,48 In particular, multifunctional poly(ethylene glycol) covalently crosslinked or modified using thiol-ene reactions has found wide use for the formation of hydrogels in the presence of cells and proteins. 49–51 Maleimide conjugation has been pursued as a rapid crosslinking reaction for the creation of PEG-based hydrogels, partly due to mild reaction conditions that are free of catalyst or initiator.19,20 An interesting property of maleimide-based hydrogels is they can be tuned to degrade in a thiol-rich environment, providing the basis for materials that can mediate drug release.52–54 As a complementary approach, we envisioned the use of cyclopropenyl ketones as crosslinkers for hydrogel formation that would retain high degree of stability even in the presence of thiols.
As a monomer for hydrogel formation, the doubly-functionalized cyclopropenyl ketone 9 was synthesized by reacting NHS ester 1 with O,O′-bis-(2-aminoethyl)octadecaethylene, followed by oxidation of the resulting alkenol with Dess Martin periodinane (Scheme 5A). For each set of polymerization experiments conducted, 9 was freshly prepared due to the hygroscopic nature of the PEGylated monomer. On mixing stoichiometric amounts of biscyclopropenyl ketone 9 and a PEG-tetra-thiol crosslinker53 (20 kDa) in PBS buffer, the gel point, as indirectly measured by G′ > G", was reached within 30 seconds – faster than the first rheological measurement that could be recorded (Scheme 5B,C). Complete gelation, with no measurable change in G′ over time, was observed within 30 minutes. While the gels formed rapidly upon mixing, the reaction resulted in the consistent formation of hydrogels with uniform properties. Further experiments showed that the cyclopropenyl ketone derived hydrogels incubated in PBS containing 10 mM GSH are are stable for greater than 6 days based on rheological measurements: specifically, the moduli of equilibrium-swollen gels statistically were the same at early and late times (24 hours, G′eq = 278.6 ± 76.6 Pa; 144 hours, G′eq = 292.7 ± 59.2 Pa), demonstrating the utility of this approach for the formation of robust hydrogels.
Scheme 5. Formation and stability of hydrogels formed via thiol−ene click chemistry.

A) Hydrogels were formed via Michael-type thiol−ene addition between PEG tetra-thiol and PEG-bis-cyclopropenyl ketone to form a stable network. B) Monomers were briefly mixed and placed between parallel plates on a rheometer to monitor gel formation (tstart = 30-35 s after mixing). Gels began to form in under 30 s, indicated by a storage modulus (G′) greater than loss modulus (G″) at the start of measurements, and reached complete gelation within 30 min. The storage modulus of gels was recorded after complete gelation (t = 30 minutes; G′o = 2814.4 ± 177.1 Pa). Gels subsequently were polymerized overnight and equilibrium swollen in PBS containing glutathione (10 mM). Stability of the resulting hydrogels was observed, where moduli of equilibrium-swollen gels statistically were the same at early and late times (24 hours, G′eq = 278.6 ± 76.6 Pa; 144 hours, G′eq = 292.7 ± 59.2 Pa; p = 0.68, two-tailed t-test).
Conclusions
In conclusion, a method for cysteine alkylation using cyclopropenyl ketones has been described. Driven by the significant release of cyclopropene strain energy, reactions of thiols with cyclopropenyl ketones are shown to be both fast and irreversible and give rise to stable conjugate addition adducts. Furthermore, the cyclopropenyl ketones have a low molecular weight and allow for simple attachment of amides via N-hydroxysuccinimide (NHS)-ester conjugation. The cyclopropenyl ketones do display slow background reactivity toward water, but labeling by thiols is much more rapid. The reaction of a cyclopropenyl ketone with glutathione (GSH) proceeds with a rate of 595 M−1s−1 in PBS at pH 7.4, which is considerably faster than α-halocarbonyl labeling reagents and competitive with maleimide/thiol couplings. The method has been demonstrated in protein conjugation, and an arylthiolate conjugate was shown to be stable upon prolonged incubation in either glutathione or human plasma. Finally, cyclopropenyl ketones were used to create PEG-based hydrogels that are stable to prolonged incubation in a reducing environment. Future efforts are being directed toward the development of cyclopropenones that display improved aqueous stability and still maintain rapid rates for irreversible thiol alkylation.
Supplementary Material
Acknowledgments
This work was supported by a NSF CHE1300329, NIH R01EB014354 and P20GM104316. Spectra were obtained with instrumentation supported by NIH grants P30GM110758, S10RR026962, S10OD016267 and NSF grants CHE-0840401, CHE-1229234, and CHE-1048367. For fellowships, LAS thanks NSF IGERT-1144726, and NJS thanks NIH CBI T32GM008550.
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare
Notes and references
- 1.Koniev O, Wagner A. Chem Soc Rev. 2015;44:5495–5551. doi: 10.1039/c5cs00048c. [DOI] [PubMed] [Google Scholar]
- 2.Boutureira O, Bernardes GJL. Chem Rev. 2015;115:2174–2195. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 3.Baslé E, Joubert N, Pucheault M. Chem Biol. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Spicer CD, Davis BG. Nat Commun. 2014;5:4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- 5.Stephanopoulos N, Francis MB. Nat Chem Biol. 2011;7:876–884. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- 6.Chalker JM, Bernardes GJLL, Lin YA, Davis BG. Chem Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- 7.Cal PMSD, Bernardes GJL, Gois PMP. Angew Chem Int Ed. 2014;53:10585–10587. doi: 10.1002/anie.201405702. [DOI] [PubMed] [Google Scholar]
- 8.Kim Y, Ho SO, Gassman NR, Korlann Y, Landorf EV, Collart FR, Weiss S. Bioconjug Chem. 2008;19:786–791. doi: 10.1021/bc7002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunnoo SB, Madder A. ChemBioChem. 2016;17:529–553. doi: 10.1002/cbic.201500667. [DOI] [PubMed] [Google Scholar]
- 10.Gregory JD. J Am Chem Soc. 1955;77:3922–3923. [Google Scholar]
- 11.Goddard DR, Michaelis L. J Biol Chem. 1935;112:361–371. [Google Scholar]
- 12.Nelson KJ, Day AE, Zeng BB, King SB, Poole LB. Anal Biochem. 2008;375:187–95. doi: 10.1016/j.ab.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aitken A, Learmonth M. In: The Protein Protocols Handbook. 2nd. Walker JM, editor. Humana Press; Totowa, NJ: 2002. pp. 455–456. [Google Scholar]
- 14.Gilbert HF. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Tsao K, De Francesco É, Keillor JW. J Org Chem. 2015;80:12182–12192. doi: 10.1021/acs.joc.5b02036. [DOI] [PubMed] [Google Scholar]
- 16.Sapra A, Thorpe C. J Am Chem Soc. 2013;135:2415–2418. doi: 10.1021/ja310553h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Bioconjug Chem. 2008;19:759–765. doi: 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- 18.Baldwin AD, Kiick KL. Bioconjug Chem. 2011;22:1946–53. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldwin AD, Kiick KL. Polym Chem. 2013;4:133–143. doi: 10.1039/C2PY20576A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kharkar PM, Kiick KL, Kloxin AM. Chem Soc Rev. 2013;42:7335–7372. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alley SC, Okeley NM, Senter PD. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 22.Lyon RP, Setter JR, Bovee TD, Doronina SO, Hunter JH, Anderson ME, Balasubramanian CL, Duniho SM, Leiske CI, Li F, Senter PD. Nat Biotechnol. 2014;32:1059–1062. doi: 10.1038/nbt.2968. [DOI] [PubMed] [Google Scholar]
- 23.Fontaine SD, Reid R, Robinson L, Ashley GW, Santi DV. Bioconjug Chem. 2015;26:145–52. doi: 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- 24.Christie RJ, Fleming R, Bezabeh B, Woods R, Mao S, Harper J, Joseph A, Wang Q, Xu ZQ, Wu H, Gao C, Dimasi N. J Control Release. 2015;220:660–670. doi: 10.1016/j.jconrel.2015.09.032. [DOI] [PubMed] [Google Scholar]
- 25.Kalia D, Malekar PV, Parthasarathy M. Angew Chem Int Ed. 2016;128:1432–1435. doi: 10.1002/anie.201508118. [DOI] [PubMed] [Google Scholar]
- 26.Akkapeddi P, Azizi SA, Freedy AM, Cal PMSD, Gois PMP, Bernardes GJL. Chem Sci. 2016;7:2954–2963. doi: 10.1039/c6sc00170j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toda N, Asano S, Barbas CF. Angew Chem Int Ed. 2013;52:12592–12596. doi: 10.1002/anie.201306241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL, Buchwald SL. Nature. 2015;526:687–91. doi: 10.1038/nature15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong V, Kislukhin AA, Finn MG. J Am Chem Soc. 2009;131:9986–9994. doi: 10.1021/ja809345d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kislukhin AA, Higginson CJ, Hong VP, Finn MG. J Am Chem Soc. 2012;134:6491–6497. doi: 10.1021/ja301491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higginson CJ, Kim SY, Peláez-Fernández M, Fernández-Nieves A, Finn MG. J Am Chem Soc. 2015;137:4984–4987. doi: 10.1021/jacs.5b02708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertran-Vicente J, Penkert M, Nieto-Garcia O, Jeckelmann JM, Schmieder P, Krause E, Hackenberger CPR. Nat Commun. 2016;7:12703. doi: 10.1038/ncomms12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards A, Rubina M, Rubin M. Curr Org Chem. 2016;20:1862–1877. [Google Scholar]
- 34.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc. 2012;134:18638–43. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 35.Patterson DM, Jones KA, Prescher JA. Mol Biosyst. 2014;10:1693. doi: 10.1039/c4mb00092g. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Šečkutė J, Cole CM, Devaraj NK. Angew Chem Int Ed. 2012;51:7476–9. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole CM, Yang J, Šečkutė J, Devaraj NK. ChemBioChem. 2013;14:205–8. doi: 10.1002/cbic.201200719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J Am Chem Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d. [DOI] [PubMed] [Google Scholar]
- 39.Sachdeva A, Wang K, Elliott T, Chin JW. J Am Chem Soc. 2014;136:7785–7788. doi: 10.1021/ja4129789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramil CP, Dong M, An P, Lewandowski TM, Yu Z, Miller LJ, Lin Q. J Am Chem Soc. 2017;139:13376–13386. doi: 10.1021/jacs.7b05674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher LA, Smith NJ, Fox JM. J Org Chem. 2013;78:3342–8. doi: 10.1021/jo302683t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravasco JMJM, Monteiro CM, Trindade AF. Org Chem Front. 2017;4:1167–1198. [Google Scholar]
- 43.Bach RD, Dmitrenko O. J Am Chem Soc. 2004;126:4444–4452. doi: 10.1021/ja036309a. [DOI] [PubMed] [Google Scholar]
- 44.Wiberg KB. Angew Chem, Int Ed Engl. 1986;25:312. [Google Scholar]
- 45.Codding JA, Israel BA, Thorpe C. Biochemistry. 2012;51:4226–4235. doi: 10.1021/bi300394w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baldwin AD, Kiick KL. Bioconjug Chem. 2011;22:1946–53. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kharkar PM, Rehmann MS, Skeens KM, Maverakis E, Kloxin AM. ACS Biomater Sci Eng. 2016;2:165–179. doi: 10.1021/acsbiomaterials.5b00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lowe AB. Polym Chem. 2014;5:4820. [Google Scholar]
- 49.Zhu J. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caliari SR, Burdick JA. Nat Methods. 2016;13:405–14. doi: 10.1038/nmeth.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fisher SA, Baker AEG, Shoichet MS. J Am Chem Soc. 2017;139:7416–7427. doi: 10.1021/jacs.7b00513. [DOI] [PubMed] [Google Scholar]
- 52.Kharkar PM, Scott RA, Olney LP, LeValley PJ, Maverakis E, Kiick KL, Kloxin AM. Adv Healthc Mater. 2017;6:1700713. doi: 10.1002/adhm.201700713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sawicki LA, Kloxin AM. Biomater Sci. 2014;2:1612–1626. doi: 10.1039/c4bm00187g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kharkar PM, Kloxin AM, Kiick KL. J Mater Chem B. 2014;2:5511–5521. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.