

Abstract
Existing oral antiretroviral (ARV) agents have been shown in human studies to exhibit limited lymph node penetration and lymphatic drug insufficiency. As lymph nodes are a reservoir of HIV, it is critical to deliver and sustain effective levels of ARV combinations in these tissues. To overcome lymph node drug insufficiency of oral combination ARV therapy (cART), we developed and reported a long-acting and lymphocyte-targeting injectable that contains three ARVs—hydrophobic lopinavir (LPV) and ritonavir (RTV), and hydrophilic tenofovir (TFV)— stabilized by lipid excipients in a nanosuspension. A single subcutaneous (SC) injection of this first-generation formulation of drug combination nanoparticles (DcNPs), named TLC-ART101, provided persistent ARV levels in macaque lymph node mononuclear cells (LNMCs) for at least 1 week, and in peripheral blood mononuclear cells (PBMCs) and plasma for at least 2 weeks, demonstrating long-acting pharmacokinetics for all three drugs. In addition, the lymphocyte-targeting properties of this formulation were demonstrated by the consistently higher intracellular drug concentrations in LNMCs and PBMCs versus those in plasma. To provide insights into the complex mechanisms of absorption and disposition of TLC-ART101, we constructed novel mechanism-based pharmacokinetic (MB PK) models. Based upon plasma PK data obtained after single administration of TLC-ART101 (DcNPs) and a solution formulation of free triple-ARVs, the models feature uptake from the SC injection site that respectively routes free and nanoparticle-associated ARVs via the blood vasculature and lymphatics, and their eventual distribution into and clearance from the systemic circulation. The models provided simultaneous description of the complex long-acting plasma and lymphatic PK profiles for all three ARVs in TLC-ART101. The long-acting PK characteristics of the three drugs in TLC-ART101 were likely due to a combination of mechanisms including: (1) DcNPs undergoing preferential lymphatic uptake from the subcutaneous space, (2) retention in nodes during lymphatic first-pass, (3) subsequent slow release of ARVs into blood circulation, and (4) limited extravasation of DcNP-associated ARVs that resulted in longer persistence in the circulation.
Keywords: mechanism-based pharmacokinetic modeling, long-acting, antiretrovirals, HIV drug combination treatment, lymphatic targeted drug delivery, lymphatic drug insufficiency
Graphical abstract

Introduction
There is growing interest in developing long-acting (LA) antiretroviral (ARV) injectable formulations that provide persistent, effective drug levels in plasma and/or HIV target cells (in the blood) for weeks to months, for HIV prevention and treatment [1]. Currently, intramuscular (IM) LA cabotegravir and LA rilpivirine are in Phase III trials; each contains one anti-HIV drug. As single agents, their use is limited to prevention. For treatment, which requires ARV combinations, the two long-acting agents (cabotegravir and rilpivirine) are currently administered in two separate injections [2]. Because LA cabotegravir and LA rilpivirine both take several days to reach maximum plasma concentrations (Tmax) (median Tmax: 9-69 days [3] or 6-11 days [4]), an additional oral lead-in or loading dose may be needed to provide effective plasma levels soon after injection to prevent the development of viral resistance from subtherapeutic concentrations. There is a need for injectables that can deliver a combination of ARVs in a single dosage form that can quickly achieve effective levels at times similar to the oral Tmax, and from thereon sustain therapeutic drug levels for extended durations.
In addition to long-acting characteristics, the combination ARV injectable should also allow targeting of HIV reservoir sites. Since HIV infects T-cells in lymph nodes throughout the body [5-7], sustained and effective ARV levels in these host cells should be a key objective of any strategy for HIV eradication. In 2003, we first proposed and verified [8], and others later confirmed in prospective clinical studies [9-11], that most orally dosed ARVs fail to achieve sufficient levels in lymph nodes [12]. This is consistent with reports showing rats and humans having limited lymphatic exposure to intravenous (IV) anti-cancer small molecules (e.g., doxorubicin, methotrexate, and carboplatin) [13-15]. To overcome this lymphatic HIV drug insufficiency, we previously developed an indinavir-lipid nanosuspension and demonstrated that it provided indinavir accumulation in lymph nodes throughout the body (in addition to local draining lymph nodes) [12, 16] and reduced lymph node viral levels in HIV-infected macaques [8]. With this drug delivery platform, we have now developed a novel long-acting and lymphocyte-targeting drug-combination injectable for subcutaneous (SC) administration. It contains multiple classes of ARVs in a single formulation that is intended to deliver the ARV combinations to HIV-infected lymph nodes and cells at much enhanced and more sustained levels of each ARV. Notably, this approach permits combining three ARV small molecules with different physicochemical properties (i.e., two hydrophobic drugs [LPV, LogD = 4.7; RTV, LogD = 5.2] and one hydrophilic drug [TFV, LogD = -3.6])–all together–into a single drug combination nanoparticle (DcNP) suspension [17, 18]. In macaques, a single SC injection produced enhanced and persistent drug levels in not only lymph node mononuclear cells (LNMCs) in lymph nodes local to the SC injection site, but in nodes throughout the body [12] for over a week [19]. Moreover, in peripheral blood mononuclear cells (PBMCs) and plasma, measurable drug levels were maintained longer than the two-week test period [19]. Plasma Tmax values occurred within hours, apparent terminal plasma half-lives for the active drugs ranged from 65 to 477 hr, and intracellular LNMC drug concentrations were consistently higher than those in PBMCs, which were consistently higher than those in plasma (i.e., drug concentrations in LNMCs > PBMCs > plasma) [19]. However, the pharmacokinetic (PK) mechanisms responsible for this enhanced, widespread lymphatic uptake and drug persistence in LNMCs, PBMCs, and plasma remained to be elucidated. Moreover, the published literature describing in general the lymphatic absorption, distribution, and elimination mechanisms of HIV drugs and drug combinations after SC dosing is limited.
To seek an understanding of the PK mechanisms governing SC absorption, distribution into lymphatics, lymph node-targeting, and sustained drug levels in lymphocytes and plasma following administration of our long-acting triple-ARV DcNP formulation—TLC-ART101 [19]—we developed compartmental PK models based on a mechanistic framework that describe the kinetic processes governing the complex plasma concentration-time course of all three ARVs following SC injection of TLC-ART101 in macaques. We leveraged plasma concentrations in macaques dosed with the solubilized (free) triple-ARV combination to inform the parameterization of the free drug component of our TLC-ART101 models. This approach not only accounted for the complex long-acting plasma drug concentration-time profiles for all three ARVs in TLC-ART101, but also allowed for prediction of first-pass drug retention in the lymphatics. These mechanistic PK models are applicable to optimizing dosing for future efficacy and safety studies of TLC-ART101 in macaques, and comparing disposition profiles between the current and future generations of long-acting drug combination formulations.
Materials and Methods
Reagents and Animals
1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG2000) were purchased from Corden Pharma (Liestal, Switzerland). Lopinavir (LPV), ritonavir (RTV), and tenofovir (TFV, PMPA) were purchased from Waterstone (Carmel, IN, USA). All other reagents used were of analytical grade or higher.
Eleven adult male macaques (Macaca nemestrina, 5.6-14.9 kg) were housed and cared for by the Washington National Primate Research Center (WaNPRC) under an approved Institutional Animal Care and Use Committee protocol.
TLC-ART101 and Free Triple-ARV Combination Formulations
The long-acting drug combination nanoparticle (DcNP) formulation (TLC-ART101) composed of DSPC and DSPE-mPEG2000 (9:1 molar ratio) that contained a combination of three antiretrovirals (ARVs) (LPV, RTV, and TFV) was prepared as previously described [8, 17]. The nanosuspension used in this study contained 18.3 mM LPV, 4.5 mM RTV, and 17.1 mM TFV The pharmacokinetics (PK) of this formulation in macaques was recently reported [19]. The mean fraction of DcNP-associated LPV, RTV, and TFV was 92.2%, 91.1 %, and 11.2%, respectively, as assessed by equilibrium dialysis using a 6-8 kDa molecular weight cut-off dialysis membrane. This mixture of lipid-associated and free ARVs constitutes the delivered drug content of TLC-ART101 [19]. Mean diameter of the DcNPs was 69.0 ± 8.3 nm, as assessed by photon correlation spectroscopy.
The solution formulation of free LPV, RTV, and TFV was prepared in 20 mM NaHCO3-buffered water (pH 7.4) with 0.7% NaCl, 8% DMSO, and 0.1% Tween20 and had the same final drug concentrations as those listed above for TLC-ART101.
Pharmacokinetic Study
Macaques were given a single subcutaneous (SC) bolus dose in the back mid-scapular region of 25.0 mg/kg LPV, 7.0 mg/kg RTV, and 10.6 mg/kg TFV, either as TLC-ART101 or free ARV formulation. In four animals dosed with TLC-ART101, blood was collected at 0, 0.25, 0.5, 1, 3, 5, 8, 24, 48, 120, 168, 192, and 336 hr for full PK profiling; in another four animals designated for lymph node biopsy, blood was only collected at 0, 0.5, and 24 hr. In three animals dosed with the free ARV formulation, blood was collected at 0, 0.5, 8, 12, 18, and 24 hr. LPV, RTV, and TFV concentrations in plasma were simultaneously analyzed using a validated LC-MS/MS method [20]. The following descriptive PK parameters were estimated using the non-compartmental analysis module in Phoenix v6.4 (Certara, Mountain View, CA, USA): AUC, area under the plasma drug concentration-time curve; CL/F, apparent total clearance; MBRT, mean body residence time; t1/2, apparent terminal plasma drug half-life.
Free ARV Compartmental Models
Structural Models and Parameter Estimation
A linear one-compartment model (1CM) with first-order absorption was fit to LPV and RTV plasma concentration-time data after SC dosing (Figure 3A). A two-compartment model (2CM) with first-order absorption was deemed to best fit the plasma concentration-time data for TFV (Figure 3B). For the 1CM analysis, the absorption rate constant k21, the elimination rate constant k02, and the apparent volume of distribution (V/F) were estimated, where F is the subcutaneous bioavailability. For the 2CM analysis, the following parameters were estimated: absorption rate constant (k21), rate constants for the exchange of drug between central and peripheral compartments (k32, k23), and the elimination rate constant from the central compartment (k02), along with the apparent volume of the central compartment (Vc/F). Secondary parameter estimates for apparent volumes in the terminal phase (Vβ/F) and at steady state (Vss/F) were computed from the primary parameter estimates [21]. It should be noted that subscript notations for all rate constants followed the engineering convention of kji with j = receiving compartment and i = originating compartment. See Supplementary Information for a description of model parameters and their interpretation.
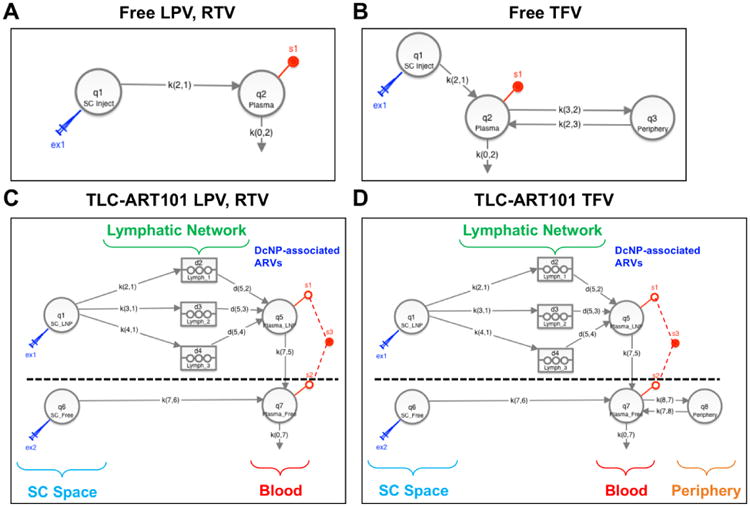
Figure 3. Structural models for free drug disposition in the systemic circulation (panels A, B) and TLC-ART101 disposition in the lymphatics and systemic circulation (panels C, D) after SC dosing.
The average or typical value of the aforementioned 1CM or 2CM parameters were estimated by subjecting the plasma concentration-time datasets from each of the macaques to an iterated two-stage (ITS) analysis using the Popkinetics module of SAAM II v2.3 (18) (The Epsilon Group, Charlottesville, VA, USA). Briefly, the first stage involves estimating the PK parameters of each macaque through nonlinear regression; the second stage involves estimating the population PK parameters across macaques (e.g., mean, variance) based upon individual estimates from the first stage. SAAM II then uses the resulting population PK parameter estimates as Bayesian priors, and iterates the two-stage process until model convergence is reached [22]. Apparent terminal half-life (t1/2) and apparent plasma clearance (CL/F) were then calculated using the population PK parameter values.
TLC-ART101 ARV Compartmental Models
Structural Models
The compartmental models for TLC-ART101 conceptualize the formulation-driven and physiological processes (i.e., mechanisms) underlying the absorption and disposition of DcNPs. As such, the mechanism-based pharmacokinetic (MBPK) models consist of two submodels: one for DcNP-associated ARV absorption and disposition (part above the dashed line in Figure 3C, 3D), and one for free ARV absorption and disposition (part below the dashed line in Figure 3C, 3D). This composite organization of two separate but linked submodels allows the accounting of differential absorption and disposition pathways between DcNP-associated ARVs and free ARVs from the subcutaneous space. In total this model structure has eleven parameters for LPV and RTV and thirteen parameters for TFV (Figure 3C, 3D; Table 2). See Supplementary Information for differential equations that define these models and a description of model parameters.
Table 2. Model-derived pharmacokinetic parameters for each drug in the triple-drug combination given as a single SC dose in either the free or TLC-ART101 formulation.
| Free Lopinavir | Free Ritonavir | Free Tenofovir | |
|---|---|---|---|
| V/F (L/kg) | 54.10 (6) | 42.41 (24) | 1.44a; 8.65b |
| k21 (1/hr) | 0.7120 (20) | 0.6242 (29) | 2.3423 (38) |
| k02 (1/hr) | 0.0784 (17) | 0.0919 (22) | 0.5896 (-) |
| k32 (1/hr) | - | - | 0.0298 (0) |
| k23 (1/hr) | - | - | 0.0738 (-) |
| t1/2 (hr) | 8.85 | 7.54 | 1.11c; 9.93d |
| CL/F (L/[hr•kg]) | 4.24 | 3.90 | 0.60 |
|
| |||
| TLC-ART101 Lopinavir | TLC-ART101 Ritonavir | TLC-ART101 Tenofovir | |
|
| |||
| k21 (1/hr) | 0.0278 (-) | 0.2361 (-) | 0.1300 (-) |
| k31 (1/hr) | 0.1236 (-) | 0.1881 (-) | 0.1588 (-) |
| k41 (1/hr) | 0.3050 (-) | 0.0198 (-) | 0.1779 (-) |
| tlag1 (hr) | 2.15 (43) | 2.13 (51) | 0.10 (-) |
| tlag2 (hr) | 10.85 (29) | 38.83 (13) | 24.04 (19) |
| tlag3 (hr) | 262.23 (12) | 181.87 (11) | 122.06 (3) |
| V5_DcNP (L/kg) | 6.53 (3) | 10.09 (27) | 0.17 (22) |
| k75 (1/hr) | 0.6836 (30) | 0.4336 (5) | 0.0164 (15) |
| k76 (1/hr) | - | - | 1.4201 (-) |
| k78 (1/hr) | - | - | 0.0738 (0) |
| k87 (1/hr) | - | - | 0.0298 (-) |
| V7_Free (L/kg) | 83.82 (1) | 67.94 (3) | 1.66 (18) |
| k07 (1/hr) | 0.0788 (3) | 0.0962 (4) | 0.5824 (2) |
Parameter value (% coefficient of variation).
Vss/F
Vβ/F
t1/2,alpha
t1/2,beta
-, not available due to being “fixed” or not applicable.
The lymphatic system draws interstitial fluid containing cells and solutes to lymph nodes and eventually drains into the thoracic lymphatic duct, which deposits its contents into the blood circulation via valves at the subclavian veins near the heart. Following SC injection, ARVs associated with DcNPs are taken up exclusively into the lymphatics (i.e., no uptake into the blood capillaries at the injection site) and transit through the lymphatic network where they may either: (1) transit as solutes through the lymph as it flows through lymph vessel and node networks before entering the systemic circulation via the thoracic lymph duct, (2) fill and become trapped in lymph node sinuses, or (3) be taken up by lymph node parenchymal cells, such as lymphocytes. This exclusive lymphatic uptake, distribution, and retention is supported by preclinical studies with SC injection of large proteins and nanoparticles in the literature [14, 15, 23-26] and confirmed by our in vivo imaging studies in mice (submitted for publication). These processes are depicted in the submodel above the dashed line in Figure 3C, 3D. In contrast, free ARVs in the subcutaneous space are absorbed entirely via blood capillaries into the circulation, which is depicted in the submodel below the dashed line in Figure 3C, 3D.
The two hydrophobic protease inhibitors (LPV, RTV) with LogP ∼5 are nearly 100% incorporated into the core of DcNPs, as evidenced by complete retention in the nanoparticles despite exposure to sink conditions for 24 hr via equilibrium dialysis at 25°C. The hydrophilic nucleotide reverse transcriptase inhibitor (TFV) with LogP -3.4 is mostly associated to the surface of DcNPs; equilibrium dialysis measurements indicate up to 90% of TFV in TLC-ART101 is effectively free under sink conditions and no significant release from the DcNPs occurs after 90% of free drug is dialyzed away, which indicates stable interactions between TFV and lipid and/or PEG in DcNPs for a minor fraction of the total amount of TFV. For DcNP-associated LPV and RTV, since 100% of the dose is assumed to be associated to DcNPs in the subcutaneous space, the entire SC dose of the protease inhibitors undergoes first-pass through the lymphatic system and is handled exclusively by the DcNP-associated ARV disposition submodel (above the dashed line in Figure 3C). For DcNP-associated TFV, since 10% of the dose is assumed to be associated to DcNPs, and 90% is assumed to be free in the subcutaneous space, these percentages of the total dose were available for each respective submodel (Figure 3D).
In the MBPK models (Figure 3C, 3D), DcNP-associated ARVs are absorbed from the SC injection site (compartment q1) into the lymphatics via three parallel first-order pathways described by rate constants k21, k31, and k41. Transit of DcNPs through the lymphatic system occurs through three parallel delay compartments (d2, d3, d4)—representing a fast, intermediate, and slow pathway, respectively. The decision to set the minimum number of delay compartments to three was based upon trial-and-error simulations to achieve reasonable visual fits of observed plasma concentration-time data. Only one or two lymphatic delay compartments, or a single uptake rate constant into one lymphatic delay compartment with three different release rate constants from this lymphatic delay compartment into the systemic circulation, were not sufficient to account for the complex and multiphasic persistent plasma levels for all three ARVs. Physiologically, the three different “waves” of drug entering the plasma from the lymph vessel and node network could result from: (1) DcNPs appearing in the plasma through more rapid flow of DcNPs and lymph through the lymphatics soon after injection, (2) DcNPs that are initially lodged in the sinus spaces in lymph nodes being eventually carried away and into the blood by slower lymph flow, and/or (3) DcNPs being taken up by lymphocytes in lymph nodes and then these lymphocytes trafficking into the blood where they could release intracellular drug into the plasma. Each delay compartment, which consisted of three sub-compartments in series, is defined by a transit time (tlag1, tlag2, or tlag3) and first-order exit rate constant (k52, k53, k54), by which DcNP- or lymphocyte-associated ARVs are directed into the central compartment (q5). Once ARVs in DcNPs or lymphocytes enter the systemic compartment, release of free ARV would commence. Free ARVs are released from DcNPs and lymphocytes in central compartment q5 by a first-order process into the free ARV central compartment (q7, described by first-order rate constant k75). We assumed free ARV PK is the same whether it is administered as such or released from DcNPs in vivo. Moreover, we assumed DcNPs do not undergo significant degradation during the two-week period (i.e., no physical clearance of DcNPs), as evidenced by their long circulation lifetimes [19]. We also assumed no significant fraction of drug is released from DcNPs during their stay in the SC injection site and while transiting through the lymphatics (i.e., there is no dissociation or release of free ARV prior to entry into the systemic circulation), as evidenced by (1) our in vitro release assays that showed stable association of both hydrophobic and hydrophilic ARVs to DcNPs under sink conditions and (2) our in vivo imaging studies that showed only DcNPs undergo preferential, rapid, and widespread lymph vessel and node distribution and retention following SC administration in mice, whereas free drug does not (personal observation, submitted for publication). Re-entry into the lymphatic system following systemic distribution of DcNPs is not explicitly recognized, as evidenced by preclinical IV studies using nanoparticles [15].
For free ARVs at the subcutaneous injection site (compartment q6), they are absorbed into the blood by a first-order process into the central compartment (q7, described by the first-order rate constant k76) (Figure 3C, 3D). Free TFV distributes in and out of the peripheral compartment q8 by first-order processes described by the rate constants k87 and k78 (Figure 3D). Free ARVs are cleared from the central compartment (q7) by a first-order process described by the rate constant k07. The entire SC dose, either in free or DcNP-associated form, is assumed to be bioavailable from the subcutaneous space.
Parameter Estimation
As with free ARV model fitting, residual error for the observed plasma concentrations were described by a proportional error model for each dependent variable. Since the LC-MS/MS assay for plasma ARV concentration quantifies both DcNP-associated, protein-bound, and free ARV, the model output is the sum of predicted concentrations in compartments 5 (DcNP-associated ARVs) and 7 (DcNP-dissociated ARVs). Free ARV PK parameter estimates, including k76, k78, k87, V7_Free, and k07, were set to Bayesian priors based upon the mean and standard deviation of the population estimates derived from modeling of free drug formulation. To establish the lag times for each ARV through each of the three different lymphatic pathways (tlag1, tlag2, tlag3), we first fixed each of the lymphatic entry rate constants (k21, k31, k41) equal to 0.1. Lag times (tlag1, tlag2, tlag3) were then set to “adjustable,” as well as well as the apparent volume of distribution of DcNPs from the blood compartment (V5_DcNP) and the rate constant for release of free ARV from DcNPs (k75). Initial estimates for “adjustable” parameters were obtained through visual fits of geometric mean data. Once lag times were established, for all subsequent model development these lag times served as Bayesian prior estimates with a 30% relative standard deviation. Next, lymphatic entry and exit rate constants (k21 = k52, k31 = k53, k41 = k54) were manually adjusted based upon trial-and-error simulations to achieve reasonable visual fits of observed plasma concentration-time data. Final parameter estimates of the lymphatic entry rate constants were “fixed” to account for the proportion of subcutaneous ARV available for the rise and fall in plasma concentrations during early, mid, and late periods. Remaining model parameters (V5_DcNP and k75) were set to “adjustable” and the model was fit to the plasma data. Finally, fits for plasma data from individual animals were loaded into the ITS SAAM II engine as described above to derive population PK parameter estimates.
Total lymphatic ARV exposure as a percentage of the subcutaneously injected dose (%ID) was estimated by adding together the ARV amounts in each of the three lymphatic delay compartments (d2, d3, d4) at each time point, and then dividing this sum by the total ARV amount administered in the SC dose.
| (14) |
Sensitivity analysis was performed on all TLC-ART101 model parameters by taking the parameter value that best fit the geometric mean plasma data, increasing and decreasing each value by 10-fold from the optimal value, and inspecting the impact on the estimated plasma ARV levels versus observed levels.
Statistical analysis
Statistical analyses were performed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). All statistical comparisons were performed with the two-tailed t-tests with significance probability α set at 0.05.
Results
Long-Acting Plasma PK of LPV, RTV, and TFV Following TLC-ART101
We reported previously that SC dosing of LPV, RTV, and TFV stabilized in a DcNP suspension in macaques exhibited plasma PK profiles markedly different than those of the free drug combination—while the same three ARVs in the free formulation cleared in ∼24 hr, DcNP-associated ARVs persisted for the 1-week duration of the experiment [18]. The plasma time course of these three ARVs in another DcNP formulation with a slight variation in the fixed dose molar ratio of LPV-to-RTV, called TLC-ART101, is presented in Figure 1. RTV is typically included in oral dosage forms to inhibit CYP3A-mediated metabolism and thus boost the oral bioavailability of protease inhibitors [27]. RTV is included in the TLC-ART101 injectable formulation containing LPV to preserve the protease inhibitor combination and possibly slow down the metabolic clearance of LPV. Measurable plasma levels of the two active ARVs of interest—LPV and TFV—persisted for over 2 weeks (Figure 1A, 1C). Compared to the free drug formulation, which cleared rapidly, the area under the plasma concentration-time curve (AUC) for LPV, RTV, and TFV in TLC-ART101 was 2.5-, 3.5-, and 28.9-fold higher, respectively (P-value = LPV, 0.0871; RTV, 0.0878; TFV, 0.0001) (Table 1). LPV and TFV in TLC-ART101 had apparent terminal half-lives (t1/2) of ∼20 and ∼3 days. In comparison to free drug, t1/2 of LPV, RTV, and TFV in TLC-ART101 was 55.1-, 5.5-, and 8.2-fold longer, respectively (P-value = LPV, 0.3814; RTV, 0.0306; TFV, <0.0001) (Table 1).
Figure 1. Plasma concentration-time profiles of lopinavir (LPV; panel A), ritonavir (RTV; panel B), and tenofovir (TFV; panel C) in macaques following a single SC dose of the triple-drug combination in either the soluble (“free”; red open circles [O] and dotted line) or TLC-ART101 (blue closed circles [•] and solid line) formulation.

The top graphs in panels A, B, and C are plasma concentration-time profiles of the first 24 hr after SC dosing, and the bottom graphs are the entire time course over 336 hr (2 weeks). Please note the differences in the y-axis scales for the top graphs over 24 hr to highlight relevant plasma concentrations for each ARV. Plasma limit of quantification (LOQ)/limit of detection (LOD) = lopinavir: 10/5, ritonavir: 50/25, tenofovir: 250/100 pg/mL. Ritonavir (intended as a PK booster for lopinavir) in plasma after TLC-ART101 dosing was <LOQ in N=2 at 192 and 336 hr. Geometric mean ± SD (N=3-8).
Table 1. Descriptive non-compartmental plasma pharmacokinetic (PK) parameters for each drug in the triple-drug combination given as a single SC dose in either the free or TLC-ART101 formulation.
| Lopinavir (LPV) | Ritonavir (RTV) | Tenofovir (TFV) | ||||
|---|---|---|---|---|---|---|
| Free | TLC-ART101 | Free | TLC-ART101 | Free | TLC-ART101 | |
| AUCa (hr•μg/mL) | 4.35 (7) | 10.98 (48) | 1.37 (9) | 4.80 (57) | 14.4 (4) | 416.55 (15) |
| Terminal t1/2 (hr) | 8.65 (28) | 476.94 (173) | 7.99 (38) | 44.06 (46) | 8.01 (51) | 65.33 (11) |
| CL/F (L/[hr•kg]) | 4.64 (14) | 0.85 (85) | 4.23 (15) | 1.43 (56) | 0.72 (4) | 0.02 (15) |
| MBRTb (hr) | 10.83 (5) | 116.09 (8) | 10.78 (5) | 24.86 (9) | 2.61 (12) | 102.68 (7) |
Geometric Mean (% coefficient of variation).
AUC0-24h for Free, AUC0-336h for TLC-ART101.
MBRT0-24h for Free, MBRT0-336h for TLC-ART101.
AUC, area under the plasma drug concentration-time curve; CL/F, apparent total clearance; MBRT, mean body residence time; t1/2, apparent terminal plasma drug half-life.
AUC and t1/2 values for TLC-ART101 only (not free drug) were recently reported elsewhere [19]. They are included here for comparison to free drug.
Following injection of the free drug formulation, the two hydrophobic protease inhibitors, LPV (LogD7.4 4.69) and RTV (LogD7.4 5.22), exhibited similar plasma PK profiles: time of maximum concentration (Tmax) occurred between 30 min and 8 hr, and plasma levels declined in a monoexponential fashion out to 24 hr (Figure 1A, 1B). Free LPV and RTV also had similar t1/2, apparent clearance (CL/F), and mean body residence time (MBRT0-24h) (Table 1). In contrast, free hydrophilic TFV exhibited a plasma PK profile resembling an IV bolus and declined in a biphasic fashion over 24 hr (Figure 1C), which was consistent with other studies in macaques given free TFV subcutaneously [28]. Initial decline of free TFV was rapid; levels fell to ∼1% of their peak by 10 hr, followed by a slower terminal phase of decline (Figure 1C). Free TFV was eliminated from the body more rapidly than free LPV and RTV, as MBRT0-24h for free TFV was ∼5-fold shorter than those of free LPV and RTV, and CL/F followed a similar trend (Table 1).
While the PK of free LPV (25 mg/kg) and RTV (7 mg/kg) were remarkably similar, the PK of an equivalent dose of LPV and RTV in TLC-ART101 were different in significant ways (Figure 1A, 1B; Table 1). Following their injection in TLC-ART101, both LPV and RTV had a very rapid initial phase of absorption that resulted in a small first peak within 0.5 to 1 hr; thereafter, plasma concentrations rose to a second peak or plateau at ∼8 hr, followed by a slow but steady decline over the next 16 to 40 hr (Figure 1A, 1B). Tmax of ∼8 hr for LPV and RTV suggested a moderate rate of absorption with the DcNP-associated dose (Figure 1A, 1B). After 48 hr, the decline of plasma LPV concentration slowed to a near plateau at ∼20 ng/mL that persisted out to 336 hr (2 weeks) (Figure 1A); RTV levels declined in a multiphasic fashion and were mostly eliminated by 2 weeks (Figure 1B). Although MBRT0-24h for free LPV and RTV were nearly identical, following TLC-ART101 administration MBRT0-336h of LPV was ∼4.6-fold longer than that of RTV (Table 1). Delivery of TFV as DcNPs resulted in remarkable transformation of its PK compared to free TFV (Figure 1C). TFV levels persisted out to 336 hr, with an initial peak within 1 hr after injection, a subsequent gradual decline, followed by a rebound at ∼8 hr that peaked at ∼2,800 ng/mL around 24-48 hr, before declining in a log-linear fashion to ∼170 ng/mL at 336 hr (Figure 1C). MBRT0-336h of TFV following injection of TLC-ART101 was ∼39-fold longer than the MBRT0-24h of free TFV, and was comparable to that of LPV (Table 1). CL/F for all three ARVs decreased in TLC-ART101, most dramatically for TFV with an ∼36-fold drop in CL/F (Table 1), reflecting its striking high and persistent plasma concentration profile (Figure 1C). In sum, these differences in plasma PK between free and TLC-ART101 ARVs illustrate the marked effect that the TLC-ART101 formulation had on plasma persistence for both the hydrophobic (LPV, RTV) and hydrophilic (TFV) ARVs.
Mechanism-Based PK (MBPK) Models
Mechanism-based compartmental PK models were developed that can provide kinetic insights into the physiological processes of subcutaneous uptake, lymphatic transit, and systemic disposition of the three ARVs delivered by the TLC-ART101 formulation. As depicted in Figure 2, we assumed the following scenario for subcutaneously administered LPV, RTV, and TFV collected into a DcNP form (69.0 ± 8.3 nm): (1) the DcNPs have appropriate particle sizes and surface properties to move via convection through the <100 nm interstitial water channels in the SC space (between the skin and muscle) [29], (2) they are too large to move paracellularly between or through the 5-12 nm intercellular gaps between capillary blood endothelial cells [30], and as a result, (3) the three drugs in DcNPs preferentially enter lymphatic capillaries, which have an architecture that promotes uptake of larger particulate matter [26]. Once in lymphatic capillaries, these DcNPs travel throughout the lymphatic vessels and nodes. A fraction of the DcNPs taken up into the lymphatics are retained in lymph node sinuses, and a fraction eventually enter the blood circulation either through lymph that flows into the venous blood through the right and left thoracic ducts, or as cargo within lymphocytes in lymph nodes that traffic out of lymph nodes into the blood via the thoracic duct or high endothelial venules (HEVs) in lymph nodes. If DcNPs prematurely released drug and disintegrated in vivo (in the lymphatics or the blood), the released ARVs would be rapidly cleared as shown with free ARVs (Figure 1, Table 1). In contrast, free drugs in the SC space are small enough to penetrate or diffuse through the capillary endothelium; while a fraction can enter the lymphatics, overall, free drug molecules are preferentially absorbed via the blood capillaries due to the ∼100-500-fold faster flow in blood compared to lymphatic capillaries [29], i.e., a more effective “sink” condition. Unlike DcNP-associated ARVs that experience retentive forces that trap them in lymph vessels and nodes, free ARVs that enter the lymph could readily diffuse out and redistribute back into the blood. As a result, free ARVs would follow drug distribution and clearance mechanisms mediated by blood carriage, whereas ARVs associated to DcNPs, following their preferential uptake into the lymphatics, would first encounter distribution and retention mechanisms within the lymphatics, before being subject to those in the blood. Taken together, DcNPs in TLC-ART101 would likely undergo first-passage through the lymphatic system before entering the blood circulation, whereas free ARVs in the SC space would be preferentially absorbed directly into the blood vasculature. These processes are schematically illustrated in Figure 2 and featured in the compartmental models in Figure 3C, 3D.
Figure 2. Schematic representation of the differential absorption and disposition pathways of ARVs associated to DcNPs in TLC-ART101, free, or in a stationary depot in the SC or IM space.
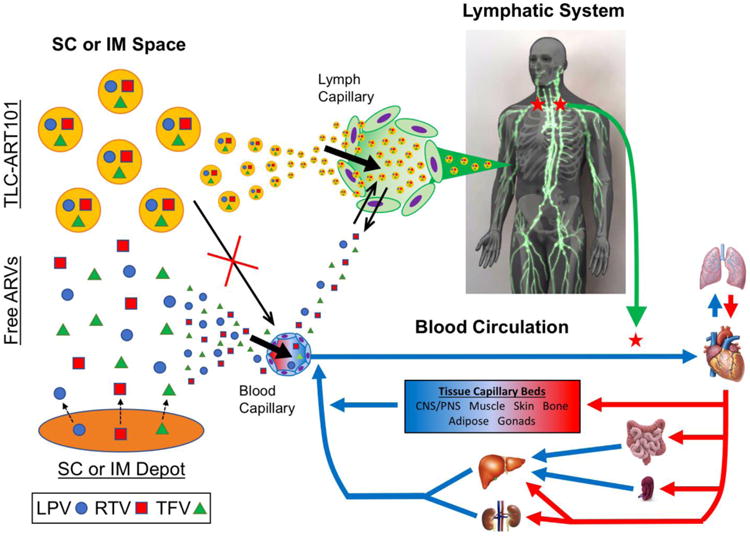
In the SC space, ARV combinations (e.g., lopinavir [LPV], ritonavir [RTV], and tenofovir [TFV]) associated to drug combination nanoparticles (DcNPs) and referred to as “TLC-ART101,” are too large to enter blood capillaries, and thus preferentially enter lymph capillaries. While free ARVs in the SC space can enter lymph capillaries initially due to a strong concentration gradient driving drug out of the SC space, free ARVs are not able to be retained in the lymphatics and can easily diffuse back into the SC space as the diffusion gradient reverses direction over time. Overall, free ARVs preferentially enter the blood from the SC space, largely due to the 100-500-fold faster blood flow via capillaries than lymph flow. A stationary SC or intramuscular (IM) depot that provides a sustained release reservoir of free drug would follow the same principles for free drug and thus would also be predominantly absorbed directly into the blood. Once in lymphatic capillaries, TLC-ART101 particles would undergo first-pass distribution throughout the lymphatic system before entering the systemic blood circulation. In contrast, free drug that mostly enters the systemic circulation would undergo distribution, metabolism, and excretion mechanisms characteristic to the free drug molecule.
These assumptions are based on in vivo animal studies with nanoparticulate drug carriers reported in the literature. For instance, in rats, a major fraction of radiolabeled liposomes ∼50-100 nm in diameter preferentially entered the lymphatics versus the blood following SC injection [31]. Also, in sheep, proteins <16-20 kDa (∼4-5 nm) in the SC space were absorbed primarily by blood capillaries draining the SC injection site [32]. Based on these observations, we propose differential absorption mechanisms between free and DcNP-associated ARVs from the SC space in accordance to their differences in physical size. Thus, we developed composite compartmental PK models that would feature parallel uptake of (1) DcNP-associated ARVs into and transit through the lymphatics, eventually making their way to the systemic circulation (submodel above the dashed line in Figure 3C, 3D) and (2) free ARVs directly into the blood for systemic distribution (submodel below the dashed line in Figure 3C, 3D). It is further assumed that once the DcNP-associated ARVs reach systemic circulation, free drug can be release from the DcNPs.
Models of Plasma PK of Free LPV, RTV, and TFV
We assumed that the model structure and parameter estimates derived from analysis of PK data obtained after SC injection of the free ARV formulation apply equally to the PK of free drug either absorbed directly or released from DcNPs following administration of TLC-ART101. This lessened the burden of parameter estimation for the full MBPK models and allayed some concerns of model complexity. Thus, for the initial step in model development, we analyzed the plasma ARV concentration-time data in macaques given a SC dose of the free ARV combination (Figure 1) according to a conventional one- or two-compartmental PK model (1CM or 2CM) with first-order absorption from the SC injection site (Figure 3A, 3B). Population estimates of free drug PK parameters obtained using an iterated two-stage approach are presented in Table 2. Excellent model fits were observed with individual and mean plasma concentration-time data from the cohort of macaques (Figure 4).
Figure 4. Observed (symbols) and fitted (curves) plasma concentration-time profiles of lopinavir (LPV, panel A), ritonavir (RTV, panel B), and tenofovir (TFV, panel C) in macaques after a single SC dose of the free and soluble triple-drug combination (N=3).
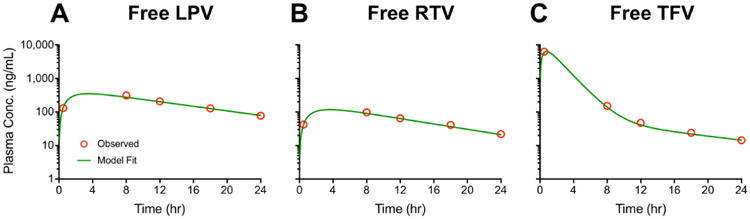
Symbols represent the geometric mean plasma concentration, and green curves represent the fits that resulted from population analysis.
All three free ARVs were assumed to have a rapid absorption following SC injection (i.e., k21 > k52, k53, and k54) (Table 2) [21]. CL/F and t1/2 values in Table 2 agreed with their corresponding estimates derived from non-compartmental analysis in Table 1. The rather high ∼4 L/(hr•kg) CL/F for free LPV and RTV (Table 2) are comparable to reported macaque liver blood flow [33], indicating a high hepatic extraction and clearance of these two protease inhibitors. The 0.60 L/(hr•kg) CL/F for free TFV (Table 2) matched macaque renal plasma flow [33], which is consistent with avid renal tubular clearance of this nucleotide reverse transcriptase inhibitor. The population parameter estimates for free ARV PK were entered as Bayesian priors in the MBPK models for TLC-ART101-associated ARVs (Figure 3C, 3D).
Models of Plasma and Lymphatic PK of DcNP-associated LPV, RTV, and TFV
As mentioned above, we designed the MBPK models for TLC-ART101 to consist of: (1) a submodel for disposition of DcNP-associated ARV following its absorption from the SC space that features first-passage through the lymphatics (lymph vessels and nodes), that is linked to (2) a submodel for disposition of free ARV either absorbed directly from the SC space or released from DcNPs during their systemic distribution phase (Figure 3C, 3D). Based on equilibrium dialysis results, the “free-to-DcNP-associated” fractions of TFV in TLC-ART101 were 90:10 (mol%/mol%); hence, 10% of the TLC-ART101 dose was assigned to the SC compartment (q1) for DcNPs and 90% was assigned to the SC compartment for free drug (q6). LPV and RTV were 100% associated to DcNPs; hence, the entire LPV and RTV dose in TLC-ART101 was assigned to the SC compartment for nanoparticles (q1).
We first characterized the lymphatic transit kinetics for each ARV using the geometric mean plasma values from N=8 animals (four of these eight only contributed blood samples at 30 min and 24 hr). Fitting the plasma concentration-time data to these models resulted in lag time estimates for the three ARVs that range from 0.1 to 2.2 hr with the fast lymphatic pathway, 10.9 to 38.8 hr with the intermediate pathway, and 122.1 to 262.2 hr for the slow pathway (Table 2). As the initial plasma levels for LPV and RTV were similar over 0-48 hr (Figure 1A, 1B), tlag1 for both of these ARVs was nearly identical at ∼2 hr (Table 2). However, because LPV plasma levels were sustained at essentially a steady-state during 48-336 hr while RTV plasma concentration declined steadily over 48-336 hr (Figure 1A, 1B), tlag3 for LPV (260 hr) was much longer than that for RTV (180 hr) (Table 2). RTV, at ¼ of the dose of LPV (i.e., LPV-to-RTV ratio of 4:1 mol%/mol%), showed a slight rebound in levels around 1 week (168 hr) (Figure 1B), which accounts for its tlag3 value (Table 2). TFV, due to its very early first peak (which likely corresponded to absorption of the free fraction in DcNPs), then second peak (which likely corresponded to the DcNP-associated fraction), and gradual but persistent terminal decline (Figure 1C), had the most rapid tlag1 (0.1 hr) and the shortest tlag3 (120 hr) amongst the three DcNP-associated ARVs (Table 2). Thus, taken together, each ARV exhibited characteristically short, medium, and long lag times which may be thought of as “waves” of DcNPs being released into the blood circulation from the lymphatics. These three different “waves” of DcNPs appearing in the plasma may occur either through a faster and a slower flow of DcNPs through the lymph into the blood (the faster flow through lymph occurring soon after injection and the slower flow through lymph resulting from DcNPs initially trapped in lymph node sinuses eventually being carried away by lymph), or through lymph node lymphocytes that have taken up DcNPs and then trafficked into the blood and slowly releasing ARVs into the plasma. These lag times were entered as Bayesian estimates for further model refinement.
Next, we performed a manual search for the guess-estimates of k21, k31, and k41, the rate constants for entry of the three ARVs into each of the three lymphatic pathways (Figure 3C, 3D). Since entry of DcNPs into the different lymphatic pathways operate in parallel, the sum total of the entry rate constants k21, k31, and k41 for DcNP-associated ARV uptake from the SC space into the respective lymphatic delay compartments d2, d3, and d4 (Figure 3C, 3D) ought to be about the same for each ARV. This is consistent with our observations that DcNPs cleared from the SC space within ∼24 hr and were predominantly taken up into lymph vessels and nodes. Through systematic adjustment of k21, k31, and k41, the minimum sum for each ARV was determined to be ∼0.45 hr-1 (i.e., k21 + k31 + k41 = ∼0.45 hr-1) and constrained to this value during regression runs (Table 2). The sum for final estimates of the entry rate constants for LPV, RTV, and TFV equaled 0.46, 0.44, and 0.47 hr-1, respectively (Table 2). However, despite constraining the sum total of lymphatic entry rate constants to be the same value for all three ARVs, the optimal estimates of k21, k31, and k41 differed among ARVs in the final model fits (Table 2). For LPV, the rank order for the lymphatic entry rate constants was k41 ≫ k31 ≫ k21; for RTV, k21 > k31 ≫ k41; and for TFV, k41 ≥ k31 ≥ k21 (Table 2), suggesting that LPV was most available for the slow lymphatic pathway (d4), RTV was most available for the fast lymphatic pathway (d2), while hydrophilic TFV entered into all three lymphatic pathways to nearly equal extent, despite the fact that all three ARVs were tightly associated with DcNPs and presumably then transit together as single entities through lymph vessels and nodes. This could be due to uneven incorporation of the three ARVs into DcNPs—creating different subpopulations of DcNPs—and retention of DcNPs within the lymphatic network being influenced by the drug payload. Alternatively, there may be different rates of intracellular metabolism/anabolism and efflux from lymphocytes for each ARV, which would impact their lymphatic transit kinetics. More investigation is required to understand the underlying mechanisms for this apparent differential lymphatic transit among each ARV, the details of which are beyond the scope of this report. In summary, although the total sum of the lymphatic entry rate constants could be set to a value comparable for all three ARVs, each DcNP-associated ARV had its own unique uptake distribution into the three lymphatic pathways (Table 2). After the best estimates of lymphatic entry rate constants were determined for each ARV using geometric mean data, they were fixed as we proceeded to the iterated two-stage population analysis of the cohort macaque data.
For all three ARVs, the TLC-ART101 MBPK model fits to observed plasma concentration-time data were excellent (Figure 5) and %CV for the regression estimates were within a reasonable range considering the limited sample size (N=8) (Table 2). Sensitivity analysis, where the optimal parameter value was increased or decreased by 10-fold, showed that tlag3, V7_DcNP, and k75 impacted the predicted plasma ARV concentration-time profile the most. We have used these MBPK models for dose selection for TLC-ART101 multi-dosing studies in healthy and SHIV-C-infected macaques, and the models predicted the observed plasma concentrations extremely well (personal observation), which demonstrates the validity and robustness of these models.
Figure 5. Observed (symbols) and fitted (curves) plasma concentration-time profiles of lopinavir (LPV, panel A), ritonavir (RTV, panel B), and tenofovir (TFV, panel C) in macaques after a single SC dose of TLC-ART101 (N=8).
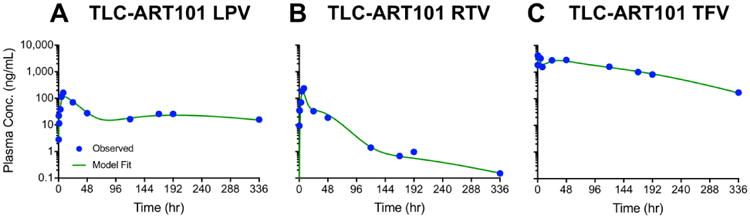
Symbols represent the geometric mean plasma concentration, and blue curves represent the fits that resulted from population analysis.
The apparent volume of distribution of DcNP-associated ARVs in the q5 plasma compartment (V5_DcNP) (Figure 3C, 3D) was ∼7-10 L/kg for the lipophilic ARVs LPV and RTV, while V5_DcNP for hydrophilic TFV was much smaller at 0.17 L/kg (Table 2). These distribution volumes were about 6.7- to 12.8-fold smaller than those of free ARVs in q7 (V7_Free) (Table 2), suggesting much more limited extravascular distribution for DcNP-associated ARVs. The rate constant for release of free ARV from DcNPs in the plasma (k75) was similar for LPV and RTV at ∼0.4-0.7 hr-1 (t1/2 ∼1.0-1.7 hr), while k75 was much slower for TFV at ∼0.02 hr-1 (t1/2 ∼35 hr) (Table 2). As expected, the final mean parameter estimates for free ARV (k76, k78, k87, V7_Free, and k07) matched the assigned Bayesian priors (Table 2). Next, we used the models to evaluate the ARV exposure in the lymphatic system.
Lymphatic Exposure to LPV, RTV, and TFV Following TLC-ART101
Based on single-photon emission computed tomography (SPECT) imaging of 111In-labeled anti-CD4 mAbs dosed by IV infusion in macaques, ∼75% of CD4+ T lymphocytes (the main target cell of HIV) are located in lymph nodes (∼15% are in the spleen, ∼10% are in Peyer's patches, and ∼0.3-0.5% are in the blood) [34]. These data are corroborated by a separate study in macaques infected with simian immunodeficiency virus (SIV) that were dosed IV with 64Cu-labeled anti-SIV Gp120 PEGylated mAbs and imaged with immunoPET (antibody-targeted positron emission tomography), which showed predominant SIV localization in lymphoid tissues in chronically viremic animals and still measurable viral levels in lymph nodes after 5 weeks of combination antiretroviral therapy [35]. Thus, it is important to target anti-HIV drugs to lymph nodes throughout the body and sustain ARV levels within lymph node cells infected by HIV. The MBPK models we developed allowed us to simulate the total lymphatic ARV exposure from TLC-ART101. Figure 6 shows the predicted amount of ARVs held in the lymphatics during first-pass over the entire 2 weeks. More than 80% of the LPV dose was held in the lymphatics at peak, and released gradually over the 2 weeks (Figure 6A). In contrast, less of the RTV dose was withheld and nearly all of it was released from the lymphatics within 96 hr (Figure 6B). With TFV, while the maximum % of dose withheld in the lymphatics was low (<10%), it accounted for nearly the entire DcNP-associated portion (10%) of the TLC-ART101 dose (Figure 6C). Subsequent release of TFV from the lymphatics was gradual, but at a faster pace compared to LPV (Figure 6A, 6C).
Figure 6. Time-course of the percentage of injected dose (%ID) of lopinavir (LPV, panel A), ritonavir (RTV, panel B), and tenofovir (TFV, panel C) in the lymphatics after a single SC dose of TLC-ART101.
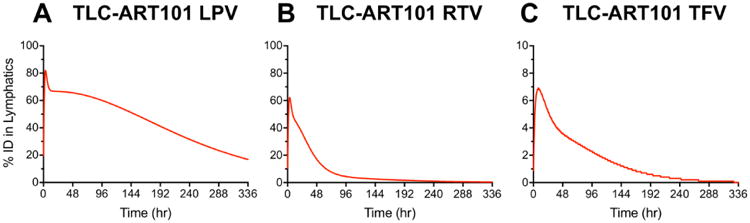
Discussion
Previously, we reported a long-acting ARV combination injectable (TLC-ART101) that provided sustained lymphocyte and plasma ARV concentrations in macaques for longer than the 2-week experiment [19]. However, detailed mechanisms leading to this lymphocyte-targeting and persistent intracellular and plasma ARV concentrations were not elucidated. In this study, PK models were developed based on consideration of physiological processes that govern the uptake of DcNPs from the SC interstitium into lymph capillaries, transit through the lymphatic network including retention by the lymph nodes, and eventual drainage into the blood circulation. The models were able to simultaneously capture the complex persistent lymphatic and plasma pharmacokinetics of three ARVs (LPV, RTV, TFV) formulated together in the TLC-ART101 lymphocyte-targeting and long-acting drug combination formulation (Figure 3C, 3D, 5, 6). Moreover, these mechanism-based models afforded mechanistic insights into the absorption and disposition of DcNP-associated ARVs and enabled prediction of overall ARV levels in the lymphatics (lymph vessels and nodes) following SC injection of TLC-ART101 (Figure 6). They can also guide the design of dosage regimens for future efficacy and safety studies of TLC-ART101.
To our knowledge, these are the first mechanistic PK models of a nanoparticle-bound drug combination that describe the time-course of plasma concentration and lymphatic retention of multiple ARVs formulated in a single nanosuspension. These models enabled, for the first time, prediction of the long-acting PK of each ARV in both the plasma and lymphatics. Our mechanism-based PK models were also able to account for the complex, multiphasic plasma-concentration time course of the three DcNP-associated ARVs (Figure 1) by having multiple (three) delay pathways through the lymphatics after SC injection of TLC-ART101 (Figure 3C, 3D). Only one or two lymphatic delay compartments were not sufficient to account for the complex plasma concentration time courses of each ARV. Previous reports on the disposition of subcutaneously injected large proteins of ∼4-10 nm in diameter [23, 36-44] (reviewed in [45]) and single, small molecule drugs in ∼10-100 nm diameter nanoparticles [15] have reported PK models featuring a lymphatic component. The few PBPK models published to date include single monoclonal antibodies (mAbs) administered SC [39, 44]. Despite evidence of significant absorption of lymphatic fluid into the blood as it flows through lymph nodes in mammalian systems (which leads to concentrating lymph solutes and decreasing the lymph flow rate) [23, 46], and substantial recirculation of mAbs via the lymphatic system after IV administration [23], these PBPK models either assumed reabsorption of drug molecules in lymph tissue and fluid back into the blood was negligible [39] or no recirculation via the lymphatic system occurred after entry of mAbs into the systemic circulation [44].
While the detailed mechanisms of how the three DcNP-associated drugs in the TLC-ART101 formulation exhibited long-lasting retention in the lymphatics, particularly in lymph nodes, and were targeted to lymphocytes, awaits further investigation; a working hypothesis is presented schematically in Figure 2. We envisioned the mechanisms that lead to the complex plasma time-course of the three ARVs in TLC-ART101 are as follows: first, in the SC space, the ARVs associated to DcNPs readily clear from the interstitium and preferentially enter lymph capillaries (as opposed to free drug with greater propensity to enter blood capillaries), followed by first-pass transit of the DcNPs through the lymphatic network. This exclusive lymphatic uptake and extensive first-pass distribution through lymph vessel and node networks has been confirmed by in vivo imaging of DcNP clearance from the SC space and lymphatic uptake, distribution, and retention in mice (personal observation, submitted for publication). This is consistent with our observation of high levels of drug distribution and accumulation in lymph nodes all throughout the body in macaques following a single SC dose of DcNPs, which was not observed with free drug [12]. This lymphatic first-pass distribution of DcNPs is also consistent with the higher drug concentrations in LNMCs versus those in PBMCs, which are both persistently higher than the drug levels in the plasma [19]. These high intracellular levels of ARVs are opposite of the higher plasma versus intracellular levels that are typically observed following oral dosing of the same ARVs in DcNPs [47-50]. Thus, as supported by our in vitro release studies under sink conditions, DcNPs must be sufficiently stable and not release a significant fraction of their ARV payloads to be able to make their extensive journey through lymph vessel and node networks, be internalized by LNMCs, and provide long-acting PK; if DcNPs readily degraded and released free ARVs, these free drugs would not be able to transit extensively through lymph vessels and nodes and be retained by the lymphatics, and they would clear from the body rapidly.
As the lymphatic network is composed of nodes with sinuses within the node tissue being able to trap ∼70 nm diameter DcNPs [51], they provide a reservoir or depot for sustained drug presence in the lymphatics. Tissue lipases in the nodes as well as those on the surface or within lymph node parenchymal cells could release the drug back into the lymphatic network, or the DcNPs could be taken up by lymphocytes trafficking in and out of the blood via high endothelial venules (HEVs) in lymph nodes. The acid- and base-stable characteristics of LPV and RTV may also contribute to the ability of these drugs to release in acidic intracellular organelles such as endosomes and lysosomes with minimal risk of degradation. Long-acting plasma PK could also arise from gradual release of DcNP- and lymphocyte-associated ARVs from the lymphatics into the blood via lymph-blood vessel anastomoses near the heart. It is likely that a combination of these processes work in concert to provide the long-acting plasma and intracellular kinetics observed for each ARV. These processes are accounted for in the three lymphatic delay compartments in the MBPK models (Figure 3C, 3D), which represent three different “waves” of release of DcNPs from the lymphatics into the systemic circulation. As discussed below, recent data collected in our laboratory and those reported by others support this integrated hypothesis (i.e., preferential DcNP uptake into lymphatics, lymphatic first-pass distribution and retention, extended release into blood, and limited extravasation in blood circulation).
Preferential Lymphatic (First-Pass) Uptake of DcNP-associated ARVs in TLC-ART101 From the SC Space
Drugs or vaccines deposited in the SC space may stay there or be taken up by lymph or blood capillaries. Free small molecules, peptides, and small proteins can permeate the ∼5-12 nm intercellular pores between capillary blood endothelial cells and/or diffuse across the blood capillary endothelium [30], while ∼70 nm DcNPs may be too large to readily enter blood capillaries. Using the near-infrared fluorescence marker indocyanine green (ICG) for real-time imaging in mice, we found that SC administered DcNP-associated ICG, but not free/soluble ICG, predominantly entered lymph vessels rather than blood vessels. Free ICG in mice penetrated into lymph as well as blood vessels, but only distributed to the first few lymph vessels and nodes draining the SC injection site; free ICG rapidly absorbed into the blood to a greater extent than DcNP-associated ICG (personal observation, submitted for publication). Due to the ∼100-500-fold faster capillary blood flow compared to lymph capillary flow [29], smaller molecules are preferentially absorbed from the SC space via the blood. This predominant route of absorption via blood for small molecules in the SC space was also observed with SC dosing of free hydrophobic doxorubicin or hydrophilic methotrexate in rats fitted with a thoracic lymph cannula, where ∼64-74% of a SC dose was recovered in the blood, while ∼0.063-2% was recovered in the lymph [14, 15]. With respect to larger particles, certain particles tend to be retained in the SC space, especially if their large size limits their convection through the ∼100 nm interstitial water channels [29]. For instance, in pigs, ∼105 nm negatively charged polymeric particles were mostly confined to the injection site 24 hr post-injection, while smaller ∼55 nm counterparts traveled extensively to draining inguinal lymph nodes ∼24 cm from the SC injection site [52]. In addition, in rats, ∼83 nm PEGylated doxorubicin liposomes (Doxil®) mostly remained at the SC injection site for an extended period [15]. This is in contrast to ∼70 nm DcNPs in TLC-ART101 that cleared from the SC injection site in mice, rats, and macaques within ∼24 hr (personal observation). Taken together, these data are consistent with free ARVs predominantly clearing from the SC space via blood capillaries, while ∼70 nm DcNPs are optimal for lymphatic uptake (Figure 2).
Lymphatic First-Passage of DcNP-associated ARVs in TLC-ART101
Since DcNP-associated ARVs cleared rapidly from the SC space and predominantly absorbed into the lymph capillaries, they are likely to first transit through the lymphatics before entering the blood. This delay in absorption into the systemic circulation is reflected in the longer Tmax for ARVs in TLC-ART101 (∼8-36 hr) versus in free formulation (∼1-3 hr) (Figures 4-5); the latter directly enter the blood from the SC space. Importantly, the Tmax values for all three ARVs in DcNPs (∼8-36 hr) were much shorter than the days to weeks it takes for long-acting (LA) cabotegravir and LA rilpivirine to reach peak levels in humans [3, 4], in which case slow absorption controls the terminal decline of LA cabotegravir and LA rilpivirine over weeks, suggesting sustained release of free drug from an IM depot, and limited lymphatic uptake, distribution, and retention (as depicted in Figure 2). This is in contrast to DcNP-associated ARVs in TLC-ART101 that cleared readily from the SC injection site, were predominantly taken into lymph capillaries, distributed through lymph vessels to lymph nodes throughout the body, and reached peak plasma levels at ∼6-8 hr. A slower rise in LA cabotegravir or LA rilpivirine plasma levels is expected due to the sustained release from slow dissolution of the nanocrystal of hydrophobic cabotegravir or rilpivirine [1]; free drug released into the extracellular fluid is then taken up immediately into blood capillaries. Thus, these formulations, by design, would require an extended time to reach Tmax and therapeutic concentrations; as a result, an initial oral (lead-in) dose supplement is essential to more quickly reach target peak levels [2], complicating these IM long-acting regimens. Such an oral lead-in is probably unnecessary for the TLC-ART101 formulation. Avoiding an oral lead-in strategy could simplify treatment and improve patient adherence.
The parallel rise and peak in plasma levels over 8 hr of the lipophilic ARVs (LPV, RTV) associated to DcNPs (Figure 1A, 1B) also suggest ARVs remain associated to DcNPs in vivo. If large amounts of ARVs released from DcNPs prematurely, they would clear in a matter of hours as free ARVs are, they would not preferentially distribute throughout and be retained within the lymphatics, and would not exhibit plasma persistence for 2 weeks or more (Figure 1). TFV in TLC-ART101 exhibited an early Tmax at ∼1 hr, which resembled the free TFV plasma PK profile (Figure 1C); this suggests a significant fraction of the TLC-ART101 formulation is present in free form (e.g., ∼90%), consistent with in vitro equilibrium dialysis results. The most remarkable finding was the persistence of plasma TFV that occurred with only ∼10% of the TFV dose associated to DcNPs; TFV must be firmly associated to sufficiently stable DcNPs with limited extravascular distribution and long blood circulation lifetimes.
Widespread lymphatic distribution and retention of ARVs in TLC-ART101
Our recent data in macaques show widespread lymph node distribution [12] and long-term LNMC retention [19] of DcNP-associated ARVs. Intracellular ARV levels in LNMCs at 1 and 8 days after a single SC dose were persistently higher than those in PBMCs, and in turn levels in PBMCs were persistently higher than those in plasma over the 2-week experiment [19], demonstrating the lymphocyte-targeting properties of the TLC-ART101 formulation. These high concentrations of ARVs in LNMCs relative to those in PBMCs and plasma suggest DcNP-associated ARVs are sequestered during lymphatic first-pass and accumulate in lymphocytes.
Such intracellular delivery performance for these ARVs in lymphocytes is unprecedented. In pharmacokinetic studies in rats and humans dosed orally with LPV and RTV, the whole blood/plasma ratio for LPV was 0.44-0.50 and for RTV was 0.26-0.68 [53, 54]. In these studies, the lymph node/plasma ratio in rats for LPV was 0.19-0.55 [54]. In HIV-infected persons, LPV and RTV concentrations in whole blood were reported to be lower than those in plasma [55]. Collectively, these studies show that LPV and RTV, administered either as a free/soluble injectable or an oral dosage form, are taken up poorly into blood cells, and LPV has limited penetration into lymph nodes. However, since erythrocytes comprise >99% of the whole blood cellular fraction, LPV and RTV penetration in PBMCs may be different than in erythrocytes. Still, in HIV-infected persons, the PBMC/plasma ratio for LPV was 0.34-0.55 and for RTV was 0.46 [47-49], indicating poor PBMC penetration. For TLC-ART101, a single SC dose in macaques exhibited PBMC/plasma ratios of 4.01 for LPV, 7.40 for RTV, and 3.01 for TFV. Thus, compared to a free/soluble injectable or an oral dosage form, association of LPV and RTV to DcNPs has increased their respective PBMC/plasma ratios by ∼7-12-fold and ∼16-fold. For hydrophilic TFV, which is negatively charged at physiological pH 7.4, a single SC dose of TLC-ART101 produced a much greater PBMC exposure (AUC) to the intracellular active metabolite of TFV, TFV-diphosphate (TFV-DP), by ∼7-fold compared to a single 1.5 mg/kg oral dose of the TFV prodrug tenofovir alafenamide (TAF) in macaques [19, 56]. The detailed mechanisms behind these strikingly enhanced intracellular ARV levels in LNMCs and PBMCs, the most commonly infected cells, remain to be elucidated. However, it is likely that the preferential lymphatic uptake of DcNPs from the SC space and the widespread lymphatic distribution and retention over long durations promote the uptake of DcNPs by lymphocytes in lymph nodes, which can traffic from lymph nodes to the blood either via lymph flow through the thoracic duct or via HEVs in lymph nodes. The MBPK models reported here could serve as a base model to account for accumulation of ARVs in target intracellular compartments, viz. PBMCs and LNMCs.
As mentioned, within the lymph node, DcNPs could access and be retained in the ∼1-500 μm sinus spaces [51], where they could readily be taken up by LNMCs. This deep lymph node retention (e.g., in LNMCs) could be a component of sequestered DcNP-associated ARVs within the lymphatics that contribute to the relatively long lag times and sustained levels in plasma (Table 2). For the models to sustain persistent plasma ARV levels, we postulated three different “waves” of release (having an early, intermediate, or late transit time) of DcNP-associated ARVs from the lymphatics into the systemic circulation (Table 2). LPV and RTV concentrations both had a rapid initial peak in the first 0.5 to 1 hr and subsequently continued to rise quickly over the first 6 hr (Figure 1A, 1B), suggesting an initial rapid wave of ARVs in DcNPs entering and exiting the lymphatics. This was followed by an intermediate wave of DcNPs between 8 to 24 hr as evidenced by the rebound in the TFV concentration (Figure 1C). Finally, there was a late and protracted emergence of ARVs in DcNPs beyond 48 hr that sustained measurable plasma levels for over 2 weeks (Figure 1). Thus, it is possible a fraction of the ARVs in DcNPs within the lymphatics moves rapidly through the major lymphatic vessels, another fraction moves less rapidly due to travel through the diffuse branched vessels and/or being trapped temporarily in the sinus spaces in lymph nodes, and a third fraction is retained in lymph nodes where they may be taken up by lymphocytes and then released slowly into the blood. The slow release of ARVs into the blood (the second and third “waves”) could occur either by (1) DcNPs being carried by lymph flow or (2) lymphocytes, which have taken up DcNPs in lymph nodes, trafficking into the blood and slowly releasing ARVs into the plasma. It is the second and third fractions that primarily contributed to persistent plasma LPV and TFV levels of TLC-ART101 by continuously supplying DcNP- and lymphocyte-associated ARVs to the blood (Figure 1A, 1C). It is noteworthy that k31 and k41 (lymphatic entry rate constants) and tlag2 and tlag3 assumed greater importance than k21 and tlag1 for LPV and TFV (Table 2), illustrating that the second and third lymphatic delay compartments contributed the most to the long-acting plasma PK of LPV and TFV.
In sum, the kinetic insights derived from the MBPK models suggest the lymphatic system itself acts as a body-wide drug depot for the DcNP-associated ARVs in the TLC-ART101 formulation. In contrast to reports on lymphatic uptake of SC proteins and nanoparticles that suggest interstitial transit being the rate-limiting step [58, 59], lymphatic transit and retention are the more prevalent rate-limiting steps for absorption of TLC-ART101 into the blood. To our knowledge, this is the first suggestion that the lymphatic system itself could serve as a distributed body-wide drug depot to enable long-acting plasma PK. Although lymphatic transit of DcNPs that are sufficiently stable to not prematurely release drug is important, there are a number of questions that remain, for example: (1) how ARVs carried by DcNPs are internalized by lymphocytes, (2) how lymphocytes in the blood release ARVs, and (3) how DcNPs in the blood release ARVs. To address these insights, additional in vitro experiments such as those investigating (1) the rate of DcNP and ARV uptake into lymphocytes, (2) the rate of ARV release from DcNPs in blood, and (3) the rate of ARV release from lymphocytes circulating in the blood could begin to discern the role and significance of each of these steps. However, these and other studies are beyond the scope of this report. Investigations are being planned to gain a deeper understanding of these and other processes (such as the impact of the biological identity and protein corona on DcNPs) that drive the long-acting intracellular and plasma PK that result from DcNPs in TLC-ART101 leveraging the lymphatic system as a body-wide drug depot.
Limited Blood Extravasation of DcNP-associated ARVs in TLC-ART101
In addition to lymphatic uptake and first-pass lymphatic distribution and retention of DcNP-associated ARVs, followed by slow release of ARVs into the blood from the lymphatics, limited blood extravasation of circulating DcNPs in blood likely contributed to the LPV and TFV persistence in the plasma (Figure 1A-1C). In Table 2, the ∼7- to 13-fold lower V5_DcNP versus V7_Free for both LPV and TFV indicated limited extravasation of DcNP-associated ARV versus free ARV.
It has been shown in rats with a thoracic lymph duct cannula and dosed IV with ∼83 nm of PEGylated liposomal doxorubicin (similar in size to DcNPs in TLC-ART101), ∼14% of the IV dose was recovered in lymph at 30 hr (i.e., ∼14% extravasated from the blood into the interstitium, and subsequently entered the lymphatics) [15]. Thus, since a minor fraction of the DcNPs circulating in the blood may extravasate and be available for re-entry into the lymphatics, this process should be considered as part of the systemic compartment (q5) for DcNP-associated ARVs (Figure 3C, 3D). Another consideration is that PEGylation of the lipid membrane surface significantly enhanced plasma circulation lifetimes of ∼110 nm liposomes [60]. Hence, it is likely that inclusion of mPEG2000 on the DcNP surface of slightly smaller particles may impart long-circulation behavior of DcNPs to the TLC-ART101 formulation.
There remains much to learn about the detailed mechanisms of the long-acting PK of TLC-ART101. It is instructive to compare the PK of lipophilic LPV and RTV between the free and TLC-ART101 formulations. While CL/F and MBRT were similar for free LPV and RTV, these statistical moment derived PK parameters were different for LPV and RTV in TLC-ART101 (Table 1), suggesting LPV and RTV, when associated to DcNPs, could have different cellular uptake and efflux, release from DcNPs, and tissue distribution profiles. Moreover, free LPV and RTV had similar t1/2 values (∼8 hr), while the t1/2 of DcNP-associated LPV was ∼11-fold longer than that of DcNP-associated RTV (Table 1), further indicating drug release from DcNPs and tissue distribution differed between these two drugs. Elucidating the details of these PK differences between LPV and RTV (as well as TFV) in TLC-ART101 requires further investigation—possibly including sampling of lymph vessel and node drug concentrations throughout the body and defining pre-systemic clearance mechanisms. Many of the uncertainties of the systemic PK of TLC-ART101 could be elucidated in further studies such as an IV study and being able to measure the time-course of free versus DcNP-associated ARV in the plasma [61]. Currently, the plasma PK of DcNP-associated ARVs after dosing TLC-ART101 is unknown.
Conclusion
In conclusion, we developed the first mechanism-based PK (MBPK) models to simultaneously describe the complex long-acting plasma and lymphatic PK profiles of three ARVs associated to DcNPs all in one SC injectable formulation called TLC-ART101. These models anchored on plasma PK data are composed of two submodels: one for disposition of ARVs incorporated in DcNPs that undergo first-pass through the lymphatics, and one for free ARVs that are routed directly into the blood circulation. Subsequent model improvements incorporating LNMC and PBMC intracellular compartments (cells in the lymph nodes and blood) would provide additional insights on ARV access to these sites. These additional data may allow for predicting the impact of TLC-ART101 dosing regimens on viral clearance in lymph nodes where ∼75% of HIV-infected cells (CD4+ T lymphocytes) reside. At present, these first-generation MBPK models suggest the lymphocyte-targeting and long-acting PK of ARVs associated to DcNPs are due to (1) preferential lymphatic uptake from the SC space, (2) first-pass transit through the lymphatics (throughout the vessels and nodes in the body), (3) lymphatic (lymph vessel and node) retention, (4) eventual release of ARVs into the blood from the lymphatics, and (5) limited extravascular distribution out of blood vessels of DcNP-associated ARVs.
Supplementary Material
Acknowledgments
We thank members of the University of Washington Targeted Long-Acting Combination Anti-Retroviral Therapy (TLC-ART) Program who contributed helpful insights during discussions. We thank Sophia Song for assistance with the preparation of TLC-ART101, as well as the Washington National Primate Research center staff (Baoping Tian, Michael Gough, Jason Ogle, Wonsok Lee, and Chris English) for their support with the macaque studies.
Funding: This work was supported by the National Institutes of Health (grant numbers UM1 AI120176, T32-GM007750, TL1-TR000422, P51OD010425, 1S10OD010652-01, and AI27757).
Footnotes
Chemical compounds studied in this article: Lopinavir (PubChem CID: 92727); Ritonavir (PubChem CID: 392622); Tenofovir (PubChem CID: 464205); 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) (PubChem CID: 94190); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-mPEG2000) (PubChem CID: 406952)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Owen A, Rannard S. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv Drug Deliv Rev. 2016;103:144–156. doi: 10.1016/j.addr.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, Eron JJ, Yazdanpanah Y, Podzamczer D, Lutz T, Angel JB, Richmond GJ, Clotet B, et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–1510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 3.Spreen W, Ford SL, Chen S, Wilfret D, Margolis D, Gould E, Piscitelli S. GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014;67(5):481–486. doi: 10.1097/QAI.0000000000000301. [DOI] [PubMed] [Google Scholar]
- 4.McGowan I, Dezzutti CS, Siegel A, Engstrom J, Nikiforov A, Duffill K, Shetler C, Richardson-Harman N, Abebe K, Back D, et al. Long-acting rilpivirine as potential pre-exposure prophylaxis for HIV-1 prevention (the MWRI-01 study): an open-label, phase 1, compartmental, pharmacokinetic and pharmacodynamic assessment. Lancet HIV. 2016;3(12):e569–e578. doi: 10.1016/S2352-3018(16)30113-8. [DOI] [PubMed] [Google Scholar]
- 5.Estes JD. Pathobiology of HIV/SIV-associated changes in secondary lymphoid tissues. Immunol Rev. 2013;254(1):65–77. doi: 10.1111/imr.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horiike M, Iwami S, Kodama M, Sato A, Watanabe Y, Yasui M, Ishida Y, Kobayashi T, Miura T, Igarashi T. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology. 2012;423(2):107–118. doi: 10.1016/j.virol.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Lederman MM, Margolis L. The lymph node in HIV pathogenesis. Semin Immunol. 2008;20(3):187–195. doi: 10.1016/j.smim.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinman L, Brodie SJ, Tsai CC, Bui T, Larsen K, Schmidt A, Anderson D, Morton WR, Hu SL, Ho RJ. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34(4):387–397. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A. 2014;111(6):2307–2312. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Kosakovsky Pond SL, Chung YS, Penugonda S, Chipman JG, Fletcher CV, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature. 2016;530(7588):51–56. doi: 10.1038/nature16933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S, Hatano H, Kashuba AD, Cottrell ML, Liegler TJ, Stephenson S, Somsouk M, Hunt PW, Deeks SG, Savic RM. Integrase and protease inhibitor concentrations in lymph node and gut mucosal tissue (Abstract #407); Conference on Retroviruses and Opportunistic Infections (CROI): 2017; Seattle, WA. 2017. [Google Scholar]
- 12.Freeling JP, Ho RJ. Anti-HIV drug particles may overcome lymphatic drug insufficiency and associated HIV persistence. Proc Natl Acad Sci U S A. 2014;111(25):E2512–2513. doi: 10.1073/pnas.1406554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Wang L, Yao Q, Ling R, Li K, Wang H. Drug concentrations in axillary lymph nodes after lymphatic chemotherapy on patients with breast cancer. Breast Cancer Res. 2004;6(4):R474–477. doi: 10.1186/bcr819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaminskas LM, McLeod VM, Ascher DB, Ryan GM, Jones S, Haynes JM, Trevaskis NL, Chan LJ, Sloan EK, Finnin BA, et al. Methotrexate-conjugated PEGylated dendrimers show differential patterns of deposition and activity in tumor-burdened lymph nodes after intravenous and subcutaneous administration in rats. Mol Pharm. 2015;12(2):432–443. doi: 10.1021/mp500531e. [DOI] [PubMed] [Google Scholar]
- 15.Ryan GM, Kaminskas LM, Bulitta JB, McIntosh MP, Owen DJ, Porter CJ. PEGylated polylysine dendrimers increase lymphatic exposure to doxorubicin when compared to PEGylated liposomal and solution formulations of doxorubicin. J Control Release. 2013;172(1):128–136. doi: 10.1016/j.jconrel.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Kinman L, Bui T, Larsen K, Tsai CC, Anderson D, Morton WR, Hu SL, Ho RJ. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr. 2006;42(2):155–161. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 17.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res Hum Retroviruses. 2015;31(1):107–114. doi: 10.1089/aid.2014.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeling JP, Koehn J, Shu C, Sun J, Ho RJ. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS. 2014;28(17):2625–2627. doi: 10.1097/QAD.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraft JC, McConnachie LA, Koehn J, Kinman L, Collins C, Shen DD, Collier AC, Ho RJ. Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. AIDS. 2017;31(6):765–770. doi: 10.1097/QAD.0000000000001405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koehn J, Ho RJ. Novel liquid chromatography-tandem mass spectrometry method for simultaneous detection of anti-HIV drugs Lopinavir, Ritonavir, and Tenofovir in plasma. Antim icrob Agents Chemother. 2014;58(5):2675–2680. doi: 10.1128/AAC.02748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibaldi M, Perrier D. Pharmacokinetics. 2nd. New York: Marcel Dekker, Inc; 1982. [Google Scholar]
- 22.Bell BM, Schumitzky A. A Generalization of the Gauss-Newton Method that Solves Extended Least Squares Problems. 1997 White Paper: https://wwwseanetcom/∼bradbell/gauss_newtonpdf.
- 23.Dahlberg AM, Kaminskas LM, Smith A, Nicolazzo JA, Porter CJ, Bulitta JB, McIntosh MP. The lymphatic system plays a major role in the intravenous and subcutaneous pharmacokinetics of trastuzumab in rats. Mol Pharm. 2014;11(2):496–504. doi: 10.1021/mp400464s. [DOI] [PubMed] [Google Scholar]
- 24.Kaminskas LM, Kota J, McLeod VM, Kelly BD, Karellas P, Porter CJ. PEGylation of polylysine dendrimers improves absorption and lymphatic targeting following SC administration in rats. J Control Release. 2009;140(2):108–116. doi: 10.1016/j.jconrel.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Oussoren C, Storm G. Liposomes to target the lymphatics by subcutaneous administration. Adv Drug Deliv Rev. 2001;50(1-2):143–156. doi: 10.1016/s0169-409x(01)00154-5. [DOI] [PubMed] [Google Scholar]
- 26.Trevaskis NL, Kaminskas LM, Porter CJ. From sewer to saviour - targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov. 2015;14(11):781–803. doi: 10.1038/nrd4608. [DOI] [PubMed] [Google Scholar]
- 27.Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, Kati W, Stewart K, Lal R, Hsu A, et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chem other. 1998;42(12):3218–3224. doi: 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Rompay KK, Babusis D, Abbott Z, Geng Y, Jayashankar K, Johnson JA, Lipscomb J, Heneine W, Abel K, Ray AS. Compared to subcutaneous tenofovir, oral tenofovir disoproxyl fumarate administration preferentially concentrates the drug into gut-associated lymphoid cells in simian immunodeficiency virus-infected macaques. Antimicrob Agents Chem other. 2012;56(9):4980–4984. doi: 10.1128/AAC.01095-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev. 2012;92(3):1005–1060. doi: 10.1152/physrev.00037.2011. [DOI] [PubMed] [Google Scholar]
- 30.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oussoren C, Zuidema J, Crommelin DJ, Storm G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II Influence of liposomal size, lipid compostion and lipid dose Biochim Biophys Acta. 1997;1328(2):261–272. doi: 10.1016/s0005-2736(97)00122-3. [DOI] [PubMed] [Google Scholar]
- 32.Supersaxo A, Hein WR, Steffen H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm Res. 1990;7(2):167–169. doi: 10.1023/a:1015880819328. [DOI] [PubMed] [Google Scholar]
- 33.Bourne GH. The Rhesus Monkey. I. New York: Academic Press; 1975. pp. 52–61. [Google Scholar]
- 34.Di Mascio M, Paik CH, Carrasquillo JA, Maeng JS, Jang BS, Shin IS, Srinivasula S, Byrum R, Neria A, Kopp W, et al. Noninvasive in vivo imaging of CD4 cells in simian-human immunodeficiency virus (SHIV)-infected nonhuman primates. Blood. 2009;114(2):328–337. doi: 10.1182/blood-2008-12-192203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santangelo PJ, Rogers KA, Zurla C, Blanchard EL, Gumber S, Strait K, Connor-Stroud F, Schuster DM, Amancha PK, Hong JJ, et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat Methods. 2015;12(5):427–432. doi: 10.1038/nmeth.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan LJ, Ascher DB, Yadav R, Bulitta JB, Williams CC, Porter CJ, Landersdorfer CB, Kaminskas LM. Conjugation of 10 kDa Linear PEG onto Trastuzumab Fab' Is Sufficient to Significantly Enhance Lymphatic Exposure while Preserving in Vitro Biological Activity. Mol Pharm. 2016;13(4):1229–1241. doi: 10.1021/acs.molpharmaceut.5b00749. [DOI] [PubMed] [Google Scholar]
- 37.Chan LJ, Bulitta JB, Ascher DB, Haynes JM, McLeod VM, Porter CJ, Williams CC, Kaminskas LM. PEGylation does not significantly change the initial intravenous or subcutaneous pharmacokinetics or lymphatic exposure of trastuzumab in rats but increases plasma clearance after subcutaneous administration. Mol Pharm. 2015;12(3):794–809. doi: 10.1021/mp5006189. [DOI] [PubMed] [Google Scholar]
- 38.Chen SA, Sawchuk RJ, Brundage RC, Horvath C, Mendenhall HV, Gunther RA, Braeckman RA. Plasma and lymph pharmacokinetics of recombinant human interleukin-2 and polyethylene glycol-modified interleukin-2 in pigs. J Pharmacol Exp Ther. 2000;293(1):248–259. [PubMed] [Google Scholar]
- 39.Gill KL, Gardner I, Li L, Jamei M. A Bottom-Up Whole-Body Physiologically Based Pharmacokinetic Model to Mechanistically Predict Tissue Distribution and the Rate of Subcutaneous Absorption of Therapeutic Proteins. AAPS J. 2016;18(1):156–170. doi: 10.1208/s12248-015-9819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krzyzanski W, Jusko WJ, Wacholtz MC, Minton N, Cheung WK. Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after multiple subcutaneous doses in healthy subjects. Eur J Pharm Sci. 2005;26(3-4):295–306. doi: 10.1016/j.ejps.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Landersdorfer CB, Caliph SM, Shackleford DM, Ascher DB, Kaminskas LM. PEGylated interferon displays differences in plasma clearance and bioavailability between male and female mice and between female immunocompetent C57Bl/6J and athymic nude mice. J Pharm Sci. 2015;104(5):1848–1855. doi: 10.1002/jps.24412. [DOI] [PubMed] [Google Scholar]
- 42.McLennan DN, Porter CJ, Edwards GA, Martin SW, Heatherington AC, Charman SA. Lymphatic absorption is the primary contributor to the systemic availability of epoetin Alfa following subcutaneous administration to sheep. J Pharmacol Exp Ther. 2005;313(1):345–351. doi: 10.1124/jpet.104.078790. [DOI] [PubMed] [Google Scholar]
- 43.Ramakrishnan R, Cheung WK, Farrell F, Joffee L, Jusko WJ. Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after intravenous and subcutaneous dose administration in cynomolgus monkeys. J Pharmacol Exp Ther. 2003;306(1):324–331. doi: 10.1124/jpet.102.047191. [DOI] [PubMed] [Google Scholar]
- 44.Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol. 2013;53(3):314–325. doi: 10.1002/jcph.4. [DOI] [PubMed] [Google Scholar]
- 45.Kagan L. Pharmacokinetic modeling of the subcutaneous absorption of therapeutic proteins. Drug Metab Dispos. 2014;42(11):1890–1905. doi: 10.1124/dmd.114.059121. [DOI] [PubMed] [Google Scholar]
- 46.Aukland K, Reed RK. Interstitial-lymphatic mechanisms in the control of extracellular fluid volume. Physiol Rev. 1993;73(1):1–78. doi: 10.1152/physrev.1993.73.1.1. [DOI] [PubMed] [Google Scholar]
- 47.Colombo S, Beguin A, Telenti A, Biollaz J, Buclin T, Rochat B, Decosterd LA. Intracellular measurements of anti-HIV drugs indinavir, amprenavir, saquinavir, ritonavir, nelfinavir, lopinavir, atazanavir, efavirenz and nevirapine in peripheral blood mononuclear cells by liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;819(2):259–276. doi: 10.1016/j.jchromb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 48.Colombo S, Telenti A, Buclin T, Furrer H, Lee BL, Biollaz J, Decosterd LA Swiss HIVCS. Are plasma levels valid surrogates for cellular concentrations of antiretroviral drugs in HIV-infected patients? Ther Drug Monit. 2006;28(3):332–338. doi: 10.1097/01.ftd.0000211807.74192.62. [DOI] [PubMed] [Google Scholar]
- 49.van Kampen JJ, Reedijk ML, Burgers PC, Dekker LJ, Hartwig NG, van der Ende IE, de Groot R, Osterhaus AD, Burger DM, Luider TM, et al. Ultra-fast analysis of plasma and intracellular levels of HIV protease inhibitors in children: a clinical application of MALDI mass spectrometry. PLoS One. 2010;5(7):e11409. doi: 10.1371/journal.pone.0011409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D'Avolio A, Simiele M, Calcagno A, Siccardi M, Larovere G, Agati S, Baietto L, Cusato J, Tettoni M, Sciandra M, et al. Intracellular accumulation of ritonavir combined with different protease inhibitors and correlations between concentrations in plasma and peripheral blood mononuclear cells. J Antimicrob Chemother. 2013;68(4):907–910. doi: 10.1093/jac/dks484. [DOI] [PubMed] [Google Scholar]
- 51.Drinker CK, Field ME, Ward HK. The Filtering Capacity of Lymph Nodes. J Exp Med. 1934;59(4):393–405. doi: 10.1084/jem.59.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khullar OV, Griset AP, Gibbs-Strauss SL, Chirieac LR, Zubris KA, Frangioni JV, Grinstaff MW, Colson YL. Nanoparticle migration and delivery of Paclitaxel to regional lymph nodes in a large animal model. J Am Coll Surg. 2012;214(3):328–337. doi: 10.1016/j.jamcollsurg.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denissen JF, Grabowski BA, Johnson MK, Buko AM, Kempf DJ, Thomas SB, Surber BW. Metabolism and disposition of the HIV-1 protease inhibitor ritonavir (ABT-538) in rats, dogs, and humans. Drug Metab Dispos. 1997;25(4):489–501. [PubMed] [Google Scholar]
- 54.Kumar GN, Jayanti VK, Johnson MK, Uchic J, Thomas S, Lee RD, Grabowski BA, Sham HL, Kempf DJ, Denissen JF, et al. Metabolism and disposition of the HIV-1 protease inhibitor lopinavir (ABT-378) given in combination with ritonavir in rats, dogs, and humans. Pharm Res. 2004;21(9):1622–1630. doi: 10.1023/b:pham.0000041457.64638.8d. [DOI] [PubMed] [Google Scholar]
- 55.Koal T, Burhenne H, Romling R, Svoboda M, Resch K, Kaever V. Quantification of antiretroviral drugs in dried blood spot samples by means of liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19(21):2995–3001. doi: 10.1002/rcm.2158. [DOI] [PubMed] [Google Scholar]
- 56.Massud I, Mitchell J, Babusis D, Deyounks F, Ray AS, Rooney JF, Heneine W, Miller MD, Garcia-Lerma JG. Chemoprophylaxis With Oral Emtricitabine and Tenofovir Alafenamide Combination Protects Macaques From Rectal Simian/Human Immunodeficiency Virus Infection. J Infect Dis. 2016;214(7):1058–1062. doi: 10.1093/infdis/jiw312. [DOI] [PubMed] [Google Scholar]
- 57.Ganusov VV, Auerbach J. Mathematical modeling reveals kinetics of lymphocyte recirculation in the whole organism. PLoS Comput Biol. 2014;10(5):e1003586. doi: 10.1371/journal.pcbi.1003586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porter CJ, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci. 2000;89(3):297–310. doi: 10.1002/(SICI)1520-6017(200003)89:3<297::AID-JPS2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 59.Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50(1-2):3–20. doi: 10.1016/s0169-409x(01)00150-8. [DOI] [PubMed] [Google Scholar]
- 60.Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066(1):29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 61.Stern ST, Martinez MN, Stevens DM. When Is It Important to Measure Unbound Drug in Evaluating Nanomedicine Pharmacokinetics? Drug Metab Dispos. 2016;44(12):1934–1939. doi: 10.1124/dmd.116.073148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.