

Abstract
Glycosylation has a pivotal role in a diverse range of biological activities, modulating the structure and function of proteins. Glycogens coupled to the nitrogen atom (N-linked) of asparagine side chains or to the oxygen atom (O-linked) of serine and threonine side chains represent the two major protein glycosylation forms. N-glycans can be released by glycosidases, whereas O-glycans are often cleaved by chemical reaction. However, it is challenging to combine these enzymatic and chemical reactions in order to analyze both N- and O-glycans. We recently developed a glycoprotei n immobilization for glycan extraction (GIG) method that allows for the simultaneous analysis of N- and O-glycans on a solid support. GIG enables quantitative analysis of N-glycans and O-glycans from a single specimen and can be applied to a high-throughput automated platform. Here we provide a step-by-step GIG protocol that includes procedures for (i) protein immobilization on an aldehyde-active solid support by reductive amination; (ii) stabilization of fragile sialic acids by carbodiimide coupling; (iii) release of N-glycans by PNGase F digestion; (iv) release of O-glycans by β-elimination using ammonia in the presence of 1-phenyl-3-methyl-5-pyrazolone (PMP) to prevent alditol peeling from O-glycans; (v) mass spectrometry (MS) analysis; and (vi) data analysis for identification of glycans using in-house developed software (GIG Tool; free to download via http://www.biomarkercenter.org/gigtool). The GIG tool extracts precursor masses, oxonium ions and glycan fragments from tandem (liquid chromatography (LC)–MS/MS) mass spectra for glycan identification, and reporter ions from quaternary amine containing isobaric tag for glycan (QUANTITY) isobaric tags are used for quantification of the relative abundance of N-glycans. The GIG protocol takes ~3 d.
INTRODUCTION
Glycosylation is one of the most common protein modifications and has crucial roles in many biological and physiological processes1. Abnormal glycosylation has been reported to be associated with a variety of diseases, such as cancer2,3, Alzheimer’s disease4, diabetes5 and cardiac disease6,7. It has been suggested that altered glycosylation, such as changes in core fucosylation, may contribute to cancer metastasis8,9. As a result, characterization of protein glycosylation patterns may be exploited to define disease progression10,11. It is thus important to study protein glycosylation, including glycoforms, glycosites and occupancy.
The two most commonly studied types of glycosylation are N- and O-linked glycosylation. N-linked glycosylation has a core glycan structure (GlcNAc2Man3) that conjugates to asparagine (Asn or N) residues in the consensus peptide motif of Asn-X-Ser/Thr (where X is any amino acid except proline). O-linked glycosylation contains glycans conjugated to serine (S) or threonine (T) without a clear consensus motif. The comprehensive study of glycomes (called glycomics) is challenging in comparison with genomics or proteomics because of its non-template-driven biosynthesis, in which glycan synthesis is determined by the availability of enzymes such as glycosidases and glycosyltransferases. The complexity of N- or O-linked glycans arises from several aspects: (i) the variable composition of different monosaccharides; (ii) glycan branching; (iii) complicated linkages, including α, β and carbon positions; and (iv) the presence of different glycan isomers. Furthermore, the analysis of glycans has been impeded by the lack of a robust and high-throughput analytical method.
Glycan isolation can be achieved using enzymatic and/or chemical treatment. N-glycans are digested primarily by enzymes such as PNGase F (which digests virtually all N-glycans except for core-a(1,3)-fucose), PNGase A (which cleaves N-glycans with a core-a(1,3)-fucose, but is ineffective on sialylated N-glycans), endoglycosidase H (which cleaves the chitobiose core of highmannose oligosaccharides) or endoglycosidase F (which digests N-glycans between the two N-acetylglucosamine residues). However, no universal O-glycosidase has been discovered for the removal of O-glycans, except for disaccharides of core 1 (Gal-GalNAc) or core 3 (GlcNAc-GalNAc) glycans. Chemical removal of O-glycans is usually performed by alkali treatment (also called β-elimination)12,13 or by hydrazinolysis at elevated temperatures (50-90°C)14,15. The concern with these methods is the sequential degradation of reducing-end monosaccharide units by consecutive β-elimination, also known as ‘peeling’16,17. To prevent a peeling reaction on the reducing end (alditol), O-glycans are usually released in a mild medium such as ammonium hydroxide (in comparison with a strong basic medium such as sodium hydroxide) in the presence of an aldehyde-reactive reagent for alditol capping18. Several chemical compounds have been exploited for the labeling of O-glycan alditol after deactivation of alditol groups by β-elimiation. Among these compounds, PMP has been widely used, not only for stabilization of the reducing end but also to enhance the hydrophobicity of O-glycans for direct analysis by HPLC and LC-electrospray ionization (ESI)–MS19.
Development of the protocol
To comprehensively profile protein glycosylation patterns in biological samples, a platform has been sought for the sequential release of N-glycans and O-glycans. Many studies on N-glycans have shown that unmodified sialic acids are fragile and easily lost during sample preparation and ionization in MALDI or even ESI20–24. Chemical modification of sialic acids, such as amidation22,25, methyl esterification26,27 and perbenzolylation28, is commonly used for sialic acid stabilization. However, it is difficult to stabilize sialic acid residues using an in-solution chemical reaction because of the similarities between the chromatographic properties of these chemicals and those of the modified glycans. It is thus difficult to remove these excess chemicals from the modified glycans20. Permethylation of the released glycans can protect sialic acids on both N- and O-glycans29,30. However, the permethylated glycans can lose reactivity on their reducing ends, consequently preventing their further use for fluorophore (UV detection), chromophore (chromatographic separation) or isobaric tag labeling (MS-based relative quantification)31. In a recent review, researchers suggested that the analysis of glycolipids, N-glycans and O-glycans will move in the direction of solid-phase extraction (SPE) methods for the sequential release of such modifications32. One such technique has been developed that immobilizes proteins through the use of protein G, or polyvinylidene fluoride (PVDF)33 or lectin affinity columns34, allowing sequential analysis of N-glycans and O-glycans. Several other approaches have been successfully developed for the analysis of N- and O-glycosylation35,36.
Our group has recently developed the GIG approach for the sequential release of N- and O-linked glycans20. GIG allows for the molecular stabilization of glycans on a solid support via chemoenzy-matic treatments. This protocol provides a step-by-step description of the procedures required for protein extraction, immobilization, glycan modification, N-glycan release, O-glycan release, LC–MS or MALDI–MS, and data analysis (Fig. 1a). One of the key steps of this protocol is the covalent conjugation of proteins or peptides to a solid support. Once immobilized (Fig. 1b), the glycans attached to amino acids are modified by carbodiimide coupling (e.g., coupling of p-toluidine (pT) in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC))—for stabilization of sialic acids), glycosyltransferase (e.g., fucosyltransferase—for synthesis of specific glycans) or glycosidase (e.g., neuraminidase—for the study of linkage). N-glycans are first released from the solid support via PNGase F or other N-glycosidase-mediated cleavage (Fig. 1b). The released N-glycans can be directly analyzed by MS. To quantify the relative abundance of N-glycans in biological samples, N-glycans are conjugated with QUANTITY via reductive amination (Fig. 1c)37. The labeled N-glycans are quantitatively analyzed by LC-MS/MS. After N-glycan removal, the solid phase still contains O-glycans for subsequent analysis. The O-glycans are released from the solid phase by β-elimination using ammonia in the presence of PMP (this reacts with O-glycan alditols in order to prevent hydrolysis of glycosidic bonds) before analysis by MS (Fig. 1b)38.
Figure 1.
Schematic diagram of sequential release of N-linked and O-linked glycans via treatment. (a) Flowchart of the protocol, including references to the steps in the PROCEDURE. (b) Chemoenzymatic method for modification and release of glycans: immobilization of glycoproteins on GIG resin by reductive amination; denaturing of glycoproteins using buffer (0.5% SDS, 50 mM DTT) at 100°C for 10 min; modification of sialic acid residues by carbodiimide coupling by 1 M pT in 1 M HCl in the presence of EDC (pH 4-6) at room temperature for 3 h; enzymatic release of N-glycans using 2 μl of PNGase F and 40 μl of GlycoBuffer (10×, 500 mM sodium phosphate) in 358 μl of DI water, followed by chemical release of O-glycans by treatment of GIG resin with 200 μl of ammonium hydroxide (NH4OH; 26-28%) and 500 mM PMP in 300 μl of methanol and incubation at 55°C for 24 h. The released O-glycans are purified using a C18 cartridge, and N-glycans are purified using a Carbograph SPE column20,37,60. (c) Molecular structure of QUANTITY, consisting of a reporter, a balancer and a reactive group. The reporter has four tags with molecular weights ranging from 176 to 179. N-glycans are labeled by QUANTITY using acetic acid (30%) and DMSO (70%) in the presence of 1 M NaCNBH3 via reductive amination. The reporter ions can be detected in their fragmentation of the precursors. Image adapted with permission from ref. 60, BioMed Central (open-access license: https://creativecommons.org/licenses/by/4.0/).
Advantages and limitations
The GIG method is based on the solid support conjugation of glycoproteins, which allows for different glycan treatments and mitigating chromatographic purification steps that are required by in-solution approaches. GIG offers several advantages over the traditional in-solution methods: (i) GIG allows for the sequential analysis of N-linked and O-linked glycans from the same specimen in a step-by-step release, (ii) sialic acid residues can be stabilized on the solid phase, (iii) GIG integrates all required steps in a high-throughput platform and (iv) samples can easily be cleaned by various washing steps without sample loss. As GIG depends on the conjugation of glycoproteins to a solid support, a high degree of conjugation efficiency is a prerequisite for quantitative analysis of glycans from samples. In general, the immobilization efficiency can be substantially improved (up to 95%) by adjusting the pH of the buffers20,31.
The GIG approach is based on a chemical coupling of glycoproteins to a solid support, followed by enzymatic digestion. As a result, GIG can be applied to high-throughput automation for biological and clinical applications. Analysis of large numbers of biological samples and/or clinical specimens requires the generation of highly reproducible data in a cost-effective manner. It is therefore undesirable to rely heavily on the manual preparation of samples. High-throughput assays, such as 96-well formats39,40 or microchips41, have been successfully demonstrated with improved efficiency as compared with that of manual procedures. The solidphase technique can be favorably adapted to automated platforms such as microfluidic devices and 96- or 384-channel automated liquid handlers. Similarly, resins, functionalized by different chemical groups (for example, photocleavable linkers for phosphorylation42 or acetylation) can be used to study other post-translational modifications on these proteins, using similar high-throughput platforms. This large-scale method is a practical approach for the screening of glycans from a large number of samples43.
Applications of the method
This protocol enables researchers to analyze both N-glycans and O-glycans from glycoproteins or glycopeptides. In our laboratory, GIG has been used for studies of N-glycan profiling in tissue or blood samples derived from patients with prostate cancer44, pancreatic cancer25, ovarian cancer45 and cardiac hypertrophy46, as well as samples from glycoengineered Chinese hamster ovary (CHO) cells47, and for studies of glycoforms of HIV gp 120 (ref. 48), glycoengineered sialylation of CHO cells49 and N-gly-cosylation in cockroach allergen regulation of human basophil function50. GIG has been used for the extraction of N-glycans for isobaric labeling using iARTs51 or QUANTITY37. The fact that GIG takes advantage of a solid phase for sample preparation substantially reduces the loss of sample and eliminates the requirement for multiple chromatography for sample cleanup after each step20,31. The solid-phase resin can easily be packed into an AutoTip (pipette tip that holds packed beads for use in an automated liquid handler) for automated sample preparation (data not shown). This means that GIG can be scaled up for high-throughput analysis of glycans for quick screening of glycan biomarkers in clinical specimens. In addition, the N-glycans released by this high-throughput platform can be rapidly labeled with a fluorescent marker or an MS-active labeling reagent such as Rapifluor52 for concurrent fluorescent detection (absolute quantification of glycans) and MS. As a variety of additional treatments can be incorporated into the procedure, GIG provides a platform for studying the structures of glycoproteins and glycans by corresponding glycosidases and glycosyltransferases.
Experimental design
Modification and detection of sialylated O-glycans
Fragile sialic acids are easily lost during sample preparation or ionization. Sialic acid is negatively charged and hydrophilic, which hinders its identification by MS in the positive ionization mode. The negative ionization mode has been well developed for the analysis of intact sialic acids, but the interpretation of MS data is different from that of spectra that are acquired in the positive ionization mode. Neutral glycans are, however, not detectable in the negative ionization mode. The positive ion mode is therefore more compatible with standard data analysis for glycans. Modification of sialic acid provides several advantages: (i) it stabilizes sialic acids for sample preparation and ionization, (ii) it neutralizes negative charge and (iii) as a result of pT hydrophobicity, the modification enhances sialic acid hydrophobicity. As occurs with the modification of N-glycans20, the sialic acid residues of O-glycans are labeled with pT by carbodiimide coupling (Fig. 1b).
Sequential release of N- and O-glycans
We have previously demonstrated the ability to release N-glycans or O-glycans from the solid phase20,48. However, the order of release of N- and O-glycans has not yet been assessed. Hydrazine hydrolysis has been an effective method for sequentially releasing O-glycans (60°C) and N-glycans (95°C)15,53. This type of chemical release is cost-effective and can be ubiquitously applied for different types of glycans; however, it may cause artifacts, and N-glycans can be released even at a relatively lower temperature (60°C). Alternatively, it has been reported that O-glycans are released during mild β-elimination (using ammonium hydroxide)54.
Sensitivity of the method
In this protocol, we work with 1 mg of protein or 200 μg of peptides as starting material. These quantities have often been used for the study of protein glycosylation. This does not, however, mean that the GIG approach cannot be used with lower amounts of starting material. We have done limitation tests on the minimum amount of glycoprotein or glycopeptide required for this approach. When 1 μg of a standard sialylglycopeptide is used for immobilization, satisfactory LC-MS signals can be obtained using 2% of the eluate after glycan modification and release by PNGase F. We therefore expect that this approach can be used to identify glycan patterns from hundreds of nanograms of glycoproteins.
MATERIALS
REAGENTS
Cells OVCAR-3 cell line (American Type Culture Collection, cat. no. HTB-161) ! CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
Mouse heart tissue ! CAUTION Any experiments involving mice must conform to relevant institutional and national regulations. The procedures in this protocol were approved by the Johns Hopkins Medical Institutions Animal Care and Use Committee.
Aminolink Plus resin (Thermo Fisher Scientific, cat. no. 20501)
2,5-Dihydroxybenzoic acid (DHB; Sigma-Aldrich, cat. no. 85707)
Acetic acid (AcOH; Sigma-Aldrich, cat. no. 320099)
Acetonitrile (ACN; Fisher Scientific, cat. no. A998) ! CAUTION ACN is corrosive and should be handled in a hood using gloves.
Ammonium bicarbonate (NH4HCO3; Sigma-Aldrich, cat. no. 379999)
Ammonium hydroxide (NH4OH; 28-30% (vol/vol); Sigma-Aldrich, cat. no. 320145) ! CAUTION NH4OH is hazardous and flammable, and should be handled in a hood using gloves.
Angiotensin I human acetate salt hydrate (Sigma-Aldrich, cat. no. A9650)
BCA protein assay reagent (Thermo Fisher Scientific, cat. no. 23225)
Chloroform (J.T. Baker, cat. no. 9180)
Deionized (DI) water (laboratory-made, or Thermo Fisher Scientific, cat. no. 1523001)
Fetuin from FBS (Sigma-Aldrich, cat. no. F2379)
Formic acid (FA; Sigma-Aldrich, cat. no. 1002640100)
HPLC-grade water, for MS sample preparation (Fisher Scientific, cat. no. W5-4)
Hydrochloric acid (HCl; 36.5-38.0% (vol/vol); Sigma-Aldrich, cat. no. H1758) ! CAUTION HCl is a highly corrosive and strong mineral acid. Concentrated HCl forms acidic mists; both solution and mist have a corrosive effect on human tissue, with the potential to damage respiratory organs, eyes, skin and intestines irreversibly. When preparing HCl solution, the concentrated HCl should be added to water slowly. HCl must be handled in a hood with personal protective equipment, including rubber or PVC gloves, eye goggles and chemical-resistant clothing and shoes.
Maltoheptaose (DP7; Sigma-Aldrich, cat. no. M7753)
Mucin from bovine submaxillary glands (MSB; Sigma-Aldrich, cat. no. M3895)
Mucin from porcine stomach (Type 3) (MPS3; Sigma-Aldrich, cat. no. M1778)
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC; Sigma-Aldrich, cat. no. 39391)
Sodium chloride solution (NaCl; 5 M; ChemCruz Biochemicals, cat. no. SC-203274)
Neuraminidase (α2-3, α2-6, α2-8) (New England BioLabs, cat. no. P0720L)
Neurotensin (Sigma-Aldrich, cat. no. N6383)
N,N-dimethylaniline (DMA; Sigma-Aldrich, cat. no. 515124)
p-Toluidine (pT; Sigma-Aldrich, cat. no. 236314)
1-Phenyl-3-methyl-5-pyrazolone (PMP; Sigma-Aldrich, cat. no. M70800)
Peptide-N-glycosidase F (PNGase F; New England BioLabs, cat. no. P0705L)
PBS (10×; Sigma-Aldrich, cat. no. 79383)
Protein denaturing buffer (10×; New England BioLabs, supplied with PNGase F) ▲ CRITICAL This buffer can also be prepared by dissolving 400 mM DTT (Sigma-Aldrich, cat. no. D0632) and 5% (wt/vol) SDS (Sigma-Aldrich, cat. no. 74255) in DI water (1×).
Protein inhibitor cocktail (Roche, cat. no. 04693116001)
Reaction buffer for neuraminidase (G1; 10×; New England BioLabs, supplied with reagent) ▲ CRITICAL This buffer can also be prepared by mixing 50 mM CaCl2 (Sigma-Aldrich, cat. no. 793639) and 500 mM sodium acetate (Sigma-Aldrich, cat. no. 791741) at pH 5.5.
Reaction buffer for PNGase F(GlycoBuffer; 10×; New England BioLabs, supplied with reagent) ▲ CRITICAL This buffer can also be prepared by dissolving sodium phosphate in DI water to a 500 mM concentration (Sigma-Aldrich, cat. no. 342483) at pH 7.5.
RIPA lysis buffer (10×; Millipore, cat. no. 20-188) ▲ CRITICAL This buffer can be prepared by mixing 1% NP-40 (Thermo Fisher Scientific, cat. no. 85124), 0.5% sodium deoxycholate (Sigma-Aldrich, cat. no. 30970), 0.1% SDS, 2 mM EDTA (Sigma-Aldrich, cat. no. 798681), 50 mM sodium fluoride (Sigma-Aldrich, cat. no. 215309) and 25 mM Tris-HCl.
Sialylglycopeptide (SGP; Fushimi Pharmaceutical, cat. no. 189035-43-6)
Sodium borocyanohydride (NaCNBH3; Sigma-Aldrich, cat. no. 156159)
Sodium carbonate (Na2CO3; Sigma-Aldrich, cat. no. 451614)
Sodium citrate (Na3C6H5O7; Sigma-Aldrich, cat. no. W302600)
Trizma hydrochloride (Tris-HCl; 1 M; Sigma-Aldrich, cat. no. 93313)
Trifluoroacetic acid (TFA; Sigma-Aldrich, cat. no. 302031) ! CAUTION TFA is a corrosive acid, and it should be handled in a hood using protective goggles and gloves.
Human serum (from healthy men or women; collected from Johns Hopkins Hospital with the approval of the institutional review board of Johns Hopkins University)
EQUIPMENT
Eppendorf Research Plus pipette (Sigma-Aldrich, cat. no. Z683884-1EA)
μ-Focus MALDI plate (384 circles; Hudson Surface Technology, cat. no. PFS2407000)
BD Falcon tube (15 ml; BD Biosciences, cat. no. 352095)
Zeba spin desalting column (2 ml; Thermo Fisher Scientific, cat. no. 89877)
Sep-Pack C-18 cartridge (Waters, cat. nos. WAT054955 (1 cc; 50-mg sorbent); WAT020805 (3 cc; 500-mg sorbent))
Carbograph Extract-Clean columns (Carbograph; Grace, cat. no. 210142)
PGC ziptip (Glygen, cat. no. NT2CAR)
Snap-cap spin column (1 ml; Thermo Fisher Scientific, cat. no. 69725)
230-mm Pasteur pipette, type I (Wheaton, cat. no. 357335)
Corning Costar 96-well cell culture plate (Corning, cat. no. 3598)
Latex dropper bulb (Fisher Scientific, cat. no. S32324)
Barnstead/Thermolyne LabQuake tube shaker (Barnstead International, cat. no. 4002110)
Allegra 6R centrifuge (Beckman Coulter, cat. no. 366816)
Centrifuge 5415 D (Eppendorf, cat. no. 2262140-8)
Boekel 13300 oven (Boekel Scientific, cat. no. 41104424)
μQuant Microplate Spectrophotometer (Biotek Instruments)
Innova 4000 incubator shaker (New Brunswick Scientific, cat. no. 1510611)
Revco freezers (Ultima II; Thermo Fisher Scientific, cat. no. 2586-9SI-V)
Lab Dancer Mini Vortexer (VWR International, cat. no. 10153)
Thermo Savant SpeedVac SPD121P Centrifugal Evaporator (Thermo Fisher Scientific, cat. no. SPD121PP1-115)
Thermo Savant Refrigerated Vapor Traps (Thermo Fisher Scientific, cat. no. RVT400-115)
EURO DPC MicroMix 5 Shaker (Conquer Scientific, cat. no. 5957)
MALDI plate holder (Hudson Surface Technology, cat. no. PFS2407000)
MALDI-QIT-TOF mass spectrometer equipped with a controllable MSn fragmentation, high-resolution precursor ion selection (Axima Resonance; Shimadzu)
Orbitrap Velos LC-MS equipped with Dionex UltiMate 3000 HPLC (Thermo Fisher Scientific)
Software
MSConvert spectrum-converting software (can be freely downloaded from Proteowizard at http://proteowizard.sourceforge.net/downloads.shtml)
Python 2.7 (can be downloaded from https://www.python.org/download/releases/2.7/)
Anaconda 4.2.0 for Python 2.7 version (can be downloaded from https://www.continuum.io/downloads) ▲ CRITICAL Anaconda should be installed in ‘c:\Program Files\Anaconda2’.
Pymzml package (can be downloaded from https://pypi.python.org/pypi/pymzML)
GIG Tool (freely available from http://www.biomarkercenter.org/gigtool)
GlycoWorkBench (free download from https://code.google.com/archive/p/glycoworkbench/downloads)
REAGENT SETUP
Resin (or Aminolink) Store the resin in 0.02% sodium azide at 4°C for up to 1 year.
Lysis buffer Dilute the protein inhibitor cocktail at a ratio of 1:100 with 1× RIPA lysis buffer. This buffer can be stored at 4°C for up to 1 year.
Binding buffer (pH 10) Dissolve 2.94 g of sodium citrate (100 mM) and 0.53 g of sodium carbonate (50 mM) in HPLC water to a final volume of 10 ml. Measure and adjust the buffer pH (if necessary) by adding either sodium citrate (Na3C6H5O7) or sodium carbonate (Na2CO3). Store the buffer at room temperature (20-25°C) for up to 6 months.
Reducing buffer (20×, pH 7.4) Dissolve 62.84 mg of NaCNBH3 in 1 ml of HPLC water to make a 1 M solution. Store the buffer at room temperature for up to 2 weeks.
PNGase F (glycerol-free) Add PNGase F to the resin solution (see release of glycan for solution used) right before release of N-glycans. 1-2 μl of PNGase F is added to 80-160 μl of solution for 20 μl of serum sample. Store the solution at 4°C. Expiration date is according to the manufacturer’s specifications.
QUANTITY reagent Add 5 mg of QUANTITY (C12N3OH26) and 11.72 mg of NaCNBH3 to 189 μl of labeling solution to make a 100 mM QUANTITY solution. Store it at 4°C for up to 6 months.
PMP solution (0.5 M) Dissolve 87.1 mg of PMP in 1 ml of methanol for a final concentration of 0.5 M. Prepare the solution fresh.
O-glycan reaction solution Dissolve 300 μl of PMP solution with 200 μl of concentrated NH4OH. Prepare the solution fresh.
2.5-Dihydroxybenzoic acid matrix solution Dissolve 100 mg of 2.5-dihydroxybenzoic acid (DHB) per ml in 50% (vol/vol) CH3OH in the presence of 0.1 mM NaCl. Store the solution in the dark at room temperature for up to 2 weeks.
Formic acid solution Dissolve formic acid (FA) in HPLC water to a 0.2% (vol/vol) concentration. Store the solution at 4°C for up to 6 months.
Cells and tissues Use 1×106 cells or 1-5 mg of tissue for each sample.
NaCl (1 M) Add 10 ml of 5 M NaCl to 40 ml of DI water. Store the solution at room temperature for up to 1 year.
HCl (1 N) Add 833 μl of concentrated HCl (36-38%) to 9.167 ml of DI water to create 1 N HCl. Gently vortex the solution. ! CAUTION Always add HCl to water slowly. Overheating or even an explosion may occur when water is added to concentrated HCl. Store the solution at room temperature for up to 6 months.
10% Formic acid Add 25 ml of 100% FA to 225 ml of DI water. Store the solution at room temperature for up to 12 months.
0.1% Trifluoroacetic acid Add 1 ml of 100% trifluoroacetic acid (TFA) to 1,000 ml of HPLC water. Store the solution at room temperature for 12 months.
80% ACN in 0.1% TFA Mix 800 ml of 100% acetonitrile (ACN) and 200 ml of HPLC water. Add 1 ml of 100% TFA. Store the solution at room temperature for up to 12 months.
10% ACN in 0.1% TFA Add 100 ml of 100% ACN to 900 ml of 0.1% TFA. Store the solution at room temperature for up to 12 months.
1% Acetic acid Add 1 ml of 100% acetic acid (AcOH) to 99 ml of DI water. Store the solution at room temperature for up to 12 months.
PROCEDURE
Cell or tissue protein extraction and denaturation ●TIMING 2 h
-
1| Disrupt and homogenize cells (OVCAR-3; 106) or tissues (1-5 mg) first before sonication; add 1 ml of RIPA lysis buffer to the sample, followed by four to six rounds of sonication (sonicate for 30 s, cool the sample in an ice container for 30 s, centrifuge the sample at 2,000g for 2 min at room temperature and then discard the supernatant).
▲ CRITICAL STEP Check the sample solution under white light for protein extraction; complete extraction is achieved when the sample solution is transparent.
▲ CRITICAL STEP Sonication conditions must be determined empirically for different cell lines or tissues, as well as for different sonication equipment.
-
2| Centrifuge the lysate at 16,000g for 10 min at 4°C. Collect the supernatant.
■ PAUSE POINT The supernatant can be stored at −80° for at least 12 months.
-
3| Buffer exchange. Remove the desalting column's bottom closure (rubber) and loosen the cap. Place the column into a 15-ml conical collection tube.
▲ CRITICAL STEP Use a 2-ml microcentrifuge tube for collection when a 0.5-ml desalting column is used (<2 mg of protein); if >2 mg of protein is immobilized, a 4-ml centrifuge should be used; centrifuge the column at 2,000g for 2 min at room temperature. Note: All centrifugation is conducted at room temperature unless stated otherwise.
▲ CRITICAL STEP This step can exchange an aldehyde-reactive buffer such as Tris-HCl that would interfere with protein immobilization.
-
4| Discard the flow-through; add up to 500 μl of binding buffer or DI water to the column. Centrifuge at 2,000g for 2 min at room temperature. Repeat this step two to three times.
▲ CRITICAL STEP Mark the tube direction for the centrifugation. Keep the mark at the same position during all subsequent centrifugation steps to ensure complete buffer exchange.
-
5| Place the column in a new collection tube and slowly apply the cell lysate to the center of the compact resin. Wait until the sample penetrates the resin completely. Centrifuge the column at 2,000g for 2 min at room temperature. Collect the flow-through.
▲ CRITICAL STEP Do not use any buffer containing amines, which react with aldehyde resin and compete with protein-resin conjugation.
? TROUBLESHOOTING(/P)(P)■ PAUSE POINT The supernatant can be stored at −80° for at least 12 months.
-
6| Sample concentration measurement. First, prepare the BCA solution by diluting BCA reagent B at a 1:50 ratio with reagent A. Prepare a series of solutions (0.0625, 0.125, 0.25, 0.5, 1 and 2 mg/ml BSA in BCA) as a reference. Make 200 μl for each concentration in a 96-well cell culture plate. Mix 20 μl of the desalted sample with 200 μl of BCA solution in the 96-well cell culture plate. Briefly mix the plate using a DPC shaker and incubate it at 37°C for 30 min. Measure the protein concentration using μQuant Microplate Spectrophotometer.
▲ CRITICAL STEP Calculate the amount of material according to its concentration and volume.
Protein/peptide immobilization ● TIMING 9 h
- 7| For sample preparation for peptides, follow option A, and for proteins follow option B.
- peptide sample preparation
- Digest the proteins using trypsin according to the protocols described in the literature55.
- Dry the sample (use 200 μg of peptides) using a SpeedVac (1-2 h at 37°C).
-
Resuspend the sample in binding buffer (pH 10) (90 μl is recommended; maximum 500 μl owing to the volume limitation of the spin column).? TROUBLESHOOTING
- protein sample preparation
- Dissolve the protein sample in 180 μl of binding buffer (pH 10) in a 1.5-ml microcentrifuge tube (Reagent Setup).
-
Add 20 μl of 10× protein denaturing buffer to alkylate/reduce the proteins, then briefly vortex and centrifuge the sample (10 s at 2,000g) at room temperature. Close the microcentrifuge tube cap and incubate the sample in a heat block at 100°C for 10 min. Cool the mixture at room temperature for 10 min.▲ CRITICAL STEP Do not use a large volume of sample (>500 μl) in this step, in order to prevent explosion of the microcentrifuge tube and loss of sample when denaturing at 100°C. Open and close the microcentrifuge tube cap every 2-3 min.
-
8| Resin preparation (Steps 8-12). Equilibrate the resin (or Aminolink) to room temperature before use; vortex the resin slurry thoroughly before transferring the resin using a pipette.
▲ CRITICAL STEP Use an appropriate amount of resin based on the sample amount (1-10 μg minimum). ~1 ml of resin can immobilize 10-20 mg of proteins.
9| Resin preconditioning. Cap the bottom of a snap-cap spin column (using a rubber cap provided by Thermo Fisher Scientific), place the resin slurry into the column, remove the bottom cap and centrifuge the column at 2,000g for 30 s at room temperature.
10| Discard the flow-through; close the bottom cap, add 500 μl of binding buffer (pH 10) to the resin, close the top cap, vortex briefly and remove both top and bottom caps.
11| Place the spin column into a new microcentrifuge tube. Centrifuge the column at 2,000g for 30 s. Discard the flow-through and repeat these steps twice.
-
12| Add 100 μl of binding buffer to the resin, close the top and bottom caps and briefly vortex to resuspend the resin.
▲ CRITICAL STEP Resin may aggregate after being conditioned with the binding buffer. If resins still aggregate after vortexing, use a pipette tip to break up aggregates.
13| Protein immobilization (Steps 13-19). Add the denatured samples to the preconditioned resin.
-
14| Wash the sample tube using 100 μl of binding buffer and transfer this to the preconditioned resin as well. Repeat this step once.
▲ CRITICAL STEP The total amount should be 500 μl The snap-cap spin column can contain up to 600 μl Reducing buffer will be added to this volume. You may need to concentrate the protein or peptide if the volume of the required samples is >500 μl in Step 6.
-
15| Vortex the sample-resin mixture for 30 s. Place the mixture on a tube rotator for end-over-end mixing. Incubate the mixture at room temperature for 4 h.
▲ CRITICAL STEP To achieve efficient protein immobilization for the resin, constant and efficient mixing is required. Resin precipitation may cause incomplete sample conjugation and reduce the yield.
? TROUBLESHOOTING
-
16| Add 25 μl of 20× reducing buffer to the sample-resin mixture. Briefly vortex the mixture and incubate it at room temperature for 4 h on a tube rotator.
▲ CRITICAL STEP This step reduces imide groups, which allows the subsequent formation of amides (reductive amination). A greater concentration of amines will allow more reactions with the resin and will improve overall immobilization.
-
17| Remove the spin-column cap. Centrifuge the column at 2,000g for 30 s and collect the flow-through.
▲ CRITICAL STEP The efficiency of protein immobilization can be determined by measuring the protein concentration in the flow-through and comparing it with the BCA measurements in Step 6. If sample recovery is <90%, more resin may be required.
? TROUBLESHOOTING
18| Wash the resin using 500 μl of 1× PBS by vortexing (cap the top and bottom of the spin column), remove the caps and centrifuge the column at 2,000g for 30 s. Discard the flow-through and repeat this step once.
19| Mix 475 μl of 1× PBS with 25 μl of 1 M NaCNBH3 and add this to the resin. Incubate the sample at room temperature for 4 h with end-over-end mixing.
20| Resin active site blocking (Steps 20-23). Wash the resin using 500 μl of 1 M Tris-HCl, discard the flow-through and repeat this step twice.
-
21| Mix 475 μl of 1 M Tris-HCl and 25 μl of 1 M NaCNBH3 and add this to the resin. Vortex the mixture and incubate the sample at room temperature for 30 min with end-over-end mixing.
▲ CRITICAL STEP It is important to block active aldehyde sites on the resin after sample immobilization to prevent their reacting with chemicals or enzymes that have primary amines, such as pT or PNGase F. Active aldehyde sites are blocked by reacting with amines of Tris-HCl buffer (reductive amination).
22| Wash the resin using 500 μl of 1 M NaCl; discard the flow-through and repeat this step twice.
-
23| Wash the resin using 500 μl of DI water; discard the flow-through and repeat this step twice.
■ PAUSE POINT The sample -resin mixture can be stored at 4°C for at least 2 weeks.
Glycoprotein treatment and modification
- 24| The immobilization of glycoproteins can be readily performed by the use of appropriate chemicals or enzymes. The advantages of GIG are demonstrated on sialic acid stabilization (option A) and desialylation (option B). Other treatments and modifications, such as galactose oxidase, galactosyltransferase and fucosidase, may be applied. If the sample does not contain sialic acids, then directly go to the ‘N-Glycan release’ section (Step 25). For sialic acid protection, use option A; for desialylation, use option B.
- sialic acid protection ● TIMING 5 h
- Weigh out 1.072 g of pT and add it to 1 N HCl solution, resulting in 1 M pT (400-μl solution of pT is applied to each spin column. Prepare 400 μl if there is only one sample). Vortex the solution and heat it to 65°C in an oven for 10 min.
-
Vortex the solution and centrifuge it (2,000g for 30 s at room temperature). If the pT does not dissolve completely, heat the solution to 65°C in an oven for another 5 min. Add 40 μl of EDC to a 400-μl solution of pT.▲ CRITICAL STEP EDC may be viscous and it requires thawing at room temperature for 20 min. Cut the tip of a pipette to facilitate pipetting of EDC.? TROUBLESHOOTING
-
Add 40 μl of EDC to a 400-μl solution of pT, and add 25 μl of HCl (36.5-38.0%) to the solution in order to adjust the pH to 4.0-6.0.▲ CRITICAL STEP Check the pH of the sample. If the pH is not in the range of 4-6, adjust it by adding either HCl (to decrease the pH) or EDC (to increase the pH).
- Add 465 μl of pT-EDC solution to the sample-resin mixture. Briefly vortex and incubate the mixture at room temperature for 4 h.
-
Wash the sample-resin mixture using 500 μl of 10% formic acid. Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.? TROUBLESHOOTING
- Wash the sample-resin mixture using 500 μl of 10% ACN (0.1% TFA). Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.
- Wash the sample-resin mixture using 500 μl of 1 M NaCl Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.
-
Wash the sample-resin mixture using 500 μl of HPLC water. Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.▲ CRITICAL STEP EDC-pT should be effectively removed by Steps v-viii. Without these washing steps or with incomplete washing, EDC-pT will contaminate the final product.■ PAUSE POINT The sample -resin mixture can be stored at 4°C for at least 2 weeks.
- Desialylation ● TIMING 2 h
- Add 100 μl of DI water per 100 μl of resin slurry; add 4 μl of G1 (reaction buffer) to the sample-resin mixture in a 100μl solution.
- Add 1-2 μl of neuraminidase per 1 μg of glycoprotein to the sample-resin mixture.
- Gently vortex the sample-resin mixture. Incubate it at 37°C for 1 h.
- Wash the resin using 500 μl of 1 M NaCl. Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.
-
Wash the resin using 500 μl of HPLC water. Centrifuge the column at 2,000g for 30 s at room temperature and discard the flow-through. Repeat this step twice.■ PAUSE POINT The sample-resin mixture can be stored at 4°C for at least 2 weeks.
N-glycan release ● TIMING 6 h
- 25| In contrast to in-solution release of glycans, both N- and O-glycans can be released sequentially using this protocol. If N-glycans are released using a volatile buffer—e.g., ammonium bicarbonate (NH4HCO3; option A) or in salt-free buffers (option B)—additional purification steps are not required.
- N-glycan release using a volatile buffer
-
Add 38 μl of 10 mM NH4HCO3 for every 100 μl of resin slurry and 2 μl of PNGase F for every 2 mg of glycoprotein to the resin slurry from Step 25. Gently vortex the mixture and incubate it at 37°C for 2 h.▲ CRITICAL STEP Use freshly prepared NH4HCO3 and check the pH. A pH of 7.5-8.5 is required for optimal enzyme activity. Overnight incubation is recommended for complex biological samples.
-
- N-glycan release in salt-free buffers
- Add to the resin sample mixture from Step 25 268 μl of DI water, 30 μl of G7 (reaction buffer for PNGase F) and 2 μl of PNGase F per 100 μl of resin slurry. Incubate the mixture at 37°C for 2 h while mixing with a Barnstead/Thermolyne LabQuake tube shaker.
26| Remove both the top and bottom caps and place the column into a clean 2-ml microcentrifuge tube. Centrifuge the column at 2,000g for 30 s at room temperature and collect the flow-through.
27| Add 500 μl of HPLC water, vortex and centrifuge at 2,000g for 30 s. Collect the flow-through. Repeat this step twice.
-
28| Combine all flow-through fractions (1,800 μl in total). Add 5 μl of 100% FA to the sample to make the pH < 3.0.
▲ CRITICAL STEP Use 100% FA to adjust the sample pH to <3.0. The positive-ion mode is expected for glycan MS analysis.
-
29| Dry the sample using a SpeedVac at 37°C (this takes 2-3 h). Resuspend the sample in 40 μl of HPLC water.
■ PAUSE POINT The glycan is stable for at least 12 months when stored at −20°C.
(Optional) N-glycan purification and labeling ● TIMING 13 h—5 h for purification and 8 h for labeling
▲ CRITICAL When N-glycans are further modified such as with reducing-end QUANTITY or fluorescence labeling, use G7 (reaction buffer for PNGase F); samples must be purified before labeling. After release of N-glycans, O-glycans are chemically released by β-elimination.
30| N-glycan purification (Steps 30-38). Add 3 ml of ACN (100%) to the Carbograph solid-phase extraction (SPE) column (Carbograph Extract-Clean column).
31| Add 3 ml of 1% TFA; repeat this step once. Use an inert gas or a latex dropper bulb to push the liquid through the cartridge if it is too slow.
32| Load the sample onto the SPE Carbograph column; allow the sample solution to penetrate the cartridge by gravity. Reload the flow-through onto the SPE Carbograph column an additional time.
33| Wash the cartridge with 3 mi of 0.1% TFA; repeat this step four times.
34| Wash the cartridge with 3 mi of 0.1% TFA in 10% ACN.
35| Elute the sample with 400 μl of 0.1% TFA in 80% ACN; repeat this step once.
36| Dry the sample solution using a SpeedVac at 37°C (this takes 1-2 h).
37| Resuspend the sample in 40 μl of HPLC water.
38| (Optional) To study Linkage, the released N-glycans can be permethylated using established methods56.
39| N-glycan isobanc labeling (Steps 39-44). Using a SpeedVac (37°C for 1-2 h), dry the N-glycans from individual samples in 2-ml microcentrifuge tubes with glass inserts (capacity 700 μl).
40| Add 20 μl of 200 mM QUANTITY labeling reagent to each sample. Each sample should be coupled to one of the 4-plex QUANTITY tags.
41| Vortex the samples in order to completely dissolve them. Incubate the mixtures at 65°C for 4 h with gentle mixing.
42| Cool the samples to room temperature and pool the labeled samples after diluting them at least 20-fold by adding 480 μl of 0.2% FA.
43| Clean up the pooled sample using a SPE Carbograph column as described in Steps 30-37.
-
44| Resuspend the sample in 40 μl of 0.2% FA, vortex the mixture and centrifuge briefly (2,000g for 30 s).
? TROUBLESHOOTING
■ PAUSE POINT Samples can be stored at −20°C for up to 6 months before LC-MS analysis in Step 60.
O-glycan release ● TIMING 32 h
45| Wash the resin from Step 27 twice with 500 μl of 1 M NaCl, followed by two washes with 500 μl of NH4OH.
46| Centrifuge the resin at 2,000g for 30 s.
47| Transfer the resin from the spin column to a 2-ml microcentrifuge tube by adding 500 μl of NH4OH to the sample-resin mixture. Vortex the sample and transfer it to the tube. Repeat this step twice for complete transfer. Centrifuge the column at 6,000g for 10 min and remove the NH4OH using a 1-ml pipette.
-
48| Add 500 μl of O-glycan reaction solution to the sample, and incubate it at 55°C for at least 24 h
▲ CRITICAL STEP Make sure to mix the resin gently. The reaction tube can easily break or burst, owing to volatile NH4OH.
▲ CRITICAL STEP Another 24 h may be required to complete the release of O-glycans from complex samples. In this case, first remove the O-glycan reaction solution from the sample using a 1-ml pipette and then add 500 μl of fresh O-glycan reaction solution. Combine these samples.
49| Transfer the resin back to the spin column, wash the microcentrifuge tube with 100 μl of DI water and transfer this to the spin column as well.
50| Centrifuge the sample at 2,000g for 30 s and collect the flow-through.
-
51| Wash the resin using 300 μl of DI water, centrifuge the sample at 2,000g for 30 s at room temperature and collect the flow-through. Repeat this step twice and combine all flow-through fractions. Dry the supernatant completely using a SpeedVac (37°C for 2-3 h).
▲ CRITICAL STEP The solution must be completely dried in order to evaporate the NH4OH. Any remaining solution will probably comprise basic reagents, which will confound the chloroform extraction in the following steps.
52| Resuspend the sample in 200 μl of 1% AcOH.
-
53| Add 400 μl of chloroform and vortex the samples at high speed. Remove the chloroform layer from the water layer and repeat this step three times.
▲ CRITICAL STEP Samples in AcOH-chloroform must be thoroughly mixed in order to completely dissolve them. In this step, the excess PMP is dissolved in chloroform, whereas the labeled O-glycans separate into the 1% AcOH layer. Sonication (~10 min at room temperature) may be used to facilitate dissolving.
-
54| Dry the aqueous layer (this contains the PMP-labeled O-glycans) using a SpeedVac (37°C for 2-3 h).
■ PAUSE POINT The dried sample is stable for at least 12 months when stored at −20°C.
55| Resuspend the sample in 1 ml of DI water.
56| Clean up the labeled O-glycans using an SPE C18 cartridge and the following procedure: add 1 ml of 100% ACN twice to the C18 column, wash the cartridge three times with 1 ml of water, load the sample onto the cartridge and collect and reload the flow-through onto the cartridge. Wash the samples four times using 1 ml of HPLC water and elute the PMP-labeled O-glycans with 200 μl of 50% ACN.
57| Transfer the eluate to a glass insert and dry the samples using a SpeedVac (37°C for 1-2 h).
-
58| Resuspend the samples in 100 μl of 0.2% FA.
? TROUBLESHOOTING
59| (Optional) To study the linkage, the released O-glycans can also be permethylated using the established methods56.
LC-MS analysis ● TIMING 3 h
-
60| Use an internal standard to estimate the concentration of glycans extracted from the biological sample. The total amount of glycans injected for LC-MS should be 10 ng-1 μg.
▲ CRITICAL STEP Maltoheptaose (DP7) is usually used for the internal standard, as it has ionization behavior similar to that of glycans. DP7 has a molecular weight (MW) of 1153.00 Da, which is used for absolute quantification of glycans.
61| Add the glycans to a sample vial to a final volume of 6-12 μl and place the sample vial in the LC autosampler.
-
62| Set LC and MS settings as listed below:
Item Parameter Value
LC pump Loading pump (μl/min) 5 Separation (Nanoflow pump) (μl/min) 0.25 LC gradient Linear gradient of 4-50% ACN (0.1% TFA) (min) 70 MS1 MS1 mass range (Da) 400-1,800 MS2 Collision energy (%) 29 Isolation width (m/z) 2 Activation time (ms) 0.2 High-energy collision dissociation (HCD) On Repeat count (dynamic exclusion) 2 Repeat duration (dynamic exclusion) (s) 25 Exclusion list size (dynamic exclusion) 500 Exclusion duration (dynamic exclusion) (s) 5
▲ CRITICAL STEP The MS2 parameters should be optimized if you are using a different mass spectrometer (we use the Axima Resonance from Shimadzu): the collision energy usually ranges from 20 to 40%; the isolation width can be increased.
-
63| Separate the glycans by one of the following methods: (i) hydrophilic interaction liquid chromatography (HILIC; apply a linear LC gradient from 90% ACN in 0.1% TFA to 0% within 90 min); (ii) use of a porous graphitized carbon (PGC; apply a linear LC gradient from 4 to 50% within 90 min) for intact glycans; and (iii) use of a C18 SPE trap column (apply a linear LC gradient from 4 to 50% within 90 min) for permethylated or isobaric labeled glycans.
▲ CRITICAL STEP Different analytical columns have specific properties on the separation of glycans. PGC may be the best for separation of isomers of glycans without permethylation. C18 can separate permethylated glycans well.
MALDI-MS ●TIMING 1 h
-
64| Add 1 μl of DHB matrix solution in the MALDI plate and immediately add 1 μl of sample from Step 58; dry the DHB sample at 37°C in an oven for 10 min.
▲ CRITICAL STEP Uniform crystals are usually formed when the sample is clean. When the sample contains contaminants, it usually forms a gel; it is recommended to use a PGC ziptip for quick cleanup.
-
65| Set the laser power to 100 (maximum = 180), two shots per location and 100 locations for a total of 200 profiles.
▲ CRITICAL STEP Optimize the laser power using internal standards (both DP7 and angiotensin or neurotensin).
66| Acquire MS spectra and analyze the MALDI data using Shimadzu Biotech Launchpad (2.9.3). Alternatively, the MALDI data can be exported to an mzXML or mzData file.
Data analysis ● TIMING 2 h
67| Convert the MS spectra (Xcalibur Raw File) to mzXML using the spectrum-converting software (MSConvert).
68| Establish a glycan database for N-glycans or O-glycans that includes glycan composition and precursor mass. Format templates for this database file are provided as supplementary information to this protocol (Supplementary Software 1 and 2). If no predefined database is available, a hypothetical database can be automatically generated using GIG Tool (version 0.0.1) by setting the upper limits of the number of monosaccharides in the composition—e.g., HexNAc (N) ≤ 20, Hexose (H) ≤ 20, Fucose (F) ≤ 10 and Neu5Ac (S) ≤ 10 for N-glycans; and N ≤ 10, H ≤ 10, F ≤ 10 and S ≤ 10 for O-glycans. The hypothetical database is used for precursor matching on the oxonium-containing spectra to identify matching glycan spectra.
-
69| Create a list of oxonium ions for data searching, including an essential oxonium list and a nonessential oxonium list. The list of oxonium ions is given below:
Glycan (N = N-linked; O = O-linked) m/z Essential
N 204.0866 Yes N 176.1167 No N 177.120055 No N 178.12341 No N 179.126765 No N 186.0761 No N 366.14 No N 138.055 No N 168.0655 No O 138.0544 No O 175.0865 No O 378.1656 No O 552.2455 No O 204.0857 No O 363.1543 No
-
70| Start a glycan search in the GIG Tool based on the precursor matching: use ‘N-linked’ for N-glycan searching and ‘O-linked’ for O-glycan searching (Supplementary Note).
▲ CRITICAL STEP The output file from the glycan search lists the MS scan number, charge, precursor mass, calculated mass, coverage, intensity of oxonium ions, reporter ions for QUANTITY-labeled N-glycans, candidate count, match count and all matched glycan compositions within the precursor mass tolerance.
? TROUBLESHOOTING
-
71| Determine the glycan structure from the MS/MS fragments using GIG Tool or commercial software such as GlycoWorkBench.
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 5 | Low yield of protein or peptide immobilization | The buffer contains amine groups, because a high concentration of amine-containing buffer was used before buffer exchange | Redo buffer exchange to reduce amine groups, minimizing amine reaction with aldehyde resin |
| 7A(iii) | Low solubility of peptides | Peptides are hydrophobic | Add ACN (10% in final solution; e.g., 10 μl in 90 μl of binding buffer) to dissolve the peptides if the binding buffer does not completely dissolve them |
| 15 | The resin aggregates after samples are added | The sample cannot interact with the surface of the resin | Use a pipette tip to disrupt aggregation; vortex and/or sonicate the resin for uniform suspension |
| 17 | Low immobilization of proteins | An insufficient amount of resin was used, the conjugation time was too short or the pH was too low (<7) | The maximum capacity of the resin is 20 mg/ml; increase the amount of resin, prolong the reaction time or adjust pH up to 10 |
| 24A(ii) | It is difficult to adjust pH | Some EDC is very viscous, even at elevated temperatures. This makes it difficult to add the exact amount of EDC to the solution | Use EDC from Sigma-Aldrich (cat. no. 39391). This EDC has low viscosity even at −20°C |
| 24A(v) | EDC-pT remains bound to the resin | The resin was washed or vortexed insufficiently | Add additional washing steps for each solution; vortex the resin in solution for up to 1 min |
| 44 | No N-glycans were identified | The sample contains salts or the aldehydes on the resin have not been blocked effectively | Use a PGC ziptip for glycan cleanup; if the resin has active sites on aldehydes, reblock the resin before PNGase F digestion |
| 58 | No O-glycans were detected | The pH of the solution was too high, so that O-glycan was dissolved in chloroform instead of in water | Check the pH of the resuspended O-glycan sample; if the pH is high (>7), use 100% TFA to lower the pH (<3) |
| 70 | An error occurred when selecting ‘run’ | The file directory was incorrect | Make sure that the right file directory is used for each Python command |
| 71 | The glycan structure cannot be determined by MS/MS | An insufficient number of MS/MS fragments were provided | In GlycoWorkBench, search for glycan structures with known modifications, based on accurate precursor m/z values. List the potential glycans from the database in GlycoWorkBench |
● TIMING
Steps 1-6, protein extraction-denaturation: 2 h
Steps 7-23, protein/peptide immobilization: 9 h
Step 24A, glycan protection: 5 h
Step 24B, glycan desialylation: 2 h
Steps 25-29, N-glycan release: 6 h
Steps 30-38, N-glycan purification: 5 h
Steps 39-44, N-glycan isobaric labeling: 8 h
Steps 45-59, O-glycan release: 32 h
Steps 60-63, LC-MS: 3 h
Steps 64-66, MALDI-MS: 1 h
Steps 67-71, data analysis (one raw data): 2 h
ANTICIPATED RESULTS
We have previously characterized steps for protein extraction, immobilization, modification, N-glycan release and labeling20,37. The release of O-glycans, modification of O-glycan sialic acids and sequence of release of N- and O-glycans were confirmed by the experiments below.
Proof of concept for GIG O-glycan analysis
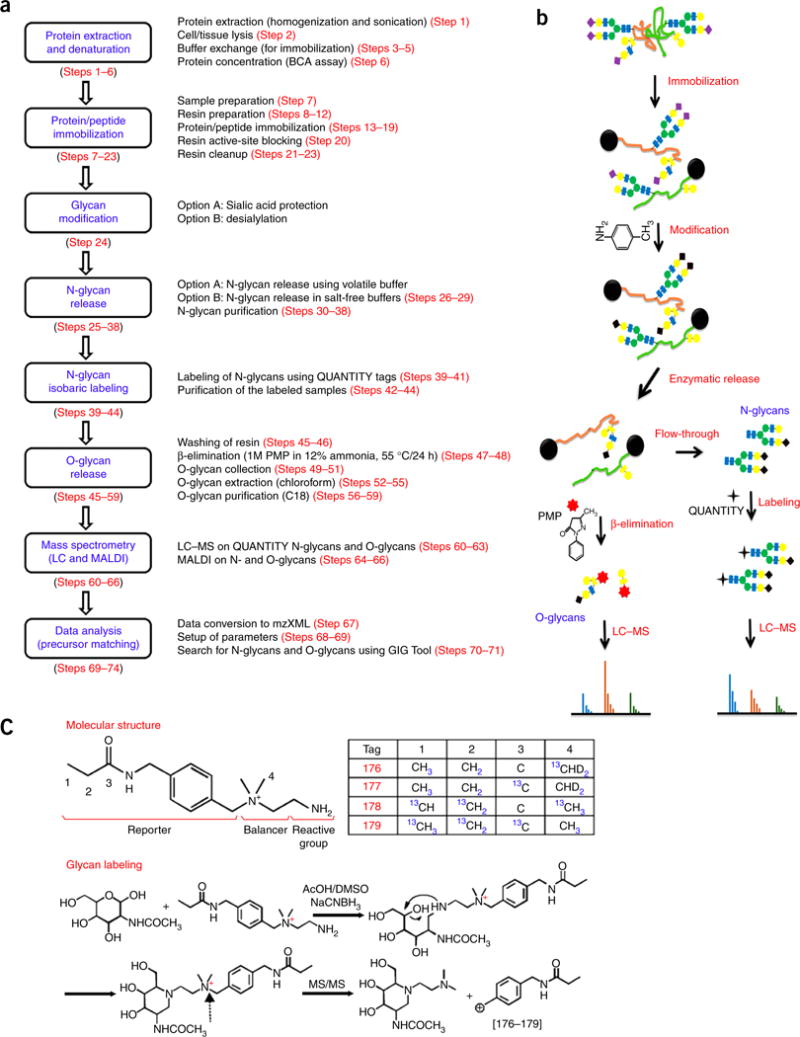
To study the release of O-glycans using the GIG approach, we used mucin from porcine stomach (Type III) (MPS3) for GIG O-glycan isolation. The O-glycans released from MPS3 that is covalently conjugated on the resin are depicted in Figure 2. Eighty O-glycans from MPS3 were detected by ESI-MS (Supplementary Table 1). The identified MPS3 O-glycans are also in agreement with reports in the literature38,57. MALDI-TOF MS analysis of this sample detected 41 O-glycans; however, none of them were sialylated38. In another study, >30 O-glycans were profiled using ESI-MS without SPE cleanup after chloroform extraction, with no sialic acids observed57. We think that the most probable explanation is that sialic acid is lost during MALDI ionization (see Introduction). Sialylated O-glycans were, however, detected by ESI-MS in a recent study that uses C18 for O-glycan cleanup58. On the basis of the manufacturer’s specifications for MPS3 (Sigma), we indeed detected additional sialylated O-glycans, including NH2S, N2H2S and FN2H2S, using GIG (where N = HexNAc, H = Hexose, S = Neu5Ac and F = Fucose). These results are evidence of the efficacy of identifying low-abundant sialylated O-glycans by chemical modification on a solid support.
Figure 2.
O-glycan profiling of mucin from porcine stomach (MPS) by ESI-MS. The MPS type III (MPS3) contains 0.5-1.5% sialic acid. A total of 80 O-GalNAc glycans were identified from MPS3. The identified O-glycans are consistent with what has been reported in the literature60. Image adapted with permission from ref. 37, Nature Publishing Group.
Sequential release of N-glycans and O-glycans
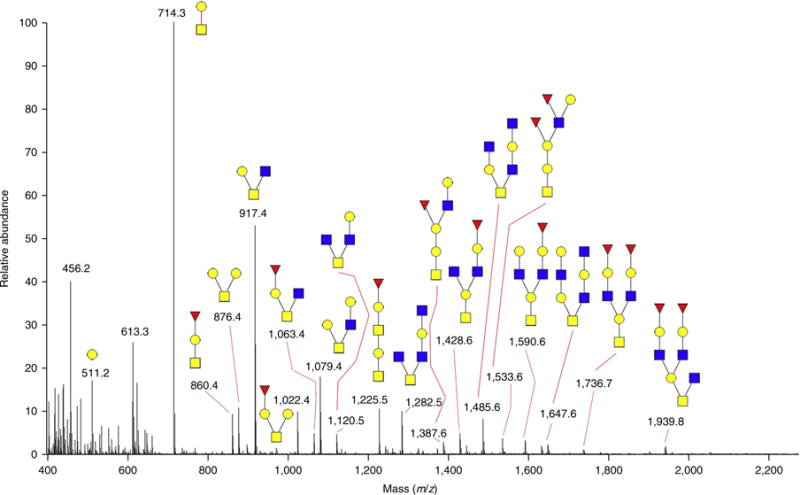
A protein sample from fetuin from bovine serum was conjugated on GIG resin to assess the release of N- and O-glycans by (i) β-elimination after PNGase F digestion, (ii) β-elimination without PNGase F digestion and (iii) PNGase F digestion after β-elimination. DP7 was spiked in as an internal standard for quantitative comparison of the three approaches.
As shown in Figure 3(i), fetuin N-glycans can efficiently be released by PNGase F. Further, O-glycans can be released by β-elimination in the presence of PMP57. Sialylated NHS is one of the most obvious O-glycans detected by MALDI or ESI with pT protection (Fig. 3(ii)), which is consistent with the results from recent chromatographic analysis59. Further digestion by PNGase F shows no discernible signal, suggesting the complete release of N-glycans in Figure 3(iii). Without PNGase F digestion, N-glycans were detected after 24-h β-elimination; N-glycans were also identified after 24-h β-elimination followed by PNGase F digestion. Therefore, N-glycans and O-glycans can be sequentially analyzed by PNGase F and β-elimination.
Figure 3.

Chemoenzymatic sequential release of N-glycans and O-glycans from bovine-serum-derived fetuin using GIG. (i) N-glycans are released from the solid phase by PNGase F. (ii) O-glycans are cleaved using ammonium hydroxide (mild β-elimination) in the presence of 0.5 M PMP (1-phenyl-3-methyl-5-pyrazolone). (iii) Any remaining N-glycans are released by further PNGase F digestion60. Image adapted with permission from ref. 60, BioMed Central (open-access license: https://creativecommons.org/licenses/by/4.0/).
Sialylated O-glycans
To demonstrate sialic acid modification on the GIG solid phase, proteins derived from a mucin sample from bovine submaxillary glands were immobilized on the solid phase20,21. MALDI-MS profiles (Fig. 4) were used to compare the most dominant sialylated O-glycans identified with sialic acid modification (Fig. 4a) and without modification (Fig. 4b). To estimate the signal between Figure 4a and Figure 4b, an internal peptide standard (neurotensin, Sigma) was spiked into the MALDI matrix (20 μM/1 μl). The intensity of neurotensin was approximately the same (2,000 mV) in Figure 4a and Figure 4b. As shown in Figure 4a, four major sialylated O-glycans were identified after pT modification, including NS, NG, N2S and N2G, which are listed in order of decreasing relative abundance. These results are consistent with findings reported in the literature17, and a similar protocol has recently been applied for analysis of glycosylation in ovarian cancer cells60.
Figure 4.
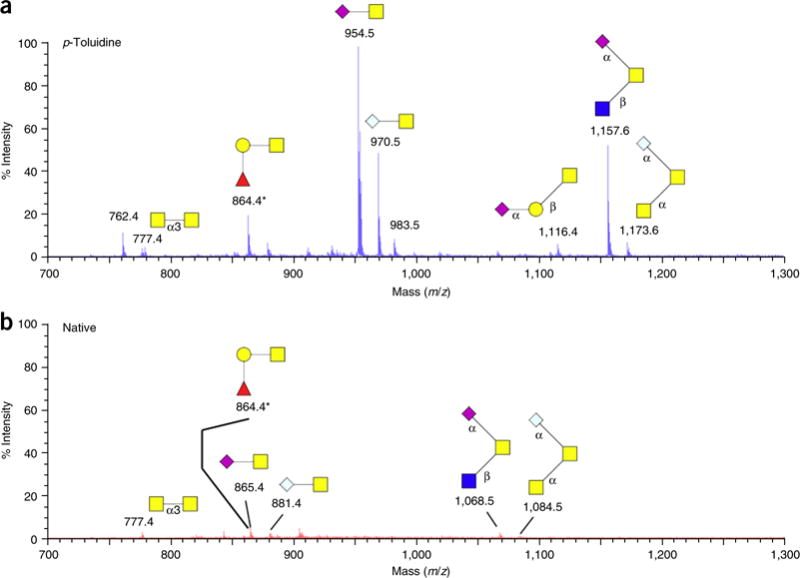
Sialylated O-glycans of mucin from bovine submaxillary glands by MALDI-MS. (a) The sialic acids that were stabilized by carbodiimide coupling have a substantially increased MS signal. (b) The sialic acids without modification have low intensity in MALDI-MS. An internal standard (neurotensin (20 μM/1 μl) was spiked into the sample. The sialic-acid-modified glycans have one sodium adduct [Na]+, whereas native glycans have an extra sodium adduct per sialic acid60. Image adapted with permission from ref. 60, BioMed Central (open-access license: https://creativecommons.org/licenses/by/4.0/).
Supplementary Material
Acknowledgments
We thank T. Stefani and P. Shah from Johns Hopkins University for help with LC-MS and Shimadzu for providing the instrument for MALDI-MS. This work was supported by the National Institutes of Health, National Cancer Institute, the Early Detection Research Network (EDRN; U01CA152813), the Clinical Proteomic Tumor Analysis Consortium (CPTAC; U24CA160036) and the National Institutes of Health, National Heart Lung and Blood Institute Programs of Excellence in Glycosciences (P01HL107153) and the Johns Hopkins Proteomics Center (N01-HV-00240).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper
AUTHOR CONTRIBUTIONS S.Y. and H.Z. designed the research. H.Z. and L.S. directed the project. S.Y. developed the experimental protocol and conducted the experiments. S.Y. wrote the manuscript. Y.H. developed the data analysis tools and provided support with the data analysis.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kellokumpu S, Sormunen R, Kellokumpu I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS Lett. 2002;516:217–224. doi: 10.1016/s0014-5793(02)02535-8. [DOI] [PubMed] [Google Scholar]
- 3.Dube DH, Bertozzi CR. Glycans in cancer and inflammation—potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 4.Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat Med. 1996;2:871–875. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- 5.Itoh N, et al. Analysis of N-glycan in serum glycoproteins from db/db mice and humans with type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;293:E1069–E1077. doi: 10.1152/ajpendo.00182.2007. [DOI] [PubMed] [Google Scholar]
- 6.Montpetit ML, et al. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc Natl Acad Sci USA. 2009;106:16517–16522. doi: 10.1073/pnas.0905414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang S, et al. Glycoproteins identified from heart failure and treatment models. Proteomics. 2015;15:567–579. doi: 10.1002/pmic.201400151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, et al. Overexpression of α (1, 6) fucosyltransferase associated with aggressive prostate cancer. Glycobiology. 2014;24:935–944. doi: 10.1093/glycob/cwu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peracaula R, Barrabés S, Sarrats A, Rudd PM, de Llorens R. Altered glycosylation in tumours focused to cancer diagnosis. Dis Markers. 2008;25:207–218. doi: 10.1155/2008/797629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci USA. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamid UMA, et al. A strategy to reveal potential glycan markers from serum glycoproteins associated with breast cancer progression. Glycobiology. 2008;18:1105–1118. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 12.Iyer R, Carlson DM. Alkaline borohydride degradation of blood group H substance. Arch Biochem Biophys. 1971;142:101–105. doi: 10.1016/0003-9861(71)90263-3. [DOI] [PubMed] [Google Scholar]
- 13.Cummings R, et al. Biosynthesis of N-and O-linked oligosaccharides of the low density lipoprotein receptor. J Biol Chem. 1983;258:15261–15273. [PubMed] [Google Scholar]
- 14.Takasaki S, Mizuochi T, Kobata A. Hydrazinolysis of asparagine-linked sugar chains to produce free oligosaccharides. Methods Enzymol. 1981;83:263–268. doi: 10.1016/0076-6879(82)83019-x. [DOI] [PubMed] [Google Scholar]
- 15.Patel T, et al. Use of hydrazine to release in intact and unreduced form both N-and O-linked oligosaccharides from glycoproteins. Biochemistry. 1993;32:679–693. doi: 10.1021/bi00053a037. [DOI] [PubMed] [Google Scholar]
- 16.Merry AH, et al. Recovery of intact 2-aminobenzamide-labeled O-glycans released from glycoproteins by hydrazinolysis. Anal Biochem. 2002;304:91–99. doi: 10.1006/abio.2002.5620. [DOI] [PubMed] [Google Scholar]
- 17.Zauner G, Koeleman CA, Deelder AM, Wuhrer M. Mass spectrometric O-glycan analysis after combined O-glycan release by beta-elimination and 1-phenyl-3-methyl-5-pyrazolone labeling. Biochem Biophys Acta. 2012;1820:1420–1428. doi: 10.1016/j.bbagen.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Mechref Y, Novotny MV. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Anal Chem. 2001;73:6063–6069. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
- 19.Honda S, et al. High-performance liquid chromatography of reducing carbohydrates as strongly ultraviolet-absorbing and electrochemically sensitive 1-phenyl-3-methyl5-pyrazolone derivatives. Anal Biochem. 1989;180:351–357. doi: 10.1016/0003-2697(89)90444-2. [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Li Y, Shah P, Zhang H. Glycomic analysis using glycoprotein immobilization for glycan extraction. Anal Chem. 2013;85:5555–5561. doi: 10.1021/ac400761e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S, Zhang H. Glycomic analysis of glycans released from glycoproteins using chemical immobilization and mass spectrometry. Curr Protoc Chem Biol. 2014;6:191–208. doi: 10.1002/9780470559277.ch140085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekiya S, Wada Y, Tanaka K. Derivatization for stabilizing sialic acids in MALDI-MS. Anal Chem. 2005;77:4962–4968. doi: 10.1021/ac050287o. [DOI] [PubMed] [Google Scholar]
- 23.Weiskopf AS, Vouros P, Harvey DJ. Electrospray ionization-ion trap mass spectrometry for structural analysis of complex N-linked glycoprotein oligosaccharides. Anal Chem. 1998;70:4441–4447. doi: 10.1021/ac980289r. [DOI] [PubMed] [Google Scholar]
- 24.Yang S, Zhang H. Glycan analysis by reversible reaction to hydrazide beads and mass spectrometry. Anal Chem. 2012;84:2232–2238. doi: 10.1021/ac202769k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah P, et al. Mass spectrometric analysis of sialylated glycans with use of solid-phase labeling of sialic acids. Anal Chem. 2013;85:3606–3613. doi: 10.1021/ac3033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powell AK, Harvey DJ. Stabilization of sialic acids in N-linked oligosaccharides and gangliosides for analysis by positive ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 1996;10:1027–1032. doi: 10.1002/(SICI)1097-0231(19960715)10:9<1027::AID-RCM634>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 27.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal Chem. 2014;86:5784–5793. doi: 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- 28.Chen P, Werner-Zwanziger U, Wiesler D, Pagel M, Novotny MV. Mass spectrometric analysis of benzoylated sialooligosaccharides and differentiation of terminal α2→ 3 and α2→ 6 sialogalactosylated linkages at subpicomole levels. Anal Chem. 1999;71:4969–4973. doi: 10.1021/ac990674w. [DOI] [PubMed] [Google Scholar]
- 29.North SJ, Hitchen PG, Haslam SM, Dell A. Mass spectrometry in the analysis of N-linked and O-linked glycans. Curr Opin Struct Biol. 2009;19:498–506. doi: 10.1016/j.sbi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marino K, Bones J, Kattla JJ, Rudd PM. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 31.Yang S, Rubin A, Eshghi ST, Zhang H. Chemoenzymatic method for glycomics: isolation, identification, and quantitation. Proteomics. 2016;16:241–256. doi: 10.1002/pmic.201500266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinhold V, Zhang H, Hanneman A, Ashline D. Toward a platform for comprehensive glycan sequencing. Mol Cell Proteomics. 2013;12:866–873. doi: 10.1074/mcp.R112.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Royle L, et al. HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal Biochem. 2008;376:1–12. doi: 10.1016/j.ab.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Young NM, et al. Structure of the N-linked glycan present on multiple glycoproteins in the Gram-negative bacterium, Campylobacter jejuni. J Biol Chem. 2002;277:42530–42539. doi: 10.1074/jbc.M206114200. [DOI] [PubMed] [Google Scholar]
- 35.Lauc G, Wuhrer M. High-Throughput Glycomics and Glycoproteomics: Methods and Protocols (Methods in Molecular Biology) Humana Press; 2017. [Google Scholar]
- 36.Stavenhagen K, Plomp R, Wuhrer M. Site-specific protein N-and O-glycosylation analysis by a C18-porous graphitized carbon-liquid chromatography-electrospray ionization mass spectrometry approach using pronase treated glycopeptides. Anal Chem. 2015;87:11691–11699. doi: 10.1021/acs.analchem.5b02366. [DOI] [PubMed] [Google Scholar]
- 37.Yang S, et al. QUANTITY: an isobaric tag for quantitative glycomics. Sci Rep. 2015;5:17585. doi: 10.1038/srep17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furukawa J, et al. A versatile method for analysis of serine/threonine posttranslational modifications by β-elimination in the presence of pyrazolone analogues. Anal Chem. 2011;83:9060–9067. doi: 10.1021/ac2019848. [DOI] [PubMed] [Google Scholar]
- 39.Huang D, Ou B, Hampsch-Woodill M, Flanagan JA, Prior RL. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J Agric Food Chem. 2002;50:4437–4444. doi: 10.1021/jf0201529. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Shah P, Zhang H. Solid phase extraction of N-linked glycopeptides using hydrazide tip. Anal Chem. 2013;85:10670–10674. doi: 10.1021/ac401812b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mach AJ, Kim JH, Arshi A, Hur SC, Di Carlo D. Automated cellular sample preparation using a centrifuge-on-a-chip. Lab Chip. 2011;11:2827–2834. doi: 10.1039/c1lc20330d. [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Ranish JA, Watts JD, Aebersold R. Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat Biotechnol. 2002;20:512–515. doi: 10.1038/nbt0502-512. [DOI] [PubMed] [Google Scholar]
- 43.Stöckmann H, Adamczyk B, Hayes J, Rudd PM. Automated, high-throughput IgG-antibody glycoprofiling platform. Anal Chem. 2013;85:8841–8849. doi: 10.1021/ac402068r. [DOI] [PubMed] [Google Scholar]
- 44.Shah P, et al. Integrated proteomic and glycoproteomic analyses of prostate cancer cells reveal glycoprotein alteration in protein abundance and glycosylation. Mol Cell Proteomics. 2015;14:2753–2763. doi: 10.1074/mcp.M115.047928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun S, et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat Biotechnol. 2015;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang S, et al. Integrated glycoprotein immobilization method for glycopeptide and glycan analysis of cardiac hypertrophy. Anal Chem. 2015;87:9671–9678. doi: 10.1021/acs.analchem.5b01663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin B, et al. Glycoengineering of Chinese hamster ovary cells for enhanced erythropoietin N-glycan branching and sialylation. Biotechnol Bioeng. 2015;112:2343–2351. doi: 10.1002/bit.25650. [DOI] [PubMed] [Google Scholar]
- 48.Yang W, et al. Glycoform analysis of recombinant and human immunodeficiency virus envelope protein gp120 via higher energy collisional dissociation and spectral-aligning strategy. Anal Chem. 2014;86:6959–6967. doi: 10.1021/ac500876p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung CY, et al. Integrated genome and protein editing swaps α-2, 6 sialylation for α-2, 3 sialic acid on recombinant antibodies from CHO. Biotechnol J. 2016;12:1600502. doi: 10.1002/biot.201600502. [DOI] [PubMed] [Google Scholar]
- 50.Do DC, et al. N-glycan in cockroach allergen regulates human basophil function. J Allergy Clin Immunol. 2017;139:AB167. doi: 10.1002/iid3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S, et al. Glycan analysis by isobaric aldehyde reactive tags and mass spectrometry. Anal Chem. 2013;85:8188–8195. doi: 10.1021/ac401226d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lauber MA, et al. Rapid preparation of released N-glycans for HILIC analysis using a labeling reagent that facilitates sensitive fluorescence and ESI-MS detection. Anal Chem. 2015;87:5401–5409. doi: 10.1021/acs.analchem.5b00758. [DOI] [PubMed] [Google Scholar]
- 53.Harvey DJ. Proteomic analysis of glycosylation: structural determination of N-and O-linked glycans by mass spectrometry. Expert Rev Proteomics. 2005;2:87–101. doi: 10.1586/14789450.2.1.87. [DOI] [PubMed] [Google Scholar]
- 54.Brockhausen I, Schachter H, Stanley P. Essentials of Glycobiology. 2nd. Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 55.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 56.Morelle W, Michalski JC. Analysis of protein glycosylation by mass spectrometry. Nat Protoc. 2007;2:1585–1602. doi: 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- 57.Wang C, Fan W, Zhang P, Wang Z, Huang L. One-pot nonreductive O-glycan release and labeling with 1-phenyl-3-methyl-5-pyrazolone followed by ESI-MS analysis. Proteomics. 2011;11:4229–4242. doi: 10.1002/pmic.201000677. [DOI] [PubMed] [Google Scholar]
- 58.Sic S, Maier NM, Rizzi AM. Quantitative fingerprinting of O-linked glycans released from proteins using isotopic coded labeling with deuterated 1-phenyl-3-methyl-5-pyrazolone. J Chromatogr A. 2015;1408:93–100. doi: 10.1016/j.chroma.2015.06.065. [DOI] [PubMed] [Google Scholar]
- 59.Windwarder M, Altmann F. Site-specific analysis of the O-glycosylation of bovine fetuin by electron-transfer dissociation mass spectrometry. J Proteomics. 2014;108:258–268. doi: 10.1016/j.jprot.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 60.Yang S, et al. Simultaneous analyses of N-linked and O-linked glycans of ovarian cancer cells using solid-phase chemoenzymatic method. Clin Proteomics. 2017;14:1–12. doi: 10.1186/s12014-017-9137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.