

Abstract
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. Neuronal apoptosis in the hippocampus has been detected after TBI. The hippocampal dysfunction may result in cognitive deficits in learning, memory, and spatial information processing. Our previous studies demonstrated that a p53 inhibitor, pifithrin-α oxygen analogue (PFT-α (O)), significantly reduced cortical cell death, which is substantial following controlled cortical impact (CCI) TBI, and improved neurological functional outcomes via anti-apoptotic mechanisms. In the present study, we examined the effect of PFT-α (O) on CCI TBI-induced hippocampal cellular pathophysiology in light of this brain region’s role in memory. To investigate whether p53-dependent apoptosis plays a role in hippocampal neuronal loss and associated cognitive deficits and to define underlying mechanisms, SD rats were subjected to experimental CCI TBI followed by the administration of PFT-α or PFT-α (O) (2 mg/kg, i.v.) or vehicle at 5 h after TBI. Magnetic resonance imaging (MRI) scans were acquired at 24 h and 7 days post-injury to assess evolving structural hippocampal damage. Fluoro-Jade C was used to stain hippocampal sub-regions, including CA1 and dentate gyrus (DG), for cellular degeneration. Neurological functions, including motor and recognition memory, were assessed by behavioral tests at 7 days post injury. p53, p53 upregulated modulator of apoptosis (PUMA), 4-hydroxynonenal (4-HNE), cyclooxygenase-IV (COX IV), annexin V and NeuN were visualized by double immunofluorescence staining with cell-specific markers. Levels of mRNA encoding for caspase-3, p53, PUMA, Bcl-2, Bcl-2-associated X protein (BAX) and superoxide dismutase (SOD) were measured by RT-qPCR. Our results showed that post-injury administration of PFT-α and, particularly, PFT-α (O) at 5 h dramatically reduced injury volumes in the ipsilateral hippocampus, improved motor outcomes, and ameliorated cognitive deficits at 7 days after TBI, as evaluated by novel object recognition and open-field test. PFT-α and especially PFT-α (O) significantly reduced the number of FJC-positive cells in hippocampus CA1 and DG subregions, versus vehicle treatment, and significantly decreased caspase-3 and PUMA mRNA expression. PFT-α (O), but not PFT-α, treatment significantly lowered p53 and elevated SOD2 mRNA expression. Double immunofluorescence staining demonstrated that PFT-α (O) treatment decreased p53, annexin V and 4-HNE positive neurons in the hippocampal CA1 region. Furthermore, PUMA co-localization with the mitochondrial maker COX IV, and the upregulation of PUMA were inhibited by PFT-α (O) after TBI. Our data suggest that PFT-α and especially PFT-α (O) significantly reduce hippocampal neuronal degeneration, and ameliorate neurological and cognitive deficits in vivo via antiapoptotic and antioxidative properties.
Keywords: Traumatic brain injury (TBI), p53, Pifithrin-α (PFT-α), PFT-α oxygen analogue (PFT-α (O)), Apoptosis, Motor and cognitive deficits, Puma, Controlled cortical impact (CCI)
1. Introduction
Traumatic brain injury (TBI) is a leading cause of death and long-term disability, particularly in children and young adults as well as in the elderly (Hyder et al., 2007; Rutland-Brown et al., 2006). Worldwide, in excess of 10 million suffer a TBI event making it a major public health problem consequent to a range encompassing physical disabilities long-term cognitive, behavioral dysfunction, psychological and social defects it causes (Rutland-Brown et al., 2006; Zaloshnja et al., 2008). This translates within the US, alone, to an estimated 1.4 million people that suffer a TBI annually, resulting in 235,000 hospitalizations and 50,000 deaths. In Taiwan, N100,000 people suffer a TBI annually, costing the economy some US $350 million (Chiu et al., 2011; Lin et al., 2008).
It is well known that TBI induces both short and long-term cognitive dysfunction resulting from neuronal loss within the hippocampus, which has been identified as a critical brain region involved in the biological basis of learning and memory (Scoville and Milner, 1957). Such injury is often sub-divided into two fundamental phases. An initial primary damage ensues at the moment of insult that includes contusion and laceration, diffuse axonal injury and intracranial hemorrhage, and causes immediate (necrotic) cell death, which is followed by an extended second phase. This involves cascades of biological processes initiated at the time of injury that may persist over later days, weeks and perhaps, months, resultant to neuroinflammation, glutamate toxicity, ischemia, oxidative stress, astrocyte reactivity, blood-brain barrier changes, cellular dysfunction and apoptosis (Diaz-Arrastia et al., 2014; Greig et al., 2014). Previous studies found that hippocampal-associated learning and memory impairment were particularly vulnerable to secondary injury following TBI (Ariza et al., 2006; Compagnone et al., 2009; Hicks et al., 1993). A diverse array of animal TBI models, including controlled cortical impact (CCI) injury (Anderson et al., 2005; Hall et al., 2005; Myer et al., 2006), lateral fluid percussion (LFP) (Lowenstein et al., 1992; Thompson et al., 2005) and weight drop-induced closed diffuse injury (Golarai et al., 2001; Isaksson et al., 2001; Rachmany et al., 2013), have shown that TBI induces neuronal apoptosis in hippocampus. One study reported that hippocampal CA1, CA3 and dentate gyrus (DG) neurons were significantly decreased in the ipsilateral injury site at 7 days after TBI (Myer et al., 2006). They found severe CCI injury caused extensive neuronal death, rapid loss of cortical tissue and consistent degeneration of hippocampal regions. Currently, there is no effective treatment to restore cognitive functions.
It is important to understand the mechanism of hippocampal neuronal apoptosis underlying learning and memory impairment following TBI. Apoptosis in neurons requires PUMA translocation to mitochondria and binding to anti-apoptotic Bcl-2 family members, thus freeing Bax or Bak to ultimately cause mitochondrial dysfunction. Subsequently, cytochrome c is released from mitochondria to assemble in apoptosomes with apoptotic protease activating factor 1 (APAF1), leading to caspase-dependent cell death (Culmsee and Mattson, 2005). For the initiation of apoptosis in neurons, the tumor suppressor and transcription factor p53 is an important upstream molecule for this mitochondrial mechanism (Culmsee and Mattson, 2005). A number of in vivo and in vitro studies have reported a relationship between increased protein/mRNA levels of p53 and neuronal damage. In this regard, neuronal p53 expression was induced in rodent models of mild cerebral concussion injury (Tashlykov et al., 2009), CCI injury (Plesnila et al., 2007), neonatal ischemic injury (Nijboer et al., 2011), and focal reversible cerebral ischemia (Leker et al., 2004). In addition, an elevation of p53 mRNA/protein levels within hippocampal regions has been reported in TBI and transient global ischemic brain injury animal models (Muir et al., 1999; Napieralski et al., 1999; Niizuma et al., 2009; Schober et al., 2010). Notably, in a mouse model of global cerebral ischemia, there was less degeneration of CA1 neurons in p53-deficient mice (p53−/−) than in wild type mice (p53+/+) (Yonekura et al., 2006), indicating that p53 is a gatekeeper in the biochemical cascade leading to neuronal death. In this regard, Hong et al. reported that TBI-induced phosphorylation of p53 in hippocampus results in the initiation of the process leading to apoptosis and cognitive deficits (Hong et al., 2012).
A stable, water-soluble p53 inhibitor, pifithrin alpha (PFT-α) has been isolated from a chemical library in a screen to reversibly block p53-dependent transcriptional activation and apoptosis associated with anticancer drug treatment in a mouse ConA cell line containing a lacZ reporter gene under the control of a p53-responsive promoter (Komarov et al., 1999). Translation of this approach to cultured neuronal cells demonstrated that PFT-α could mitigate p53-dependent death in neural cells, including that due to glutamate excitotoxicity, amyloid-β peptide and hypoxia, and resulted in the generation of yet more potent tetrahydrobenzothiazole-based analogues (Zhu et al., 2002; Culmsee et al., 2001; Greig et al., 2004). Studies subsequently have reported that p53 inhibition by PFT-α suppressed p53-regulated apoptosis genes, including PUMA and BAX, and reduced neuronal dysfunction and loss in ischemic reperfusion injury and stroke (Leker et al., 2004; Gupta et al., 2007; Luo et al., 2009), TBI (Plesnila et al., 2007; Yang et al., 2015), Huntington’s disease (Bae et al., 2005) and Parkinson’s disease (Duan et al., 2002) models. Moreover, a significant increase in the number of surviving neurons in the hippocampal CA1 region was observed in ischemic animals treated with PFT-α (Gupta et al., 2007). Likewise, PFT-α administration an hour following injury ameliorated cognitive impairments following mild TBI-induced secondary injury expansion after weight drop (Rachmany et al., 2013).
The stability and activity of the novel p53 inhibitor, PFT-α oxygen analogue (PFT-α (O)) has been found to be more effective than PFT-α in cellular studies (Greig et al., 2004; Zhu et al., 2002). Our previous study demonstrated that in a well-characterized CCI rat model of TBI that resulted in substantial primary injury to the nearby cerebral cortex, PFT-α (O) significantly reduced cortical cell death and improved neurological functional outcome via anti-apoptotic mechanisms (Yang et al., 2015). In the present study, in light of the role of the hippocampus on cognition and memory we compared the activity of PFT-α (O) with PFT-α on measures of cognitive functional recovery and hippocampal neuronal damage in the same CCI model of TBI, to evaluate whether blocking p53 transcriptional actions could reduce hippocampal neuronal apoptosis and, thereby, mitigate cognitive deficits resulting from moderate TBI. Our hypothesis is that the vast majority of TBI-induced neuronal cell death occurring within the hippocampus results from secondary apoptotic, rather than primary necrotic cell death, and is thereby amenable to rescue by inactivating the gatekeeper, p53, to the apoptotic cascade.
2. Methods
2.1. Animals
Male Sprague-Dawley (SD) rats (250–300 g, body weight) were used in accordance with the international guidelines for animal research. The study design was approved by the Animal Ethics Committee (Approval number LAC-2015-0051) of Taipei Medical University. All animals were kept three per cage under a constant 12-h light/dark cycle, at room temperature (21–25°C) and humidity (45–50%). Food and water were available ad libitum.
2.2. Animal model of traumatic brain injury (TBI)
The CCI injury procedure was performed as described previously (Chen et al., 2008; Yang et al., 2015). Rats were randomly placed into 4 groups ((i) sham injury, (ii) CCI+ vehicle, (iii) CCI + PFT-α and (iv) CCI+ PFT-α (O)). A CCI instrument with a rounded metal tip (5 mm diameter), an impact velocity of 4 m/s and a deformation depth of 2 mm below the dura was used. Body temperature was monitored throughout surgery by using a rectal probe; the temperature was maintained at 37.0 ± 0.5° C using a heated pad during recovery from anesthesia. Thereafter, animals were euthanized 24 h or 7 days after TBI and their brains were processed for the following experiments to examine the neuroprotective effects of p53 inhibitors. These analysis times are in accord with peak levels of apoptosis, as well as behavioral and imaging abnormalities and allow comparison with other studies (Obenaus et al., 2007; Plesnila et al., 2007; Luo et al., 2009; Yang et al., 2015).
2.3. Drug administration
Synthesis of the p53 inhibitors, PFT-α (1-(4-methylphenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzothiazolyl)ethanone hydrobromide salt) and the oxygen-containing analogue PFT-α (O) (1-(4-methylphenyl)-2-(4,5,6,7-tetrahydro-2-imino-.3(2H)-benzoxazolyl)ethanone hydrobromide salt) was achieved by a published synthetic route (Zhu et al., 2002) (Fig. 1A). Chemical characterization confirmed the structures of the final products with a high purity (>99.5%) (for information on PFT-α (O), contact NHG <greign@mail.nih.gov>). Animals were given PFT-α or PFT-α (O) (2 mg/kg, i.v.) or vehicle (10% DMSO in saline) at 5-h post-injury. No temperature changes were observed after PFT-α or PFT-α (O) administration. The time window for administration of PFT-α or PFT-α (O) was selected on the basis of a previous study (Yang et al., 2015), in which times of 5-h and 7-h post-injury administration were tested; the 5-h administration showed maximal neuroprotective actions.
Fig. 1.
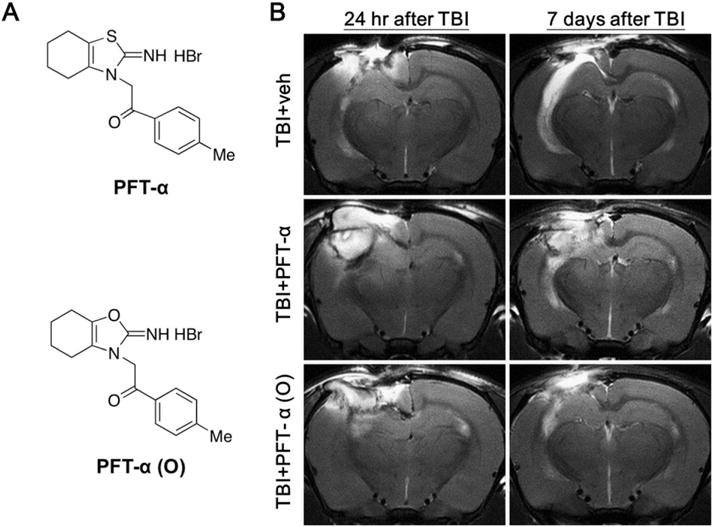
PFT-α (O) treatment substantially reduced TBI induced hippocampal injury. (A) Chemical structures of PFT-α and PFT-α (O). (B) Representative spin-spin relaxation time (T2)-weighted MRI images from rats in the TBI + veh, TBI + PFT-α and TBI + PFT-α (O) groups evaluated at 24 h and 7 days post-injury (sham animals, as anticipated, lacked hyperintensities or damage and are not shown). As illustrated in the two top images, vehicle-treated TBI animals had severe ipsilateral hippocampal tissue damage, as evidenced by hyperintensity abnormalities at 24 h to 7 days following CCI. In comparison at 7 days after TBI, a single post-treatment at 5 h with PFT-α (center panel) and, in particular, PFT-α (O) (lower panel) resulted in less damage to the ipsilateral hippocampus versus TBI + veh.
2.4. Cognitive behavioral tests
Cognitive behavioral testing was performed in rats before CCI and at 7 days after CCI. The effects of TBI on cognitive performance were assessed using the following behavioral paradigms: novel object recognition (NOR) and an open field test (OFT). These assessments were conducted once per day at approximately the same time during the light phase of the cycle. All tests were performed by an observer blinded to the experimental groups.
2.4.1. Novel object recognition paradigm
The NOR task was used to evaluate short-term recognition memory, based upon the original Ennaceur and Delacour procedure (Ennaceur and Delacour, 1988). The NOR procedure consists of three phases: habituation, familiarization, and a test phase. The NOR testing chamber was an open-field black plexiglass arena (60 × 60 × 100 cm in dimension). A video camera fixed on the wall above the chamber recorded the test phase for analysis. In the habituation phase, each rat was allowed 10 min to freely explore the empty arena over each of three days. After 3 days, in the familiarization phase, each rat was permitted to explore and become familiar with two objects (A + A) that were placed in symmetrical positions within the arena for 10 min. A test trial was then performed 1 h following the familiarization phase. During this test trial, one of the now familiar objects (A) was replaced by a novel one (B), and each animal was again placed within the arena and allowed to explore for 10 min with the two objects (A + B). A tracking system (EthoVision XT 11.0) was used to quantify the duration of time spent with each object. A rat was scored as exploring an object when its nose approached an object at a distance of <2 cm. Grounded on the innate tendency of rodents to be inquisitive, a normal (uninjured) animal will spend more time exploring a novel object than a familiar one. The calculation of a discrimination index has been described in previous studies (Mugwagwa et al., 2015), where the discrimination index % = [(time (in seconds) spent exploring the novel object)/(total time (in seconds) spent exploring both objects)] × 100%. An uninjured normal rat should be able to discriminate between the old and novel objects and perform better than 50%. The arena and all objects were thoroughly cleansed with 70% ethanol between each evaluation to prevent odor recognition impacting the discrimination index.
2.4.2. Open field test
The OFT is often used to measure general locomotor activity and anxiety-like behavior (Prut and Belzung, 2003). In the OFT, each rat was monitored by a video camera in an open field black plexiglass arena (60 × 60 × 100 cm in dimension). The total distance traveled and the movement time of each animal was recorded during a 10 min testing period at 7 days after CCI. Each trial was recorded and analyzed using the EthoVision XT 11.0 tracking system. The testing arena was thoroughly cleansed with 70% ethanol between each testing period for each rat.
2.5. Brain magnetic resonance imaging (MRI) analysis
Animals from each treatment group were evaluated by MRI at 24 h and 7 days post TBI. Each rat was anesthetized with 2.5% isoflurane by mechanical ventilation and placed in a dedicated holder and positioned at the isocenter of a 7.0 Tesla small animal MRI scanner (70/16 PharmaScan, Bruker Biospin GmbH, Germany) with a 72-mm volume coil as the transmitter and a rat surface coil as the receiver. The brain was scanned from the brain stem to the olfactory bulb with fast spin-echo T2-weighted imaging pulse sequences (TR/TE, 3000/37 ms). All images were multi-slice images acquired with a field of view of 20 × 20 mm and with a slice thickness of 1 mm with no gap. The pixel matrix was 256 × 256. These original Bruker images were then converted to DICOM format with the software program (Paravision 6.0.1) included with the scanner.
2.6. Brain tissue preparation and staining
Following terminal anesthesia, depending on whether biochemical or histological analyses were to be performed, select areas of fresh brain tissue were rapidly removed without fixation after cervical dislocation. These were snap-frozen in liquid nitrogen and stored at −70°C until use. Alternatively, rats were transcardially perfused initially with phosphate-buffered saline (PBS) and then with 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight and transferred to PBS containing 30% sucrose and 0.1% sodium azide (Sigma Chemical Co., St Louis, MO, USA) for cryoprotection (with all solutions maintained at pH 7.4 and 4C.) and were routinely processed and paraffin embedded for histological evaluation.
2.6.1. Hematoxylin and eosin staining
Before Fluoro Jade C (FJC) or double immunofluorescence staining, paraffin embedded brain sections were de-waxed and rehydrated, and stained with hematoxylin and eosin (HE), according to standard protocols. The histological results were blindly examined under light microscopy and used to locate the hippocampal regions for observation.
2.6.2. Fluoro Jade C staining
To visualize degenerating neurons, FJC staining was performed as previously described (Yang et al., 2015). Specifically, a modified FJC ready-to-dilute staining kit was used according to the manufacturer’s instructions (Biosensis, TR-100-FJ, Thebarton, South Australia). De-waxed slides were incubated in a potassium permanganate solution (1:15) for 10 min, rinsed in distilled water for 2 min, and then incubated in FJC solution (1:25) for 30 min. The slides were thereafter washed and mounted on coverslips with Vecta-shield mounting medium (Vector, Burlingame, CA, USA). To quantify FJC-positive cells, three sections from each animal were evaluated and representative images were captured using a fluorescence microscope with a blue (450–490 nm) excitation light.
2.7. Double immunofluorescence staining
To evaluate co-localization of neuronspecific nuclear protein (NeuN) with p53, annexin V, or 4-hydroxynonenal (4-HNE) and PUMA with COX IV, we performed double immunofluorescence staining. For immunofluorescence staining, adjacent sections to those stained with HE were then blocked for 60 min in 5% BSA (Sigma Chemical Co., St Louis, MO, USA). Thereafter, the following primary antibodies were used and incubated overnight at 4°C: (i) mouse monoclonal anti-NeuN (Millpore; 1:500), (ii) rabbit polyclonal anti-p53 (GeneTex; 1:500), (iii) rabbit polyclonal anti-annexin V (Abcam; 1:500), (iv) rabbit polyclonal anti-4-HNE (Abcam; 1:250), (v) rabbit polyclonal anti-PUMA (Abcam; 1:1000), and (vi) mouse monoclonal anti-COX IV (Abcam; 1:250) antibody. Following incubation with the primary antibody, the sections were washed and incubated with Alexa Fluor® 488 goat anti-rabbit IgG (Jackson; 1:200) and Alexa Fluor® 594 anti-mouse IgG (Jackson; 1:200) at room temperature for 2 h. Sections were then mounted with Mounting Medium H-1000 (Vector Laboratories). The numbers of NeuN/p53-, annexin V-, 4-HNE and PUMA-COX IV positive cells were counted in three sections by means of SPOT image analysis software (Diagnostic Instruments, Sterling Heights, MI). Control samples were generated by omitting the respective primary antibodies.
2.8. Quantitative reverse transcription-PCR (qRT-PCR)
Approximately 20 mg of fresh hippocampal tissue was immediately collected from the ipsilateral (injured) hemisphere of euthanized rats, and processed for real-time quantitative RT-PCR. Total RNA was extracted by using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). The purity and quality of RNA were confirmed by defining the ratio of absorbance at 260 and 280 nm wavelengths (NanoDrop® ND-1000, Thermo Scientific). A sample of 3 μg total RNA was treated with ReverTra Ace set (PU-TRT-100; Purigo) and reverse transcribed into cDNA. For mRNA measurement; diluted cDNA was amplified using the Rotor-Gene SYBR Green PCR Kit (Qiagen) in a Rotor-Gene Q 2plex HRM Platform (Qiagen). Reaction conditions were carried out for 35–40 cycles (5 min at 95°C, 5 s at 95°C and 10 s at 60°C). The primers used for the qRT-PCR assay were the following:
caspase-3, 5′-AATTCAAGGGACGGGTCATG-3′ (forward) and 5′-GCTT GTGCGCGTACAGTTTC-3′ (reverse);
p53, 5′-CCA TGG CCA TCT ACA AGA AGT-3′ (forward) and 5′-TGTC GTCCAGATACTCAGCATA-3′ (reverse);
PUMA, 5′-CATGGGACTCCTCCCCTTAC-3′ (forward) and 5′-CACCTA GTTGGGCTCCATTT-3′ (reverse);
BAX, 5′-GTGAGCGGCTGCTTGTCT-3′ (forward) and 5′-GTGGGGGTCC CGAAGTAG-3′ (reverse);
Bcl-2, 5′-GTACCTGAACCGGCATCTG-3′ (forward) and 5′-GGGGCCAT ATAGTTCCACAA-3′ (reverse);
SOD1, 5′-CGGCTTCTGTCGTCTCCTTGC-3′ (forward) and 5′-GTTCAC CGCTTGCCTTCTGC-3′ (reverse);
SOD2, 5′-ACGCGACCTACGTGAACAATCT-3′ (forward) and 5′-CAGT GCAGGCTGAAGAGCAA-3′ (reverse);
β-actin, 5′-GACCCAGATCATGTTTGAGACCTTC-3′ (forward) and 5′-GGTGACCGTAACACTACCTGAG-3′ (reverse). The comparative threshold cycle (Ct) value of the β-actin gene was used as reference. Relative transcript expression of mRNA levels was calculated by the Ct method.
2.9. Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Between-group comparisons were made by one-way analysis of ANOVA with a post hoc test (Bonferroni) to determine individual group differences. Differences between means were assessed at the probability level of P ≤ 0.05, 0.01, and 0.001. Sigma Plot and Stat version 2.0 were used for all analyses and generation of bar graphs.
3. Results
3.1. Post-injury treatment with PTF-α and the PFT-α (O) analogue reduced hippocampal injuries caused by CCI (TBI)
MRI images were acquired at 24 h and 7 days from rats to assess progressive hippocampal tissue damage induced by CCI (TBI). Representative T2-weighted MRI images from animals with TBI and treated with vehicle showed clearly visualized brain injuries by the apparent hyperintensities in their ipsilateral hippocampus at 24 h following injury; sham animals, as expected, lacked hyperintensities or damage. The injury area in vehicle treated TBI rats was further increased at 7 days following TBI, when compared to contralateral hippocampus (Fig. 1B). By contrast, rats challenged with TBI and treated with PFT-α and, particularly, with PFT-α (O) had a comparatively smaller injury volume in ipsilateral hippocampus versus vehicle-treated TBI animals (Fig. 1B). This qualitative visual finding provided a basis for the subsequent quantitative evaluation of PFT-α (O) and PFT-α treatment on TBI induced damage.
On histological evaluation of areas within the hippocampus (Fig. 2 A and B), FJC-positive cells with neuronal morphology were evident 24 h after CCI in the CA1 and DG regions (Fig. 2 C and D). Notably, there was a significant decrease in the number of FJC-positive cells in both TBI + PFT-α and TBI + PFT-α (O) groups in the CA1 region (Fig. 2 E; P < 0.001). However, only PFT-α (O), but not PFT-α treatment, significantly decreased FJC-positive cells within the DG region of hippocampus (Fig. 2 F; P < 0.01). Remarkably, FJC number within both CA1 and DG regions in the TBI + PFT-α (O) group was not significantly different from shams without TBI challenge.
Fig. 2.
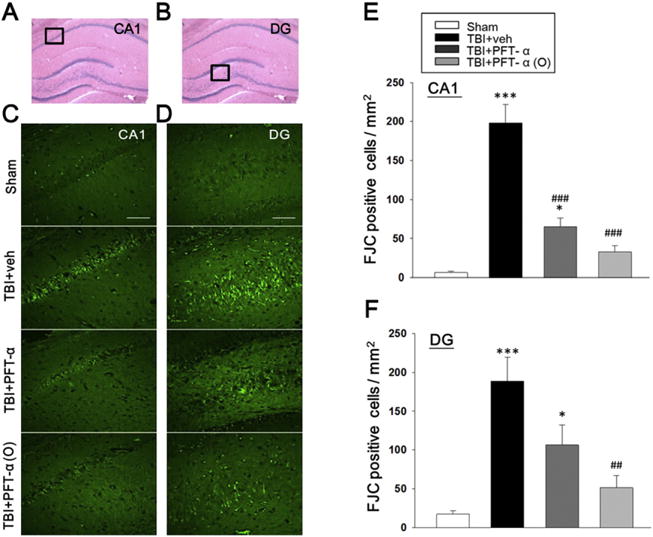
Post-injury administration of PFT-α (O) at 5 h after TBI significantly decreased FJC positive cells in the CA1 and dentate gyrus (DG) regions of the hippocampus at 24 h. (A, B) Representative HE-stained coronal sections showing the hippocampal CA1 and DG areas, respectively, as indicated by the black square boxes (the brain sections shown are from a sham control). These two areas were further evaluated to compare the fluorescent signals between the 4 groups of rats in FJC-stained sections. (C) Photomicrographs of FJC-stained CA1 regions of interest in the four groups. (D) Photomicrographs of FJC-stained DG regions. (E) There was a significant decrease in the number of FJC-positive cells in both TBI + PFT-α and TBI + PFT-α (O) groups in CA1 regions. (F) PFT-α (O) treatment significantly decreased FJC-positive cells in the DG region of hippocampus. The total number of FJC-positive cells was expressed as the mean number per field of view (0.259 mm2). Data are expressed as mean ± SEM. *p < 0.05, ***p < 0.001 versus sham group; ## p < 0.01, ### p < 0.001 versus TBI + veh group, analyzed by one-way ANOVA. Bar = 100 μm. (n = 5 in each group).
3.2. Post-injury treatment of PFT-α or PFT-α (O) at 5 h ameliorated cognitive deficits and anxiety-like behaviors after TBI
The NOR paradigm was used to examine visual recognition memory of rats in our study (Fig. 3A). A cognitive deficit, measured by the NOR test after CCI, was evident in vehicle-treated animals at 7 days. Specifically, 7 days after the TBI event the TBI + vehicle group exhibited impairments in visual memory as reflected in a discrimination index that was significantly lower than the sham group (discrimination indexes ~62% vs. ~48%, P < 0.05, Fig. 3B). In contrast, TBI rats treated with PFT-α (O) 5 h post injury demonstrated complete mitigation of any loss of visual memory, as indicated by the higher percentage of time devoted to the exploration of the novel object in comparison to injured rats treated with vehicle (discrimination index ~60% vs. ~48%, P < 0.05, Fig. 3B). The open field activity for sham injury, TBI+ vehicle, TBI + PFT-α and TBI + PFT-α (O) rats are illustrated in Fig. 4. Behavioral measures reported include movement pathway traveled within the chamber on day 7 (Fig. 4A). Rats in the TBI-challenged vehicle and PFT-α groups were significantly less active than the sham group in relation to locomotor activity and total distance traveled in the arena (Fig. 4B; P < 0.05). This TBI-induced deficit was ameliorated by administration of the PFT-α (O) 5 h post injury (Fig. 4B). In addition, post-injury treatment with PFT-α (O) produced a statistically significant increase in the time spent exploring compared to the vehicle and PFT-α group (Fig. 4C; P < 0.05).
Fig. 3.
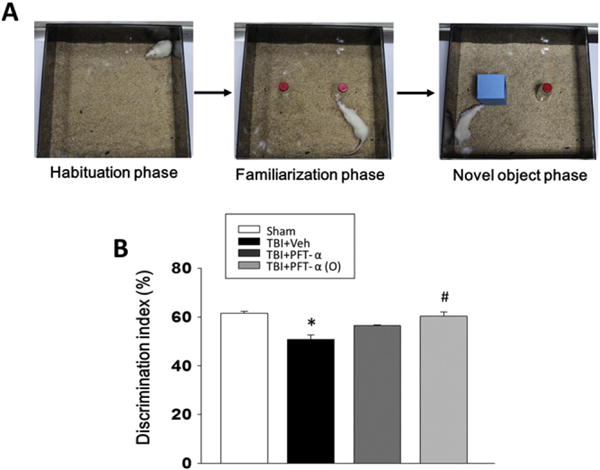
Post-injury administration of PFT-α (O) at 5 h after TBI prevents object recognition memory loss at 7 days after TBI. (A) The novel object recognition (NOR) task consists of three phases: habituation, familiarization, and a novel object test phase. In the habituation phase, each rat was allowed 10 min to freely explore the empty arena for 3 days. In the familiarization phase, two alike objects were assessed for 10 min. One hour later, one of the initial (old) objects was randomly replaced by a novel one, and the rat exploratory behavior was analyzed over a 10 min period. (B) PFT-α (O) treatment reverses cognitive impairment as indicated by spending more time devoted to the exploration of the novel object in comparison to rats treated with vehicle. Data are expressed as mean ± SEM. *p < 0.05 versus sham group; # p < 0.05 versus TBI + veh group, analyzed by one-way ANOVA (n = 5 in each group).
Fig. 4.
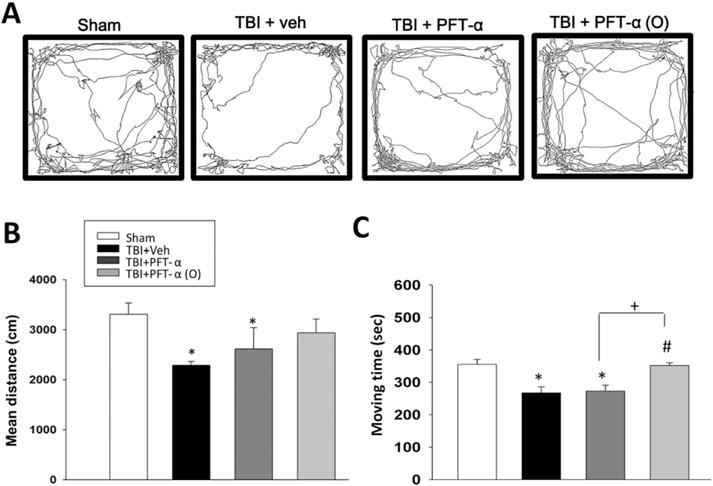
Post-injury administration of PFT-α (O) at 5 h mitigates anxiety-like behavior at 7 days after TBI. The open field test (OFT) is often used to measure general locomotor activity and anxiety-like behavior. (A) Representative traces of rat movement during performance of an OFT. (B) The mean overall distance traveled by the TBI vehicle (Veh) rats was significantly lower than the sham group. However, the mean distance was not different between sham and PFT-α (O)-treated TBI groups. (C) Notably, the PFT-α (O) treated group showed a significantly increased moving time as compared to the TBI + Veh and TBI + PFT-α groups. Data are expressed as mean ± SEM. (n = 5 in each group). *p < 0.05 versus sham group; # p < 0.05 versus TBI + Veh group; +p < 0.05 vs. TBI + PFT-α group.
3.3. Post-injury PFT-α (O) treatment downregulated caspase-3, p53, and PUMA, and upregulated SOD2 gene mRNA expressions in the hippocampus
To examine whether PFT-α or PFT-α (O) would inhibit transcriptional activity of p53, we compared caspase 3, p53 and p53-regulated pro-apoptosis gene mRNA levels (BAX, PUMA and BCL-2) in sham, and TBI-challenged vehicle, PFT-α and PFT-α (O) treated groups by reverse transcription followed by real-time PCR (quantitative RT-PCR). Our results indicated that post-injury treatment with PFT-α or PFT-α (O) reduced caspase3 and PUMA mRNA expression in rat hippocampus following CCI (Fig. 5A and C). Post-injury treatment with PFT-α (O) but not PFT-α, additionally, reduced p53 and increased SOD2 mRNA expression in rat hippocampus following CCI (Fig. 5B and D; P < 0.05). Indeed, PFT-α (O) elevated SOD2 mRNA versus the PFT-α group (P < 0.05), further differentiating these two treatments.
Fig. 5.
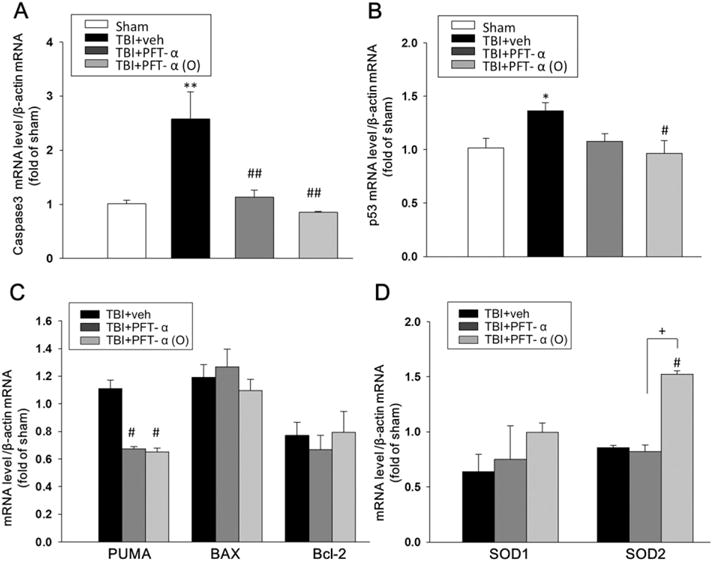
Treatment of PFT-α (O) at 5 h after TBI significantly decreased caspase-3, p53, PUMA and increased SOD2 mRNA expression in the hippocampus at 24 h. The mRNA levels of (A) caspase-3, (B) p53, (C) PUMA, BAX, Bcl-2 (D) SOD1 and SOD2 in hippocampal tissue from sham and TBI challenged vehicle (Veh) treated, and PFT-α or PFT-α (O) groups were analyzed by RT-PCR. Results are expressed as a fold change relative to that of sham animals. PFT-α (O) significantly reduced TBI-induced caspase-3, p53 and PUMA, and elevated SOD2 mRNA expressions in the ipsilateral hemisphere compared with vehicle-treated rats. However, PFT-α (O) did not change the level of the BAX, Bcl-2 and SOD1 genes. Data are expressed as means ± SEM (n = 5 in each group). * p < 0.05,**p < 0.01 vs. sham; #p < 0.05, ##p < 0.05vs. TBI + veh group; + p < 0.05 vs. TBI + PFT-α group.
3.4. Post-injury PFT-α or PFT-α (O) treatment reduced p53 and annexin V protein expression in hippocampal neurons
To examine the effect of PFT-α or PFT-α (O) on p53 protein expression, immunohistochemistry was used to evaluate gene translation 24 h after injury in the hippocampal region. Our results indicated that p53 positive cells did not co-localize with astrocytes (figure not shown). In light of this, we evaluated whether the p53 protein was expressed in neurons (Fig. 6A). We found that p53 co-localized to neurons and that p53-positive staining neuron levels in the hippocampus were significantly elevated at 24 h following CCI injury (Fig. 6C; P < 0.001). We additionally assessed annexin V staining to examine apoptosis in neurons, which was largely absent in sham but clearly evident in vehicle-treated TBI challenged animals (Fig. 6B). Quantitative analysis revealed a 25% and 70% reduction in annexin V-positive neurons in PFT-α and PFT-α (O) treated TBI groups, respectively, as compared to TBI + vehicle, which represented a significant decrease (Fig. 6D; P < 0.001). Notably, treatment with PFT-α (O) showed fewer apoptotic neurons than the PFT-α treated group (P < 0.001).
Fig. 6.
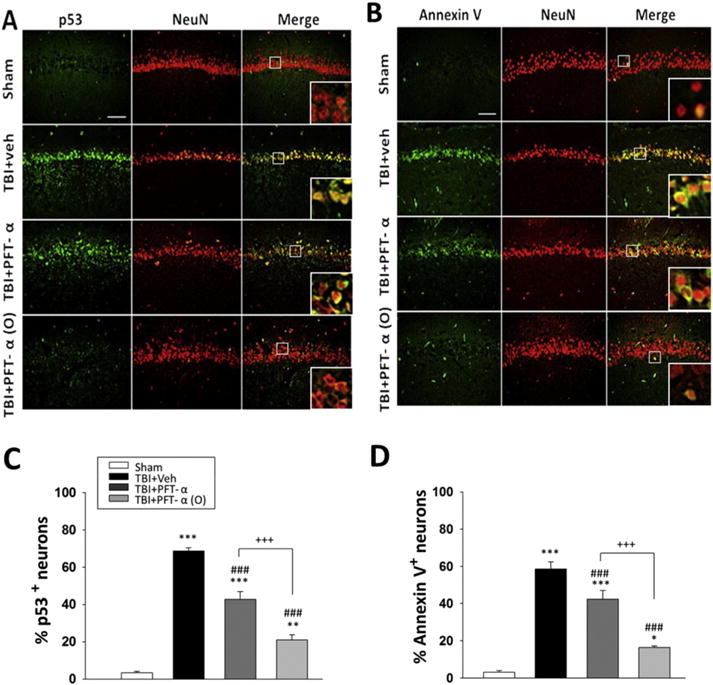
Post-injury administration PFT-α (O) at 5 h after TBI decreased p53 and annexin V positive neurons in the hippocampal CA1 region at 24 h. (A) Co-immunohistochemistry of p53-positive cells colocalized with NeuN positive cells in hippocampal CA1 region. (B) Co-immunohistochemistry of Annexin V and NeuN in hippocampal CA1 region. p53 or annexin V immunoreactivity is shown in green, and NeuN (a marker for neurons) is shown in red. Yellow labelling indicates colocalization. (B, D) There was a significant decrease in the number of p53 and Annexin V positive neurons in TBI + PFT-α and TBI + PFT-α (O) groups, respectively. Data represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 versus the sham group; ###p < 0.001 versus the TBI + veh group; +++p < 0.001 versus the TBI + PFT-α group. Scale bar = 100 μm. (n = 5 for each group).
3.5. Post-injury PFT-α or PFT-α (O) treatment reduced PUMA/COX IV and 4-HNE expression in hippocampal neurons
We evaluated 4-HNE staining to determine whether CCI TBI provokes oxidative stress, to which neurons are highly vulnerable, and whether this could be suppressed by drug treatment (Fig. 7A). We found that TBI induced 4-HNE protein expression within hippocampal CA1 neurons, and that the number of 4-HNE-expressing cells was significantly decreased in PFT-α as well as PFT-α (O) treated animals (Fig. 7C; P < 0.001). In this regard, PFT-α (O) treatment again proved more effective than PFT-α (P < 0.001). To further characterize the effect of PFT-α or PFT-α (O) on the inhibition of p53 transcriptional activity, immunohistochemistry was used to evaluate PUMA/COX IV expression within the hippocampal region (Fig. 7B). We found that PUMA co-localized with the mitochondrial marker COX IV following CCI, and that TBI-induced PUMA/COX IV co-expressing cells were significantly decreased by PFT-α and, in particular, by PFT-α (O) treatment (Fig. 7D; P < 0.05 and b0.001, respectively). Indeed, PFT-α (O) provided significantly greater action than did PFT-α (P < 0.01).
Fig. 7.
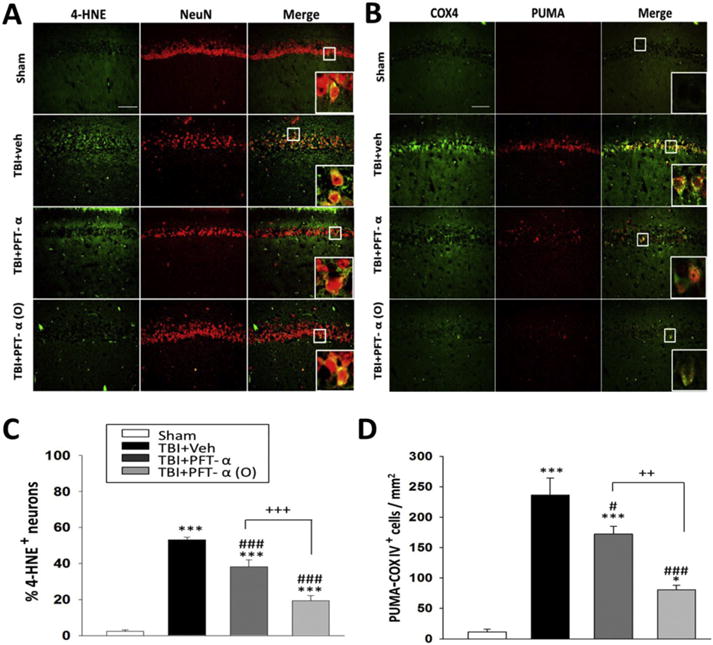
Post-injury administration PFT-α (O) at 5 h after TBI decreased 4-HNE/NeuN positive neurons and PUMA-COX IV positive cells in the hippocampal CA1 region at 24 h. (A) Co-immunohistochemistry of 4-HNE positive cells colocalized with NeuN positive cells in hippocampal CA1 region. (B) Co-immunohistochemistry of PUMA and COX IV in hippocampal CA1 region. 4-HNE or COX IV immunoreactivity is shown in green; NeuN and PUMA is shown in red. Yellow labelling indicates colocalization. (C, D) There was a significant decrease in the number of 4-HNE positive neurons and PUMA-COX IV positive cells in TBI + PFT-α (O) group, respectively. The total number of PUMA-COX IV positive cells was expressed as the mean number per field of view (0.259 mm2). Scale bar = 100 μm. Data represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 versus the sham group; ###p < 0.001 versus the TBI + veh group; +++p < 0.001 versus the TBI + PFT-α group. Scale bar = 100 μm. (n = 5 for each group).
4. Discussion
Our data demonstrates that post-trauma administration of the p53 inactivator PFT-α (O) reduces hippocampal neuronal loss and improves cognitive deficits after CCI-induced experimental TBI. PFT-α (O) and to a lesser extent PFT-α also reduced injury-induced elevations of caspase3 and p53 expression in the neurons, and the former augmented SOD 2 levels. CCI resulted in substantial upregulation of PUMA mRNA and protein, and PFT-α (O) or PFT-α attenuated PUMA mRNA transcription and protein synthesis within the hippocampus. Our study hence supports a primary role for p53 in the delayed neuronal death that occurs in hippocampus following a TBI incident.
Previous studies have reported that edema and hemorrhage are associated with structural brain alterations on MRI (Graham et al., 2000; Iwamoto et al., 1997). TBI-induced elevated water content and decreased cell density may result in prolonged T2 relaxation times, and thereby provide hyperintense signals on T2 weighted imaging. In this regard, the hippocampal T2 relaxation time in the brain of animals subjected to CCI has been reported to be significantly increased during the first 7 days post injury (Obenaus et al., 2007). In line with this time course, we performed longitudinal T2 MRI on rats challenged with CCI and without drug treatment, and verified substantial injury localized to the site of CCI, and progressive injury expansion within the hippocampus between 24 h and 7 days post-injury. Our T2 weighted images additionally demonstrated that a single PFT-α (O) treatment at 5 h following CCI dramatically decreased hippocampal injury volumes at 7 days following injury (Fig. 1B). This provided a basis to quantitatively evaluate whether or not PFT-α (O) treatment had potential to preserve the integrity of the hippocampus and improve experimental TBI behavioral outcomes.
Previous research has demonstrated that increased severity of impact to the head in a preclinical animal closed head TBI model positively correlates with an elevation in the number of pyknotic and apoptotic neurons within the hippocampus and cerebral cortex both ipsi- and contralateral to injury (Tashlykov et al., 2009). In the present study, we verified the development of diffuse neuronal cell death by the use of FJC, an anionic fluorescein analogue that has the highest resolution of the currently available Fluoro-Jade dyes and hence is broadly used in the ex vivo identification of degenerating neurons in the brain as it can be combined with other stains (Schmued et al., 2005). Its use with the neuronal lineage marker NeuN permits quantification of degeneration within the total neuron pool by immunostaining (Yang et al., 2015). Moreover, we linked this p53-dependent neuronal cell death to subsequent cognitive impairment, assessed at 7 days, by the use of p53 inactivators, PFT-α and PFT-α (O).
Our current study followed our previous research (Yang et al., 2015) and used 2 mg/kg PFT-α and PFT-α (O) doses within their previously ascertained therapeutic window to appraise the role of p53 in the diffuse neuronal cell death that occurs following a moderate TBI induced by CCI. A prior evaluation of PFT-α (8 mg/kg i.p.) in mice subjected to CCI (Plesnila et al., 2007) found that elevated p53 expression was evident as early as 15 min and persisted for 24 h in CCI challenged vehicle-treated animals. Nuclear translocation of p53 was evident and occurred predominantly in neurons, with increases in p53 levels strongly correlating to cell death (Plesnila et al., 2007); PFT-α mitigated these actions with a window of opportunity of 6 h. In our study, we therefore evaluated neuronal degeneration at 24 h post-TBI by FJC/NeuN double-staining to define neuron loss at a time of peak neuronal apoptosis (Zhou et al., 2012). Such staining with Fluoro Jade dyes compares favorably with Tunnel staining (Chidlow et al., 2009), and hence has been extensively used to identify neurodegeneration across multiple models of acute and chronic neural injury (Ehara and Ueda, 2009).
In accord with previous studies evaluating PFT-α and analogues in the area of contusion following CCI (Plesnila et al., 2007; Yang et al., 2015), PFT-α (O) administration within 5 h effectively inhibited neuronal apoptosis in hippocampus, and proved to be more potent than equimolar PFT-α. Among the key target genes regulated by p53 is PUMA (p53-up-regulated modulator of apoptosis), a member of the Bcl-2 homology domain 3 (BH3)-only subgroup of Bcl-2 family proteins (Han et al., 2001). PUMA is considered one of the most potent pro-apoptotic proteins, as it avidly binds and inhibits all pro-survival Bcl-2 family proteins (Willis and Adams, 2005) and can directly activate Bax in mitochondria (Cartron et al., 2004). Consequently, the pro-apoptotic actions of p53 in neurons are considered to derive from transcriptional activation of PUMA (Engel et al., 2010), in accord with the over expression of PUMA alone in the absence of other insults being able to induce neuronal apoptosis (Cregan et al., 2004). In the present study, in accord with prior reports in TBI models (Plesnila et al., 2007; Sabirzhanov et al., 2016; Yang et al., 2015), PUMA was found up regulated in neurons and followed p53 expression. PFT-α (O) and PFT-α effectively suppressed this TBI-induced neuronal PUMA mRNA expression (Fig. 5). To determine the involvement of mitochondria in TBI-induced neuronal death, the mitochondrial marker COX IV was evaluated and found to co-localize with PUMA (Fig. 7). TBI-induced mitochondrial dysfunction is recognized by elevated levels of oxidative damage, a loss of respiratory function, a reduced ability to buffer cytosolic calcium, and results in lipid peroxidation-mediated oxidative damage. 4-HNE, one of the toxic aldehydic byproducts of lipid peroxidation (Esterbauer et al., 1991), has been shown to covalently bind to mitochondrial proteins and, thereby, further impair their function and induce neuronal cell death (Mustafa et al., 2010). In line with this, 4-HNE levels were elevated and co-localized with NeuN in hippocampus following TBI (Fig. 7). Notably, this action was also mitigated by PFT-α (O) and PFT-α.
Although within the present study we focused on neuronal dysfunction and apoptosis, TBI injury is multifactorial and involves death of multiple cellular types including glial and endothelial cells (Bralic and Stemberga, 2012). p53 plays an essential role in the regulation of cell death across cell types. In accord with studies of PFT-α analogues, p53−/− knockout mice are reported to exhibit better motor function at 7 days after injury (Tomasevic et al., 2010), which further supports that p53 is an important factor for functional outcome in TBI. However, due to loss of p53 function across all cell types in traditional p53−/− mice and the potential confounding factor of tumor development in these mice, the precise role of the p53 gene in different cell types during TBI-induced cell death remains unclear. An important question as to whether p53 function determines the outcome of TBI by either directly regulating the cell death of neurons or through indirect modulation of the microenvironment in TBI injury remains to be answered. Transgenic mice carrying neuronal-specific ablation of the p53 gene would therefore be useful in future studies to further elucidate the role of p53 in neuronal death and functional outcome after ‘neuronal specific’ p53 deletion in TBI models.
Over recent years it has been increasingly recognized that p53-dependent apoptosis can also occur without p53 transcriptional activity, which involves a direct action at the mitochondria (Vaseva and Moll, 2009). Characterized predominantly in tumor and non-neuronal cells, this involves a cytoplasmic p53 pool that, in response to a stress, rapidly translocates to the outer surface of the mitochondrial membrane. Here, it functions like a BH3-only protein by interacting with pro-apoptotic (PUMA, Bax, Bak) and anti-apoptotic (Bcl-xL, Bcl-2) members of the Bcl-2 family to support Bax/Bak-mediated permeabilization of the outer mitochondrial membrane. This results in the release of cytochrome c and the activation of caspase 3-dependent apoptosis (Wang et al., 2014). Although it has been reported that the apoptotic activity of p53 in postnatal cortical neurons does not rely on its direct action at the cytosol/mitochondria but, rather, is fully mediated through its transcription-dependent functions (Uo et al., 2007), several studies have subsequently suggested a role for mitochondrial p53 in mediating neuronal apoptosis in cellular and in vivo models of ischemia, excitoxicity and DNA damage (Dong et al., 2012; Martin et al., 2009; Nijboer et al., 2008; Nijboer et al., 2011). An increasing number of target proteins have been described for p53, and its context-dependent action appears to be governed by its levels of expression and activity, its subcellular localization, and the accessibility of its interacting partners (Wang et al., 2014). Many of these factors remain unknown in TBI, and thus warrant further research.
A relationship between TBI and the increased likelihood of developing dementia later in life has been suggested from clinical studies. While the proposed link between TBI and the subsequent development of dementia is still controversial, an ever expanding number of epidemiological studies points towards a strong association between TBI and the development of dementia syndromes (Abner et al., 2014; Barnes et al., 2014; Gardner et al., 2014; Lee et al., 2013; Nordstrom et al., 2014; Seichepine et al., 2013; Wang et al., 2012) as well as Parkinson’s disease later in life (Gardner et al., 2015). Furthermore, it is becoming clearer that multiple mild TBIs associate with a high risk of chronic traumatic encephalopathy (CTE), a dementia with distinctive clinical and pathologic features (Shively et al., 2012; Gardner and Yaffe, 2015). Additionally, there is evidence that sub-concussive head blows can induce acute brain changes as evaluated by imaging (Breedlove et al., 2012; Bazarian et al., 2014; Merchant-Borna et al., 2016), changes in neurochemistry (Lin et al., 2015) and cognitive function (McAllister et al., 2012), although the long-term consequences of these changes remain to be determined. Data from our gene array studies in rodents support a link between mild concussive and blast TBI and dementia as well as Parkinson’s disease, illustrated by the identification of gene sets and pathways associated with AD and Parkinson’s disease observed by Day 3 and 14 after injury (i.e., Blalock a1 Alzheimer’s disease incipient; Alzheimer’s disease dn and up and Parkin pathway (Tweedie et al., 2013a; Tweedie et al., 2016; Tweedie et al., 2013b). Parkin was recently identified as a p53 target gene, as human and mouse Parkin genes contain functional p53 responsive elements, and p53 increases the transcription of Parkin in both human and mouse cells (Zhang et al., 2011). Hence, long-term studies may be warranted to evaluate whether p53 inactivators have the potential to turn off TBI-triggered cascades, whether single or repetitive, leading to later degenerations, as well as long term events deriving from sub-concussive injuries.
In conclusion, the neuroprotective effects of the transcriptional p53 inhibitor PFT-α (O) appear to be significantly better than those of PFT-α. Our studies support the notion that a substantial neural population within the hippocampus is amenable to rescue following TBI, and add to the increasing weight of evidence that inhibition of p53-induced apoptosis by a well-tolerated PFT-α analogue may have the potential to develop into a novel therapeutic strategy for TBI and possibly other tissue injuries as well.
Acknowledgments
This study was supported in part by grants from (i) the Ministry of Science and Technology (MOST104-2923-B-038-001-MY3 (1–3 & 2–3) (ii) National Institutes of Health NINDS grant R01NS094152 and by (iii) the Intramural Research Program, National Institute on Aging, National Institutes of Health NINDS grant R01NS094152, USA. The authors thank the Translational Imaging Research Center at Taipei Medical University for technical support in relation to animal setup and MRI imaging. For information relating to PFT-α (O) contact NHG <Greign@mail.nih.gov.
Footnotes
Financial disclosure
No competing financial interest exists.
Conflict of interest
The authors have no conflicts of interest relevant to this article to disclose.
References
- Abner EL, Nelson PT, Schmitt FA, Browning SR, Fardo DW, Wan L, Jicha GA, Cooper GE, Smith CD, Caban-Holt AM, Van Eldik LJ, Kryscio RJ. Self-reported head injury and risk of late-life impairment and AD pathology in an AD center cohort. Dement Geriatr Cogn Disord. 2014;37:294–306. doi: 10.1159/000355478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KJ, Miller KM, Fugaccia I, Scheff SW. Regional distribution of fluoro-jade B staining in the hippocampus following traumatic brain injury. Exp Neurol. 2005;193:125–130. doi: 10.1016/j.expneurol.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Ariza M, Serra-Grabulosa JM, Junque C, Ramirez B, Mataro M, Poca A, Bargallo N, Sahuquillo J. Hippocampal head atrophy after traumatic brain injury. Neuropsychologia. 2006;44:1956–1961. doi: 10.1016/j.neuropsychologia.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology. 2014;83:312–319. doi: 10.1212/WNL.0000000000000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarian JJZT, Zhong J, Janigro D, Rozen E, Roberts A, Jaiven H, Merchant-Borna K, Abar B, Blackman EG. Persistent, long-term cerebral white matter changes after sports-related repetitive head impacts. PLoS One. 2014;9:e94734. doi: 10.1371/journal.pone.0094734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bralic M, Stemberga V. Calpain expression in the brain cortex after traumatic brain injury. Coll Antropol. 2012;36:1319–1323. [PubMed] [Google Scholar]
- Breedlove EL, Robinson M, Talavage TM, Morigaki KE, Yoruk U, O’Keefe K, King J, Leverenz LJ, Gilger JW, Nauman EA. Biomechanical correlates of symptomatic and asymptomatic neurophysiological impairment in high school football. J Biomech. 2012;45:1265–1272. doi: 10.1016/j.jbiomech.2012.01.034. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Chen SF, Hsu CW, Huang WH, Wang JY. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br J Pharmacol. 2008;155:1279–1296. doi: 10.1038/bjp.2008.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow G, Wood JP, Sarvestani G, Manavis J, Casson RJ. Evaluation of Fluoro-Jade C as a marker of degenerating neurons in the rat retina and optic nerve. Exp Eye Res. 2009;88:426–437. doi: 10.1016/j.exer.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Chiu WT, Chu SF, Chang CK, Lui TN, Chiang YH. Implementation of a motorcycle helmet law in Taiwan and traffic deaths over 18 years. JAMA. 2011;306:267–268. doi: 10.1001/jama.2011.989. [DOI] [PubMed] [Google Scholar]
- Compagnone C, d’Avella D, Servadei F, Angileri FF, Brambilla G, Conti C, Cristofori L, Delfini R, Denaro L, Ducati A, Gaini SM, Stefini R, Tomei G, Tagliaferri F, Trincia G, Tomasello F. Patients with moderate head injury: a prospective multicenter study of 315 patients. Neurosurgery. 2009;64:690–696. doi: 10.1227/01.NEU.0000340796.18738.F7. discussion 696–7. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, Loh LT, Adam LT, Oskvig D, Curley KC, Salzer W. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31:135–158. doi: 10.1089/neu.2013.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XX, Wang YR, Qin S, Liang ZQ, Liu BH, Qin ZH, Wang Y. p53 mediates autophagy activation and mitochondria dysfunction in kainic acid-induced excitotoxicity in primary striatal neurons. Neuroscience. 2012;207:52–64. doi: 10.1016/j.neuroscience.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- Ehara A, Ueda S. Application of Fluoro-Jade C in acute and chronic neurodegeneration models: utilities and staining differences. Acta Histochem Cytochem. 2009;42:171–179. doi: 10.1267/ahc.09018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Murphy BM, Hatazaki S, Jimenez-Mateos EM, Concannon CG, Woods I, Prehn JH, Henshall DC. Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic PUMA. FASEB J. 2010;24:853–861. doi: 10.1096/fj.09-145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(Pt B):75–80. doi: 10.1016/j.mcn.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014;71:1490–1497. doi: 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. 2015;77:987–995. doi: 10.1002/ana.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001;21:8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DI, Raghupathi R, Saatman KE, Meaney D, McIntosh TK. Tissue tears in the white matter after lateral fluid percussion brain injury in the rat: relevance to human brain injury. Acta Neuropathol. 2000;99:117–124. doi: 10.1007/pl00007414. [DOI] [PubMed] [Google Scholar]
- Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann N Y Acad Sci. 2004;1035:290–315. doi: 10.1196/annals.1332.018. [DOI] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 2014;10(1 Suppl):S62–S75. doi: 10.1016/j.jalz.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Gupta YK, Sharma SS. Protective effect of pifithrin-alpha on brain ischemic reperfusion injury induced by bilateral common carotid arteries occlusion in gerbils. Indian J Physiol Pharmacol. 2007;51:62–68. [PubMed] [Google Scholar]
- Hall ED, Gibson TR, Pavel KM. Lack of a gender difference in post-traumatic neurodegeneration in the mouse controlled cortical impact injury model. J Neurotrauma. 2005;22:669–679. doi: 10.1089/neu.2005.22.669. [DOI] [PubMed] [Google Scholar]
- Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci U S A. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J Neurotrauma. 1993;10:405–414. doi: 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- Hong MY, Gao JL, Cui JZ, Wang KJ, Tian YX, Li R, Wang HT, Wang H. Effect of c-Jun NH(2)-terminal kinase-mediated p53 expression on neuron autophagy following traumatic brain injury in rats. Chin Med J. 2012;125:2019–2024. [PubMed] [Google Scholar]
- Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- Isaksson J, Hillered L, Olsson Y. Cognitive and histopathological outcome after weight-drop brain injury in the rat: influence of systemic administration of monoclonal antibodies to ICAM-1. Acta Neuropathol. 2001;102:246–256. doi: 10.1007/s004010100361. [DOI] [PubMed] [Google Scholar]
- Iwamoto Y, Yamaki T, Murakami N, Umeda M, Tanaka C, Higuchi T, Aoki I, Naruse S, Ueda S. Investigation of morphological change of lateral and midline fluid percussion injury in rats, using magnetic resonance imaging. Neurosurgery. 1997;40:163–167. doi: 10.1097/00006123-199701000-00036. [DOI] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8:e62422. doi: 10.1371/journal.pone.0062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol. 2004;187:478–486. doi: 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Lin JW, Lin CM, Tsai JT, Hung KS, Hung CC, Chiu WT. Neurotrauma research in Taiwan. Acta Neurochir Suppl. 2008;101:113–117. doi: 10.1007/978-3-211-78205-7_19. [DOI] [PubMed] [Google Scholar]
- Lin AP, Ramadan S, Stern RA, Box HC, Nowinski CJ, Ross BD, Mountford CE. Changes in the neurochemistry of athletes with repetitive brain trauma: preliminary results using localized correlated spectroscopy. Alzheimers Res Ther. 2015;7:13. doi: 10.1186/s13195-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann Neurol. 2009;65:520–530. doi: 10.1002/ana.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex. 2009;19:1273–1293. doi: 10.1093/cercor/bhn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister TW, Flashman LA, Maerlender L, Greenwald RM, Beckwith JG, Tosteson TD, Crisco JJ, Brolinson PG, Duma SM, Duhaime AC, Grove MR, Turco JH. Cognitive effects of one season of head impacts in a cohort of collegiate contact sport athletes. Neurology. 2012;78:1777–1784. doi: 10.1212/WNL.0b013e3182582fe7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant-Borna K, Asselin P, Narayan D, Abar B, Jones CM, Bazarian JJ. Novel method of weighting cumulative helmet impacts improves correlation with brain white matter changes after one football season of sub-concussive head blows. Ann Biomed Eng. 2016 doi: 10.1007/s10439-016-1680-9. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Mugwagwa AT, Gadaga LL, Pote W, Tagwireyi D. Antiamnesic effects of a hydroethanolic extract of Crinum macowanii on scopolamine-induced memory impairment in mice. J Neurodegener Dis. 2015;2015:242505. doi: 10.1155/2015/242505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir JK, Raghupathi R, Emery DL, Bareyre FM, McIntosh TK. Postinjury magnesium treatment attenuates traumatic brain injury-induced cortical induction of p53 mRNA in rats. Exp Neurol. 1999;159:584–593. doi: 10.1006/exnr.1999.7187. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Napieralski JA, Raghupathi R, McIntosh TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain Res Mol Brain Res. 1999;71:78–86. doi: 10.1016/s0169-328x(99)00155-2. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008;39:2129–2137. doi: 10.1161/STROKEAHA.107.504175. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, van der Kooij MA, Zijlstra J, van Velthoven CT, Culmsee C, van Bel F, Hagberg H, Kavelaars A. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol. 2011;70:255–264. doi: 10.1002/ana.22413. [DOI] [PubMed] [Google Scholar]
- Nordstrom P, Michaelsson K, Gustafson Y, Nordstrom A. Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann Neurol. 2014;75:374–381. doi: 10.1002/ana.24101. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Robbins M, Blanco G, Galloway NR, Snissarenko E, Gillard E, Lee S, Curras-Collazo M. Multi-modal magnetic resonance imaging alterations in two rat models of mild neurotrauma. J Neurotrauma. 2007;24:1147–1160. doi: 10.1089/neu.2006.0211. [DOI] [PubMed] [Google Scholar]
- Plesnila N, von Baumgarten L, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007;14:1529–1541. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Yu QS, Li Y, Wang JY, Pick CG, Greig NH. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53-dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT-alpha. PLoS One. 2013;8:e79837. doi: 10.1371/journal.pone.0079837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544–548. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- Sabirzhanov B, Stoica BA, Zhao Z, Loane DJ, Wu J, Dorsey SG, Faden AI. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016;23:654–668. doi: 10.1038/cdd.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Schober ME, Block B, Beachy JC, Statler KD, Giza CC, Lane RH. Early and sustained increase in the expression of hippocampal IGF-1, but not EPO, in a developmental rodent model of traumatic brain injury. J Neurotrauma. 2010;27:2011–2020. doi: 10.1089/neu.2009.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seichepine DR, Stamm JM, Daneshvar DH, Riley DO, Baugh CM, Gavett BE, Tripodis Y, Martin B, Chaisson C, McKee AC, Cantu RC, Nowinski CJ, Stern RA. Profile of self-reported problems with executive functioning in college and professional football players. J Neurotrauma. 2013;30:1299–1304. doi: 10.1089/neu.2012.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively S, Scher AI, Perl DP, Diaz-Arrastia R. Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol. 2012;69:1245–1251. doi: 10.1001/archneurol.2011.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Volkov A, Gazit V, Schreiber S, Zohar O, Pick CG. Minimal traumatic brain injury induce apoptotic cell death in mice. J Mol Neurosci. 2009;37:16–24. doi: 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Tomasevic G, Raghupathi R, Scherbel U, Wieloch T, McIntosh TK. Deletion of the p53 tumor suppressor gene improves neuromotor function but does not attenuate regional neuronal cell loss following experimental brain trauma in mice. J Neurosci Res. 2010;88:3414–3423. doi: 10.1002/jnr.22491. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, Perez E, Miller J, Hoffer BJ, Greig NH, Pick CG. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp Neurol. 2013a;239:170–182. doi: 10.1016/j.expneurol.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis. 2013b;54:1–11. doi: 10.1016/j.nbd.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Rachmany L, Rubovitch V, Li Y, Holloway HW, Lehrmann E, Zhang Y, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH. Blast traumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimers Dement. 2016;12:34–48. doi: 10.1016/j.jalz.2015.07.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J Neurosci. 2007;27:12198–12210. doi: 10.1523/JNEUROSCI.3222-05.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HK, Lin SH, Sung PS, Wu MH, Hung KW, Wang LC, Huang CY, Lu K, Chen HJ, Tsai KJ. Population based study on patients with traumatic brain injury suggests increased risk of dementia. J Neurol Neurosurg Psychiatry. 2012;83:1080–1085. doi: 10.1136/jnnp-2012-302633. [DOI] [PubMed] [Google Scholar]
- Wang DB, Kinoshita C, Kinoshita Y, Morrison RS. p53 and mitochondrial function in neurons. Biochim Biophys Acta. 2014;1842:1186–1197. doi: 10.1016/j.bbadis.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LY, Chu YH, Tweedie D, Yu QS, Pick CG, Hoffer BJ, Greig NH, Wang JY. Post-trauma administration of the pifithrin-alpha oxygen analog improves histological and functional outcomes after experimental traumatic brain injury. Exp Neurol. 2015;269:56–66. doi: 10.1016/j.expneurol.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura I, Takai K, Asai A, Kawahara N, Kirino T. p53 potentiates hippocampal neuronal death caused by global ischemia. J Cereb Blood Flow Metab. 2006;26:1332–1340. doi: 10.1038/sj.jcbfm.9600293. [DOI] [PubMed] [Google Scholar]
- Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J Head Trauma Rehabil. 2008;23:394–400. doi: 10.1097/01.HTR.0000341435.52004.ac. [DOI] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A. 2011;108:16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Chen L, Gao X, Luo B, Chen J. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol. 2012;71:348–359. doi: 10.1097/NEN.0b013e31824ea078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Yu QS, Cutler RG, Culmsee CW, Holloway HW, Lahiri DK, Mattson MP, Greig NH. Novel p53 inactivators with neuroprotective action: syntheses and pharmacological evaluation of 2-imino-2,3,4,5,6,7-hexahydrobenzothiazole and 2-imino-2,3,4,5,6,7-hexahydrobenzoxazole derivatives. J Med Chem. 2002;45:5090–5097. doi: 10.1021/jm020044d. [DOI] [PubMed] [Google Scholar]