

Abstract
Orthologues and paralogues are types of homologous genes that are related by speciation or duplication, respectively. Orthologous genes are generally assumed to retain equivalent functions in different organisms and to share other key properties. Several recent comparative genomic studies have focused on testing these expectations. Here we discuss the complexity of the evolution of gene–phenotype relationships and assess the validity of the key implications of orthology and paralogy relationships as general statistical trends and guiding principles.
Walter Fitch1,2 introduced the concepts of orthology and paralogy to distinguish between two fundamentally distinct types of homologous relationships between genes according to the mode of descent from their common ancestor. Orthologues (‘ortho’ meaning ‘exact’) are genes that are derived by speciation, whereas paralogues (‘para’ meaning ‘beside’ or ‘next to’) are genes that evolved through duplication. After the advent of comparative genomics in the late 1990s, orthology and paralogy as concepts and terms have pervaded biological research3. Clear delineation of orthologous relationships between genes is obviously indispensable for the reconstruction of the evolution of species and their genomes. Indeed, species phylogenies aim at representing the course of past speciation events, and hence only relationships among orthologous genes are expected to serve that purpose4. Furthermore, orthology is the most accurate way of describing differences and similarities in the composition of genomes from different species, because orthologues by definition trace back to an ancestral gene that was present in a common ancestor of the compared species. Of even more immediate importance to biologists is the common, even if often implicit, reliance on orthology for the transfer of functional information from experimentally characterized genes in model organisms to uncharacterized genes in newly sequenced genomes5–7. The validity of such transfer of functional annotation is predicated on the ‘orthology–function conjecture’: orthologues carry out identical, or more precisely biologically equivalent, functions in different organisms; by contrast, the functions of paralogues typically diverge after duplication6,8. Recently, however, this conjecture has been seriously challenged by a report claiming that paralogues within the same organism are more closely related functionally than are orthologues in different organisms at the same level of divergence9.
Methods for inferring orthology and paralogy relationships from sequence data have been topics of intense research for the past 15 years or so, and there is currently a plethora of practical approaches and databases. The methodological aspects of orthology and paralogy inference, as well as the most popular methods, have been extensively reviewed elsewhere6,10–14. Moreover, a recent international initiative, called the Quest for Orthologs, consolidates the efforts of many research groups in setting common standards and benchmarking various methods for orthology identification15,16. In this Perspective, we do not focus on specific methods but rather investigate the implications of the orthology and paralogy concepts and assess different facets of the ‘generalized orthology conjecture’: that is, the set of key implications of the orthologous and paralogous relationships between genes for sequence, structural and functional similarity. We believe that a reassessment of the entire concept of orthology is timely because, should the orthology conjecture indeed prove to be false9, the implications both for our fundamental understanding of the evolutionary process and for functional annotation of genomes would be dramatic.
Gene homology relationships
The original definition of orthology refers to two modes of divergence from a common ancestral gene and purely rests on evolutionary grounds1,2,6. This definition is simple in principle, but complex combinations of lineage-specific gene duplications, losses and horizontal gene transfer events often give rise to intricate evolutionary scenarios and complicated relationships when considering more than a pair of genes (that is, when multiple paralogues and/or multiple species are involved)6,14,17–19 (BOX 1). This inherent complexity of the evolutionary scenarios drove the adoption of additional definitions to account for particular situations when comparing two or more genomes (BOX 1). In particular, the term co-orthologue indicates that a gene (or several genes) has more than one orthologue in a given genome. The related concept of orthologous groups refers to a set of homologous genes that evolved from a single ancestral gene after a given speciation event20. Orthologous groups thus include orthologues and co-orthologues but also paralogues that evolved by lineage-specific duplication after the relevant speciation event. The terms in-paralogues and out-paralogues were introduced to distinguish between paralogous genes that duplicated, respectively, after or before a given speciation event5,21.
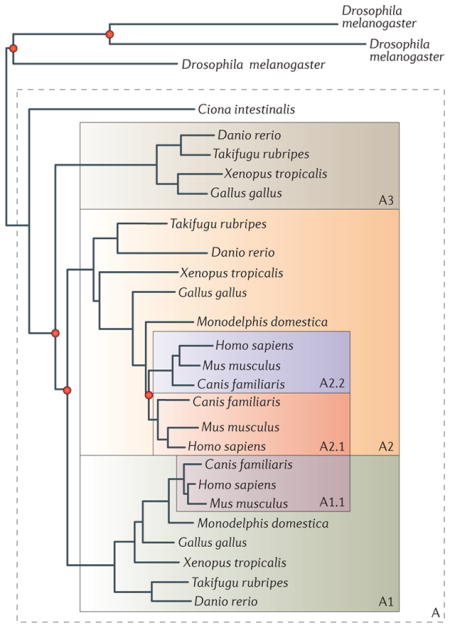
Box 1. Orthology and paralogy.
The evolutionary diversification of gene families through speciation but also through duplication and loss can give rise to intricate scenarios. The figure represents the evolutionary relationships of diverse members of the transferrin family, which is a group of proteins involved in iron transport and homeostasis55. It was derived using information from the Phy0008E70_HUMAN entry in PhylomeDB56 and a previous phylogenetic analysis of this family55. The transferrin family underwent a series of gene duplications (indicated as red nodes in the figure) and gene loss events at different evolutionary times, thus creating distinct subgroups of genes that derived from a single ancestral gene (genes in shaded boxes). Each shaded area encompasses genes that derive from a single ancestral gene in the chordate (A), vertebrate (A1, A2 and A3) or eutherian (A2.1, A2.2 and A1.1) ancestors.
According to the original definition of orthology, the type of homologous relationship between any pair of related genes can be established on the basis of the history of divergence from their common ancestor. In the example shown in the figure, orthologous pairs of genes can be recognized as those for which the last common ancestor is represented as a speciation node (for example, Danio rerio A3 and Gallus gallus A3, or D. rerio A3 and Ciona intestinalis A). Conversely, paralogous genes would trace back to a duplication node as their last common ancestor (for example, D. rerio A3 and D. rerio A1, or D. rerio A3 and G. gallus A2). These straightforward pairwise relationships yield more complex correspondence structures when considering more than a pair of genes or multiple species. For instance, D. rerio A2 has two orthologues (that is, co-orthologues) in humans (Homo sapiens A2.1 and H. sapiens A2.2), constituting a one-to-many relationship. Over a greater evolutionary distance — for example, between H. sapiens and Drosophila melanogaster — even more complicated, many-to-many orthology relationships are observed within this family. The three D. melanogaster genes are co-orthologous to the three H. sapiens genes in the family, but this relationship cannot be specified further because the duplications that yielded the paralogues in each lineage occurred after the speciation event that led to their radiation. D. melanogaster and D. rerio also have a three-to-three orthology relationship, but the correspondence between D. rerio and H. sapiens is not always one-to-one. Instead, the orthology relationships between D. rerio and H. sapiens are one-to-zero (for the A3 subgroup), one-to-two (for the A2 subgroup) and one-to-one (for the A1 subgroup) owing to a mammal-specific gene loss (shown in A3) and a eutherian-specific duplication (shown in A2).
Orthologous groups (that is, groups of genes that descend from a single ancestral gene) are convenient to describe evolutionary relationships across species. Orthologous groups (also known as orthogroups) must be defined in relation to a given ancestral species. Thus, the same family can be subdivided into different orthologous groups (corresponding to subfamilies), depending on the desired level of resolution. In the transferrin family, a single orthologous group (A) is defined if the ancestral chordate is taken as a reference, whereas three orthologous groups (A1, A2 and A3) would have to be considered at the level of vertebrates. A given orthologous group may contain genes that are paralogous to each other as a result of lineage-specific duplications that occurred after the reference ancestral species. Paralogues that emerged from such recent duplications are denoted ‘in-paralogues’, in contrast to ‘out-paralogues’, which emerge from duplications predating the reference ancestor. Considering the vertebrate ancestor as a reference, human A2.1 and human A2.2 are in-paralogues, whereas human A2.1 and human A1.1 are out-paralogues. However, all of these genes would be in-paralogues and members of the same orthologous group (A), if the ancestral chordate was taken as the reference.
This example demonstrates the inherently hierarchichal nature of the orthology and paralogy relationships. Such hierarchical relationships are naturally represented with the bifurcating structure of a phylogenetic tree but can also be projected into simplified units (orthologous groups) if a given ancestral species is chosen as a reference. The choice of the appropriate level of abstraction and resolution depends on the purpose of the study, but the researcher has to be aware of the assumptions and implications of the chosen model.
Orthology beyond the gene
The original definition of orthology and paralogy is genocentric: evolutionary biologists traditionally speak of orthologous and paralogous genes. However, the advances of comparative genomics have made it clear that the exclusive focus on genes as units of evolution is an over-simplification of the actual evolutionary relationships. Differences in domain architectures among proteins encoded by genes that are deemed orthologous or paralogous are common and functionally important22,23, particularly in multicellular eukaryotes, in which domain accretion often leads to lineage-specific, highly complex domain architectures22–24. Furthermore, alternative splicing and alternative transcription, which are pervasive in multicellular eukaryotes25,26, additionally complicate the notions of orthology and paralogy.
These findings seem to require re- conceptualization of orthology, whereby the unit of orthology is changed from a gene (and thereby a protein or a non-coding RNA) to an evolutionarily stable domain or possibly even smaller units6. Between-species differences in domain architectures of homologous proteins can lead to complex networks of relationships that cannot be disambiguated under a genocentric definition of orthology (BOX 2). Moreover, when repetitive, small, promiscuous (that is, occurring in numerous unrelated proteins) domains — such as ankyrin repeats or tetratricopeptide repeats that are extremely common in eukaryotes and some bacteria and viruses — are involved, the concept of orthology seems to break down even with a domain-based definition27. In principle, the correct description of the evolutionary process can be obtained only by tracing the fates of individual nucleotides. Realistically, however, identification in genomes of stable evolving units, to which the concepts of orthology and paralogy most appropriately apply, is becoming one of the key goals of evolutionary genomics.
Box 2. Units of orthology.
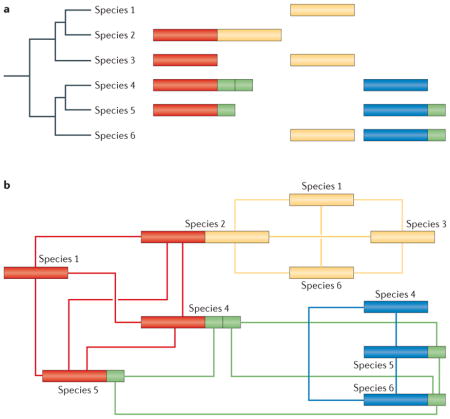
The genocentric definition of orthology becomes problematic when homologous proteins in different species differ in domain architecture. The figure is a schematic illustration of the impact of differences in domain architecture across homologous genes on the definition and identification of orthology. Part a shows four homologous functional domains (red, green, yellow and blue) that are present in some of the six species, sometimes combined into multi-domain proteins. Part b is a network representation of the homology relationships among the different domains in different species. Although all four genes coding for proteins with a red domain could be considered to be orthologues, and this would be the outcome of many prediction methods, the variation in domain composition creates conceptual problems. For example, the gene in species 2 probably evolved by fusion between two genes that are independent in the other species; in species 2, this gene is homologous to the other members of the red group only for the first half of the sequence, whereas the second half is homologous to the genes that encode the yellow domain in the other species. Thus, the gene-based definition of orthology does not apply when genes evolve in pieces. A further problem is presented by promiscuous domains (the green domain in the figure), which combine with many other domains, resulting in intricate networks of homology relationships between parts of proteins. An extra layer of complexity is added by lineage-specific domain duplications (for example, as in the protein containing red and green domains in species 4); this creates a situation in which two segments of a given gene are paralogous. To accommodate such complex situations, orthology and paralogy relationships would have to be determined at the level of the smallest observed evolutionary unit, which may correspond to structural domains, exons or even smaller regions.
The use and abuse of orthology
Despite efforts to capture the complexity of the evolutionary process, the characteristic penchant for simplicity in the descriptions of evolutionary scenarios in non-specialist scientific literature has given rise to widespread misuse and abuse of the orthology concept17,19,28,29. It remains rather common to equate orthology to functional equivalence and thus seek ‘functional orthologues’30,31 or to imply the necessary existence of a single ‘true orthologue’ of a given gene in any species. Here we attempt to clear these misconceptions by examining the genuine and false implications of the original definitions of orthology and paralogy (BOX 3).
Box 3. Genuine and false implications of orthology and paralogy relationships.
Orthologues form a clade (that is, they are monophyletic) in an accurate phylogenetic tree. This is a necessary corollary of the orthology definition (BOX 1).
Orthology does not imply a one-to-one relationship between genes from different organisms. Lineage-specific gene duplications often lead to one-to-many and many-to-many co-orthology relationships (BOX 1).
The molecular clock is not implicit in the definition of orthology: orthologues in different lineages may evolve at different (in principle, arbitrarily different) rates (BOX 1).
Conservation of sequence, structure or genomic context is not implicit in the definition of orthology.
Given the above, orthology does not necessarily imply that orthologous genes (even in the absence of lineage-specific duplications) are the most similar sequences or structures in compared genomes.
The converse is not necessarily true either: genes that are most similar to each other in compared genomes (often denoted bidirectional best hits (BBHs)) might not be orthologous. The BBHs may represent cryptic paralogy after differential loss of ancestral paralogues in compared lineages or xenologues, whereby one of the genes in a BBH pair was acquired by horizontal gene transfer.
Orthology does not necessarily imply conservation of gene function.
The converse is not necessarily true either: genes with equivalent functions are not necessarily orthologous.
All of the above caveats notwithstanding, the generalized orthology conjecture predicts that, as a genome-wide statistical trend, orthologues are the most similar genes in different species, in terms of sequence, structure and function.
Paralogy applies to genes not only within species (as often assumed) but also between species; in cases of differential gene loss and complex evolutionary scenarios, distinguishing orthology and paralogy may be non-trivial (BOX 1).
Paralogy does not necessarily imply functional divergence (as is often assumed): for instance, paralogy may contribute to protein dosage modulation.
Nevertheless, the generalized orthology conjecture implies that, as a general trend, paralogues are more functionally different than orthologues at the same level of sequence divergence.
Sequence conservation
The original definition of orthology focuses only on the mode of evolution from a common ancestral sequence, not on the level of sequence conservation. However, a lower sequence divergence between orthologues as compared to paralogues in the same pair of compared genomes is implied by this definition because the speciation event separating orthologues is inevitably more recent than the duplication events that give rise to out-paralogues in the same two species. This principle is the basis of the popular bidirectional best hit (BBH) approach to orthology identification, which is based on the key assumption that genes in different genomes that are reciprocally the best hit of each other in a sequence similarity search are orthologues20,32. However, violations of the molecular clock — the assumption that orthologous genes evolve at characteristic, gene-specific rates — are common33,34 and can clearly affect the relationships between divergence time and sequence similarity.
Thus, a pertinent and still debated question is whether the expectation that BBHs constitute orthologues — arguably the most straightforward implication of orthology — holds in actual comparative genomic analysis. Indeed, it has been reported that the proteins that show the highest sequence similarity to each other in database searches are often not the closest neighbours in phylogenetic trees: that is, they are not orthologues35. Several benchmarking studies have shown that BBHs are orthologues, and conversely orthologues identified from independent evidence form BBHs in the overwhelming majority of cases, at least at short to moderate evolutionary distances12,36,37. Importantly, however, the BBH approach (at least in its straightforward implementation) fails to capture complex relationships of co-orthology. This aspect of orthologous relationships is highly relevant for comparative genomics because the number of BBHs notably drops with the increase of evolutionary distance between compared genomes37 (FIG. 1); this is apparently to a large extent due to lineage-specific gene loss and non-orthologous gene displacement38–40. In addition, the high prevalence of duplications in the evolutionary histories of genes in complex eukaryotes40,41, together with the fact that the evolutionary rates in duplicated genes can greatly vary 42, adds an extra layer of complexity to the analysis of homologous relationships. In such contexts, the use of more sophisticated methods for orthology and paralogy detection, such as those based on phylogenetic analysis, have a clear advantage over BBH11,13.
Figure 1. Decay of the number of one-to-one orthologues with the increase of the intergenomic evolutionary distance.

Here we use bidirectional best hits (BBHs) as a proxy for one-to-one orthologues. The dependency of the fraction of genes that can be assigned a BBH on the evolutionary distance between genomes is shown for two genomes: the bacterium Escherichia coli K12 (a); and the archaeon Haloarcula marismortui (b). In this analysis, these are termed the ‘master’ genomes. For all proteins encoded in each of the master genomes, a BLASTP search57 was carried out against the protein sequences from 573 representative bacterial and archaeal genomes, and for the most similar proteins (that is, the best hits), a reciprocal BLASTP search was carried out to identify BBHs. The BLASTP score for each BBH was normalized by the self-hit score in the master genome and converted into distance using the formula distance = –ln(score). The distance between the genomes that were compared was estimated as the median distance between BBH pairs. Please see REF. 37 for further details. The figure is modified, with permission, from REF. 37 © (2012) Oxford Univ. Press.
Conservation of protein structure
Directly related to the higher level of sequence conservation among orthologues as compared to paralogues that are diverged to a similar extent at the sequence level is the expectation that orthologues would retain higher levels of similarity at the structural level. Structural conservation can be investigated either at the level of the organization of structural domains along a sequence (that is, the domain architecture) or at the level of the three-dimensional structure of individual domains. Two independent studies have yielded results that are compatible with these expectations. A genome-wide benchmarking analysis showed a significantly greater similarity of domain architectures among orthologous proteins compared with paralogous proteins that are similarly diverged at the sequence level43. However, detailed analysis of domain architectures within orthologous protein sets from diverse organisms reveals a more complex picture22,44. Especially in eukaryotes, orthologous proteins often show differences in domain architectures, and there is a clear trend towards domain accretion in complex, multicellular forms22,44.
Another study directly compared available crystal structures to test whether proteins encoded by orthologous genes were more highly structurally conserved than proteins encoded by similarly diverged paralogous genes45. The results of this analysis again are compatible with the hypothesis, demonstrating that orthologous proteins indeed possess slightly, but consistently and statistically significantly, more similar structures than do paralogous proteins at the same evolutionary distances.
Orthology–function conjecture
The aspect of orthology that is most immediately important for genome researchers and biologists in general is the expectation that orthologous genes are responsible for equivalent functions in different organisms. This orthology–function conjecture (which is a key facet of the generalized orthology conjecture discussed above) constitutes the conceptual basis of functional annotation of sequenced genomes that is essential for experimental biology in the post-genomic era6,17. The strongest form of the orthology conjecture would also encompass the converse hypothesis: namely, that functionally equivalent genes in organisms are orthologous. This proposition has been directly falsified: functional equivalency does not imply orthology. Indeed, many cases of non-orthologous gene displacement were already discovered in the early days of comparative genomics when it was shown that various biological functions, including central ones, such as DNA replication, are carried out by unrelated or at least non-orthologous proteins in evolutionarily distant organisms38,46. A systematic survey of the evolution of enzymes has shown that up to 10% of enzymatic reactions are catalysed by non-homologous and isofunctional enzymes in different organisms47,48. Thus, evolution of independent solutions for the same molecular function is an exception in biology but not a rare one.
The ‘forward’ orthology conjecture — namely, that orthologues carry out equivalent functions, whereas paralogues undergo functional diversification — is often taken more or less for granted. However, anecdotal evidence has been presented that the functional differences between orthologues are in some cases greater than expected and might be about the same as the difference between paralogues at a similar level of sequence divergence8. For example, it has been shown that orthologous transcription factors do not necessarily share specificity49. Moreover, a recent benchmarking study9 unexpectedly has suggested that, at the same level of sequence divergence, orthologous genes are significantly more functionally divergent than are paralogues within the same species. This finding was congruently supported by comparison of the functional annotation of genes according to Gene Ontology (GO) and by comparison of expression profiles across a wide range of animal tissues9.
The iconoclastic conclusion of this study — namely, that intra-organismal environment is of greater importance as a determinant of gene function than is orthology — has attracted broad attention50 and has triggered several independent reanalysis efforts51,52. These studies seem to converge on the conclusion that the perceived greater functional similarity between paralogues can primarily be explained by biases in functional annotation and gene expression measurements within and between species. In particular, it appears that both functional annotations and expression measurement are biased towards a greater similarity in within-species compared with between-species comparisons. When the measurements are carefully controlled, orthologues appear to be more functionally similar than paralogues, albeit not necessarily by a wide margin51,52 (FIG. 2). This difference is more pronounced for the GO category ‘cellular component’ than in, for instance, the ‘biological process’ category, and it is as of yet unclear whether this reflects actual differences in the process of functional adaptation or rather differences in how these two GO categories are structured. Indeed, the ‘biological process’ category has a complex structure with boundaries between terms that are more difficult to assign than the more clear-cut concept of the subcellular localization of a protein. These results based on GO term analyses are in line with earlier analyses showing that orthologues have more similar patterns of tissue expression than across-species paralogues with a similar level of sequence divergence53 and are also in line with a recent study of tissue expression in eight mammalian species that arrived to the same conclusion54. However, this study in mammalian species detected no significant differences in the functional similarity between orthologues and paralogues, presumably owing to the insufficient accuracy of GO annotations54. Clearly, the quality of functional information is a major factor in assessing the validity of the orthology–function conjecture, and decisive tests require substantial improvements over the current models.
Figure 2. Functional divergence versus sequence divergence for orthologues and paralogues.

Plotted is the excess of functional similarity between orthologous pairs of genes as compared to paralogues for different degrees of sequence divergence and for different types of functional ontologies. The data are from REF. 52, and further details of the analysis can be found in that paper. Functional similarities52 are averaged over homologous gene pairs (as predicted by the OMA method) from 13 model species (namely, Homo sapiens, Mus musculus, Rattus norvergicus, Drosophila melanogaster, Danio rerio, Caenorhabditis elegans, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Candida albicans, Dictyostelium discoideum, Arabidopsis thaliana, Escherichia coli and Pseudomonas aeruginosa). They are based on comparisons of Gene Ontology annotations backed by experimental evidence extracted from publications sharing no common authors. In addition, this measure takes into account annotation biases in the different species and other possible confounding factors. Nonsignificant differences between orthologues and paralogues (P value >0.001, using a Mann–Whitney U test) are indicated in grey.
On the whole, the orthology conjecture appears to hold, at least as a statistical trend. Having stated this central conclusion, it is useful to note that another outcome of all these analyses is the rather weak (on average) functional similarity between orthologous genes (FIG. 2). It seems certain that this limited congruence between orthologues depends both on the imprecise methods that are currently used for comparing gene functions in different organisms (with respect to expression analysis and especially to gene ontologies) and on actual differences caused by distinct, organism-specific environments and the general ambiguity of the genotype-to-phenotype mapping. The relative contributions of these factors are expected to be the subject of many future studies.
Conclusions
Orthology and paralogy as concepts and terms are central to comparative and functional genomics owing to their several major, biologically important implications. Some of these implications directly follow from the original definitions, whereas others rely on additional observations, such as the association of gene duplication with functional divergence (BOX 3). Each of the implications of orthology and paralogy can be formulated as a falsifiable hypothesis, and as discussed here several focused efforts have aimed at testing the predictions of these hypotheses. In general, the results of comparative genomic studies appear to be compatible with the generalized orthology conjecture: orthologues typically are the most similar genes in the respective species in terms of sequence, structure, domain architecture and function, the reservations regarding the current state of functional annotation notwithstanding. Despite many exceptions, this conjecture appears to hold as a general, statistical trend. Although the basic definitions are straightforward, it has to be emphasized that orthology and paralogy are simplifications that are used to dissect extremely complex processes of evolution, so extensive further research is required to refine and to optimize the application of these concepts.
Acknowledgments
T.G. is supported by funds from the European Research Council and the Spanish Ministry of Economy and Competitiveness. E.V.K. is supported by intramural funds of the US Department of Health and Human Services (to the US National Library of Medicine).
Glossary
- Alternative transcription
The expression of multiple transcripts with different structures from the same gene locus.
- Bidirectional best hit (BBH)
A pair of genes that show the greatest sequence similarity to each other in a complete, reciprocal comparison of the gene (protein) sequences from a pair of compared genomes.
- Co-orthologue
A gene in a species (or group of species) that is jointly orthologous to the same gene (or genes) in another species (or group of species).
- Domain accretion
In evolution, the addition of sequences encoding extra structural domains to protein-coding genes.
- Gene Ontology (GO)
A collaborative bioinformatic project aiming at providing an ontology of defined terms representing gene product properties.
- In-paralogues
Paralogous genes that originate from a lineage-specific duplication that postdates that reference ancestral species.
- Non-homologous and isofunctional
When referring to proteins, these are proteins that in different species carry out equivalent biological functions but are not homologous.
- Orthologues
Homologous genes related by speciation.
- Orthologous groups
Sets of genes that are inferred to have evolved from a single ancestral gene in the reference ancestral species.
- Out-paralogues
Paralogous genes that originate from a duplication that antedates that reference ancestral species.
- Paralogues
Homologous genes related by duplication.
- Xenologues
Homologous genes that originate from horizontal gene transfer.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Toni Gabaldón’s laboratory: http://gabaldonlab.crg.es
Eugene V. Koonin’s laboratory: http://www.ncbi.nlm.nih.gov/CBBresearch/Koonin
eggNOG: http://eggnog.embl.de/version_3.0
The Gene Ontology: http://www.geneontology.org
Inparanoid: Eukaryotic Ortholog Groups: http://inparanoid51.sbc.su.se/cgi-bin/index.cgi
OMA browser — Orthology prediction algorithm: http://omabrowser.org/Algorithm.html
PhylomeDB: http://phylomedb.org
Quest for Orthologs initiative: http://questfororthologs.org
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Toni Gabaldón, Bioinformatics and Genomics Programme, Centre for Genomic Regulation (CRG) and Universitat Pompeu Fabra (UPF), Dr. Aiguader 88, 08003 Barcelona, Spain.
Eugene V. Koonin, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, 8600 Rockville Pike, Bethesda, Maryland 20894, USA
References
- 1.Fitch WM. Distinguishing homologous from analogous proteins. Systemat Zool. 1970;19:99–106. [PubMed] [Google Scholar]
- 2.Fitch WM. Homology a personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- 3.Koonin EV. Walter Fitch and the orthology paradigm. Brief Bioinform. 2011;12:377–378. doi: 10.1093/bib/bbr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldauf SL. Phylogeny for the faint of heart: a tutorial. Trends Genet. 2003;19:345–351. doi: 10.1016/S0168-9525(03)00112-4. [DOI] [PubMed] [Google Scholar]
- 5.Sonnhammer EL, Koonin EV. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/s0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- 6.Koonin EV. Orthologs, paralogs and evolutionary genomics. Annu Rev Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 7.Dolinski K, Botstein D. Orthology and functional conservation in eukaryotes. Annu Rev Genet. 2007;41:465–507. doi: 10.1146/annurev.genet.40.110405.090439. [DOI] [PubMed] [Google Scholar]
- 8.Studer RA, Robinson-Rechavi M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009;25:210–216. doi: 10.1016/j.tig.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Nehrt NL, Clark WT, Radivojac P, Hahn MW. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput Biol. 2011;7:e1002073. doi: 10.1371/journal.pcbi.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuzniar A, van Ham RC, Pongor S, Leunissen JA. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet. 2008;24:539–551. doi: 10.1016/j.tig.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Gabaldón T. Large-scale assignment of orthology: back to phylogenetics? Genome Biol. 2008;9:235. doi: 10.1186/gb-2008-9-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trachana K, et al. Orthology prediction methods: a quality assessment using curated protein families. Bioessays. 2011;33:769–780. doi: 10.1002/bies.201100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. Computational methods for Gene Orthology inference. Brief Bioinform. 2011;12:379–391. doi: 10.1093/bib/bbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabaldón T, et al. Joining forces in the quest for orthologs. Genome Biol. 2009;10:403. doi: 10.1186/gb-2009-10-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dessimoz C, Gabaldón T, Roos DS, Sonnhammer EL, Herrero J. Toward community standards in the quest for orthologs. Bioinformatics. 2012;28:900–904. doi: 10.1093/bioinformatics/bts050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Descorps-Declere S, Lemoine F, Sculo Q, Lespinet O, Labedan B. The multiple facets of homology and their use in comparative genomics to study the evolution of genes, genomes, and species. Biochimie. 2008;90:595–608. doi: 10.1016/j.biochi.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Mahmood K, Webb GI, Song J, Whisstock JC, Konagurthu AS. Efficient large-scale protein sequence comparison and gene matching to identify orthologs and co-orthologs. Nucleic Acids Res. 2012;40:e44. doi: 10.1093/nar/gkr1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roux J, Robinson-Rechavi M. An ontology to clarify homology-related concepts. Trends Genet. 2010;26:99–102. doi: 10.1016/j.tig.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 21.Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 22.Koonin EV, Aravind L, Kondrashov AS. The impact of comparative genomics on our understanding of evolution. Cell. 2000;101:573–576. doi: 10.1016/s0092-8674(00)80867-3. [DOI] [PubMed] [Google Scholar]
- 23.Sjolander K, Datta RS, Shen Y, Shoffner GM. Ortholog identification in the presence of domain architecture rearrangement. Brief Bioinform. 2011;12:413–422. doi: 10.1093/bib/bbr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forslund K, Sonnhammer EL. Evolution of protein domain architectures. Methods Mol Biol. 2012;856:187–216. doi: 10.1007/978-1-61779-585-5_8. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann B, Valcarcel J. Decrypting the genome’s alternative messages. Curr Opin Cell Biol. 2009;21:377–386. doi: 10.1016/j.ceb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Irimia M, Blencowe BJ. Alternative splicing: decoding an expansive regulatory layer. Curr Opin Cell Biol. 2012;24:323–332. doi: 10.1016/j.ceb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Basu MK, Poliakov E, Rogozin IB. Domain mobility in proteins: functional and evolutionary implications. Brief Bioinform. 2009;10:205–216. doi: 10.1093/bib/bbn057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouzounis C. Orthology: another terminology muddle. Trends Genet. 1999;15:445. doi: 10.1016/s0168-9525(99)01845-4. [DOI] [PubMed] [Google Scholar]
- 29.Theissen G. Birth, life and death of developmental control genes: new challenges for the homology concept. Theory Biosci. 2005;124:199–212. doi: 10.1016/j.thbio.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Bandyopadhyay S, Sharan R, Ideker T. Systematic identification of functional orthologs based on protein network comparison. Genome Res. 2006;16:428–435. doi: 10.1101/gr.4526006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh R, Xu J, Berger B. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc Natl Acad Sci USA. 2008;105:12763–12768. doi: 10.1073/pnas.0806627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynen MA, Bork P. Measuring genome evolution. Proc Natl Acad Sci USA. 1998;95:5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bromham L, Penny D. The modern molecular clock. Nature Rev Genet. 2003;4:216–224. doi: 10.1038/nrg1020. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S. Molecular clocks: four decades of evolution. Nature Rev Genet. 2005;6:654–662. doi: 10.1038/nrg1659. [DOI] [PubMed] [Google Scholar]
- 35.Koski LB, Golding GB. The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001;52:540–542. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- 36.Hulsen T, Huynen MA, de Vlieg J, Groenen PM. Benchmarking ortholog identification methods using functional genomics data. Genome Biol. 2006;7:R31. doi: 10.1186/gb-2006-7-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf YI, Koonin EV. A tight link between orthologs and bidirectional best hits in bacterial and archaeal genomes. Genome Biol Evol. 2012:1286–1294. doi: 10.1093/gbe/evs100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nature Rev Microbiol. 2003;1:127–136. doi: 10.1038/nrmicro751. [DOI] [PubMed] [Google Scholar]
- 39.Snel B, Huynen MA, Dutilh BE. Genome trees and the nature of genome evolution. Annu Rev Microbiol. 2005;59:191–209. doi: 10.1146/annurev.micro.59.030804.121233. [DOI] [PubMed] [Google Scholar]
- 40.Blomme T, et al. The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006;7:R43. doi: 10.1186/gb-2006-7-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makarova KS, Wolf YI, Mekhedov SL, Mirkin BG, Koonin EV. Ancestral paralogs and pseudoparalogs and their role in the emergence of the eukaryotic cell. Nucleic Acids Res. 2005;33:4626–4638. doi: 10.1093/nar/gki775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huerta-Cepas J, Gabaldón T. Assigning duplication events to relative temporal scales in genome-wide studies. Bioinformatics. 2011;27:38–45. doi: 10.1093/bioinformatics/btq609. [DOI] [PubMed] [Google Scholar]
- 43.Forslund K, Pekkari I, Sonnhammer EL. Domain architecture conservation in orthologs. BMC Bioinformatics. 2011;12:326. doi: 10.1186/1471-2105-12-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koonin EV, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson ME, et al. Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci. 2009;18:1306–1315. doi: 10.1002/pro.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA. 1996;93:10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galperin MY, Walker DR, Koonin EV. Analogous enzymes: independent inventions in enzyme evolution. Genome Res. 1998;8:779–790. doi: 10.1101/gr.8.8.779. [DOI] [PubMed] [Google Scholar]
- 48.Omelchenko MV, Galperin MY, Wolf YI, Koonin EV. Non-homologous isofunctional enzymes: a systematic analysis of alternative solutions in enzyme evolution. Biol Direct. 2010;5:31. doi: 10.1186/1745-6150-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch VJ, Wagner GP. Resurrecting the role of transcription factor change in developmental evolution. Evolution. 2008;62:2131–2154. doi: 10.1111/j.1558-5646.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 50.Casci T. Functional genomics: Degrees of similarity. Nature Rev Genet. 2011;12:522. doi: 10.1038/nrg3038. [DOI] [PubMed] [Google Scholar]
- 51.Thomas PD, Wood V, Mungall CJ, Lewis SE, Blake JA. On the use of gene ontology annotations to assess functional similarity among orthologs and paralogs: a short report. PLoS Comput Biol. 2012;8:e1002386. doi: 10.1371/journal.pcbi.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altenhoff AM, Studer RA, Robinson-Rechavi M, Dessimoz C. Resolving the ortholog conjecture: orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput Biol. 2012;8:e1002514. doi: 10.1371/journal.pcbi.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huerta-Cepas J, Dopazo J, Huynen MA, Gabaldón T. Evidence for short-time divergence and long-time conservation of tissue-specific expression after gene duplication. Brief Bioinform. 2011;12:442–448. doi: 10.1093/bib/bbr022. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Zhang J. The ortholog conjecture is untestable by the current gene ontology but is supported by RNA sequencing data. PLoS Comput Biol. 2012;8:e1002784. doi: 10.1371/journal.pcbi.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohd-Padil H, Mohd-Adnan A, Gabaldón T. Phylogenetic analyses uncover a novel clade of transferring in non-mammalian vertebrates. Mol Biol Evol. 2013;30:894–905. doi: 10.1093/molbev/mss325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huerta-Cepas J, et al. PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011;39:D556–D560. doi: 10.1093/nar/gkq1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]