

Abstract
Various organisms exist in the oceanic environment. These marine organisms provide an abundant source of potential medicines. Many marine peptides possess anticancer properties, some of which have been evaluated for treatment of human cancer in clinical trials. Marine anticancer peptides kill cancer cells through different mechanisms, such as apoptosis, disruption of the tubulin-microtubule balance, and inhibition of angiogenesis. Traditional chemotherapeutic agents have side effects and depress immune responses. Thus, the research and development of novel anticancer peptides with low toxicity to normal human cells and mechanisms of action capable of avoiding multi-drug resistance may provide a new method for anticancer treatment. This review provides useful information on the potential of marine anticancer peptides for human therapy.
Keywords: peptide, anticancer, marine organism, antiproliferative, therapeutic agents
1. Introduction
The oceans are a diverse environment comprising about 75% of living organisms [1]. The oceanic environment is an abundant source of nutraceuticals and potential candidates with therapeutic functions [2].
Cancer is among the major causes of death with high morbidity and mortality [3]. Cell division that occurs within the cell tissue is a normal process. Under normal conditions, apoptosis constantly maintains the equilibrium between proliferating cells and programmed cell death. Additionally, DNA mutations cause cancer by interruption of the regulating programs. In carcinogenesis, normal cells are transformed into cancer cells [4,5]. In recent decades, marine compounds were studied for the treatment of cancer. Various types of marine products, such as alkaloids, polyketides, terpenes, peptides, and carbohydrates, have the potential to prevent and cure cancer [6]. Therefore, these products may be important in the development of anticancer drugs [7].
In recent years, novel peptides with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities have been found in marine organisms and studied [2]. Many marine peptides also possess anticancer activity. Some of these peptides have been studied as cancer treatment in human clinical trials [8,9,10]. Marine anticancer peptides can kill cancer cells through different mechanisms, such as apoptosis, affecting the tubulin-microtubule imbalance, and inhibition of angiogenesis [11]. Understanding the structure and function of pharmacologically active marine-derived anticancer peptides will enhance the development of lead drug candidates.
This review focuses on the function and structure of pharmacologically active marine peptides with anticancer activity. Research on peptides derived from marine sources is underway to develop new potential cancer treatments.
2. Marine Organisms that are Sources of Anticancer Peptides
Many researchers have isolated anticancer peptides from various marine sources, such as, cyanobacteria, fungi, sponges, tunicates, ascidians, mollusks, and fish. Anticancer peptides cause cell death through a unique mechanism involving selectivity to cancer cells and membrane breakdown. The outer portion of the cell membrane of cancer cells has a higher concentration of anionic phosphatidylserine than found in normal cells, resulting in a total negative charge on the cell surface. As a result, the cationic anticancer peptide is attracted to the surface of the cancer cell and binds to the cell membrane, resulting in the collapse of the membrane and rapid cell death [12,13]. The structural properties of these peptides are attributable to diverse unusual amino acid residues exhibiting anticancer activity [14].
2.1. Cyanobacteria
Cyanobacteria (marine blue-green algae) are an abundant source of established and novel bioactive compounds with powerful therapeutic applications [15,16]. The cyanobacteria population has considerable diversity, comprising 150 genera and about 2000 species [17]. Most studies involve anticancer agents derived from marine cyanobacteria, which kill cancer cells by inducing apoptotic cell death or affecting cell signaling [5]. In addition to their cytotoxic effect on human cancer cell lines, some peptides have been found as templates for the development of new anticancer drugs. Moreover, these anticancer peptides possess anti-inflammatory and antibacterial properties [18].
2.2. Fungi
Marine fungi produce various secondary metabolites with biological activity and may be structurally unique [19,20]. Increasing attention regarding the pharmaceutical potential of marine fungi and screening for novel compounds with anticancer properties have led to the discovery of several related marine products. In recent years, some marine fungi-derived compounds have shown potent anticancer activity through different mechanisms, such as cell death induced via multiple cell death pathways and stimulation of cell cycle arrest [21]. Facultative marine fungi have been researched since they are known to produce novel compounds having anticancer activities [18].
2.3. Sponges
One of the most abundant sources of therapeutically effective products in marine organisms are marine sponges [22]. Every year, around 5300 different new compounds are being found from sponges [22]. Among the sponge-derived peptides reported so far, peptides having anticancer activity are mainly produced in Haliclona, Petrosia, and Discodemia [22]. Sponge-derived peptides have broad biological activities [22,23,24]. Among these peptides, dolastatins, didemnin B and aplidine have been recently reported in clinical trials [25]. Natural products isolated from sponges are of interest to the pharmaceutical industry, but most are cyclodepsipeptides, secondary metabolites which contain specific amino acids and non-amino acid moieties. For this reason, natural products isolated from sponges are difficult to isolate sufficiently in order to conduct pharmacological testing. Nevertheless, a great deal of research has been done on these products because of their various biological activities.
2.4. Tunicates and Ascidians
The novel structured peptides found in tunicates and ascidians often possess physiological activity. This is also the case with sack-like sea squirts that produces complex antitumor compounds. Only a small number of these are currently being used in cancer treatment [11]. Tunicates are a group of marine organisms that belong to Tunicata, a subphylum of Chordata. Although ascidians were considered as tunicates, recent studies have shown that ascidians and tunicates are actually a different species [7]. Many of these ascidian species produce anticancer peptides, steroids and antioxidants in novel structures that have biological activity [18].
2.5. Mollusks and Fish
Mollusks are organisms with a broad range of pharmacological uses. Mollusks are a major source of primary metabolites because they produce a variety of peptides, enzymes, polysaccharides, and lipids. In the case of a Sea hare, a shelled organism, it produces a variety of peptides in linear, cyclic and conjugated forms, with potent anticancer activity [26].
Marine fish are an important source of bioactive peptides and proteins. For this reason, the importance of fish producing novel bioactive materials is rapidly increasing. Fish-derived peptides are involved in various pharmacological effects, including antihypertensive, immunomodulatory, antioxidant, antitumor, and antimicrobial activities [18,27,28].
3. Bioactive Peptides with Anticancer Potential Isolated from Marine Organisms
Most of the commercially useful anticancer drugs are naturally derived compounds [25]. The suitability of novel marine-derived anticancer compounds has been determined through discovery, development, and marketing approval. These results were used for the prediction and development of potential marine-derived compounds for cancer chemotherapy [6]. Most of the marine natural products used for research are secondary metabolites. However, primary metabolites such as various types of peptides are of growing interest to the pharmaceutical industry [11].
This review describes the development of marine anticancer peptides derived from different organisms. Furthermore, a list of marine-derived anticancer peptides and their mode of action are summarized in Table 1.
Table 1.
List of marine-derived anticancer peptides and their mode of action.
| Name of Peptide | Natural Sources | Class/Types | Mode of Action/Investigative Status | Growth Inhibition Concentration (Cell Line) | Ref. |
|---|---|---|---|---|---|
| Apratoxin A–D (1–4) |
Cyanobacteria: Lyngbya majuscula, L. sordida | Cyclic depsipeptide |
Induction of G1 phase cell cycle arrest and apoptosis/in vitro only | IC50: 0.36 nM (LoVo), 0.52 nM (KB), 2.6 nM (H-460) |
[29,30,31,32,33,34,35] |
| Aurilide (5), B (6), C (7) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide |
Cytotoxicity /in vitro only |
LC50: 40–130 nM (NCI-H460), 10–50 nM (neuro-2a), GI50 < 10 nM (NCI 60 cell line panel) |
[36,37] |
| Bisebromoamide (8) | Cyanobacteria: Lyngbya majuscula | Linear peptide | Activation of ERK pathway/in vitro only | IC50: 0.04 μg/mL (HeLa S3), GI50 = 40 nM (JFCR39 cell line panel) |
[38] |
| Coibamide A (9) | Cyanobacteria: Leptolyngbya sp. | Cyclic depsipeptide |
Cancer cell proliferation inhibition/in vitro only | LC50 < 23 nM (NCI-H460, neuro-2a) GI50 < 7.6 nM (NCI 60 cell line panel) |
[39] |
| Cryptophycin (10) | Cyanobacteria: Nostoc sp. | Depsipeptide | Apoptosis and microtubule inhibition /in vitro only (Cryptophycin-52: Phase II human clinical trial) |
IC50 < 50 pM (MDR tumor cell lines), | [40,41,42,43,44] |
| Desmethoxymajusculamide C (11) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide |
Tubulin polymerization Inhibition/in vitro only |
IC50: 20 nM (HCT-116), | [45] |
| Grassypeptolide A–E (12–16) | Cyanobacteria: Lyngbya confervoides | Cyclic depsipeptide |
Induction of G2/M phase cell cycle arrest /in vitro only |
IC50: 192–335 nM (HeLa), 407–599 nM (neuro-2a) | [46,47] |
| Hantupeptin A (17) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide |
Cytotoxicity /in vitro only |
IC50: 32 nM (MOLT-4), 4 μM (MCF-7) | [48,49,50] |
| Hectochlorin (18) | Cyanobacteria: Lyngbya majuscula | Lipopeptide | Hyperpolymerization /in vitro only |
IC50: 20 nM (CA46), 300 nM (PtK2) | [51] |
| Hormothamnin A (19) | Cyanobacteria: Hormothamnion enteromorphoides | Cyclic undecapeptide | Cytotoxicity /in vitro only |
IC50: 0.13–0.72 μg/mL (SW1271, A529, B16-F10, HCT-116)) | [52] |
| Itralamide A (20), B (21) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide |
Antiproliferative activity/in vitro only | IC50: 6 μM (HEK293) | [53] |
| Lagunamide A–C (22–24) | Cyanobacteria: Lyngbya majuscula | Cyclic depsipeptide |
Antiproliferative activities and apoptosis/in vitro only | IC50: 6.4–24.4 nM (P388) | [54,55] |
| Largazole (25) | Cyanobacteria: Symploca sp. | Cyclic depsipeptide |
Stimulation of histone hyperacetylation in the tumor/in vitro only | GI50: 7.7 nM (MDA-MB-231), 122 nM (NMuMG), 55 nM (U2OS), 480 nM (NIH3T3) | [56,57,58,59,60] |
| Laxaphycin A (26), B (27) | Cyanobacteria: Lyngbya majuscula | Cyclic peptide | Antiproliferative activities/in vitro only |
IC50 < 2 μM (CEM-WT) | [61,62,63] |
| Lyngbyabellin A (28), E (29), B (30) | Cyanobacteria: Lyngbya majuscula | Lipopeptides | Cytoskeletal actin disruption/in vitro only | IC50: 0.03–0.1 μg/mL (KB), IC50: 0.5–0.83 μg/mL (LoVo), LC50: 0.4–1.2 μg/mL (NCI-H460, neuro-2a) |
[64,65] |
| Lyngbyastatin 4–7 (31–34) | Cyanobacteria: Lyngbya sp. | Depsipeptide | Porcine pancreatic elastase inhibition /in vitro only |
IC50: 120–210 μM (elastase inhibition) | [66,67] |
| Symplocamide A (35) | Cyanobacteria: Symploca sp. | Cyclic depsipeptide | Proteasome inhibition/in vitro only | IC50: 40 nM (NCI-H460), 29 nM (neuro-2a) | [68] |
| Tasiamide (36), B (37) | Cyanobacteria: Symploca sp. | Linear peptide | Cytotoxicity /in vitro only |
IC50: 0.48 μg/mL (KB), 3.47 μg/mL (LoVo) | [69,70,71] |
| Veraguamide A (38), D (39), E (40) | Cyanobacteria: Oscillatoria margaritifera, Symploca cf. Hydnoides sp. | Cyclic depsipeptide | Cytotoxicity /in vitro only |
LC50: 141 nM (H-460), IC50: 0.5–1.5 μM (HT29, HeLa) | [72,73] |
| Azonazine (41) | Fungus: Aspergillus insulicola | Hexacyclic dipeptide | Cytotoxicity /in vitro only |
IC50 < 15 ng/mL (HCT-116) | [74] |
| Sansalvamide A (42) | Fungus: the genus Fusarium | Cyclic depsipeptide |
Apoptosis and inhibition of topoisomerase I /in vitro only |
IC50: 4.5 μg/mL (HT29) | [75,76] |
| Scopularides A (43), B (44) | Fungus: Scopulariopsis brevicaulis | Cyclic depsipeptide |
Cytotoxicity /in vitro only |
IC50: 10 μg/mL (Colo357, Panc89, HT29) | [77] |
| Arenastatin A (45) | Sponge: Dysidea arenaria | Cyclic depsipeptide |
Inhibition of microtubule assembly /in vitro only |
IC50: 5 pg/mL (KB) | [78,79,80,81,82,83] |
| Discodermin A–H (46–53) | Sponge: Discodermia kiiensis | Tetra- decapeptide |
Membrane permeabilization /in vitro only |
IC50: 0.02–20 μg/mL (P388, A549) | [84] |
| Geodiamolide H (54) | Sponge: Geodia corticostylifera | Cyclic depsipeptide |
Antiproliferative activity /in vitro only |
G100: 18.6 nM (OV Car-4), | [85,86,87] |
| Hemiasterlin (55), A (56), C (57) | Sponge: Auletta sp., Siphonochalina sp. | Linear tripeptide |
Inhibition of tubulin polymerization /Phase I human clinical trial, (HT1286: Phase I) |
IC50: 0.0484–0.269 nM (PC3), 0.404–10.3 nM (NFF), | [88,89,90,91,92,93,94,95] |
| Homophymine A–E (58–62), A1–E1 (63–67) | Sponge: Homophymia sp. |
Cyclic depsipeptide |
Activation of caspase-3 and 7/in vitro only | IC50: 2–100 nM (MCF7/MCF7R, HCT116/HCT15, HL60/HL60R) | [96] |
| Jaspamide (68) | Sponge: Jaspis johnstoni | Cyclic depsipeptide |
Activation of caspase-3, depression of Bcl-2 protein expression /in vitro only |
IC50 : 0.04 ng/mL (P388) | [97,98,99] |
| Koshikamide B (69), F–H (70–72) | Sponge: Theonella sp. | Peptide lactone | Cytotoxicity /in vitro only |
IC50: 0.45–2.3 μM (P388), 5.5–10 μM (HCT-116) | [100,101] |
| Microcionamide A (73), B (74) | Sponge: Clathria (Thalysias) abietina | Cyclic heptapeptide |
Cytotoxicity /in vitro only |
IC50: 125–177 nM (MCF-7), 98–172 μM (SKBR-3) | [102] |
| Orbiculamide A (75) | Sponge: Theonella sp. | Cyclic peptide | Cytotoxicity /in vitro only |
IC50: 4.7 μg/mL (P388) | [103] |
| Papuamide A–F (76–81) | Sponge: Theonella mirabilis, T. swinhoei | Cyclic depsipeptide | Cytotoxicity and inhibition of infection /in vitro only |
IC50: 0.75 ng/mL (human cell line panel) | [83,104,105] |
| Phakellistatin 1 (82), 13 (83) | Sponge: Phakellia carteri | Cyclic heptapeptides | Antiproliferative activity/in vitro only |
ED50: 7.5 ug/mL (P388), IC50: 0.75 ng/mL (BEL-7404) | [106,107,108] |
| Rolloamide A (84) | Sponge: Eurypon laughlini | Cyclic heptapeptides | Tubulin polymerization inhibition/in vitro only |
IC50: 0.4–5.8 μM (SKBR3, A2780) | [109] |
| Scleritodermin A (85) | Sponge: Scleritoderm nodosum | Cyclic peptide | Microtubule assembly inhibition/in vitro only |
IC50 < 2 μM (HCT116, SKBR3, A2780) | [110,111] |
| Aplidin (plitidepsin, 86) |
Tunicate: Aplidium albicans | Cyclic depsipeptide |
Activation of JNK and p38 MAPK/Phase III human clinical trial | IC50: 0.2–27 nM (CFU-GEMM, CFU-GM, BFU-E) | [112,113,114,115,116,117,118,119,120,121] |
| Didemnin B (87) | Tunicate: Trididemnum solidum | Cyclic depsipeptide |
Apoptosis/Phase II human clinical trial |
IC50: 2 ng/mL (L1210) | [122,123,124] |
| Cycloxazoline (88) | Ascidia: Lissoclinum bistratum |
Cyclic hexapeptide |
Apoptosis/in vitro only | IC50: 0.5 μg/mL (MRC5CV1, T24) | [125] |
| Diazonamide A (89) | Ascidia: Diazona angulata | Macrocyclic peptide |
Tubulin polymerization inhibition/in vitro only |
IC50: 2–5 nM (CA46, MCF7, PC3, A549) | [126,127] |
| Mollamide B (90), C (91) | Ascidia: Didemnum molle | Cyclic depsipeptide | Antiproliferative activity/in vitro only |
IC50: 1 μg/mL (P388), 1 μg/mL (A549, HT29) | [128,129,130] |
| Tamandarin A (92), B (93) | Ascidia: Didemnum sp. | Cyclic depsipeptide |
Cytotoxicity /in vitro only |
IC50: 1.79 μg/mL (BX-PC3), 1.36 μg/mL (DU145), 0.99 μg/mL (UMSCC10b) | [131] |
| Trunkamide A (94) | Ascidia: Lissoclinum sp. |
Cyclic peptide | Cytotoxicity /in vitro only |
IC50: 0.5 μg/mL (P388, A549, HT29), 1.0 μg/mL (MEL-28) | [132] |
| Virenamides A–C (95–97) | Ascidia: Diplosoma virens |
Linear tripeptides | Apoptosis /in vitro only |
IC50: 5–10 μg/mL (P388, A549, HT29, CV1) | [133] |
| Vitilevuamide (98) | Ascidia: Didemnum cuculiferum, Polysyncranton lithostrotum |
Bicyclic depsipeptide | Tubulin polymerization inhibition/in vitro only |
IC50: 6–311 nM (P388, A549, HT29) | [122,134] |
| Dolastatin 10 (99), 15 (100) | Mollusk: Dolabella auricularia | Linear peptide | Microtubule assembly inhibition and Bcl-2 phosphorylation /Human clinical trial Dolastatin 10: phase II (TZT-1027: Phase III) Dolastatin 15: preclinical (ILX651: Phase II, LU-103793: Phase I) |
IC50: 50–5000 pM (CA46), 0.5–3 nM (L1210) | [135,136,137,138,139,140,141] |
| Kahalalide F (101) | Mollusk: Elysia rufescens | Depsipeptide | ErbB3 protein and PI3K-Akt pathway involved in necrosis induction, apoptosis /Phase I human clinical trial (Elisidepsin: phase II) |
IC50: 0.162–0.288 μM (colon), 0.135 μM (A549), 0.162 μM (H5578T), 0.479 μM (HS-578T) | [142,143,144,145,146,147,148,149,150,151,152,153,154,155] |
| Keenamide A (102) | Mollusk: Pleurobranchus forskalii | Cyclic hexapeptide | Cytotoxicity /in vitro only |
IC50: 2.5 μg/mL (P388, A549, MEL-20), 5 μg/mL (HT29) | [156] |
| Kulokekahilide-2 (103) | Mollusk: Philinopsis speciosa | Cyclic depsipeptide | Cytotoxicity /in vitro only |
IC50: 4.2–59.1 nM (P388, SKOV-3, MDA-MB-435, A-10) | [157] |
| Ziconotide (104) | Mollusk: Conus magus | Linear peptide | Selective N-type calcium channel blocker/FDA approved | IC50: 100 nM (HEK), 10 nM (IMR32) | [158,159,160,161] |
| Pardaxin (105) | Fish: Pardachirus marmoratus | Linear peptide | Caspase-dependent and ROS-mediated Apoptosis /active in animal |
IC90: 13 μg/mL (NH-11) | [162,163] |
| YALRAH (106) | Fish: Setipinna taty | Linear peptide | Antiproliferative activity/in vitro only |
IC50: 11.1 μM (PC3) | [164] |
3.1. Cyanobacteria-Derived Peptides
Cyanobacteria are regarded as a significant source of various biologically and chemically active natural peptides. Various types of peptides derived from cyanobacteria (such as linear or cyclic peptide, lipopeptides, and linear or cyclic depsipeptides) are able to induce anticancer effects on various animal and human tumor cell lines.
3.1.1. Apratoxin A–D
Apratoxins are cyclic depsipeptides with cytotoxic activity. They were isolated from a variety of Lyngbya sp. found in Papua New Guinea, Palau and Guam (Figure 1) [29,30]. Apratoxins are metabolites of high cytotoxicity and have a novel skeleton composed of peptides and polyketide fragments [31]. Apratoxin A (1) was mainly discovered in the cyanobacterium Lyngbya majuscula. It showed potent cytotoxic activity in human tumor cell lines and showed half-maximal inhibitory concentration (IC50) values ranging from 0.36 nM and 0.52 nM in LoVo colon carcinoma cancer cells and epidermal KB carcinoma cancer cells, respectively [32]. On comparison with other natural analogs, such as apratoxin B (2) and C (3), it was observed to be structurally biologically active due to the N-methylated Ile residue and the hydroxyl group at C-35 [32]. In the case of apratoxin D (4), IC50 value for H-460 lung cancer cells was 2.6 nM, indicating strong in vitro cytotoxicity [33,34].
Figure 1.

Dose-dependent DNA content analysis and gene expression profiling of apratoxin A revealed critical inhibition of cell division, delay in the G1 stage cell cycle arrest, and apoptosis-induced cell death. Apratoxin A was found to inhibit fibroblast growth factor receptor (FGFR) signaling pathway, which is generally associated with cancer [35]. Also, it prevented phosphorylation or activation of signal transducer and activator of transcription 3 (STAT3), a downstream transcriptional effector of FGFR signaling, in the functional genome approach [35]. STAT3 is expressed and activated in a broad range of cancers, which makes it an important anticancer drug target [35]. Because FGF is important for cell proliferation and angiogenesis, the inactivation of STAT3 occurs through angiogenesis mediated by the FGF signaling pathway and initiates the apoptotic cascade [35]. On the basis of proteomic studies, apratoxin A inhibits the N-glycosylation of endoplasmic reticulum receptors, thereby depleting cancer-associated receptor tyrosine kinases only in cancer cells [29].
3.1.2. Aurilides
The aurilides (5–7) are cyclic depsipeptides containing a polyketoide that is part of a macrocyclic carbon skeleton and six amino acid-derived moieties (Figure 2) [36,37]. Aurilide (5) was isolated from the Dolabella auricularia in Japanese sea hare, whereas aurilide B (6) and C (7) were isolated from the oceanic cyanobacterium Lyngbya majuscula in the Papua New Guinea collection [36,37]. Aurilide B and C exhibited in vitro cytotoxicity against NCI-H460 (LC50 of 40 and 130 nM, respectively) and the neuro-2a mouse neuroblastoma cell line (LC50 of 10 and 50 nM, respectively) [37]. The effect of aurilide B on the NCI 60 cell line panel was also analyzed and it was found to exhibit high cytotoxicity with a 50% growth inhibition (GI50) value of less than 10 nM in the leukemia cell line and renal and prostate cancer cell lines [37]. The net tumor cell killing activity of aurilide B was confirmed by the National Cancer Institute (NCI)’s hollow fiber assay for preliminary in vivo screening of novel anticancer drugs.
Figure 2.

Structures of aurilide (5), aurilide B (6), and aurilide C (7) [36,37].
3.1.3. Bisebromoamide
Bisebromoamide (8) is a linear peptide that is a marine toxic substance isolated from Lyngbya sp. in the Okinawan collection (Figure 3) [38]. Bisebromoamide consists of the N-pivaloyl-alanine, N-methyl-3-bromo-tyrosine, 4-methylproline, 2-(1-oxo-propyl)-pyrrolidine and 2-methylcystine, leucine, N-methylphenyl-alanine.
Figure 3.

Structure of bisebromoamide (8) [38].
Bisebromoamide is a strong cytotoxin with an IC50 value of 40 ng/mL in HeLa S3 cells. Also, for a panel of 39 human cancer cell lines (JFCR39), it showed an average GI50 value of 40 nM. Moreover, a wealth of experimental data demonstrated that the extracellular signal-regulated protein kinase (ERK) signaling cascade recognized its targets through interaction with bisebromoamide [38].
3.1.4. Coibamide A
Coibamide A (9) was isolated from Leptolyngbya sp., a Panamanian marine cyanobacterium, which is a cyclic depsipeptide (Figure 4) [39]. Testing of coibamide A in the NCI-H460 lung cancer cells and mouse neuro-2a cells showed strong cytotoxicity with LC50 values of less than 23 nM. Furthermore, coibamide A, a powerful cancer cell toxin with an unique selectivity for the NCI 60 cancer cell line panel, showed significant cytotoxicity against HL-60 human myeloid cells, LOX IMVI human melanoma cell, MDA-MB-231 breast cancer cells and SNB-75 cells with low nM potency. In addition, powerful anti-proliferative activity of the cancer cell was found via a novel target or mechanism of action using COMPARE assays [39].
Figure 4.

Structure of coibamide A (9) [39].
3.1.5. Cryptophycin
Cryptophycin (10) was isolated from the marine cyanobacteria Nostoc sp. ATCC 53789 and GSV 224 and is a depsipeptide with potent antifungal activity (Figure 5) [40]. Cryptophycin bound strongly to the microtubule ends at the vinca-binding site and inhibited the microtubule polymerization. It showed marked cytotoxicity with an IC50 value of less than 50 pM for multidrug-resistant (MDR) tumor cell lines [41].
Figure 5.

Cryptophycin (10) isolated from the cyanobacterium Nostoc sp. [35].
The synthetic derivative of cryptophycin, cryptophycin-52 (LY355703), is produced by total synthesis. Cryptophycin-52 induced apoptosis, which is confirmed through the hyperphosphorylation of Bcl-2, cell cycle arrest, and growth inhibition in preclinical trials for the in vitro human non-small cell lung carcinoma (NSCLC) cell line [42]. A clinical phase II study of cryptophycin-52 revealed the antitumor effect in advanced NSCLC and platinum-resistant advanced ovarian cancer in patients [43,44].
3.1.6. Desmethoxymajusculamide C
Desmethoxymajusculamide C (11) is a cyclic depsipeptide produced by the L. majuscula. It has strong and selective antitumor activity when tested against HCT-116 human colon carcinoma cells as demonstrated by an IC50 of 20 nM and the destruction of cell microfibrils networks (Figure 6) [45]. In the disk diffusion assay, linear desmethoxymajusculamide C maintained powerful actin depolymerization ability as well as solid tumor selectivity [45].
Figure 6.

Structure of desmethoxymajusculamide C (11) [45].
3.1.7. Grassypeptolides
Grassypeptolides A–C (12–16) are cyclic depsipeptides containing bis-thiazoline and were isolated from Lyngbya confervoides [46]. Grassypeptolides also contains an unusual β-amino acid (2-methyl-3-aminobutyric acid), many d-amino acids, and a number of N-methylated amino acids. When the ethyl substituent of Grassypeptolide A (12) (IC50 = 1.22 and 1.01 μM against HT29 and HeLa cancer cell lines, respectively) turned to the methyl group in grassypeptolide B (13), the activity decreases to an extent (3–4-fold; IC50 = 4.97 and 2.93 μM), while the reversal of the Phe unit adjacent to the bis-thiazoline moiety (grassypeptolide C (14)) results in 16–23 fold greater efficacy (IC50 = 76.7 and 44.6 nM) (Figure 7) [46]. Grassypeptolide A and C induce G1 cell cycle arrest at lower concentrations and induce G2/M cell-cycle arrest at higher concentrations in HeLa cells [46].
Figure 7.

Grassypeptolide D (15) and E (16) exhibit potent cytotoxicity against HeLa (IC50 = 335 and 192 nM, respectively) and mouse neuro-2a blastoma cells (IC50 = 599 and 407 nM, respectively) (Figure 7) [47].
3.1.8. Hantupeptin A
Hantupeptin A (17) is a cyclodepsipeptide with a potent cytotoxicity, isolated from Lyngbya majuscula (Figure 8) [48]. Hantupeptin A is a 19-membered cyclic tetrapeptide, which consists of α-amino acids, α-hydroxy acid residue (either phenyl lactic acid, proline, N-methylvaline, valine, or N-methylisoleucine), and an α-methyl-β-hydroxy acid unit with an alkyne at the C-terminal end. The hydroxyl group is attached on the carbon (C-35) of an unusual hydroxy acid, 3-hydroxy-2-methyloctynoic acid [49,50]. Hantupeptin A has shown cytotoxicity with IC50 values of 32 nM and 4.0 μM when tested against MOLT-4 leukemia and MCF-7 breast cancer cells, respectively [49,50].
Figure 8.

Hantupeptin A (17) isolated from cyanobacterium Lyngbya majuscula [48].
3.1.9. Hectochlorin
Hectochlorin (18) was identified from L. majuscula and showed potent activity in promoting actin polymerization (Figure 9) [51].
Figure 9.

Structure of hectochlorin (18) [51].
The pharmacological target for hectochlorin has been proposed as actin microfilaments owing to the cumulation of CA46 cells in the G2/M phase. Subsequently, hectochlorin induced actin polymerization with a half-maximal effective concentration (EC50) value of 20 μM in PtK2 cells. Among the NCI 60 cancer cell lines, 23 cancer cell lines such as colon melanoma, ovarian tumor, and renal cells showed strong cytotoxicity. Hectochlorin (18) showed cytotoxic activity with IC50 values of 20 and 300 nM for CA46 human Burkitt lymphoma cell and PtK2 cells, respectively [51]. However, the dose-response curve of hectochlorin was flat, suggesting that hectochlorin is more antiproliferative than cytotoxic [51].
3.1.10. Hormothamnin A
Hormothamnin A (19) isolated from Hormothamnion enteromorphoides is a cyclic undecapeptide containing six common and five uncommon or novel amino acid residues (Figure 10) [52]. Hormothamnin A (19) was significantly cytotoxic in a variety of solid cancer cell lines, including human lung cell (SW1271 and A529), murine melanoma cell (B16-F10), and human colon cell (HCT-116) [47] with IC50 values ranging from 0.13 to 0.72 μg/mL.
Figure 10.

Hormothamnin A (19) isolated from the cyanobacterium Hormothamnion enteromorphoides [52].
3.1.11. Itralamide A and B
Caribbean sources of L. majuscula yielded two cytotoxic cyclodepsipeptides: itralamide A (20) and B (21) (Figure 11) [53]. Itralamide B (21) showed a higher activity than the former, in addition to displaying moderate activity against human embryonic kidney cells 293 (HEK293, IC50 = 6 μM). Itralamide A (20) is structurally similar to itralamide B, but has a much lower activity [53].
Figure 11.

Itralamide A (20) and B (21) isolated from cyanobacterium Lyngbya majuscula [53].
3.1.12. Lagunamides
The cyclodepsipeptides lagunamide A (22) and B (23) were isolated from the filamentous marine cyanobacterium L. majuscule. These compounds were cytotoxic against P388 murine leukemia cell lines with IC50 values of 6.4–20.5 nM, respectively (Figure 12) [54]. Furthermore, a biochemical analysis using HCT8 and MCF7 cancer cells indicated that lagunamide A and B exhibit cytotoxicity by inducing mitochondria-mediated apoptosis [54].
Figure 12.

Structures of Lagunamide A (22), B (23), and C (24) [54,55].
Lagunamide C (24) exhibited strong cytotoxicity against P388, A549, PC3, HCT8, and SK-OV3 cell lines with IC50 values ranging from 2.1–24.4 nM [55].
3.1.13. Largazole
Largazole (25) is a cytotoxic cyclic depsipeptide isolated from Symploca sp. (Figure 13) [56]. Largazole is a potent growth inhibitor against transformed human mammary epithelial cells (MDA-MB-231, GI50 of 7.7 nM) over non-transformed murine mammary epithelial cells (NMuMG, GI50 of 122 nM) [56]. These molecules also showed strong antiproliferative activity of the transformed U2OS fibroblastic osteosarcoma cells (GI50 = 55 nM). However, it was less active in non-transformed NIH3T3 fibroblasts (GI50 = 480 nM) than in transformed U2OS cells (GI50 = 55 nM). Due to its biological activity and selectivity, several research groups investigated the total chemical synthesis of largazole [57,58,59,60].
Figure 13.

Largazole (25) isolated from cyanobacterium Symploca sp. [56].
3.1.14. Laxaphycin A and B
Laxaphycins (26, 27) are cyclic peptides isolated from Anabaena laxa. Laxaphycins A (26) and B (27) were re-isolated from an assemblage of L. majuscule, together with Anabaena sp. [61,62]. Laxaphycin A is a cyclic undecapeptide containing a 3-aminooctanoic acid residue, and laxaphycin B has a 3-aminodecanoid acid moiety (Figure 14). Laxaphycin A exhibited weak cytotoxicity on a panel of solid cancer cell lines. These include A549 (human lung adenocarcinoma epithelial cell), MCF7 (human breast adenocarcinoma cell), PA1 (human ovarian teratocarcinoma cell), PC3 (human prostate cancer cell), DLD1 (human colorectal adenocarcinoma cell), and M4Beu (human pigmented malignant melanoma) cell lines [63]. Laxaphycins A and B also showed synergistic inhibitory effects on cancer cells in an antifungal assay [63]. Furthermore, laxaphycin B exhibited strong anticancer activity against both sensitive and resistant cancer cell lines [63].
Figure 14.

Structures of laxaphycin A (26) and laxaphycin B (27) [61,62].
The mode of action of laxaphycin B (27) alone may be different from that of laxaphycin A in combination with laxaphycin B (26) [63]. Therefore, laxaphycins A and B could offer new visions into the clinical use of combinatorial drugs in the treatment of cancer.
3.1.15. Lyngbyabellin A, E, and B
Lyngbyabellin A (28) and E (29) isolated from L. majuscula are hectochlorin-related lipopeptides with potent actin polymerization activity, and lyngbyabellin A (28) showed moderate cytotoxicity in KB (IC50 values of 0.03 μg/mL) and LoVo (IC50 = 0.5 μg/mL) cells (Figure 15) [64,65]. It disrupted the cellular actin microfilament network in A10 cells at 0.01–7.2 μM. Furthermore, lyngbyabellin A was fatal to mice when injected at a median lethal dose (LD50) value of 1.2–1.5 mg/kg [64].
Figure 15.

Structures of Lyngbyabellin A (28), E, (29) and B (30) [64,65].
Lyngbyabellin E (29) also exhibited actin polymerization ability at 60 nM; it completely blocked the cellular microfilaments in A10 cells, forming binucleated cells [65]. Furthermore, it showed cytotoxic activity against NCI-H460 (human lung carcinoma cell, LC50 values of 0.4 μM) and neuro-2a (mouse neuroblastoma cell (LC50 = 1.2 μM) [65].
Lyngbyabellin B (30) showed effects on A10 human Burkitt lymphoma cells [64]. Furthermore, the cytotoxic effect of lyngbyabellin B was weaker than that of lyngbyabellin A against KB (IC50 values of 0.1 μg/mL) and LoVo (IC50 = 0.83 μg/mL) cells [64].
3.1.16. Lyngbyastatin 4–7
Many cyclic depsipeptides isolated from marine cyanobacteria show strong inhibition of serine proteases such as pepsin, trypsin, α-chymotrypsin, and elastase. Serine protease is involved in a variety of disease states, including extermination of the extracellular surface of the cartilage covering bones (articular cartilage) in arthritic refractory, emphysema, and inflammatory infections. For this reason, inhibition of serine proteases will be available as a new potential drug target for cancer therapy [66]. Lyngbyastatin 4–7 were depsipeptides isolated from Lyngbya (31–34) (Figure 16) [67].
Figure 16.

Structures of lyngbyastatin 4–7 (31–34) [67].
Lyngbyastatins showed in vitro inhibitory effect against elastase, chymotrypsin, and trypsin. Lyngbyastatins 4–7 are potent inhibitors of elastase with IC50 values between 120 and 210 nM [67].
3.1.17. Symplocamide A
Symplocamide A (35), isolated from Symploca sp. is a cyclodepsipeptide that showed potent cytotoxicity to non-small cell lung cancer cells H-460 (IC50 = 40 nM) and to neuro-2a neuroblastoma cells (IC50 = 29 nM) (Figure 17) [68]. Symplocamide A also inhibited serine proteases, and its inhibitory activity on chymotrypsin was 200-fold higher than trypsin (IC50 = 0.38 μM) [68].
Figure 17.

Structure of Symplocamide A (35) [68].
3.1.18. Tasiamides
Tasiamide (36) and tasiamide B (37) are linear peptides isolated from the cyanobacterium Symploca sp. (Figure 18) [69]. Tasiamide (36) showed strong toxicity to human nasopharyngeal carcinoma (KB) and human colon carcinoma (LoVo) cells, with IC50 values of 0.48 and 3.47 μg/mL, respectively [69].
Figure 18.

Structures of tasiamide (36) and tasiamide B (37) [69,70,71].
Tasiamide B (37) (an octapeptide) contains 4-amino-3-hydroxy-5-phenylpentanoic acid, a unique amino acid moiety, and have cytotoxity against KB cell lines [70]. The synthetic analogues of tasiamide also inhibited KB and non-small cell lung tumor cell lines [71].
3.1.19. Veraguamide A, D, and E
Veraguamides, a family of cyclodepsipeptides, were isolated from Oscillatoria margaritifera (Figure 19) [72]. Veraguamide A (38) was highly toxic in the H-460 human lung cancer cell line (LD50 = 141 nM) [72]. In addition, veraguamides A–G were isolated from Symploca cf. hydnoides. Veraguamides D (39) and E (40) showed cytotoxicity against both HT 29 and HeLa cell lines (IC50 = 0.5–1.5 μM) [73]. The increase in the hydrophobicity of specific units in veraguamides results in their increased activity.
Figure 19.

Veraguamide A (38), D (39), and E (40), isolated from Oscillatoria margaritifera [72,73].
3.2. Fungi-Derived Peptides
Marine fungi produce biologically active and chemically solitary peptides that have offered research opportunities for effective cancer treatments. However, fungi-derived peptides, like sponge-derived peptides, are mostly secondary metabolites that are difficult to isolate. For this reason, the fungi-derived peptides are less studied than other marine peptides [11].
3.2.1. Azonazine
Azonazine (41) is a hexacyclic dipeptide from Aspergillus insulicola, a Hawaiian marine sediment-derived fungus (Figure 20) [74]. Azonazine showed cytotoxicity with IC50 values < 15 ng/mL against HCT-116. This peptide showed anti-inflammatory activity by inhibition of NF-κB luciferase (IC50 = 8.37 μM) and nitrite production (IC50 = 13.70 μM).
Figure 20.
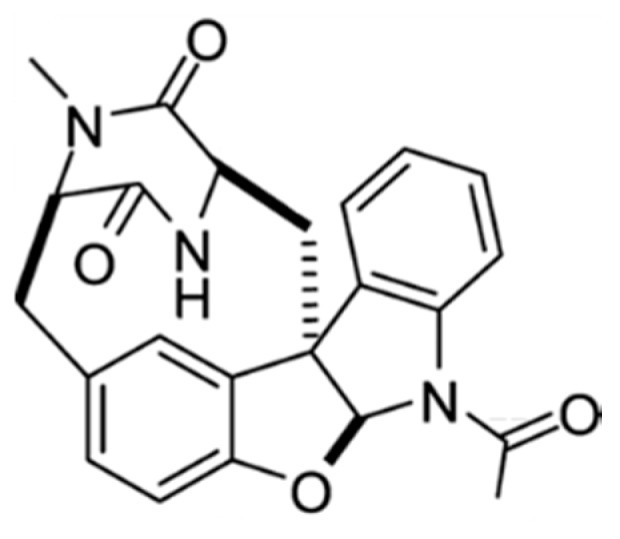
Azonazine (41) isolated from Aspergillus insulicola [74].
3.2.2. Sansalvamide A
The cyclic depsipeptide, Sansalvamide A (42), was isolated from Fusarium sp. living on the marine plant Halodule wrightii (Figure 21) [75]. This peptide showed cytotoxicity against different cell lines such as pancreatic, colon, breast, prostate sarcoma and melanoma cancer cell lines, which suggests it is a potent chemotherapeutic agent (cytotoxicity against the HT29 cell was IC50 = 4.5 μg/mL) [76]. Until now, the exact mechanism of sansalvamide A is unknown. A recent research showed interaction between the HSP90 heat shock protein and client cancer proteins in mammalian cell lines. Sansalvamide A-amid is a synthetic peptide that has similar action to Sansalvamide A [76]. Sansalvamide A-amid was reported to bind to the N-middle domain of HSP90 and allosterically inhibits the formation of protein complex needed to promote tumor growth [76]. In addition, Sansalvamide A-amid caused G1 phase cell cycle arrest in the AsPC-1 and CD18 human pancreatic cancer cell lines. Ssansalvamide A acts as an inhibitor of topoisomerase I by inducing cell death by negligible apoptosis in several cancer cell lines [76].
Figure 21.
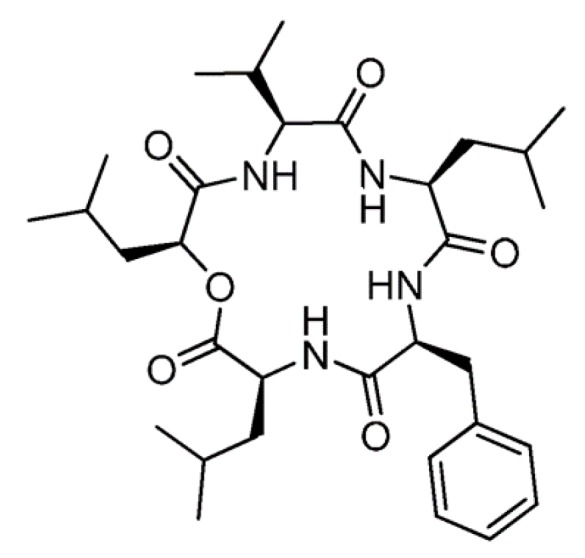
Structure of sansalvamide A (42) [75].
3.2.3. Scopularide A and B
Scopularide A (43) and B (44) are cyclodepsipeptides isolated from the marine fungi Scopulariopsis brevicaulis and the marine sponge Tethya aurantium (Figure 22) [77]. Both compounds have the ability to inhibit the growth of pancreatic and colon tumor cell lines [77]. These peptides have no antimicrobial activity against gram-negative bacteria and have weak activity against gram-positive bacteria [77]. However, the cytotoxicity of various tumor cell lines including the Colo357 and Panc89 pancreatic tumor cell lines and the HT29 colon tumor cell line was confirmed at the concentration of 10 μg/mL (IC50) [77].
Figure 22.

Scopularide A (43) and B (44) isolated from fungi Scopulariopsis brevicaulis [77].
3.3. Sponge-Derived Peptides
Anticancer peptides from sponges are mostly cyclodepsipeptides with unusual amino acids or non-amino acid parts. These sponge-derived peptides showed a broad range of anticancer or antitumor activity [78].
3.3.1. Arenastatin A
Arenastatin A (45), a macrocyclic depsipeptide, was isolated from Dysidea arenaria with extremely strong cytotoxic activity (IC50 = 5 pg/mL) against a human nasopharyngeal carcinoma (KB) cell line (Figure 23) [79]. Arenastatin A inhibits microtubule assembly due to the binding rhizoxin/maytansine sites of tubulin, resulting in cytotoxicity [80,81]. However, arenastatin A is unstable in mouse serum; there is little in vivo antitumor activity. The high sensitivity of the ester linkage to hydrolysis and degradation in blood is a disadvantage [82,83]. The synthesized 15-tert-butyl derivative of arenastatin A overcomes this disadvantage and exhibits improved serum stability as well as in vivo antitumor activity.
Figure 23.

Structure of arenastatin A (45) [79].
3.3.2. Discodermin A–H
Discodermins are cytotoxic tetradecapeptides obtained from Discodermia sp. containing a macrocyclic ring formed by lactonization of a threonine amino acid unit at the C-terminal and 13–14 common and unusal amino acids-linked structures [84].
Discodermin A–H (46–53) are all cytotoxic; tests against murine leukemia cell line (P388), human lung cell line (A549), and the inhibition of starfish development resulted in IC50 values of 0.02 to 20 μg/mL [84]. Among these, discodermin A (46) was shown to permeabilize the plasma membrane of tissues and vascular smooth muscle cells (Figure 24) [84].
Figure 24.

Structure of discodermin A–H (46–53) [84].
3.3.3. Geodiamolide H
Geodiamolides were originally isolated from Geodia sp. as cyclic forms of peptides consisting of three amino acid residues with a common polyketide unit [85,86]. Geodiamolide H (54) is a cyclic depsipeptide isolated from G. corticostylifera, which showed in vitro cytotoxicity against several human cancer cell lines such as ovarian cancer, OV Car-4 (G100 = 18.6 nM). Also, it has antiproliferative activity against the human breast cancer cells by altering the actin cytoskeleton (Figure 25) [87].
Figure 25.
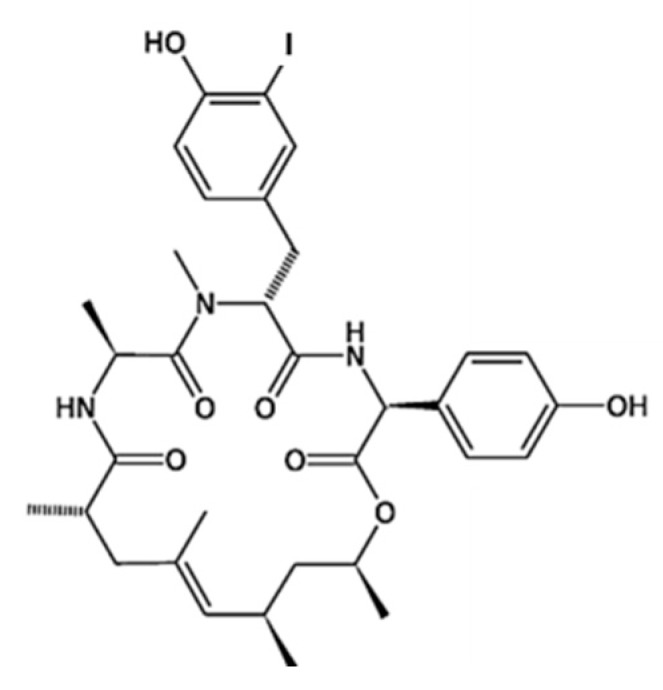
Geodiamolide H (54) isolated from Discodermia sp. [85,86,87].
3.3.4. Hemiasterlin
Hemiasterlin (55), a linear tripeptide consisting of three sterically congested amino acids, was isolated from Hemiasterella minor [88]. In particular, Hemiasterlin, hemiasterlin A and hemiasterlin C (55–57) showed strong cytotoxic activity against the P388 leukemia cell (Figure 26) [88,89,90]. Hemiasterlins show inhibition with IC50 ranges of 0.0484–0.269 nM and 0.404–10.3 nM in PC3 and NFF cells, respectively. They block mitotic spindle formation and induce mitotic arrest and apoptosis, resulting in tubulin depolymerization by binding to the Vinca alkaloid binding site [91]. Hemiasterlin is in phase I clinical trials for patients with solid malignancies [92].
Figure 26.

Hemiasterlin (55), hemiasterlin A (56), and hemiasterlin C (57) isolated from the marine sponge Hemiasterella minor [88,89,90].
HT1286 (also called SPA-110 or taltobulin), a synthetic derivative of hemiasterlin, showed stronger cytotoxicity against human cancer cell lines than hemiasterlin, although both hemiasterlin and HT1286 have a similar mechanism of action. HT1286 has been shown to inhibit the growth of human tumor xenografts in mice, in a preclinical study [93]. The observed side effects, including alopecia, pain, and nausea, resulted in the cessation of HT1286 phase I clinical trials. However, HT1286 has high potential in the treatment of solid tumors in humans. Therefore, the development of new synthetic derivatives with fewer side effects is an important step toward the use of hemiasterlin as an anticancer drug [94,95].
3.3.5. Homophymine A–E and A1–E1
Homophymine A–E (58–62) and A1–E1 (63–67), isolated from Homophymia sp. are cyclodepsipeptides with very strong cytotoxicity against tumor cell lines (Figure 27) [96]. Homophymines (58–67) showed strong cytotoxicity with IC50 values of 2–100 nM against a panel of human cancer cell lines. They exhibited the highest activity against human prostate adenocarcinoma (PC3) and ovarian adenocarcinoma (SK-OV3) cell lines. Additional research on homophymines (58–67) was conducted to determine whether these were toxic or antiproliferative. They have been shown to induce apoptosis on the part of a caspase-independent mechanism and cytotoxicity through non-specific mechanism [96]. Homophymine A1–E1 (63–67) differ from homophymine A–E (58–62) owing to an amide residue instead of the carboxylic acid in the 4-amino-2,3-dihydroxy-1,7-heptanedioic acid residue and exert stronger potency than their corresponding homophymine A–E structures (58–62) [96].
Figure 27.

Structures of homophymine A–E (58–62) and A1–E1 (63–67) [96].
3.3.6. Jaspamide
Jaspamide (Jasplakinolide, 68) was isolated from Jaspis johnstoni as a cyclic depsipeptide with a 15-carbon macrocyclic ring of three amino acid residues (Figure 28) [97].
Figure 28.
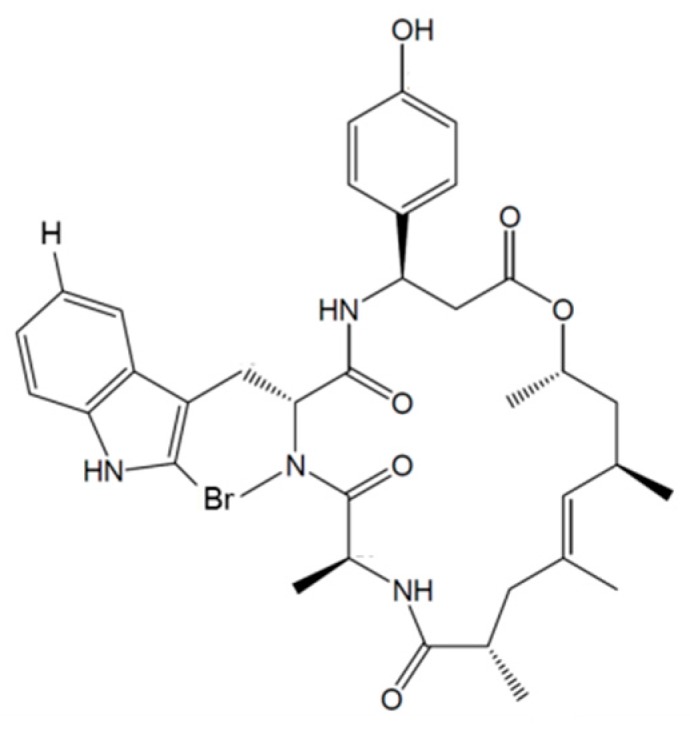
Structure of jaspamide (68) [97].
Jaspamide was found to be highly cytotoxic with an IC50 value of 0.04 ng/mL in P388 murine leukemia cells. It is a bioactive peptide that induces apoptosis in human leukemia cell lines and brain tumor Jurkat T cells by activation of caspase-3 protein expression and decrease in Bcl-2 levels [97,98,99].
In particular, transformed cell lines were more sensitive to jaspamide-induced cell death (apoptosis) than normal non-transformed cells [98].
Apoptosis induced by Jaspamide was associated with caspase-3 activation, decreased Bcl-2 protein expression, and increased Bax levels, suggesting that jaspamide induced a caspase-independent cell death pathway for cytosolic and membrane changes in apoptosis cells, and a caspase-dependent cell death pathway for PARP protein degradation [99].
3.3.7. Koshikamide B and F–H
Koshikamide B (69) has been identified as the major cytotoxic constituent in two separate collections of the Theonella sp. Koshikamide B is a peptide lactone of 17-residue consisting of 6 proteinogenic amino acids, 2 D-isomers of proteinogenic amino acids, 2 unusual amino acids, and 7 N-methylated amino acids (Figure 29) [100]. It was confirmed that the IC50 values for human leukemia cells (P388) and human colon tumor (HCT-116) cell lines were 0.22 and 3.7 μM, respectively [100].
Figure 29.

Koshikamide F–H (70–72) are 17-residue depsipeptides with macrolactone, their IC50 values were inhibited entry from 2.3 to 5.5 μM. (Figure 29) [101]. Koshikamide H (72) was found to have moderate cytotoxicity against the HCT-116 colon cancer cell line with IC50 value of 10 μM. However, koshikamides F–H did not inhibit the growth of Candida albicans, indicating that it is cytotoxic without antifungal activity [101].
3.3.8. Microcionamide A and B
Microcionamide A (73) and B (74) isolated from the Clathria (Thalysias) abietina are linear peptides cyclized via cyclic hexapeptides. Microcionamide A (A) and B (B) showed significant cytotoxicity with IC50 ranges of 125 (A) and 177 nM (B) and 98 (A) and 172 nM (B) in MCF-7 and SKBR-3 cells, respectively (Figure 30) [102]. In addition, these compounds were found to induce apoptosis in MCF-7 cells within 24 h at 5.7 μM. Microcionamides A and B showed cytotoxicity for Mycobacterium tuberculosis H37Ra with MIC values of 5.7 μM [102].
Figure 30.

Structures of microcionamide A (73) and B (74) [102].
3.3.9. Orbiculamide A
Orbiculamide A (75) is a cyclic peptide from Theonella sp. with reported cytotoxicity in P388 murine leukemia cells (IC50 = 4.7 μg/mL) and several other melanoma cell lines (Figure 31) [103].
Figure 31.

Structure of orbiculamide A (75) [103].
3.3.10. Papuamides
Papuamide A–D (76–79), cyclic depsipeptides isolated from Theonella mirabilis and T. swinhoei, are known as the first marine-derived peptides to effectively inhibit the infection of human T-lymphoblastoid cells by HIV-1 in vitro (Figure 32) [83,104]. Especially, papuamides A (76) and B (77) blocked the infection with an EC50 value of about 4 ng/mL and papuamide A has an average IC50 of 75 ng/mL, indicating cytotoxicity against a panel of human cancer cell lines. Papuamide E (80) and F (81) were isolated from Melophlus and LD50 values for brine shrimp were 104 and 106 μg/mL, respectively, confirming cytotoxicity [105].
Figure 32.

3.3.11. Phakellistatins
Proline rich-cyclic heptapeptides, Phakellistatins, are isolated from Phakellia carteri and these compounds inhibit leukemia cell growth (Figure 33) [106]. Phakellistatin 1 (82) significantly inhibited growth of the P388 lymphocytic leukemia (50% effective dose, ED50 = 7.5 μg/mL) and other different melanoma cell lines [107]. Phakellistatin 13 (83) isolated from Phakellia fusca showed cytotoxicity against the BEL-7404 human hepatoma cell line (IC50 < 10 ng/mL). When synthetic phakellistatins were tested, they were inactive, unlike natural products. This may be explained by conformational differences, especially around the proline residue [108]. In other words, the biological properties of synthetic products were found to be significantly different from the products isolated from nature.
Figure 33.

Structures of phakellistatin 1 (82) and 13 (83) [106].
3.3.12. Rolloamide A and B
Rolloamide A (84) and B are two cyclic heptapeptides contained in the Dominican sponge Eurypon laughlini. Rolloamide A (84) showed antiproliferative activity against a panel of histologically diverse cancer cells and showed moderate activity in prostate (LNCap and DU145), breast (MDA468, MDA435 and so on), ovarian (OVCAE3 and SKOV3) and diverse cell lines with IC50 value range from 0.4 to 5.8 μM, while rolloamide B was largely inactive (Figure 34) [109]. Rolloamide B has been shown to have potent activity against several gram-negative bacteria along with high antifungal potency [109].
Figure 34.
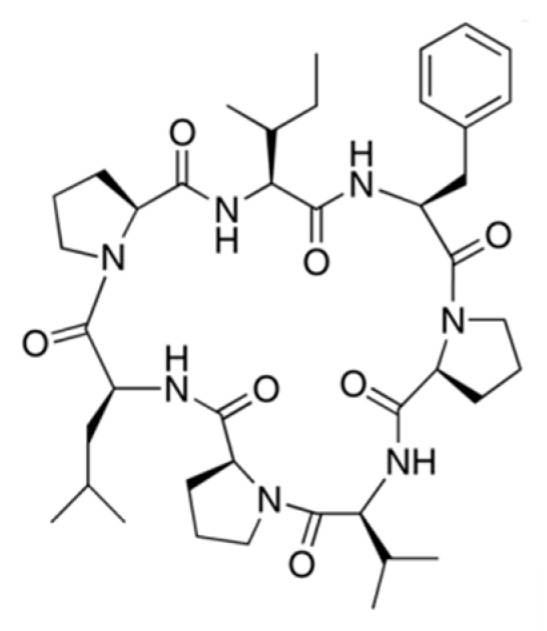
Rolloamide A (84) isolated from the Dominican sponge Eurypon laughlini [109].
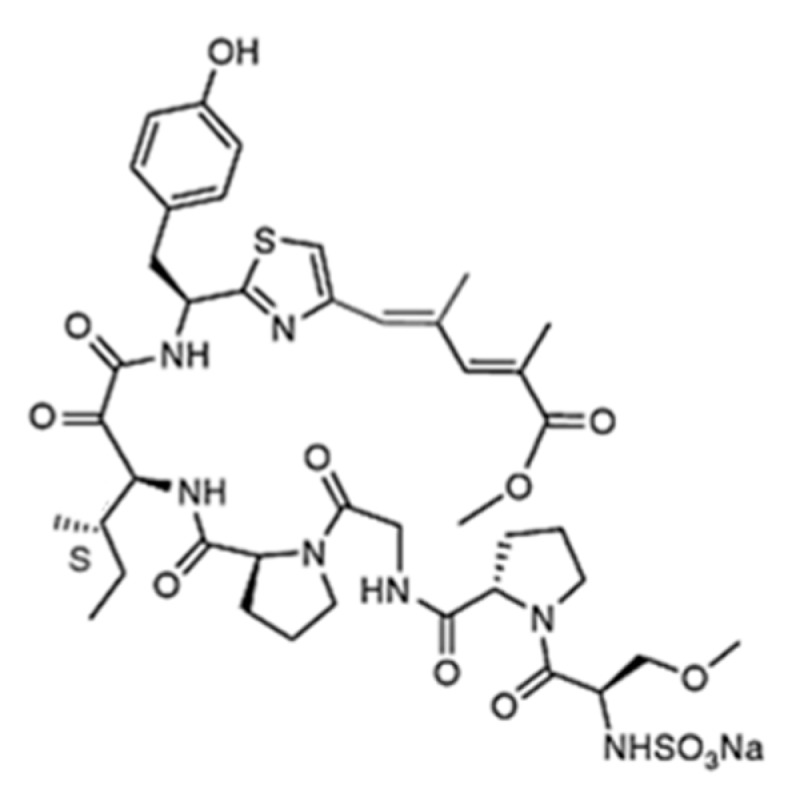
3.3.13. Scleritodermin A
Scleritodermin A (85) isolated from Scleritoderma nodosum is a cyclic peptide and showed in vitro cytotoxicity against human tumor cell lines, including HCT116 colon carcinoma (IC50 = 1.92 μM), A2780 ovarian carcinoma (IC50 = 0.94 μM), and SKBR3 breast carcinoma (IC50 = 0.97 μM) (Figure 35) [110,111]. Scleritodermin A has a structure that combines the keto-Ile-Pro-Ser-Pro-SerOMe portion and the conjugated thiazole moiety. Scleritodermin A (85) has been observed to potently inhibit tubulin polymerization in different cancer cells because of the cell cycle arrest in the G2/M phase [110,111].
Figure 35.
3.4. Tunicate and Ascidia-Derived Peptides
Marine tunicates and ascidiae produce many biologically active peptides; they produce more antitumor and anticancer peptides than any other group of marine organisms [7,11].
3.4.1. Aplidin
Aplidin, the trade name for dehydrodidemnin B/plitidepsin (86), is a cyclic depsipeptide isolated from the ascidian Aplidium albicans (Figure 36) [112]. It has shown strong anticancer activity against a variety of human cancer cell lines including melanoma, breast and lung [113,114,115]. The toxicity of aplidine in normal hematopoietic tissue (IC50 = 150–2250 nM) was lower than that in tumor cells (IC50 = 0.2–27 nM).
Figure 36.

Structure of aplidin (86) [112].
The mechanism of action of aplidin involves cell cycle arrest at the G1-G2 phase, inhibition of protein synthesis, and induction of cancer cell death through induction of apoptosis [115]. Aplidin (86) induced early oxidative stress and then, the activation of both JNK and p38 mitogen-activated protein kinases (MAPK) occurred rapidly and steadily. Phosphorylation by the activation of both kinases occurred rapidly and fully in the drug treatment of HeLa human tumor cells. Activation of JNK and p38 MAPK resulted in downstream of cytochrome c release, upstream of caspase-9 and caspase-3 activation, and PARP cleavage, indicating the strong suppressor of the mitochondrial apoptosis pathway [115,116]. The two mechanisms (apoptosis via caspase-dependent and -independent mechanisms) by which aplidin activated JNK are rapid induction of Rac1 small GTPase and downregulation of MKP-1 phosphatase. Furthermore, aplidin induced other kinases such as the epidermal growth factor receptor (EGFR), the non-receptor protein-tyrosine kinase Src, and the serine/threonine kinases JNK and p38 MAPK in the MDA-MB-231 breast cancer cells.
The availability of the natural marine resource of aplidin is limited due to the difficulty of collecting A. albicans. The limited availability has led to the development of the synthetic analogues.
In patients with advanced melanoma, multiple myeloma, non-Hodgkin’s lymphoma, advanced medullary thyroid carcinoma, or urothelium carcinoma, relevant anticancer activity of plitidepsin has been shown in Phase III clinical studies [117,118,119,120].
Aplidin also inhibited the expression of the vascular endothelial growth factor gene, resulting in biological effects such as angiogenesis, hematopoiesis, and vascular homeostasis [121]. However, aplidin was more effective than didemnin B in preclinical models and, to date, there is no evidence of toxicity to life-threatening neuromuscular disease [112,115].
3.4.2. Didemnin B
Some peptides derived from natural marine resource are shown to induce apoptosis through various mechanisms including cell membrane blebbing, nuclear condensation, and DNA fragmentation. However, the actual mechanism of action for this cytotoxic activity is still unclear. The cycle depsipeptides didemnins were first isolated from Trididemnum solidum, which showed strong antitumor, immunosuppressive, and antiviral properties [8,83].
Didemnin B (87) showed the highest anticancer activity against human prostatic cancer cell lines (IC50 = 2 ng/mL in L1210 leukemia cell) and antiproliferative activity against P388 leukemia as well as B16 melanoma (Figure 37) [83]. For this reason, didemnin B (87) became the first ascidiacea cytotoxin to enter into clinical trials as a potent anticancer drug [122].
Figure 37.

Structure of didemnin B (87) [123].
Didemnin B has shown impressive anti-cancer activity via the inhibition of protein synthesis [123]. Didemnin B has been approved for clinical use as an anticancer agent, but its use is limited due to toxicity and lack of efficacy during a phase II study and an unclear mechanism of action [124]. Furthermore, its low solubility and a short lifespan led to the termination of didemnin B phase II clinical trials [122]. Dehydrodidemnin B (aplidin), an oxidative derivatve of didemnin B, was isolated from A. albicans and exhibits more potent anticancer activity than didermin B [25]. Currently dehydrodidemnin B is in phase II clinical trials in the USA and Europe [25].
3.4.3. Cycloxazoline
Cycloxazoline (88), a cyclic hexapeptide found from Lissoclinum bistratum, showed potent anti-apoptotic activity in various cancer cell lines but the exact target is presently unknown (Figure 38) [124,125]. The compound displayed cytotoxicity against MRC5CV1 and T24 cells (IC50 = 0.5 μg/mL). Cycloxazoline caused accumulation of HL-60 leukemia cells in the G2/M phase and interference with cytokinesis by phosphorylation of cellular proteins involved in cell-cycle control [125].
Figure 38.
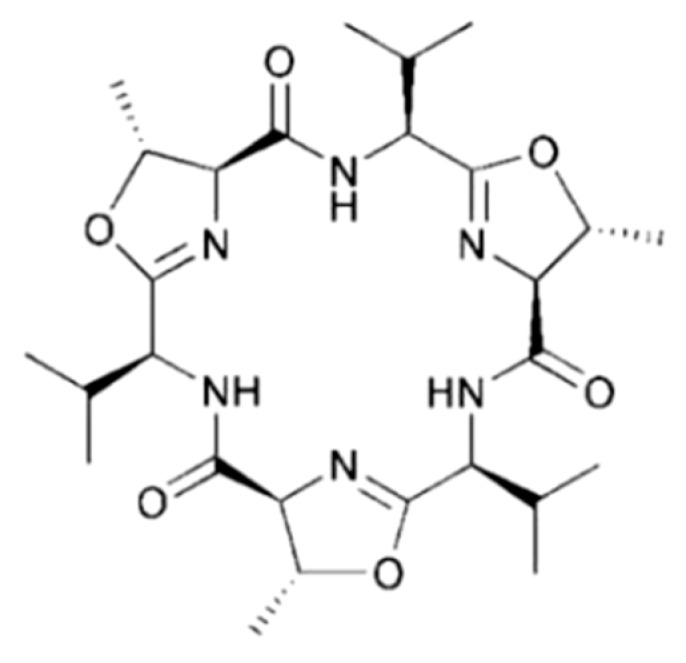
Cycloxazoline (88) isolated from ascidian Lissoclinum bistratum [124,125].
3.4.4. Diazonamide A
Diazonamide A (89), a complex cytotoxic peptide was isolated from Diazona angulate, and displayed potent inhibition of tubulin polymerization in several cancer cells (Figure 39) [126,127]. It showed strong antitumor activity with an IC50 value of 2–5 nM against four human cancer cell lines (CA46, MCF7, PC-3, and A549). Furthermore, diazonamide A has a unique binding site for tubulin that differs from the vinca alkaloid or dolastatin 10 binding sites. So, it weakly inhibits the polymerization of tubulin into strong microtubules ends [126,127].
Figure 39.

Diazonamide A (89) isolated from ascidian Diazona angulata [126,127].
3.4.5. Mollamide B and C
Mollamide from Didemnum molle was cyclodepsipeptide and showed cytotoxicity in P388 murine leukemia cells (IC50 values of 1 μg/mL) and in A549 human lung carcinoma and HT29 human colon carcinoma cells (2.5 μg/mL) (Figure 40) [128,129]. Among these, mollamide B (90) showed significant percent growth inhibition at 100 μM in the H460 non-small cell lung cancer cell, MCF7 breast cancer cell, and SF-268 CNS cancer cell line, but the National Cancer Institute (NCI) 60-cell line panel lacked tumor cell selectivity [130]. In contrast to mollamide B, mollamide C (91) showed strong in vitro cytotoxicity against leukemias (L1210 and CCRF-CEM), solid tumors (38, HCT-116, H125, MCF-7, and LNCaP), and a human normal cell (hematopoietic progenitor cell, CFU-GM); this was observed using a disk diffusion assay. The results showed that the leukemia and normal cells were slightly toxic but showed a larger difference in the solid tumor group [130].
Figure 40.

Mollamides B (90) and C (91) isolated from the ascidian Didemnum molle [128,129].
3.4.6. Tamandarin A and B
Tamandarin A (92) and B (93) are cyclodepsipeptides associated with didemnins known as highly active antiviral, antitumor, and immunosuppressive peptides [131]. Tamandarin A (92) and B (93) were isolated from Trididemnum solidum, Trididemnum cyanophorum, Aplidium albicans, and an unidentified ascidian (family Didemnidae) (Figure 41) [131]. Tamandarin A (92) and B (93) showed strong cytotoxicity against human cancer cell lines, and appeared to be a more potent inhibitor of protein synthesis in comparison with dedermin B [131]. However, their molecular mechanism of action is still unclear. Tamandarin A (92) was found to show cytotoxic activity on three cell lines, BX-PC3 pancreatic carcinoma, DU145 carcinoma, and UMSCC10b head and neck carcinoma with IC50 values of 1.79, 1.36, and 0.99 ng/mL, respectively [131].
Figure 41.

Chemical structure of tamandarin A (92) and tamandarin B (93) [131].
3.4.7. Trunkamide A
Trunkamide A (94) is a cyclopeptide with a triazoline ring similar to mollamide, obtained from Lissoclinum (Figure 42) [132]. Trunkamide A exhibited cytotoxicity with an IC50 of 0.5 μg/mL against P388 mouse lymphoma, A-549 human lung carcinoma, and HT29 human colon cell lines, and 1.0 μg/mL for MEL-28 human melanoma cell lines.
Figure 42.
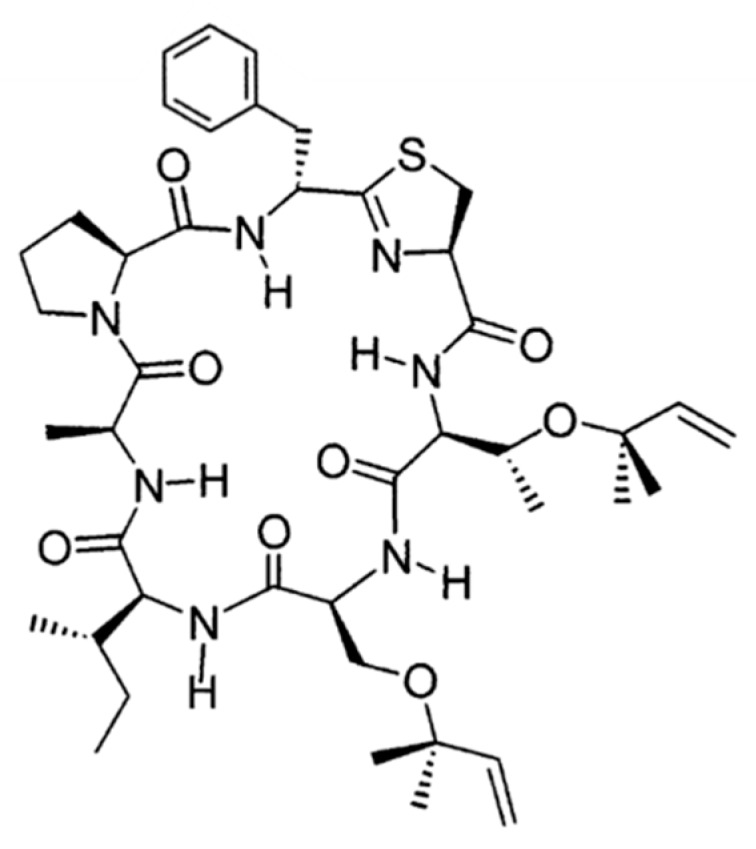
Trunkamide A (94) isolated from ascidians of the genus Lissoclinum [132].
3.4.8. Virenamide A–C
The linear cytotoxic tripeptides virenamide A–C (95–97) were isolated from Diplosoma virens and showed strong anti-apoptotic activity in several cancer cell lines (Figure 43) [133]. Virenamide A (95) had an IC50 of 2.5 μg/mL for P388 and 10 μg/mL for A549, HT29, and CV1 cells, and inhibited topoisomerase II activity. Both virenamide B (96) and C (97) had an IC50 of 5 μg/mL against P388, A549, HT29, and CV1 cells [133].
Figure 43.

Structures of virenamide A–C (95–97) [133].
3.4.9. Vitilevuamide
Vitilevuamide (98), a bicyclic depsipeptide isolated from Didemnum cuculiferum and Polysyncranton lithostrotum, is a tubulin-interactive agent, and potentially positive in a cell-based screening for tubulin polymerization inhibitors of purified tubulin (IC50 = 2.5 μM) (Figure 44) [134]. This demonstrates that vitilevuamide (98) showed non-competitive inhibition of vinblastine binding to tubulin.
Figure 44.

Structure of vitilevuamide (98) [134].
The colchicine binding to tubulin was tested at concentrations up to 80 μM, and was found to be stabilized in the presence of vitilevuamide. It has also been shown to be weakly affected by the presence of GTP-binding in the presence of vitilevuamide, suggesting that vitilevuamide may inhibit tubulin polymerization through interaction at a unique site [134].
Vitilevuamide inhibited the growth of various human cancer cell lines with IC50 values in the range of 6–311 nM, confirming that they were associated with G2/M cell cycle arrest. In mice implanted with P388 lymphocytic leukemia, the intraperitoneal administration of vitilevuamide at doses of 12–30 mg/kg on days 1.5 and 9 after administration resulted in increased life span [122,134].
3.5. Mollusk and Fish-Derived Anticancer Peptides
Marine mollusks and fish-derived peptides are pharmacologically active products and often have activity in cancerous tumors. For this reason, many research groups have been interested in identifying anticancer peptides from fish for many years. However, there are few reports of marine mollusks and fish-derived anticancer peptides compared to other marine resources. Therefore, further research is needed to develop mollusks and fish-derived anticancer peptide and other physiologically active peptides, and it is expected to be a biomass in the future.
3.5.1. Dolastatins
Dolastatins are cytotoxic linear or cyclic peptides isolated from Dolabella auricularia, among the dolastatins, dolastatin 10 (99) and 15 (100) showed the most promising antiproliferative action (Figure 45) [135]. Dolastatin 10 is a linear pentapeptide that contains several unique amino acid residues and is the most potent antiproliferative dolastatin. It inhibited the cell growth of L1210 murine leukemia (IC50 = 0.5 nM) and human bucket lymphoma CA46 cells (IC50 = 50 pM). For this reason, dolastatin 10 (99) has been selected for clinical trials because of its favorable preclinical advantage and is currently on clinical trials in Phase I as an anticancer drug [136].
Figure 45.

Chemical structure of dolastatin 10 (99) and dolastatin 15 (100) [137,138].
The depsipeptide dolastatin 15 (100) inhibited the growth of L1210 cells (IC50 = 3 nM) and CA46 cells (IC50 = 5 nM) [137]. Dolastatin 10 was also extremely potent in vitro. Microtubule assembly and tubulin polymerization were inhibited, causing the accumulation of cells that were arrested in metaphase [138,139]. Dolastatin 10 is a promising candidate with an antineoplastic profile against various cancer cell lines. It has been investigated in various phase I clinical studies, which reported good tolerability and identified bone marrow depression as dose-limiting toxicity. However, dolastatin 10 has been found to cause myelosuppression, peripheral sensory neuropathy, and local irritation at the drug injection site. Structural complexity, low synthetic yield and low water solubility of dolastatins are critical barriers to their broadly clinical evaluation as anticancer drugs [137].
Based on the structural model of dolastatin 10, various analogs were synthesized. Among these analogs, TZT-1027 is a drug with antivascular activity that depolarizes microtubules and disrupts newly formed tumor vasculature. TZT-1027 was found to possess antiangiogenic activity in chorioallantoic membrane embryo (CAM) and human umbilical vein endothelial cells (HUVECs) in vivo [140]. However, it did not show anticancer activity in a phase II clinical trials in patients with advanced non-small cell lung cancer treated with platinum-based chemotherapy [141].
Dolastatin 15 (100) isolated from D. auricularia has not yet been clinically studied, but water-soluble derivates LU-103793 (cematodin) and ILX651 (synthadotin) were developed as cancer drug candidates for clinical studies. LU-103793 was successful in a phase I clinical trial for the treatment of several cancers. The phase II trial was interrupted by unexpected research results. ILX-651 successfully completed the phase I clinical trial and a phase II trial has been recommended owing to its good tolerability with no cardiotoxicities, unlike LU-103793. ILX651 has finished at the least three rounds of phase II clinical trials in patients with advanced and/or metastatic hormone-refractory prostate cancer [29].
3.5.2. Kahalalides
Kahalalides are peptides isolated from Elysia rufescens. Among the kahalalides, kahalalide F (101) is a depsipeptide [142], displaying both in vitro and in vivo antitumor activity in colon (IC50 = 0.162–0.288 μM), A549 breast (IC50 = 0.135 μM), H5578T non-small cell lung (IC50 = 0.162 μM), and HS-578T-particular prostate cancer (IC50 = 0.479 μM) (Figure 46) [142,143,144].
Figure 46.

Structure of kahalalide F (101) [142].
Kahalalide F exhibits various mechanisms, such as lysosomal membrane, inhibition of ErbB (HER3) receptor tyrosine kinase, induction of oncosis, influence on the cell membrane permeability, and mediation of necrosis-like apoptosis [142,143,144,145,146,147,148]. Furthermore, kahalalide F protects tumor cell growth and spreading by inhibiting the expression of specific genes that trigger DNA replication and cell proliferation [149]. Extensive data indicated that kahalalide F exhibits strong cytotoxicity against lung non-small cell carcinoma (A549), melanoma, androgen-independent prostate cancer, hepatocellular carcinoma (HepG2), colon cancer (LoVo), and breast cancer (SKBR-3, BT474, MCF7, and MDA-MB-231) cell lines [149]. Kahalalide F is currently in phase I clinical trials for advanced malignant melanoma patients [150].
A synthetic cyclic depsipeptide kahalalide F derivative, elisidepsin (PM02734, also known as Irvalec®) has potential antineoplastic activity similar to kahalalide F. Elisidepsin shows anti-proliferative activity in a broad variety of cancer cell types [151,152,153]. The action of elisidepsin appears to be as a result of the induction of oncolysis rather than cell death by apoptosis in cancer cells. Elisidepsin is also in phase II clinical trials on metastatic or advanced gastroesophageal cancer [154]. These results were promising and presented a rational basis for further investigations and clinical trials for cancer treatment [155].
3.5.3. Keenamide A
Keenamide A (102) is a cyclic hexapeptide isolated from Pleurobranchus forskalii, which has been shown to cause antitumor activity through undiscovered mechanisms (Figure 47) [156]. Keenamide A (102) showed anticancer activity against several tumor cell lines such as P-388, A-549, MEL-20 (IC50 = 2.5 μg/mL) and HT29 (5.0 μg/mL). In addition, the anti-malarial activity for D6 and W2 clones of Plasmodium falciparum was tested using keenamide A but showed no significant activity [156].
Figure 47.
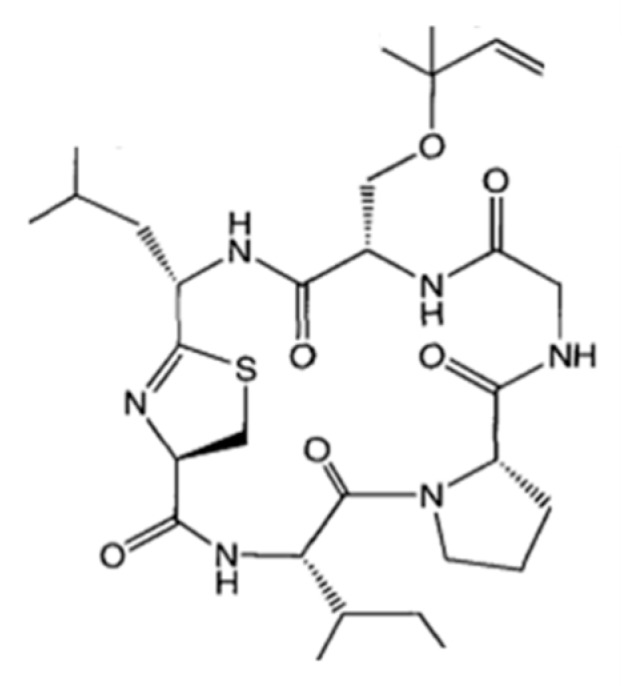
Structure of keenamide A (102) [156].
3.5.4. Kulokekahilide-2
The cyclic depsipeptide, kulokekahilide-2 (103), is the potent aurilide-related substance first isolated from Philinopsis speciosa and it is biosynthesized by marine cyanobacteria owing to its structural similarities with the aurilides (Figure 48) [157]. Kulokekahilide-2 (103) showed strong antitumor activity against various cell lines, such as P388, SKOV-3, MDA-MB-435, and A-10 (IC50 = 4.2~59.1 nM) [157].
Figure 48.

Structure of kulokekahilide-2 (103) [157].
3.5.5. Ziconotide
Ziconotide (104) present in the venom of Conus magus, has a structure in which 25 amino acid peptides linked by three disulfide bonds (Figure 49) [158]. Ziconotide has been developed as an atypical analgesic to relieve severe and chronic pain by acting as an optional N-type voltage gated calcium channel blocker. This action has been shown to relieve pain by inhibiting the release of glutamate, calcitonin gene related peptide and pro-invasive neurochemicals such as substance P in the brain and spinal cord [158,159].
Figure 49.

Primary structure of ziconotide (104) [158]. Ziconotide has six cysteine residues, forming three disulfide bonds.
It has been shown to have a 1000-fold higher activity than morphine in animal models of nociceptive pain and has a remarkable analgesic character [158]. Ziconotide (trade name Prialt®) was the first marine peptide to receive FDA approval for analgesic use in 2004 and another marine peptide Brentuximab vedotin (trade name Adcetris®) was approved by the FDA in 2011 for drugs that are effective against cancer [159,160]. Various marine peptides are now entering phases of clinical trials in the United States and Europe [161].
3.5.6. Pardaxin
Pardaxin (105, GFFALIPKIISSPLFKTLLSAVGSALSSSGGQE-NH2) is an antimicrobial peptide consisting of 33 amino acids, isolated from the fish Pardachirus marmoratus [77]. This peptide showed antitumor activity in human fibrosarcoma (HT1080) cells and HeLa cells, which inhibited proliferation in a dose-dependent manner in HT1080 cells and induced programmed cell death in HeLa cells [77]. In HT1080 cells, pardaxin induced apoptosis by causing caspase-dependent and ROS-mediated cell death [162].
Pardaxin plays important roles in the scavenging of reactive oxygen species (ROS) to alleviate c-Jun activation. Small interfering RNA-mediated knockdown of c-Jun requiring ROS and c-Jun in pardaxin-induced apoptosis signaling as it abrogates pardaxin-induced caspase activation and cell death [162].
Pardaxin, unlike HT1080 cells, causes caspase-dependent and caspase-independent apoptosis in human cervical cancer cells. Pardaxin also induces >90% inhibition of colony formation in MN-11 cells derived from MC1A fibrosarcoma in male C57BL/10 mice at a concentration of 13 μg/mL [163]. In addition, pardaxin has a potential veterinary application because it has a lytic action with potent activity against canine perianal gland adenomas [163].
3.5.7. YALRAH
Tyr-Ala-Leu-Pro-Ala-His (106), an anticancer peptide consisting of six residues derived from the half-fin anchovy (Setipinna taty), has been shown to inhibit the proliferation of prostate cancer cells [164]. YALPAH and three analogs (YALRAH, YALPAR and YALPAG) exhibited anti-proliferative activity in PC3 cells. Among them, the modified peptide YALRAH showed the strongest activity (IC50 value of 11.1 μM). It has been confirmed that arginine (R) is an important residue for anticancer activity, but the mechanism for this is still unclear [164].
4. Anticancer Peptide-Based Drug Therapeutics Developed from Marine Organisms and Future Prospects
Many factors are involved in the discovery and development of anticancer drugs from marine natural products. However, there are various reasons that have ensured the use of marine peptides in the search for anticancer drugs. Marine-derived peptides are chemically diverse, have a wide range of therapeutic activities, and are highly specific to cells or tissues. These peptides can act specifically against cancer cells by either membranolytic mechanisms or mitochondrial disruption [165]. Generally, the negative net charge of the cancer membrane is an important factor for peptide selectivity and toxicity, especially relative to the typically zwitterionic properties of normal cell membranes [166]. Amphiphilicity levels and hydrophobic arc size allow the penetration of these peptides through cancerous cell membranes, leading to destabilization of membrane integrity [167,168]. Since they are composed of metabolically and allergenically tolerable amino acids, the risk of undesirable destructive side-reactions is reduced, and they are usually safe and non-toxic compounds. Furthermore, anticancer peptides are also used as vehicles in drug formulations for the improvement of biological properties, targeted drug delivery, or transport through target cell membranes [169]. Several potential anticancer peptides, such as stimuvax, primovax, melanotan, and cilengitide, are in clinical trials [170]. Combination therapy has emerged as an important strategy for fighting cancer in recent years because a single method may not be enough to completely cure the disease and prevent recurrence [159]. For the purpose of synergistic effects, combinations of anti-angiogenic compounds and traditional therapies are currently being investigated in clinical trials and will make anticancer peptide discovery more interesting in the next few years.
Author Contributions
All authors contributed equally to the manuscript preparation and design.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Burgess J.G. New and emerging analytical techniques for marine biotechnology. Curr. Opin. Biotechnol. 2012;23:29–33. doi: 10.1016/j.copbio.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Lin X. Discovery of antitumor agents with peptides from marine sources. JSM Clin. Oncol. Res. 2014;2:1034. [Google Scholar]
- 3.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 4.Suarez-Jimenez G.M., Burgos-Hernandez A., Ezquerra-Brauer J.M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs. 2012;10:963–986. doi: 10.3390/md10050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sithranga Boopathy N., Kathiresan K. Anticancer Drugs from Marine Flora: An Overview. J. Oncol. 2010;2010:214186. doi: 10.1155/2010/214186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruiz-Torres V., Encinar J.A., Herranz-López M., Pérez-Sánchez A., Galiano V., Barrajón-Catalán E., Micol V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules. 2017;22:1037. doi: 10.3390/molecules22071037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordaliza M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007;9:767–776. doi: 10.1007/s12094-007-0138-9. [DOI] [PubMed] [Google Scholar]
- 8.Blunden G. Biologically active compounds from marine organisms. Phytother. Res. 2001;15:89–94. doi: 10.1002/ptr.982. [DOI] [PubMed] [Google Scholar]
- 9.Molina-Guijarro J.M., García C., Macías Á., García-Fernández L.F., Moreno C., Reyes F., Martínez-Leal J.F., Fernández R., Martínez V., Valenzuela C., et al. Elisidepsin interacts directly with glycosylceramides in the plasma membrane of tumor cells to induce necrotic cell death. PLoS ONE. 2015;10:e0140782. doi: 10.1371/journal.pone.0140782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitsiades C.S., Ocio E.M., Pandiella A., Maiso P., Gajate C., Garayoa M., Vilanova D., Montero J., Mitsiades N., McMullan C.J., et al. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008;68:5216–5225. doi: 10.1158/0008-5472.CAN-07-5725. [DOI] [PubMed] [Google Scholar]
- 11.Malaker A., Shah A.I.A. Therapeutic potency of anticancer peptides derived from marine organisms. Int. J. Eng. Appl. Sci. 2013;2:53–65. [Google Scholar]
- 12.Riedl S., Zweytick D., Lohner K. Membrane-active host defense peptides-challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids. 2011;164:766–781. doi: 10.1016/j.chemphyslip.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Q.Z., Wang C., Lang L., Zhou Y., Wang H., Shang D.J. Design of potent, non-toxic anticancer peptides based on the structure of the antimicrobial peptide, temporin-1CEa. Arch. Pharm. Res. 2013;36:1302–1310. doi: 10.1007/s12272-013-0112-8. [DOI] [PubMed] [Google Scholar]
- 14.Cheung R.C.F., Ng T.B., Wong J.H. Marine peptides: Bioactivities and applications. Mar. Drugs. 2015;13:4006–4043. doi: 10.3390/md13074006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thajuddin N., Subramanian G. Cyanobacterial biodiversity and potential applications in biotechnology. Curr. Sci. 2005;89:47–57. [Google Scholar]
- 16.Jha R.K., Zi-Rong X. Biomedical compounds from marine organisms. Mar. Drugs. 2004;2:123–146. doi: 10.3390/md203123. [DOI] [Google Scholar]
- 17.Sainis I., Fokas D., Vareli K., Tzakos A.G., Kounnis V., Briasoulis E. Cyanobacterial cyclopeptides as lead compounds to novel targeted Cancer drugs. Mar. Drugs. 2010;8:629–657. doi: 10.3390/md8030629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshmukh S.K., Prakash V., Ranjan N. Marine fungi: A source of potential anticancer compounds. Front. Microbiol. 2018;8:2536. doi: 10.3389/fmicb.2017.02536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cragg G.M., Grothaus P.G., Newman D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 20.Bhatnagar I., Kim S.K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs. 2010;8:2673–2701. doi: 10.3390/md8102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evidente A., Kornienko A., Cimmino A., Andolfi A., Lefranc F., Mathieu V., Kiss R. Fungal metabolites with anticancer activity. Nat. Prod. Rev. 2014;31:617–627. doi: 10.1039/C3NP70078J. [DOI] [PubMed] [Google Scholar]
- 22.Laport M.S., Santos D.C., Muricy G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 2009;10:86–105. doi: 10.2174/138920109787048625. [DOI] [PubMed] [Google Scholar]
- 23.Sagar S., Kaur M., Minneman K.P. Antiviral lead compounds from marine sponges. Mar. Drugs. 2010;8:2619–2638. doi: 10.3390/md8102619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang H.K., Seo C.H., Park Y. Marine peptides and their anti-infective activities. Mar. Drugs. 2015;16:618–654. doi: 10.3390/md13010618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain M.S., Fareed S., Ansari S., Khan M.S. Marine natural products: A lead for anti-cancer. Indian J. Geo-Mar. Sci. 2012;41:27–39. [Google Scholar]
- 26.Chakraborty S., Ghosh U. Oceans: A store of house of drugs-A review. J. Pharm. Res. 2010;3:1293–1296. [Google Scholar]
- 27.Venkatesan J., Anil S., Kim S.K., Shim M.S. Marine fish proteins and peptides for cosmeceuticals: A review. Mar. Drugs. 2017;15:143. doi: 10.3390/md15050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khora S.S. Marine fish-derived bioactive peptides and proteins for human therapeutics. Int. J. Pharm. Pharm. Sci. 2013;5:31–37. [Google Scholar]
- 29.Tan L.T. Filamentous tropical marine cyanobacteria: A rich source of natural products for anticancer drug discovery. J. Appl. Phycol. 2010;22:659–676. doi: 10.1007/s10811-010-9506-x. [DOI] [Google Scholar]
- 30.Ma D., Zou B., Cai G., Hu X., Liu J.O. Total synthesis of the cyclodepsipeptide apratoxin a and its analogues and assessment of their biological activities. Chemistry. 2006;12:7615–7626. doi: 10.1002/chem.200600599. [DOI] [PubMed] [Google Scholar]
- 31.Nagarajan M., Maruthanayagam V., Sundararaman M. A review of pharmacological and toxicological potentials of marine cyanobacterial metabolites. J. Appl. Toxicol. 2012;32:153–185. doi: 10.1002/jat.1717. [DOI] [PubMed] [Google Scholar]
- 32.Luesch H., Yoshida W.Y., Moore R.E., Paul V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002;10:1973–1978. doi: 10.1016/S0968-0896(02)00014-7. [DOI] [PubMed] [Google Scholar]
- 33.Gutierrez M., Suyama T.L., Engene N., Wingerd J.S., Matainaho T., Gerwick W.H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from papua new guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008;71:1099–1103. doi: 10.1021/np800121a. [DOI] [PubMed] [Google Scholar]
- 34.Liu L., Rein K.S. New peptides isolated from Lyngbya species: A review. Mar. Drugs. 2010;8:1817–1837. doi: 10.3390/md8061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leusch H., Chanda S.K., Raya R.M., deJesus P.D., Orth A.P., Walker J.R., Belmont J.C.I., Schultz P.G. A functional genomics approach to the mode of action of Apratoxin A. Nat. Chem. Biol. 2006;2:158–166. doi: 10.1038/nchembio769. [DOI] [PubMed] [Google Scholar]
- 36.Suenaga K., Mutou T., Shibata T., Itoh T., Fujita T., Takada N., Hayamizu K., Takagi M., Irifune T., Kigoshi H., et al. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron. 2004;60:8509–8527. doi: 10.1016/j.tet.2004.06.125. [DOI] [Google Scholar]
- 37.Han B., Gross H., Goeger D.E., Mooberry S.L., Gerwick W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006;69:572–575. doi: 10.1021/np0503911. [DOI] [PubMed] [Google Scholar]
- 38.Teruya T., Sasaki H., Fukazawa H., Suenaga K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009;11:5062–5065. doi: 10.1021/ol9020546. [DOI] [PubMed] [Google Scholar]
- 39.Medina R.A., Goeger D.E., Hills P., Mooberry S.L., Huang N., Romero L.I., Ortega-Barria E., Gerwick W.H., McPhail K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008;130:6324–6325. doi: 10.1021/ja801383f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz R., Hirsch C., Sesin D., Flor J., Chartrain M., Fromtling R., Harris G., Salvatore M., Liesch J., Yudin K. Pharmaceuticals from cultured algae. J. Ind. Microbiol. 1990;5:113–123. doi: 10.1007/BF01573860. [DOI] [Google Scholar]
- 41.Smith C.D., Zhang X., Mooberry S.L., Patterson G.M., Moore R.E. Cryptophycin: A new antimicrotubule agent active against drug-resistant cells. Cancer Res. 1994;54:3779–3784. [PubMed] [Google Scholar]
- 42.Lu K., Dempsey J., Schultz R.M., Shih C., Teicher B.A. Cryptophycin-induced hyperphosphorylation of Bcl-2, cell cycle arrest and growth inhibition in human H460 NSCLC cells. Cancer Chemother. Pharmacol. 2001;47:170–178. doi: 10.1007/s002800000210. [DOI] [PubMed] [Google Scholar]
- 43.D’Agostino G., del Campo J., Mellado B., Izquierdo M.A., Minarik T., Cirri L., Marini L., Perez-Gracia J.L., Scambia G. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. Int. J. Gynecol. Cancer. 2006;16:71–76. doi: 10.1111/j.1525-1438.2006.00276.x. [DOI] [PubMed] [Google Scholar]
- 44.Edelman M.J., Gandara D.R., Hausner P., Israel V., Thornton D., DeSanto J., Doyle L.A. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer. 2003;39:197–199. doi: 10.1016/S0169-5002(02)00511-1. [DOI] [PubMed] [Google Scholar]
- 45.Simmons T.L., Nogle L.M., Media J., Valeriote F.A., Mooberry S.L., Gerwick W.H. Desmethoxymajusculamide C, a cyanobacterial depsipeptide with potent cytotoxicity in both cyclic and ring-opened forms. J. Nat. Prod. 2009;72:1011–1016. doi: 10.1021/np9001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwan J.C., Ratnayake R., Abboud K.A., Paul V.J., Luesch H. Grassypeptolides A-C, cytotoxic bis-thiazoline containing marine cyclodepsipeptides. J. Org. Chem. 2010;75:8012–8023. doi: 10.1021/jo1013564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thornburg C.C., Thimmaiah M., Shaala L.A., Hau A.M., Malmo J.M., Ishmael J.E., Youssef D.T., McPhail K.L. Cyclic depsipeptides, grassypeptolides D and E and Ibu-epidemethoxylyngbyastatin 3, from a Red Sea Leptolyngbya cyanobacterium. J. Nat. Prod. 2011;74:1677–1685. doi: 10.1021/np200270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tripathi A., Puddick J., Prinsep M.R., Lee P.P.F., Tan L.T. Hantupeptin A, a cytotoxic cyclic depsipeptide from a Singapore collection of Lyngbya majuscula. J. Nat. Prod. 2009;72:29–32. doi: 10.1021/np800448t. [DOI] [PubMed] [Google Scholar]
- 49.Gerwick W.H., Tan L.T., Sitachitta N. Nitrogen-containing metabolites from marine cyanobacteria. In: Cordell G.A., editor. The Alkaloids: Chemistry and Biology. 1st ed. Volume 57. Academic; San Diego, CA, USA: 2001. pp. 75–184. [DOI] [PubMed] [Google Scholar]
- 50.Tan L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 2007;68:954–979. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 51.Marquez B.L., Watts K.S., Yokochi A., Roberts M.A., Verdier-Pinard P., Jimenez J.I., Hamel E., Scheuer P.J., Gerwick W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002;65:866–871. doi: 10.1021/np0106283. [DOI] [PubMed] [Google Scholar]
- 52.Gerwick W.H., Mrozek C., Moghaddam M.F., Agarwal S.K. Novel cytotoxic peptides from the tropical marine cyanobacterium Hormothamnion enteromorphoides. 1. Discovery, isolation and initial chemical and biological characterization of the hormothamnins from wild and cultured material. Experientia. 1989;45:115–121. doi: 10.1007/BF01954842. [DOI] [PubMed] [Google Scholar]
- 53.Jimenez J.I., Vansach T., Yoshida W.Y., Sakamoto B., Porzgen P., Horgen F.D. Halogenated fatty acid amides and cyclic depsipeptides from an eastern Caribbean collection of the cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2009;72:1573–1578. doi: 10.1021/np900173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tripathi A., Puddick J., Prinsep M.R., Rottmann M., Tan L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010;73:1810–1814. doi: 10.1021/np100442x. [DOI] [PubMed] [Google Scholar]
- 55.Tripathi A., Puddick J., Prinsep M.R., Rottmann M., Chan K.P., Chen D.Y., Tan L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry. 2011;72:2369–2375. doi: 10.1016/j.phytochem.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 56.Taori K., Paul V.J., Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 57.Salvador L.A., Park H., Al-Awadhi F.H., Liu Y., Kim B., Zeller S.L., Chen Q.Y., Hong J., Luesch H. Modulation of Activity Profiles for Largazole-Based HDAC Inhibitors through Alteration of Prodrug Properties. ACS Med. Chem. Lett. 2014;5:905–910. doi: 10.1021/ml500170r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeng X., Yin B., Hu Z., Liao C., Liu J., Li S., Li Z., Nicklaus M.C., Zhou G., Jiang S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010;12:1368–1371. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong J., Luesch H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012;29:449–456. doi: 10.1039/c2np00066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y., Salvador L.A., Byeon S., Ying Y., Kwan J.C., Law B.K., Hong J., Luesch H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010;335:351–361. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frankmolle W.P., Knubel G., Moore R.E., Patterson G.M.L. Antifungal cyclic peptides from the terrestrial blue-green alga Anabeana laxa. 2. Structures of laxaphycins A, B, C, D, and E. J. Antibiot. 1992;5:1458–1466. doi: 10.7164/antibiotics.45.1458. [DOI] [PubMed] [Google Scholar]
- 62.Bonnard I., Rolland M., Francisco C., Banaigs B. Total structure and biological properties of laxaphycins A and B, cyclic lipopeptides from the marine cyanobacterium Lyngbya majuscula. Lett. Pept. Sci. 1997;4:289–292. doi: 10.1007/BF02442891. [DOI] [Google Scholar]
- 63.Bonnard I., Rolland M., Salmon J.M., Debiton E., Barthomeuf C., Banaigs B. Total structure and inhibition of tumor cell proliferation of laxaphycins. J. Med. Chem. 2007;50:1266–1279. doi: 10.1021/jm061307x. [DOI] [PubMed] [Google Scholar]
- 64.Luesch H., Yoshida W.Y., Moore R.E., Paul V.J., Mooberry S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000;63:611–615. doi: 10.1021/np990543q. [DOI] [PubMed] [Google Scholar]
- 65.Han B., McPhail K.L., Gross H., Goeger D.E., Mooberry S.L., Gerwick W.H. Isolation and structure of five lyngbyabellin derivatives from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. Tetrahedron. 2005;61:11723–11729. doi: 10.1016/j.tet.2005.09.036. [DOI] [Google Scholar]
- 66.Drag M., Salvesen G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matthew S., Ross C., Rocca J.R., Paul V.J., Luesch H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2007;70:124–127. doi: 10.1021/np060471k. [DOI] [PubMed] [Google Scholar]
- 68.Linington R.G., Edwards D.J., Shuman C.F., McPhail K.L., Matainaho T., Gerwick W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008;71:22–27. doi: 10.1021/np070280x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams P.G., Yoshida W.Y., Moore R.E., Paul V.J. Tasiamide, a cytotoxic peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2002;65:1336–1339. doi: 10.1021/np020184q. [DOI] [PubMed] [Google Scholar]
- 70.Williams P.G., Yoshida W.Y., Moore R.E., Paul V.J. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2003;66:1006–1009. doi: 10.1021/np030114z. [DOI] [PubMed] [Google Scholar]
- 71.Zhang W., Sun T., Ma Z., Li Y. Design, synthesis and biological evaluation of tasiamide analogues as tumor inhibitors. Mar. Drugs. 2014;12:2308–2325. doi: 10.3390/md12042308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mevers E., Liu W.T., Engene N., Mohimani H., Byrum T., Pevzner P.A., Dorrestein P.C., Spadafora C., Gerwick W.H. Cytotoxic veraguamides, alkynyl bromide-containing cyclic depsipeptides from the marine cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod. 2011;74:928–936. doi: 10.1021/np200077f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salvador L.A., Biggs J.S., Paul V.J., Luesch H. Veraguamides A-G, cyclic hexadepsipeptides from a dolastatin 16-producing cyanobacterium Symploca cf. hydnoides from Guam. J. Nat. Prod. 2011;74:917–927. doi: 10.1021/np200076t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Q.X., Crews M.S., Draskovic M., Sohn J., Johnson T.A., Tenney K., Valeriote F.A., Yao X.J., Bjeldanes L.F., Crews P. Azonazine, a novel dipeptide from a Hawaiian marine sediment-derived fungus, Aspergillus insulicola. Org. Lett. 2010;12:4458–4461. doi: 10.1021/ol101396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan P.S., Vasko R.C., Lapera S.A., Johnson V.A., Sellers R.P., Lin C.C., Pan C.M., Davis M.R., Ardi V.C., McAlpine S.R. A comprehensive study of Sansalvamide A derivatives: The structure-activity relationships of 78 derivatives in two pancreatic cancer cell lines. Bioorg. Med. Chem. 2009;17:5806–5825. doi: 10.1016/j.bmc.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vasko R.C., Rodriguez R.A., Cunningham C.N., Ardi V.C., Agard D.A., McAlpine S.R. Mechanistic Studies of Sansalvamide A-Amide: An Allosteric Modulator of Hsp90. ACS Med. Chem. Lett. 2010;1:4–8. doi: 10.1021/ml900003t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Z., Lang G., Kajahn I., Schmaljohann R., Imhoff J.F. Scopularides A and B, cyclodepsipeptides from a marine sponge-derived fungus, Scopulariopsis brevicaulis. J. Nat. Prod. 2008;71:1052–1054. doi: 10.1021/np070580e. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H., Zhao Z., Wang H. Cytotoxic natural products from marine sponge-derived microorganisms. Mar. Drugs. 2017;15:68. doi: 10.3390/md15030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kobayashi M., Aoki S., Ohyabu N., Kurosu M., Wang W., Kitagawa I. Arenastatin A, a potent cytotoxic depsipeptide from the Okinawan marine sponge Dysidea arenaria. Tetrahedron Lett. 1994;35:7969–7972. doi: 10.1016/0040-4039(94)80024-3. [DOI] [Google Scholar]
- 80.Koiso Y., Morita K., Kobayashi M., Wang W., Ohyabu N., Iwasaki S. Effects of arenastatin A and its synthetic analogs on microtubule assembly. Chem. Biol. Interact. 1996;102:183–191. doi: 10.1016/S0009-2797(96)03743-X. [DOI] [PubMed] [Google Scholar]
- 81.Morita K., Koiso Y., Hashimoto Y., Kobayashi M., Wang W., Ohyabu N., Iwasaki S. Interaction of arenastatin A with porcine brain tubulin. Biol. Pharm. Bull. 1997;20:171–174. doi: 10.1248/bpb.20.171. [DOI] [PubMed] [Google Scholar]
- 82.Murakami N., Tamura S., Koyama K., Sugimoto M., Maekawa R., Kobayashi M. New analogue of arenastatin A, a potent cytotoxic spongean depsipeptide, with anti-tumor activity. Bioorg. Med. Chem. Lett. 2004;14:2597–2601. doi: 10.1016/j.bmcl.2004.02.080. [DOI] [PubMed] [Google Scholar]
- 83.Kotoku N., Kato T., Narumi F., Ohtani E., Kamada S., Aoki S., Okada N., Nakagawa S., Kobayashi M. Synthesis of 15,20-triamide analogue with polar substituent on the phenyl ring of arenastatin A, an extremely potent cytotoxic spongean depsipeptide. Bioorg. Med. Chem. 2006;14:7446–7457. doi: 10.1016/j.bmc.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 84.Aneiros A., Garateix A. Bioactive peptides from marine sources: Pharmacological properties and isolation procedures. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004;803:41–53. doi: 10.1016/j.jchromb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 85.Sonnenschein R.N., Farias J.J., Tenney K., Mooberry S.L., Lobkovsky E., Clardy J., Crews P. A further study of the cytotoxic constituents of a milnamide-producing sponge. Org. Lett. 2004;6:779–782. doi: 10.1021/ol036446c. [DOI] [PubMed] [Google Scholar]
- 86.Coleman J.E., van Soest R., Andersen R.J. New geodiamolides from the sponge Cymbastela sp. collected in Papua New Guinea. J. Nat. Prod. 1999;62:1137–1141. doi: 10.1021/np990155o. [DOI] [PubMed] [Google Scholar]
- 87.Freitas V., Rangel M., Bisson L., Jaeger R., Machado-Santelli G. The geodiamolide H, derived from Brazilian sponge Geodia corticostylifera, regulates actin cytoskeleton, migration and invasion of breast cancer cells cultured in three-dimensional environment. J. Cell. Physiol. 2008;216:583–594. doi: 10.1002/jcp.21432. [DOI] [PubMed] [Google Scholar]
- 88.Gamble W.R., Durso N.A., Fuller R.W., Westergaard C.K., Johnson T.R., Sackett D.L., Hamel E., Cardellina J.H., II, Boyd M.R. Cytotoxic and tubulin-interactive hemiasterlins from Auletta sp. and Siphonochalina spp. sponges. Bioorg. Med. Chem. 1999;7:1611–1615. doi: 10.1016/S0968-0896(99)00089-9. [DOI] [PubMed] [Google Scholar]
- 89.Molinski T.F., Dalisay D.S., Lievens S.L., Saludes J.P. Drug development from marine natural products. Nat. Rev. Drug. Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 90.Talpir R., Benayahu Y., Kashman Y., Pannell L., Schleyer M. Hemiasterlin and geodiamolide TA; two new cytotoxic peptides from the marine sponge Hemiasterella minor (Kirkpatrick) Tetrahedron Lett. 1994;35:4453–4456. doi: 10.1016/S0040-4039(00)73382-X. [DOI] [Google Scholar]
- 91.Anderson H.J., Coleman J.E., Andersen R.J., Roberge M. Cytotoxic peptides hemiasterlin, hemiasterlin A and hemiasterlin B induce mitotic arrest and abnormal spindle formation. Cancer Chemother. Pharmacol. 1997;39:223–226. doi: 10.1007/s002800050564. [DOI] [PubMed] [Google Scholar]
- 92.Rocha-Lima C.M., Bayraktar S., Macintyre J., Raez L., Flores A.M., Ferrell A., Rubin E.H., Poplin E.A., Tan A.R., Lucarelli A., et al. A phase 1 trial of E7974 administered on day 1 of a 21-day cycle in patients with advanced solid tumors. Cancer. 2012;118:4262–4270. doi: 10.1002/cncr.27428. [DOI] [PubMed] [Google Scholar]
- 93.Loganzo F., Discafani C.M., Annable T., Beyer C., Musto S., Hari M., Tan X., Hardy C., Hernandez R., Baxter M., et al. HTI-286, a synthetic analogue of the tripeptide hemiasterlin, is a potent antimicrotubule agent that circumvents p-glycoprotein-mediated resistance in vitro and in vivo. Cancer Res. 2003;63:1838–1845. [PubMed] [Google Scholar]
- 94.Hadaschik B.A., Ettinger S., Sowery R.D., Zoubeidi A., Andersen R.J., Roberge M., Gleave M.E. Targeting prostate cancer with HTI-286, a synthetic analog of the marine sponge product hemiasterlin. Int. J. Cancer. 2008;122:2368–2376. doi: 10.1002/ijc.23406. [DOI] [PubMed] [Google Scholar]
- 95.Matsui Y., Hadaschik B.A., Fazli L., Andersen R.J., Gleave M.E., So A.I. Intravesical combination treatment with antisense oligonucleotides targeting heat shock protein-27 and HTI-286 as a novel strategy for high-grade bladder cancer. Mol. Cancer Ther. 2009;8:2402–2411. doi: 10.1158/1535-7163.MCT-09-0148. [DOI] [PubMed] [Google Scholar]
- 96.Zampella A., Sepe V., Luciano P., Bellotta F., Monti M., D’Auria M., Jepsen T., Petek S., Adeline M., Laprevote O., et al. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008;73:5319–5327. doi: 10.1021/jo800583b. [DOI] [PubMed] [Google Scholar]
- 97.Braekman J.C., Daloze D., Moussiaux B., Riccio R. Jaspamide from the marine sponge Jaspis johnstoni. J. Nat. Prod. 1987;50:994–995. doi: 10.1021/np50053a048. [DOI] [Google Scholar]
- 98.Odaka C., Sanders M.L., Crews P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000;7:947–952. doi: 10.1128/CDLI.7.6.947-952.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cioca D.P., Kitano K. Induction of apoptosis and CD10/neutral endopeptidase expression by jaspamide in HL-60 line cells. Cell. Mol. Life Sci. 2002;59:1377–1387. doi: 10.1007/s00018-002-8515-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Araki T., Matsunaga S., Nakao Y., Furihata K., West L., Faulkner D.J., Fusetani N. Koshikamide B, a cytotoxic peptide lactone from a marine sponge Theonella sp. J. Org. Chem. 2008;73:7889–7894. doi: 10.1021/jo801032n. [DOI] [PubMed] [Google Scholar]
- 101.Plaza A., Bifulco G., Masullo M., Lloyd J.R., Keffer J.L., Colin P.L., Hooper J.N.A., Bell L.J., Bewley C.A. Mutremdamide A and koshikamides C–H, peptide inhibitors of HIV-1 entry from different Theonella species. J. Org. Chem. 2010;75:4344–4355. doi: 10.1021/jo100076g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davis R.A., Mangalindan G.C., Bojo Z.P., Antemano R.R., Rodriguez N.O., Concepcion G.P., Samson S.C., de Guzman D., Cruz L.J., Tasdemir D., et al. Microcionamides A and B, Bioactive Peptides from the Philippine Sponge Clathria (Thalysias) abietina. J. Org. Chem. 2004;69:4170–4176. doi: 10.1021/jo040129h. [DOI] [PubMed] [Google Scholar]
- 103.Fusetani N., Sugawara T., Matsunaga S., Hirota H. Orbiculamide A: A novel cytotoxic cyclic peptide from a marine sponge Theonella sp. J. Am. Chem. Soc. 1991;113:7811–7812. doi: 10.1021/ja00020a080. [DOI] [Google Scholar]
- 104.Ford P.W., Gustafson K.R., McKee T.C., Shigematsu N., Maurizi L.K., Pannell L.K., Williams D.E., de Silva E.P., Lassota P., Allen T.M., et al. Papuamides A-D, HIV-inhibitory and cytotoxic depsipeptides from the sponges Theonella mirabilis and Theonella swinhoei collected in Papua New Guinea. J. Am. Chem. Soc. 1999;121:5899–5909. doi: 10.1021/ja990582o. [DOI] [Google Scholar]
- 105.Prasad P., Aalbersberg W., Feussner K.D., Van Wagoner R.M. Papuamides E and F, Cytotoxic Depsipeptides from the Marine Sponge Melophlus sp. Tetrahedron. 2011;67:8529–8531. doi: 10.1016/j.tet.2011.08.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li W.L., Yi Y.H., Wu H.M., Xu Q.Z., Tang H.F., Zhou D.Z., Lin H.W., Wang Z.H. Isolation and structure of the cytotoxic cycloheptapeptide Phakellistatin 13. J. Nat. Prod. 2002;66:146–148. doi: 10.1021/np020223y. [DOI] [PubMed] [Google Scholar]
- 107.Pettit G.R., Cichacz Z., Barkoczy J., Dorsaz A.C., Herald D.L., Williams M.D., Doubek D.L., Schmidt J.M., Tackett L.P., Brune D.C., et al. Isolation and structure of the marine sponge cell growth inhibitory cyclic peptide phakellistatin 1. J. Nat. Prod. 1993;56:260–267. doi: 10.1021/np50092a011. [DOI] [PubMed] [Google Scholar]
- 108.Napolitano A., Rodriquez M., Bruno I., Marzocco S., Autore G., Riccio R., Gomez-Paloma L. Synthesis, structural aspects and cytotoxicity of the natural cyclopeptides yunnanins A, C and phakellistatins 1, 10. Tetrahedron. 2003;59:10203–10211. doi: 10.1016/j.tet.2003.10.073. [DOI] [Google Scholar]
- 109.Williams D.E., Yu K., Behrisch H.W., Van Soest R., Andersen R.J. Rolloamides A and B, cytotoxic cyclic heptapeptides isolated from the Caribbean marine sponge Eurypon laughlini. J. Nat. Prod. 2009;72:1253–1257. doi: 10.1021/np900121m. [DOI] [PubMed] [Google Scholar]
- 110.Schmidt E.W., Raventos-Suarez C., Bifano M., Menendez A.T., Fairchild C.R., Faulkner D.J. Scleritodermin A, a cytotoxic cyclic peptide from the lithistid sponge Scleritoderma nodosum. J. Nat. Prod. 2004;67:475–478. doi: 10.1021/np034035z. [DOI] [PubMed] [Google Scholar]
- 111.Liu S., Cui Y.M., Nan F.J. Total synthesis of the originally proposed and revised structures of scleritodermin A. Org. Lett. 2008;10:3765–3768. doi: 10.1021/ol801419m. [DOI] [PubMed] [Google Scholar]
- 112.Faivre S., Chieze S., Delbaldo C., Ady-Vago N., Guzman C., Lopez-Lazaro L., Lozahic S., Jimeno J., Pico F., Armand J., et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005;23:7871–7880. doi: 10.1200/JCO.2005.09.357. [DOI] [PubMed] [Google Scholar]
- 113.Depenbrock H., Peter R., Faircloth G.T., Manzanares I., Jimeno J., Hanauske A.R. In vitro activity of aplidine, a new marine-derived anti-cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. Br. J. Cancer. 1998;78:739–744. doi: 10.1038/bjc.1998.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Urdiales J.L., Morata P., de Castro I.N., Sánchez-Jiménez F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996;102:31–37. doi: 10.1016/0304-3835(96)04151-1. [DOI] [PubMed] [Google Scholar]
- 115.Garcia-Fernandez L.F., Losada A., Alcaide V., Alvarez A.M., Cuadrado A., Gonzalez L., Nakayama K., Nakayama K.I., Fernandez-Sousa J.M., Munoz A., et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene. 2002;21:7533–7544. doi: 10.1038/sj.onc.1205972. [DOI] [PubMed] [Google Scholar]
- 116.Munoz-Alonso M.J., Gonzalez-Santiago L., Martinez T., Losada A., Galmarini C.M., Munoz A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs. 2009;10:536–542. [PubMed] [Google Scholar]
- 117.Baudin E., Droz J.P., Paz-Ares L., van Oosterom A.T., Cullell-Young M., Schlumberger M. Phase II study of plitidepsin 3-hour infusion every 2 weeks in patients with unresectable advanced medullary thyroid carcinoma. Am. J. Clin. Oncol. 2010;33:83–88. doi: 10.1097/COC.0b013e31819fdf5e. [DOI] [PubMed] [Google Scholar]
- 118.Mateos M.V., Cibeira M.T., Richardson P.G., Prosper F., Oriol A., de la Rubia J., Lahuerta J.J., Garcia-Sanz R., Extremera S., Szyldergemajn S., et al. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010;16:3260–3269. doi: 10.1158/1078-0432.CCR-10-0469. [DOI] [PubMed] [Google Scholar]
- 119.Ribrag V., Caballero D., Fermé C., Zucca E., Arranz R., Briones J., Gisselbrecht C., Salles G., Gianni A.M., Gomez H., et al. Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica. 2013;98:357–363. doi: 10.3324/haematol.2012.069757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dumez H., Gallardo E., Culine S., Galceran J.C., Schoffski P., Droz J.P., Extremera S., Szyldergemajn S., Fléchon A. Phase II study of biweekly plitidepsin as second-line therapy for advanced or metastatic transitional cell carcinoma of the urothelium. Mar. Drugs. 2009;7:451–463. doi: 10.3390/md7030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Broggini M., Marchini S.V., Galliera E., Borsotti P., Taraboletti G., Erba E., Sironi M., Jimeno J., Faircloth G.T., Giavazzi R., et al. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia. 2003;17:52–59. doi: 10.1038/sj.leu.2402788. [DOI] [PubMed] [Google Scholar]
- 122.Newman D.J., Cragg G.M. Advanced preclinical and clinical trials of natural products and related compounds from marine sources. Curr. Med. Chem. 2004;11:1693–1713. doi: 10.2174/0929867043364982. [DOI] [PubMed] [Google Scholar]
- 123.Vera M.D., Joullie M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002;22:102–145. doi: 10.1002/med.10003. [DOI] [PubMed] [Google Scholar]
- 124.Hambley T.W., Hawkins C.J., Lavin M.F., van den Brenk A., Watters D.J. Cycloxazoline: A cytotoxic cyclic hexapeptide from the ascidian Lissoclinum bistratum. Tetrahedron. 1992;48:341–348. doi: 10.1016/S0040-4020(01)88146-1. [DOI] [Google Scholar]
- 125.Watters D.J., Beamish H.J., Marshall K.A., Gardiner R.A., Seymour G.J., Lavin M.F. Accumulation of HL-60 leukemia cells in G2/M and inhibition of cytokinesis caused by two marine compounds, bistratene A and cycloxazoline. Cancer Chemother. Pharmacol. 1994;33:399–409. doi: 10.1007/BF00686269. [DOI] [PubMed] [Google Scholar]
- 126.Cruz-Monserrate Z., Vervoort H.C., Bai R., Newman D.J., Howell S.B., Los G., Mullaney J.T., Williams M.D., Pettit G.R., Fenical W., et al. Diazonamide A and a synthetic structural analog: Disruptive effects on mitosis and cellular microtubules and analysis of their interactions with tubulin. Mol. Pharmacol. 2003;63:1273–1280. doi: 10.1124/mol.63.6.1273. [DOI] [PubMed] [Google Scholar]
- 127.Lachia M., Moody C.J. The synthetic challenge of diazonamide A, a macrocyclic indole bis-oxazole marine natural product. Nat. Prod. Rep. 2008;25:227–253. doi: 10.1039/b705663j. [DOI] [PubMed] [Google Scholar]
- 128.Hamada Y., Shioiri T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem. Rev. 2005;105:4441–4482. doi: 10.1021/cr0406312. [DOI] [PubMed] [Google Scholar]
- 129.Carroll A., Bowden B., Coll J., Hockless D., Skelton B., White A. Studies of Australian ascidians. Mollamide, a cytotoxic cyclic heptapeptide from the compound ascidian Didemnum molle. Aust. J. Chem. 1994;47:61–69. doi: 10.1071/CH9940061. [DOI] [Google Scholar]
- 130.Donia M.S., Wang B., Dunbar D.C., Desai P.V., Patny A., Avery M., Hamann M.T. Mollamides B and C, Cyclic hexapeptides from the indonesian tunicate Didemnum molle. J. Nat. Prod. 2008;71:941–945. doi: 10.1021/np700718p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vervoort H., Fenical W., Epifanio R.A. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000;65:782–792. doi: 10.1021/jo991425a. [DOI] [PubMed] [Google Scholar]
- 132.Wipf P., Miller C., Venkatraman S., Fritch P. Thiolysis of oxazolines-A new, selective method for the direct conversion of peptide oxazolines into thiazolines. Tetrahedron Lett. 1995;36:6395–6398. doi: 10.1016/0040-4039(95)01322-9. [DOI] [Google Scholar]
- 133.Carroll A.R., Feng Y., Bowden B.F., Coll J.C. Studies of Australian Ascidians. 5. Virenamides A–C, New Cytotoxic Linear Peptides from the Colonial Didemnid Ascidian Diplosoma virens. J. Org. Chem. 1996;61:4059–4061. doi: 10.1021/jo951379o. [DOI] [PubMed] [Google Scholar]
- 134.Edler M.C., Fernandez A.M., Lassota P., Ireland C.M., Barrows L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the Vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002;63:707–715. doi: 10.1016/S0006-2952(01)00898-X. [DOI] [PubMed] [Google Scholar]
- 135.Von Angerer E. Tubulin as a target for anticancer drugs. Curr. Opin. Drug Discov. Devel. 2000;3:575–584. [PubMed] [Google Scholar]
- 136.Poncet J. The dolastatins, a family of promising antineoplastic agents. Curr. Pharm. Des. 1999;5:139–162. [PubMed] [Google Scholar]
- 137.Bai R., Friedman S.J., Pettit G.R., Hamel E. Dolastatin-15, a potent antimitotic depsipeptide derived from Dolabella auricularia: Interaction with tubulin and effects on cellular microtubules. Biochem. Pharmacol. 1992;43:2637–2645. doi: 10.1016/0006-2952(92)90153-A. [DOI] [PubMed] [Google Scholar]
- 138.Bai R., Pettit G.R., Hamel E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the Vinca alkaloid binding domain. Biochem. Pharmacol. 1990;39:1941–1949. doi: 10.1016/0006-2952(90)90613-P. [DOI] [PubMed] [Google Scholar]
- 139.Pathak S., Multani A.S., Ozen M., Richardson M.A., Newman R.A. Dolastatin-10 induces polyploidy, telomeric associations and apoptosis in a murine melanoma cell line. Oncol. Rep. 1998;5:373–376. doi: 10.3892/or.5.2.373. [DOI] [PubMed] [Google Scholar]
- 140.Watanabe J., Endo Y., Shimada N., Natsume T., Sasaki T., Kobayashi M. Antiangiogenic activity of TZT-1027 (soblidotin) on chick chorioallantoic membrane and human umbilical vein endothelial cells. In Vivo. 2007;21:297–304. [PubMed] [Google Scholar]
- 141.Riely G.J., Gadgeel S., Rothman I., Saidman B., Sabbath K., Feit K., Kris M.G., Rizvi N.A. A phase 2 study of TZT1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer. 2007;55:181–185. doi: 10.1016/j.lungcan.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 142.García-Rocha M., Bonay P., Avila J. The antitumoral compound Kahalalide F acts on cell lysosomes. Cancer Lett. 1996;99:43–50. doi: 10.1016/0304-3835(95)04036-6. [DOI] [PubMed] [Google Scholar]
- 143.Faircloth G.T., Smith B., Grant W. Selective antitumor activity of Kahalalide F, a marine derived cyclic depsipeptide. Proc. Am. Assoc. Cancer Res. 2001;42:1140. [Google Scholar]
- 144.Rademaker-Lakhai J.M., Horenblas S., Meinhardt W., Stokvis E., de Reijke T.M., Jimeno J.M., Lopez-Lazaro L., Lopez Martin J.A., Beijnen J.H., Schellens J.H. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005;11:1854–1862. doi: 10.1158/1078-0432.CCR-04-1534. [DOI] [PubMed] [Google Scholar]
- 145.Wosikowski K., Schuurhuis D., Johnson K., Paull K.D., Myers T.G., Weinstein J.N., Bates S.E. Identification of epidermal growth factor receptor and c-erbB2 pathway inhibitors by correlation with gene expression patterns. J. Natl. Cancer Inst. 1997;89:1505–1515. doi: 10.1093/jnci/89.20.1505. [DOI] [PubMed] [Google Scholar]
- 146.Suarez Y., Gonzalez L., Cuadrado A., Berciano M., Lafarga M., Munoz A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003;2:863–872. [PubMed] [Google Scholar]
- 147.Sewell J.M., Mayer I., Langdon S.P., Smyth J.F., Jodrell D.I., Guichard S.M. The mechanism of action of kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur. J. Cancer. 2005;41:1637–1644. doi: 10.1016/j.ejca.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 148.Janmaat M.L., Rodriguez J.A., Jimeno J., Kruyt F.A., Giaccone G. Kahalalide F induces necrosis-like cell death that involves depletion of erbB3 and inhibition of akt signaling. Mol. Pharmacol. 2005;68:502–510. doi: 10.1124/mol.105.011361. [DOI] [PubMed] [Google Scholar]
- 149.Martín-Algarra S., Espinosa E., Rubió J., López J.J.L., Manzano J.L., Carrión L.A., Plazaola A., Tanovic A., Paz-Ares L. Phase II study of weekly Kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer. 2009;45:732–735. doi: 10.1016/j.ejca.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 150.Salazar R., Corte´s-Funes H., Casado E., Pardo B., Lo´pez-Martı´n A., Cuadra C., Tabernero J., Coronado C., García M., Soto Matos-Pita A., et al. Phase I study of weekly kahalalide F as prolonged infusionin patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013;72:75–83. doi: 10.1007/s00280-013-2170-5. [DOI] [PubMed] [Google Scholar]
- 151.Serova M., de Gramont A., Bieche I., Riveiro M.E., Galmarini C.M., Aracil M., Jimeno J., Faivre S., Raymond E. Predictive factors of sensitivity to elisidepsin, a novel kahalalide F-derived marine compound. Mar. Drugs. 2013;11:944–959. doi: 10.3390/md11030944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Teixido C., Arguelaguet E., Pons B., Aracil M., Jimeno J., Somoza R., Mares R., Ramon Y.C.S., Hernandez-Losa J. ErbB3 expression predicts sensitivity to elisidepsin treatment: In vitro synergism with cisplatin, paclitaxel and gemcitabine in lung, breast and colon cancer cell lines. Int. J. Oncol. 2012;41:317–324. doi: 10.3892/ijo.2012.1425. [DOI] [PubMed] [Google Scholar]
- 153.Teixido C., Mares R., Aracil M., Ramon y Cajal S., Hernandez-Losa J. Epithelial-mesenchymal transition markers and her3 expression are predictors of elisidepsin treatment response in breast and pancreatic cancer cell lines. PLoS ONE. 2013;8:e53645. doi: 10.1371/journal.pone.0053645. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 154.Petty R., Anthoney A., Metges J.P., Alsin M., Gonçalves A., Brown J., Montagut C., Gunzer K., Laus G., Iglesias-Dios J.L., et al. Phase Ib/II study of elisidepsin in metastatic or advanced gastroesophageal cancer (IMAGE trial) Cancer Chemother. Pharmacol. 2016;77:819–827. doi: 10.1007/s00280-016-2991-0. [DOI] [PubMed] [Google Scholar]
- 155.Ling Y.H., Aracil M., Jimeno J., Perez-Soler R., Zou Y. Molecular pharmacodynamics of PM02734 (elisidepsin) as single agent and in combination with erlotinib; synergistic activity in human non-small cell lung cancer cell lines and xenograft models. Eur. J. Cancer. 2009;45:1855–1864. doi: 10.1016/j.ejca.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wesson K.J., Hamann M.T. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii. J. Nat. Prod. 1996;59:629–631. doi: 10.1021/np960153t. [DOI] [PubMed] [Google Scholar]
- 157.Nakao Y., Yoshida W.Y., Takada Y., Kimura J., Yang L., Mooberry S.L., Scheuer P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004;67:1332–1340. doi: 10.1021/np049949f. [DOI] [PubMed] [Google Scholar]
- 158.Olivera B.M. ω-Conotoxin MVIIA: From Marine Snail Venom to Analgesic Drug. In: Fusetani N., editor. Drugs from the Sea. Karger; Basel, Switzerland: 2000. pp. 74–85. [Google Scholar]
- 159.Olivera B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006;281:31173–31177. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- 160.Minich S.S. Brentuximab vedotin: A new age in the treatment of Hodgkin lymphoma and anaplastic large cell lymphoma. Ann. Pharmacother. 2012;46:377–383. doi: 10.1345/aph.1Q680. [DOI] [PubMed] [Google Scholar]
- 161.Leal M., Sapra P., Hurvitz S.A., Senter P., Wahl A., Schutten M., Shah D.K., Haddish-Berhane N., Kabbarah O. Antibody-drug conjugates: An emerging modality for the treatment of cancer. Ann. N. Y. Acad. Sci. 2014;1321:41–54. doi: 10.1111/nyas.12499. [DOI] [PubMed] [Google Scholar]
- 162.Hsu J.C., Lin L.C., Tzen J.T., Chen J.Y. Pardaxin-induced apoptosis enhances antitumor activity in HeLa cells. Peptides. 2011;32:1110–1116. doi: 10.1016/j.peptides.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 163.Pan C.Y., Lin C.N., Chiou M.T., Yu C.Y., Chen J.Y., Chien C.H. The antimicrobial peptide pardaxin exerts potent anti-tumor activity against canine perianal gland adenoma. Oncotarget. 2015;10:2290–2301. doi: 10.18632/oncotarget.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Song R., Wei R.-B., Luo H.-Y., Yang Z.-S. Isolation and identification of an antiproliferative peptide derived from heated products of peptic hydrolysates of half-fin anchovy (Setipinna taty) J. Funct. Foods. 2014;10:104–111. doi: 10.1016/j.jff.2014.06.010. [DOI] [Google Scholar]
- 165.Harris F., Dennison S.R., Singh J., Phoenix D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013;33:190–234. doi: 10.1002/med.20252. [DOI] [PubMed] [Google Scholar]
- 166.Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009;625:190–194. doi: 10.1016/j.ejphar.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 167.Dennison S.R., Whittaker M., Harris F., Phoenix D.A. Anticancer α-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Curr. Protein Pept. Sci. 2006;7:487–499. doi: 10.2174/138920306779025611. [DOI] [PubMed] [Google Scholar]
- 168.Huang Y.B., Wang X.F., Wang H.Y., Liu Y., Chen Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 2011;10:416–426. doi: 10.1158/1535-7163.MCT-10-0811. [DOI] [PubMed] [Google Scholar]
- 169.Blanco-Míguez A., Gutiérrez-Jácome A., Pérez-Pérez M., Pérez-Rodríguez G., Catalán-García S., Fdez-Riverola F., Lourenço A., Sánchez B. From amino acid sequence to bioactivity: The biomedical potential of antitumor peptides. Protein Sci. 2016;25:1084–1095. doi: 10.1002/pro.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vetter I., Davis J.L., Rash L.D., Anangi R., Mobli M., Alewood P.F., Lewis R.J., King G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids. 2011;40:15–28. doi: 10.1007/s00726-010-0516-4. [DOI] [PubMed] [Google Scholar]