

Abstract
Developmental encephalopathies constitute a broad and genetically heterogeneous spectrum of disorders associated with global developmental delay, intellectual disability, frequent epilepsy, and other neurofunctional abnormalities. Here, we report a male presenting with infantile onset epilepsy and syndromic features resembling Dubowitz syndrome identified to have a de novo PLXNA1 variant by whole exome sequencing. This constitutes the second report of PLXNA1 sequence variation associated with early onset epilepsy, and the first to expand on the clinical features of this emerging disorder. This reports suggests that nonsynonymous de novo sequence variations in PLXNA1 are associated with a novel human phenotype characterized by intractable early onset epilepsy, intellectual disability, and syndromic features.
Keywords: developmental encephalopathy, epilepsy, PLXNA1
1 | INTRODUCTION
Whole exome sequencing (WES) has accelerated gene discovery for pediatric developmental disorders, particularly forms presenting with early onset epilepsy (Epi4K Consortium et al., 2013). Similar findings in individuals with autism and intellectual disability have suggested common biological pathways in the pathogenesis of these developmental brain disorders (De Rubeis et al., 2014; Lee, Smith, & Paciorkowski, 2015; O’Roak et al., 2014). Here, we report an individual with a developmental encephalopathy characterized by infantile onset intractable epilepsy, hyperkinetic movements, autism, and dysmorphic facial features identified by WES to have a heterozygous sequence variation in PLXNA1. This is the second reported individual with PLXNA1-related epilepsy, to our knowledge. Furthermore, our subject had syndromic features of growth failure, sparse hair, persistent dermatologic symptoms, and dysmorphic facial features reminiscent of Dubowitz syndrome. Our data suggest that PLXNA1 mutations are associated with a clinically recognizable phenotype associated with infantile onset developmental encephalopathy with syndromic features overlapping Dubowitz syndrome.
2 | CLINICAL REPORT
Subject DB13-040 was the male product of a full-term pregnancy complicated by maternal nephrolithiasis. Fetal ultrasounds were normal. Birth weight was 3.7 kg (the 50th percentile for gestational age and gender). Birth head circumference and length were not documented. Neonatal jaundice was treated with phototherapy.
This child presented with seizures at 15 months of age. Seizures were characterized by ictal vocalization followed by tonic posturing and upward eye deviation. He was treated with phenobarbital and was seizure free for 7 years. Phenobarbital was discontinued, and seizures recurred at age 12 years and were intractable, and included tonic-clonic seizures. Several anti-seizure medications failed to control epilepsy including acetazolamiDe, clorazepate, carbamazepine, Lamictal, valproic acid, and zonisamide. Most recently, the addition of clobazam provided decreased seizure frequency. His neurologic exam was also notable for frequent hyperkinetic movements.
Between the ages of 3 and 5 years he was treated with growth hormone for short stature. He had several persistent dermatologic problems including atopic dermatitis, seborrheic dermatitis, and angular cheilitis requiring ongoing medical therapy.
Developmentally, gross motor delays were first noted at age 7 months. He sat at 12–15 months and walked at 2.5 years. He had abnormal language and social development, and received a diagnosis of pervasive developmental disorder/autism after a formal developmental assessment. At age 16 years, he was nonverbal. He could communicate, however, with sign language for simple words such as “yes,” “more,” and “please” and follow simple commands. He required assistance with activities of daily living including self-care. He displayed stereotypies consisting of breath holding and arm-raising movements. He had not shown any developmental regression.
On examination at 15 years of age, weight was 54.1 kg (0.20 standard deviations below the mean for age and gender), height was 156 cm (1.65 standard deviations below the mean for age and gender), and the head circumference was at the 50th percentile. Physical examination revealed several dysmorphic facial features (Figure 1a–c) including sparse lateral eyebrows, hypoplastic infraorbital ridge, small uvula, and cupped, low-set ears. Hair was sparse on the temples and lateral forehead. Teeth were normal. Skin examination revealed eczematous lesions on the arms, and between the fingers and toes. He had prominent digit pads on his fingers and toes. Genitourinary examination revealed an undescended testicle, which has since resolved. It is not specified in the medical record which testicle this was. Neurologic examination was normal aside from frequent motor stereotypies.
FIGURE 1.
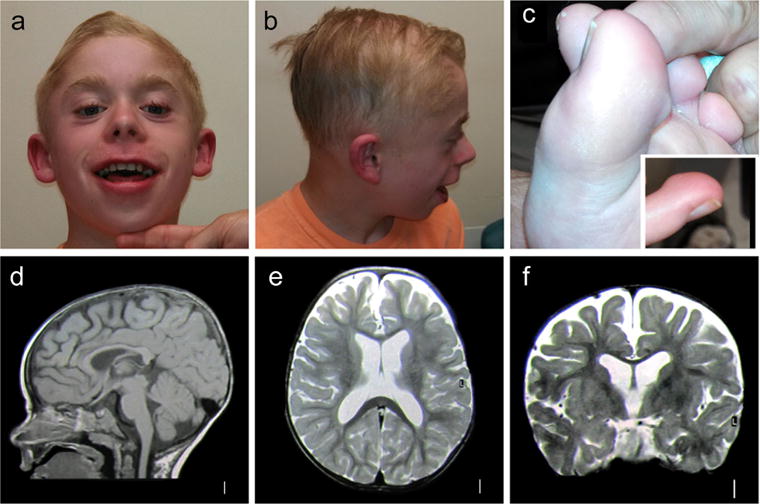
Clinical photographs and MRI images of PLXNA1 mutation-positive patient. (a) Facial photograph of the patient showing several dysmorphic features including upturned nares, prominent eyebrows and eyelashes, laterally prominent and small ears, low hairline, and angular cheilitis with dermatitis of the philtrum; (b) lateral photograph of the face showing fleshy and laterally prominent earlobes, sparse hair with abnormal hair growth pattern in the temporal areas bilaterally; (c) photograph of the foot showing prominent digit pads on fingers and toes; d–f, T1 weighted sagittal (d), T2-weighted axial (e), and coronal (f) brain MRI images showing moderately dilated third and lateral ventricles, and increased extra-axial spaces predominantly in the frontal region. The cortical gyral pattern is normal [Color figure can be viewed at wileyonlinelibrary.com]
Brain MRI at 1 year of age showed mild prominence of the ventricles and extra-axial fluid spaces, with no cortical or cerebellar malformations (Figure 1d–f). Electroencephalograms (EEGs) showed bilateral spike and wave discharges lasting up to 2 s consistent with generalized epilepsy. Previous normal genetic testing included karyotype, chromosomal microarray, chromosome 15 methylation, Fragile X syndrome testing, ELN FISH, and MECP2 sequencing.
Research WES identified a heterozygous variant in PLXNA1 (NM_032242, chr3:g.126735788, c.3184G>A, p.Gly1062Ser) that was highly conserved, determined to be pathogenic by two prediction algorithms, and was not present in either the NHLBI Exome Variant Server (EVS) or the ExAC data sets (Table 1). Sanger sequencing of the proband and parents confirmed that this variant was de novo. Our WES analysis considered several other candidate variants (Supplementary Table). However, none of these were determined to be likely pathogenic based upon multiple lines of evidence.
TABLE 1.
Clinical and neuroimaging features associated with of PLXNA1 developmental encephalopathy
| Patient | DB13-040 |
|---|---|
| Age at last evaluation | 16 years |
| Gender | Male |
| Dysmorphic facial features | Sparse lateral eyebrows, hypoplastic infraorbital ridge, small uvula, cupped, low-set ears |
| Hair findings | Sparse pattern on temples and lateral forehead |
| Dermatologic findings | Atopic dermatitis, seborrheic dermatitis, angular cheilitis |
| Seizures | |
| Onset | 15 months |
| Type | Ictal vocalization with tonic posturing and upward eye deviation, tonic clonic, behavioral arrest |
| Treatment | Multiple medications, intractable |
| Other neurological features | |
| DD/ID | Nonverbal, minimal sign language. Sat at 12–15 months, walked at 2.5 years. Autism spectrum disorder |
| Tone | Normal |
| MRI findings | Mild prominence of ventricles and extra-axial fluid spaces |
| EEG findings | Bilateral spike and wave discharges lasting up to two seconds, consistent with generalized epilepsy |
| Variant information | PLXNA1:NM_032242:chr3:g.126735788, exon16:c.G3184A:p.G1062S |
DD, developmental delay; ID, intellectual disability.
3 | METHODS
3.1 | Patient ascertainment
The subject was consented through the Genetic Studies of Developmental Brain Disorders protocol, approved by the University of Rochester Medical Center Research Subjects Review Board.
3.2 | Whole exome sequencing
The subject and both parents had research WES performed on saliva-derived DNA, using the Agilent Sure-Select 50 Mb whole exome capture kit. Paired end 100 bp reads were generated on an Illumina HiSeq2500 sequencer at the University of Rochester Genomics Research Center. Sequence was aligned to hg19 with BWA v.0.6.2 and analyzed with Picard v.1.84, SAMtools v.0.1.18, and GATK v.2.3–9. Annotation of variants was performed with Annovar, and de novo, autosomal recessive, and X-linked variants were identified, and common variants in dbSNP 137 excluded with SOLVE-Brain v.1.0.1. Common population variants were identified in the NHLBI EVS and the Exome Aggregation Consortium data sets. Ensembl’s Variant Effect Predictor provided SIFT and PolyPhen predictions of pathogenicity. Candidate sequence variants were confirmed with Sanger methods.
4 | DISCUSSION
We report an individual with a de novo variant in PLXNA1 presenting with intractable infantile onset epilepsy, and intellectual disability with autism spectrum disorder. Interestingly, this individual also had features suggestive of Dubowitz syndrome, including growth failure, dermatologic symptoms, and characteristic dysmorphic facial features.
To date, only two individuals have been reported with PLXNA1 mutations. The first was an individual with epileptic encephalopathy and very limited clinical data who had a missense PLXNA1 mutation (NM_032242.2:c.683G>A, p.Arg228His) (Epi4K Consortium et al., 2013; Oliver et al., 2016). The second was an adult with schizophrenia who had a missense PLXNA1 variant (NM_032242, c.4201A>T, p.1401M>L) (Fromer et al., 2014). Further, a small 191-Kb duplication of uncertain clinical significance of the PLXNA1 Locus (3q21.3) has been reported in an individual with congenital heart disease (Zhao et al., 2013). Our findings constitute the second report of PLXNA1 mutation with infantile onset epilepsy, and the first report to elaborate on the developmental encephalopathy phenotype and dysmorphic features associated with sequence variations in this gene.
It is noteworthy that our patient had several features overlapping with Dubowitz syndrome. Dubowitz syndrome is a highly variable clinical diagnosis characterized by the association of multiple congenital anomalies including distinctive facial features, intellectual disability, microcephaly, and eczema (Tsukahara & Opitz, 1996). Hematologic and immune system complications, as well as increased risk for malignancy have been reported (Al-Nemri, Kilani Salih, & Al-Ajlan, 2000; Gröbe, 1983; Sauer & Spelger, 1977). Some have argued that the clinical entity identified in the literature as “Dubowitz syndrome” is in fact a set of several overlapping disorders with different molecular etiologies (Stewart et al., 2014). Some have identified autosomal recessive mutations in NSUN2 in individuals with intellectual disability and facial features resembling Dubowitz syndrome (Martinez et al., 2012), however variations in this gene have not been found to date in other patients. Overlapping features with this syndrome that were present in our patient include some of the distinctive facial features (such as overall small facies, short palpebral fissures, ptosis, laterally prominent ears, and micrognathia), feeding difficulties, and dermatologic abnormalities, including eczema. One puzzling aspect to our WES findings, however, is the heterozygous nature of the PLXNA1 mutation identified, whereas Dubowitz syndrome as classically described is autosomal recessive. However, we found no compelling recessive variants in our WES data (Supplementary Table).
The missense PLXNA1 mutation identified in our patient was not present in public databases including the NHLBI EVS and the ExAc. This variant was predicted to be deleterious by SIFT, probably damaging by PolyPhen, and had a GERP score of 4.08 suggesting evolutionary conservation. This evidence supports the pathogenicity of the PLXNA1 variant in this individual, although future functional assays need to be pursued.
PLXNA1 encodes plexin a1, a transmembrane protein highly expressed in the developing nervous system. Plexin forms a complex with neuropilin and acts as a receptor for class three semaphorins, a group of chemorepellants important in axon guidance (Tamagnone et al., 1999). Plexin regulates the affinity of the receptor complex for specific semaphorins, and its cytoplasmic domain is necessary for the activation of downstream signaling events within the cytoplasm (Usui, Taniguchi, Yokomizo, & Shimizu, 2003). Overall, PLXNA1 plays a role in axon guidance, invasive growth, and cell migration (Hernandez-Enriquez et al., 2015). This available knowledge about the biological role of plexins in brain development is consistent with a mechanism associated with epilepsy and intellectual disability. Together with the previous literature, we suggest that a nonsynonymous de novo variant in PLXNA1 is associated with a novel developmental encephalopathy characterized by intractable epilepsy and intellectual disability. Additional features suggest overlap with Dubowitz syndrome.
Supplementary Material
Acknowledgments
We thank the patient’s family and care providers for their contribution to this study. This study was funded by the National Institute of Health, National Institute of Neurological Disease and Strokes (NINDS) grant K08NS092898 (to GMM) and K08NS078054 (to ARP).
Funding information
NIH, Grant number: K08NS078054; National Institute of Neurological Disease and Strokes, Grant number: K08NS092898
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Al-Nemri AR, Kilani RA, Salih MA, Al-Ajlan AA. Embryonal rhabdomyosarcoma and chromosomal breakage in a newborn infant with possible Dubowitz syndrome. American Journal of Medical Genetics. 2000;92:107–110. doi: 10.1002/(sici)1096-8628(20000515)92:2<107::aid-ajmg5>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Buxbaum JD. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium, Epilepsy Phenome/Genome Project. Allen AS, Berkovic SF, Cossette P, Delanty N, Winawer MR. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröbe H. Dubowitz syndrome and acute lymphatic leukemia. Monatsschrift Kinderheilkd. Organ Dtsch Ges Für Kinderheilkd. 1983;131:467–468. [PubMed] [Google Scholar]
- Hernandez-Enriquez B, Wu Z, Martinez E, Olsen O, Kaprielian Z, Maness PF, Tran TS. Floor plate-derived neuropilin-2 functions as a secreted semaphorin sink to facilitate commissural axon midline crossing. Genes & Development. 2015;29:2617–2632. doi: 10.1101/gad.268086.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Smith T, Paciorkowski A. Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy & Behavior. 2015;47:191–201. doi: 10.1016/j.yebeh.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Gleeson JG. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. Journal of Medical Genetics. 2012;49:380–385. doi: 10.1136/jmedgenet-2011-100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak B, Stessman H, Boyle E, Witherspoon K, Martin B, Lee C, Eichler E. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nature Communications. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KL, Lukic V, Freytag S, Scheffer IE, Berkovic SF, Bahlo M. In silico prioritization based on coexpression can aid epileptic encephalopathy gene discovery. Neurology Genetics. 2016;2:e51. doi: 10.1212/NXG.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer O, Spelger G. Dubowitz syndrome with immunodeficiency and solid malignant tumor in two siblings (author’s transl) Monatsschrift Für Kinderheilkd. 1977;125:885–887. [PubMed] [Google Scholar]
- Stewart DR, Pemov A, Johnston JJ, Sapp JC, Yeager M, He J, Savage SA. Dubowitz syndrome is a complex comprised of multiple, genetically distinct and phenotypically overlapping disorders. PloS ONE. 2014;9:e98686. doi: 10.1371/journal.pone.0098686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming G, Song H, Comoglio P. Plexins are a large family of receptors for transmembrane, secreted, and GPI-Anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Opitz JM. Dubowitz syndrome: Review of 141 cases including 36 previously unreported patients. American Journal of Medical Genetics. 1996;63:277–289. doi: 10.1002/(SICI)1096-8628(19960503)63:1<277::AID-AJMG46>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Usui H, Taniguchi M, Yokomizo T, Shimizu T. Plexin-A1 and plexin-B1 specifically interact at their cytoplasmic domains. Biochemical and Biophysical Research Communications. 2003;300:927–931. doi: 10.1016/s0006-291x(02)02966-2. [DOI] [PubMed] [Google Scholar]
- Zhao W, Niu G, Shen B, Zheng Y, Gong F, Wang X, Li S. High-resolution analysis of copy number variants in adults with simple-to-moderate congenital heart disease. American Journal of Medical Genetics Part A. 2013;161A:3087–3094. doi: 10.1002/ajmg.a.36177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.