

Abstract
Caspase-9 is a critical factor in the initiation of apoptosis, and as a result is tightly regulated by a number of mechanisms. Caspase-9 contains a Caspase Activation and Recruitment Domain (CARD), which enables caspase-9 to form a tight interaction with the apoptosome, a heptameric activating platform. The caspase-9 CARD has been thought to be principally involved in recruitment to the apoptosome, but its roles outside this interaction have yet to be uncovered. In this work we show that the CARD is involved in physical interactions with the catalytic core of caspase-9 in the absence of the apoptosome; this interaction requires a properly formed caspase-9 active site. The active sites of caspases are composed of four extremely mobile loops. When the active-site loops are not properly ordered, the CARD and core domains of caspase-9 do not interact and behave independently, like loosely tethered beads. When the active-site loop bundle is properly ordered, the CARD domain interacts with the catalytic core, forming a single folding unit. Together these findings provide mechanistic insight into a new level of caspase-9 regulation, prompting speculation that the CARD may also play a role in the recruitment or recognition of substrate.
INTRODUCTION
Apoptosis or programmed cell death is a fundamental cellular process that is paramount for cellular regeneration and tissue homeostasis in multicellular organisms. Unlike other cell death pathways, apoptosis efficiently dismantles the cell without adverse effects on neighboring cells or its environment. Its faithful execution is essential in avoiding a number of catastrophic disease states and is also critical in organismal development, so apoptosis is very tightly regulated. Caspases, aspartate-directed, cysteine proteases play prominent roles in apoptotic pathways. Initiator caspases (caspase-2, -8 and -9) function upstream of the apoptotic pathways while executioners (caspase-3, -6 and -7) mediate downstream reactions. Because highly active caspases are lethal, they are synthesized and held as inactive zymogens (procaspases) to avoid untimely activation of apoptosis. Apoptotic pathways are triggered upon receipt of a death signal, either through ligand binding (extrinsic pathway) or mitochondrial assault (intrinsic pathway). Initiator caspases are then recruited to a multi-complex activating scaffold to undergo activation. These active initiators in turn cleave and activate downstream executioners, thereby initiating the series of proteolytic reactions, which culminates in cell death. Because the caspase activation cascade signals the commitment of the cell to its demise, the activation of these enzymes is tightly controlled.
Caspase-9 is a critical initiator of the intrinsic pathway, and is responsible for activating downstream executioners caspases. Defects in the intrinsic pathway are characteristic of diseases ranging from autoimmune disorders to cancer. In these diseases, activation of caspase-9 is particularly critical (1–4), however, because of caspase-9’s upstream role in the intrinsic pathway, its regulation is quite distinct from that of the executioner caspases. Caspase-9 also has structural and functional characteristics that are distinct from other caspases. Like all caspases, the caspase-9 structure contains the highly homologous catalytic core, composed of the large and the small subunit connected by an intersubunit linker (Figure 1). While cleavage at the linker converts most executioner caspases to their active form, caspase-9 is active even prior to cleavage of the intersubunit linker, probably due to having a longer intersubunit linker (5). Caspase-9 is also predominantly monomeric in solution, unlike other executioner caspases, which are constitutively dimeric. In addition, the catalytic core of caspase-9 is preceded by a long prodomain called the Caspase Activation Recruitment Domain (CARD). A six-helix bundle with protein binding motifs, the caspase-9 CARD facilitates protein-protein interaction with the CARD in apoptotic protease activation factor 1 (Apaf-1), anchoring caspase-9 to its activation platform, the apoptosome (6–8). Caspase-9’s interaction with the apoptosome is one of the most significant unique features of this caspase.
Figure 1. Levels of caspase-9 activation.
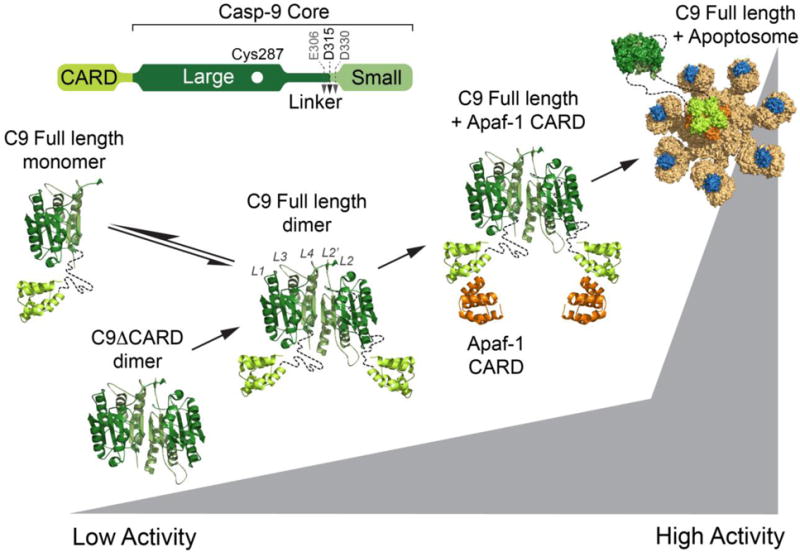
Model depicting the increase in enzymatic activity of caspase-9 (C9) in various states. Caspase-9 is predominantly monomeric (composed of one large subunit, one small subunit an the CARD), but requires oligomerization, minimally dimerization, for activity. Removing the CARD (C9ΔCARD) decreases caspase-9 activity. An increase in activity of full-length caspase-9 is observed in the presence of exogenous Apaf-1 and is highest when full-length casp-9 is bound to the apoptosome. The structure of the full-length caspase-9 was modeled using the CARD-deleted caspase-9 structure (PDB ID: 1JXQ) and caspase-9 CARD in complex with Apaf-1 CARD (PDB ID: 3YGS). The caspase-9-bound apoptosome was modeled based on the structure of the apoptosome (PDB ID: 5JUY).
The apoptosome platform is formed when an intracellular stress leads to release of cytochrome c from the mitochondria. Cytochrome c is then available to bind to Apaf-1, initiating a conformational change resulting in a dATP-dependent Apaf-1 heptamerization into the apoptosome. The apoptosome then recruits and activates uncleaved procaspase-9 (Figure 1). The molecular details of caspase-9 activation by the apoptosome have not been fully elucidated, but binding to the apoptosome increases caspase-9’s activity by approximately 2000-fold (5). Intriguingly, even in the absence of the apoptosome, the presence of individual domains influence caspase-9 activity. Although the prodomain is rarely removed intracellularly, caspase-9 was observed to be 20% more active when caspase-9’s catalytic core remains covalently linked to the CARD, as in full-length caspase-9, than when it is proteolytically removed to form the ΔCARD version of caspase-9 (9,10) (Figure 1). Later reports have shown that ΔCARD versions of caspase-9 exhibited increased activity relative to the cleaved full-length, prompting the hypothesis that CARD plays an inhibitory role in caspase-9 activity (11–13). Despite the differences in reported free caspase-9 activity, a consistent observation is that ΔCARD caspase-9 does not exhibit significant enhancement in activity in the presence of the Apaf-1 and cytochrome c. Addition of the isolated Apaf-1 CARD further enhances caspase-9 activity by five-fold, in vitro (9). The greatest increase in activity, though, occurs when full-length caspase-9, including both the CARD and core domains, binds to the apoptosome through caspase-9 CARD: Apaf-1 CARD interactions.
Early models of the apoptosome activation of caspase-9 argued that the increased activity is due to a change in the oligomeric state of the enzyme via increasing the local concentration of monomeric caspase-9. Recruitment of additional molecules of caspase-9 was hypothesized to provide partners for dimerization (14) or facilitate dimerization amongst the apoptosome-bound monomers (15). This model was supported by the evidence that enhanced activity is associated with dimerized caspase-9 molecules (16). Alternative views of the activation mechanism invoked induced conformational changes in which the apoptosome binds to the dimerization interface of caspase-9 and stabilizes the active-site region leading to a catalytically competent conformation (17). This conformational change model was supported by the evidence from high-resolution cryoelectron microscopy (cryo-EM) which shows that caspase-9 is potentially monomeric in the highly active state, bound to the apoptosome (18). Recent near atomic resolution cryo-EM structures of the human apoptosome show that a catalytic core of a caspase-9 monomer is bound to the apoptosome hub, independent of potential caspase-9 dimers undergoing activation (18,19). Additional studies of caspase-9’s activation mechanism have also suggested that cleavage of its intersubunit linker initiates dissociation from the apoptosome thus regulating the period of apoptosome activity (20).
While it is clear that caspase-9 is recruited to the apoptosome through CARD:CARD interactions, it is not clear what factors, potentially in addition to the rare CARD removal event, prompt the release of caspase-9 from the apoptosome or how the caspase-9 CARD influences caspase-9 function when not bound to the apoptosome. This is relevant due the observation that caspase-9 activation can be achieved without apoptosome formation through different pathways (21–23). If the caspase-9 CARD were predominantly involved in tethering caspase-9 to the apoptosome, then in the absence of the apoptosome removal of CARD should not decrease activity significantly. On the contrary, we observed a decrease in caspase-9 catalytic efficiency is observed when the CARD is removed. This suggests that the CARD plays another role in function and regulation of caspase-9. The most direct hypothesis is that the caspase-9 CARD interacts this the core domain directly, influencing the structure and thus the function of the enzyme. The goal of this work is to probe and uncover the nature of any existing CARD:caspase-9-core interactions.
EXPERIMENTAL
Caspase-9 expression and purification
The caspase-9 full-length gene (human sequence) construct, encoding amino acids 1-416, in pET23b (Addgene plasmid 11829 (5)) was transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB) and purified in a manner similar to that previously reported (24). The cultures were grown in 2xYT media supplemented with ampicillin (100 mg/L) at 37°C until they reached an optical density (OD) at 600 nm of 1.2. The temperature was reduced to 15°C and cells were induced with 1 mM IPTG (Anatrace) to express soluble protein. Cells were harvested after 3 h by centrifugation to obtain single-site processing at Asp315. Cell pellets stored at −20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed cells were centrifuged at 37,000 × g to remove cellular debris. The filtered supernatant was loaded onto a 5-mL HiTrap Ni-affinity column (GE Healthcare). The column was washed with a buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole until 280 nm absorbance returned to baseline. The protein was eluted using a linear imidazole gradient of 2 to100 mM. The eluted fractions containing protein of the expected molecular weight and composition were diluted 10-fold into a buffer composed of 20 mM Tris pH 8.5, 10 mM DTT to reduce the salt concentration. This protein sample was loaded onto a 5-mL Macro-Prep High Q column (Bio-Rad). The column was developed with a linear NaCl gradient and eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 5 mM DTT buffer. The eluted protein was stored in −80°C in the above buffer conditions. Purified caspase-9 was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity. Caspase-9 variants, C287A, R7E, R11E, D23E, R51E E365R, R7E/R11E, S183E, S183A, C287A/S183A, S99E, C287A/S99E, T125E, C287A/T125E and the Ser-Gly linker extension, were constructed by site-directed mutagenesis method in the full-length expression construct, and were purified by the same method (except for S183E, S183A, C287A/S183A, S99E, C287A/S99E, T125E, C287A/T125E) as described here for the wild-type protein.
Caspase-9 S183E, S183A, C287A/S183A, S99E, C287A/S99E, T125E, C287A/T125E were purified using the same method except 50 mM NaH2PO4 pH 7.0, 300 mM NaCl and 2 mM imidazole buffer was used to lyse the bacterial cells and wash the HiTrap Ni column. All proteins were purified by Ni-affinity and anion exchange chromatography as described above.
Caspase-9 ΔCARD was expressed from a two-plasmid expression system. Two separate constructs, one encoding the large subunit, residues 140-305, and the other encoding the small subunit, residues 331-416, each in the pRSET plasmid, were separately transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The recombinant large and small subunits were individually expressed as inclusion bodies for subsequent reconstitution. Cultures were grown in 2xYT media supplemented with ampicillin (100 mg/L) at 37°C until they reached an OD600 of 0.6. Protein expression was induced with 0.2 mM IPTG. Cells were harvested after 3 hrs at 37°C. Cell pellets stored at −20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 10 mM Tris pH 8.0 and 1 mM EDTA. Inclusion body pellets were washed twice in 100 mM Tris pH 8.0, 1 mM EDTA, 0.5 M NaCl, 2% Triton, and 1M urea, twice in 100 mM Tris pH 8.0, 1 mM EDTA and finally resuspended in 6 M guanidine hydrochloride. Caspase-9 large and small subunit proteins in guanidine hydrochloride were combined in a ratio of 1:2, large:small subunits, and rapidly diluted dropwise into refolding buffer composed of 100 mM Tris pH 8.0, 10% sucrose, 0.1% CHAPS, 0.15 M NaCl, and 10 mM DTT, allowed to stir for one hour at room temperature and then dialyzed four times against 10 mM Tris pH 8.5, 10 mM DTT, and 0.1mM EDTA buffer at 4°C. Typically 5 mL of mixed caspase large and small subunits was diluted to 80 mL in refolding buffer and dialyzed against 5 L of dialysis buffer. The first and last dialysis steps were allowed to proceed for 4 hours at 4°C while the second dialysis proceeded overnight at 4°C. The dialyzed protein was centrifuged for 15 minutes at 12,000 × g to remove precipitate and then purified using a HiTrap Q HP ion exchange column (GE Healthcare) with a linear gradient from 0 to 250 mM NaCl in 20 mM Tris buffer pH 8.5, with 10 mM DTT. Protein eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM DTT buffer was stored in −80°C. The identity of the purified caspase-9 ΔCARD was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity. The ΔCARD-His6 construct was generated by deleting DNA coding for the CARD (res1-138) from the caspase-9 full-length construct. ΔCARD-His6 was purified using the same method as wild-type caspase-9.
Oligomeric state determination
Caspase-9 wild-type, full-length and ΔCARD variant protein samples in 20 mM Tris pH 8.5, 110 mM NaCl, and 5 mM DTT were incubated alone (monomer) or with covalent inhibitor z-VAD-fmk (carbobenzoxy-Val-Ala-Asp-fluoromethylketone, Enzo Life Sciences) (dimer) for 2 hours at room temperature. The oligomeric state of the caspase-9 samples was determined via gel filtration. 100 μL of 0.5 mg/mL protein sample was loaded onto a Superdex 200 10/300 GL (GE Healthcare) gel-filtration column. Apo and z-VAD-fmk-incubated protein samples were eluted with 20 mM Tris pH 8.0, 100 mM NaCl, and 2 mM DTT. Eluted peaks were analyzed by SDS-PAGE to identify the eluted protein. Four different molecular weight standards from the gel-filtration calibration kit LMW (GE Healthcare) were run in the same conditions and a standard plot was generated to determine whether the peaks were caspase-9 monomer or dimer.
CARD expression and purification
The CARD only construct (amino acids 1-138) in pET23b was made by QuikChange mutagenesis (Stratagene) using the oligonucleotide primer 5`-CCCAGACCAGTGGACATT-GGTTCTGGAGGATTCGGTGATCACCACCACCACCACCACTAAGTCGGTGCTCTTGAGAGTTTGAGGGGAAATGCAGATTTGG-3`and its reverse complement on the caspase-9 full-length gene (Addgene plasmid 11829). These oligo-nucleotide primers insert a His6-tag and a stop codon after the last amino acid of the CARD domain (Asp138), leaving the remaining portion of the caspase-9 gene in the plasmid. A separate CARD construct was made to insert a human rhinovirus-3C (hRV3C) protease cleavage site (LEVLFQGP) before the His6-tag using the primers 5′-CTCGGGCTGGAAGTGCTGTTCCAGGGTCCGCACCACCACCACCACCACTAAG-CCG-3′ (forward) and 5′-ATCACCGAATCCTCCAGAACCAATGTCC-3′ (reverse). Each construct was transformed into BL21 (DE3) T7 Express strain of E. coli. The cultures were grown in 2×YT media supplemented with ampicillin (100 mg/L) at 37°C until they reached an OD600 of 0.6. The temperature was reduced to 15°C and cells were induced with 1 mM IPTG (Anatrace) to express soluble His6-tagged full-length protein. Cells were harvested after 18 hrs. Cell pellets stored at −20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 50 mM sodium phosphate pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed cells were centrifuged at 37,000 × g to remove cellular debris. The filtered supernatant was loaded onto a 5-mL HiTrap Ni-affinity column (GE Healthcare). The column was washed with a buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM imidazole until 280 nm absorbance returned to baseline. The column was washed with 50 mM phosphate pH 8.0, 300 mM NaCl, 50 mM imidazole and the protein was eluted with 50 mM phosphate pH 8.0, 300 mM NaCl, 250 mM imidazole. The eluted fraction was diluted 10-fold into Buffer A (20 mM Tris pH 8.0 and 2 mM DTT) to reduce the salt concentration. This protein sample was loaded onto a 5 mL Macro-Prep High Q column (Bio-Rad Laboratories, Inc.). The column was developed with a linear NaCl gradient. Protein eluted in 20 mM Tris pH 8.0, 2 mM DTT, and 130 mM NaCl. Eluted protein was analyzed by SDS-PAGE to assess purity and stored in −80°C. For cleavage of the His6-tag, eluted fractions from the Ni-NTA column were diluted 2x with 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA and 1 mM DTT. 100 μg of hRV3C protease was added per 1 μg of CARD-His6 and the reaction was incubated for 16h at 4°C with gentle mixing. The cleavage reaction was filtered through 0.45 μm PVDF. Filtered protein solution was diluted 6x with Buffer A and loaded onto a HiTrap Q column (GE Healthcare). The column was developed with a linear NaCl gradient. Caspase-9 CARD (no His tag) eluted in 20 mM Tris pH 8.0, 130 mM NaCl and 2 mM DTT. Full cleavage was assessed by running samples on a 16% SDS-PAGE gel.
Thermal stability and secondary structure analysis by circular dichroism
All caspase-9 variants (except for S183E, S183A, C287A/S183A, S99E, C287A/S99E, T125E, C287A/T125E) and the CARD protein were buffer exchanged via dialysis against 100 mM sodium phosphate pH 7.0, 110 mM NaCl, and 5 mM TCEP and diluted to 7 μM. The samples were split in half and incubated in the presence or absence of four molar equivalents of active-site ligand z-VAD-fmk for 3 hours at room temperature. To ensure complete binding of the active-site ligand to the protein, remaining enzymatic activity was assayed using 300 μM substrate, Ac-LEHD-afc (N-acetyl-Leu-Glu-His-Asp-7-amino-4-fluorocoumarin), (Enzo Life Sciences). Once full inhibition was achieved, samples were buffer-exchanged six times with 100 mM phosphate buffer pH 7.0, 100 mM NaCl and 5 mM TCEP using an Amicon Ultracell 3K concentrator (Millipore) to remove unbound inhibitor. For S183E, S183A, C287A/S183A, S99E, C287A/S99E, T125E, C287A/T125E variants of caspase-9, prior to thermal denaturation monitored by CD analysis, the proteins were buffer exchanged with 100 mM phosphate buffer pH 7.5, 110 mM NaCl and 5 mM TCEP using a NAP™-5 Column (GE Healthcare). For cleavage of the unprocessed caspase-9 C287A and S183E variants, 7 μM protein sample was incubated with 3% active caspase-3 protein for two hours at room temperature. Full processing of caspase-9 C287A and S183E by caspase-3 was determined by SDS-PAGE analysis.
Thermal denaturation of caspase-9 variants and CARD was monitored by loss of CD signal at 222 nm over a range of 20–90°C. The circular dichroism spectra were monitored from 250–190 nm. Both were performed on a J-720 or J-1150 CD spectrometer (Jasco) with a Peltier controller. Data were collected four separate times on different days from different batches of purified proteins. Curves were fit with Origin Software (OriginLab) using sigmoid fit to determine the melting temperature.
Caspase-3 expression and purification
Caspase-3 full-length gene (human sequence) in pET23b (Addgene plasmid 11821 (25)) was transformed into BL21 (DE3) T7 Express strain of E. coli and protein expression was induced with 1 mM IPTG at 30°C for 3 hours (26). The protein was purified as described previously for caspase-3 (27). The eluted protein was stored in −80°C in the buffer in which they eluted. The identity of purified caspase-3 was assessed by SDS-PAGE and ESI-MS to confirm mass and purity.
Native gel electrophoresis and Ni-NTA affinity isolation assay to determine in trans interactions
For native gel electrophoresis to diagnose an interaction between caspase-9 ΔCARD and caspase-9 CARD in trans, full-length caspase-9, caspase-9 ΔCARD and CARD were dialyzed twice against 100 mM phosphate pH 7.0 and 2 mM DTT for 90 minutes to rid of excess salt. Samples were incubated either alone or combined with CARD to achieve a 1:1 ratio of caspase-9 ΔCARD plus CARD. Each protein sample was diluted to a final concentration of 10 μM in the dialysis buffer. To induce dimerization samples were incubated with 5-fold excess z-VAD-fmk. All samples were allowed to incubate at room temperature for one hour. All samples were mixed with glycerol loading dye and fractionated on a 10% Tris/Glycine pH 8.3 polyacrylamide gel.
For the Ni-NTA affinity isolation assay of caspase-9 ΔCARD-His6 (ΔCARD, ΔCARD C287A, ΔCARD + z-VAD-fmk) with the CARD in trans, samples were diluted to 10 μM in binding buffer (50 mM phosphate pH 8.0, 100 mM NaCl) with 5 mM DTT. To induce dimerization, 20 μM ΔCARD was incubated with 5-fold excess z-VAD-fmk for 1 h at RT. Complete inhibition was assessed by assaying caspase-9 activity using 300 μM LEHD-afc. Excess z-VAD-fmk was removed by buffer exchanging 5x with binding buffer using Amicon Ultra centrifugal filter 10K MWCO (Millipore). Samples were incubated either alone or with CARD to achieve a 1:2 ratio of caspase-9 ΔCARD-His6 plus CARD (no His tag). 100 μL of protein sample was added to a tube containing 35 μL of HisPur Ni-NTA magnetic beads (ThermoFisher) that were washed three times in water and twice in binding buffer without DTT. Ni-NTA beads plus caspase-9 ΔCARD-His6 and CARD samples were incubated for 3 hours at 4°C with mixing using an end-to-end rotator. The supernatant (unbound fraction) was aspirated and the beads were washed three times with binding buffer to remove any unbound or weakly bound protein (wash fraction). Protein elution was then carried out by incubating the Ni-NTA beads with 50 μL elution buffer (binding buffer + 250 mM imidazole) for 30 minutes at room temperature. The supernatant (elution fraction) was collected and all fractions were subjected to SDS-PAGE analysis.
Fluorescence anisotropy
Fluorescence anisotropy was monitored using a SpectraMax M5 plate reader (Molecular Devices, Inc.) with a fixed excitation wavelength set to 485 nm and an emission wavelength set to 525 nm. Caspase-9 CARD (without the His tag) was labeled with fluorescein isothiocyanate (FITC) isomer 1 (Sigma) in labeling buffer containing 0.1 M sodium bicarbonate pH 9.0, 100 mM NaCl for 2h at RT. Unreacted FITC was removed by buffer exchange using a NAP5 column equilibrated in 50 mM Tris (pH 7.5) and 150 mM NaCl. A fixed concentration of FITC-labeled CARD (25 nM) was titrated into a serially diluted caspase-9 ΔCARD (3 nM – 25 μM). The final volume of each binding reaction is 100 μL. All measurements were taken at 25°C after a 1.5 h incubation at RT.
Activity assays
For kinetic measurements of caspase activity, 800 nM caspase-9 full-length protein was diluted in 100 mM MES pH 6.5, 10% PEG 8,000 and 5 mM DTT. Each sample was subjected to a substrate titration, performed in the range of 0–300 μM fluorogenic substrate, Ac-LEHD-afc, (Ex 365/Em 495) which was added to initiate the reaction. Assays were performed in duplicates at 37°C in 100 μL volumes in 96-well microplate format using a Molecular Devices Spectramax M5 spectrophotometer. Initial velocities versus substrate concentration were fit to a rectangular hyperbola using GraphPad Prism (Graphpad Software) to determine kinetic parameters KM and kcat. Enzyme concentrations were determined by active site titration with quantitative inhibitor z-VAD-fmk. Active site titrations were incubated over a period of 3 h in 100 mM MES pH 6.5, 10% PEG 8,000, and 5 mM DTT. 90 μL aliquots were transferred to black-well plates in duplicate and assayed with 300 μM Ac-LEHD-afc. The protein concentration was determined to be the lowest concentration at which full inhibition was observed.
To test the ability of the CARD to activate caspase-9 ΔCARD in trans, 10 μM of ΔCARD was incubated with CARD at different ratios (1x, 5x and 10x CARD) in minimal activity assay buffer (100 mM MES pH 6.5, 20% PEG 400 and 5 mM DTT) for 1h at RT. Control reactions were made using BSA in place of CARD. Samples were diluted to a final concentration of 800 nM ΔCARD using minimal activity assay buffer and LEHDase activity was measured over the course of 10 min in 100 μL volumes using a Spectramax M5 fluorescence plate reader (Molecular Devices).
Protein cleavage kinetics
Procaspase-3 C163S (2 μM) was incubated with a series of increasing concentration of caspase-9 (WT full-length or ΔCARD) in minimal activity assay buffer (100 mM MES pH6.5, 20% PEG 400 and 5 mM DTT) for 1h at 37°C. The reaction was terminated by adding SDS loading buffer and boiling the samples for 10 min. Samples were run on a 5–22% SDS-PAGE and stained with Coommassie dye. Densitometry was performed on the bands corresponding to procaspase-3 to estimate E1/2, which represents the concentration of caspase-9 (enzyme) that cleaves 50% of procaspase-3 (substrate) in time (t). The catalytic efficiency, kcat/KM, of caspase-9 was then determined using the half-life equation:
For the cleavage kinetics, 3 μM of procaspase-3 was incubated with 500 nM caspase-9 (WT full-length or ΔCARD) in minimal activity assay buffer at 37°C. Samples were taken at different time points through the course of 1h. Cleavage was stopped by adding SDS loading buffer and boiling the samples for 10 min. Samples were run on a 5–22% SDS-PAGE and cleavage of procaspase-3 was assessed by densitometry using Image Lab software. Graphpad Prism was used to generate curves, which were then fit using the above equation to estimate kcat/KM.
RESULTS
The influence of CARD on the oligomeric state of caspase-9
The catalytic core of caspase-9 (ΔCARD) has a lower catalytic efficiency than the full-length, wild-type (WT) version of the enzyme, but the fundamental structural and physical basis of these differences are not known. We interrogated the effect of the presence of the CARD on the specific biophysical properties of caspase-9 to understand why the presence of the CARD domain has a synergistic effect on function. To ensure that the caspase-9 ΔCARD and full-length reagents used for this study were comparable to the studies that show CARD’s ability to enhance caspase-9 activity, we independently measured the catalytic properties of various caspase-9 constructs. Consistent with a previous report (9), the both ΔCARD variants of caspase-9 exhibited decreased catalytic efficiency relative to the full-length version against a peptide substrate, LEHD (Figure 2A). In addition, removal of the CARD clearly attenuated caspase-9′s ability to cleave procaspase-3 (C163S), one of its physiological substrates (Figures 2B-2D). We also estimated the kcat/KM of ΔCARD-His6 against procaspase-3, and observed a similar decrease in catalytic efficiency than that of WT (Figures 2E–2G). While other studies have reported ΔCARD being more active than full-length cleaved caspase-9 (11–13), we consistently and reproducibly observed the opposite. Both the ΔCARD caspase-9 which was allowed to autoprocess at the intersubunit linker (ΔCARD-His6 in Figure 2A) and ΔCARD caspase-9 refolded from separately expressed large and small subunits (ΔCARD in Figure 2A) were less active than full-length cleaved caspase-9. We surmise that the different buffer conditions used in our study may account for the observed difference in activity.
Figure 2. Removal of CARD decreases caspase-9 activity.
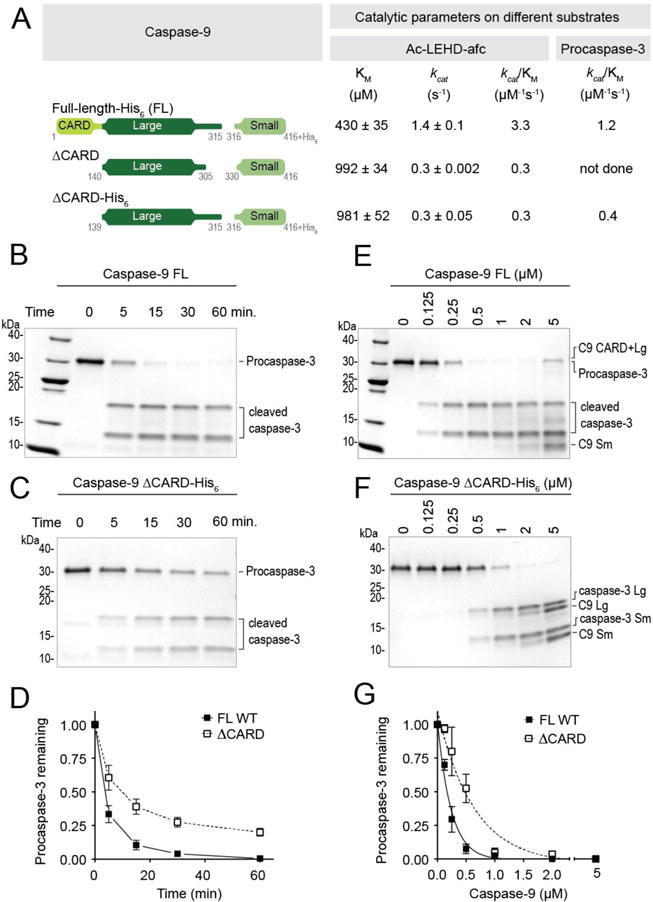
(A) Catalytic parameters of different versions of caspase-9 using Ac-LEHD-afc as a peptide substrate and procaspase-3 (C163S) as protein substrate. Two versions of ΔCARD were used in this study. Both versions exhibited very similar catalytic efficiencies. Using both substrates, ΔCARD was observed to be less active than full-length, wild-type (WT) caspase-9. Values reported are mean (+SEM) of three independent trials done on three separate days.
(B) and (C) ΔCARD has slower protein cleavage kinetics than full-length WT caspase-9. Catalytic cysteine mutant (C163S) procaspase-3 (3 μM) was incubated with 500 nM of either full-length, WT caspase-9 (B) or ΔCARD (C) for 1h at 37°C.
(D) Plot for time-course analysis of protein cleavage kinetics done on (B) and (C).
(E) and (F) Cleavage of procaspase-3 (C163S) to estimate kcat/KM of full-length WT caspase-9 and ΔCARD. 2 μM of procaspase-3 was incubated with either full-length WT (E) or ΔCARD (F) caspase-9 of different dilutions (0 - 5000 nM) for 1h at 37°C.
(G) Corresponding curves of (E) and (F) generated from the amount of procaspase-3 remaining and their non-linear fits to estimate kcat/KM.
Caspase-9 is predominantly monomeric in solution, but when subjected to size exclusion chromatography, proteolytic activity correlated with the small fraction of dimeric caspase-9 and not with monomeric caspase-9, suggesting that dimerization is required for caspase-9 activity (16). Thus, one potential reason for the increase in the activity of full-length caspase-9 could be due to an increase in the ratio of dimeric caspase-9 when the CARD is attached, as has been suggested previously (9). To assess this, full-length and ΔCARD versions of caspase-9 were subjected to size exclusion chromatography (SEC) to determine the oligomeric state of each enzyme. Both full length and ΔCARD caspase-9 were predominantly monomeric in solution and no oligomeric fraction could be observed in the chromatogram (Figure 3). Caspase-9 can be forced into a dimeric state by covalent binding of an active-site inhibitor, z-VAD-fmk, which resembles a bound substrate. The full-length and ΔCARD caspase-9 were both capable of completely converting to their respective dimeric states in the presence z-VAD-fmk (Figure 3). z-VAD-fmk appears to occupy half of the active sites under these conditions (Figure S1), consistent with previous reports and existing crystal structures (16,28). In addition, active site titration using z-VAD-fmk also suggests that all functional active sites are fully occupied by z-VAD-fmk. Strictly speaking, neither we nor others are certain about whether one or both of the caspase-9 active sites are simultaneously functional. Nevertheless, we observed that when bound to z-VAD-fmk the molecular weight of the dimeric full-length caspase-9 was larger than expected (136 kDa vs. 94 kDa expected) (Table 1), which could be due to either a significant change in the hydrodynamic radius of dimeric full-length caspase-9 or an enhanced interaction with the negatively charged matrix in the column, as we have observed with caspase-6 (29). These results suggest that the CARD does not appear to have a great influence on the oligomeric state of the enzyme and thus cannot be the cause of the increased activity observed in the presence of the CARD.
Figure 3. CARD does not influence caspase-9 oligomerization.
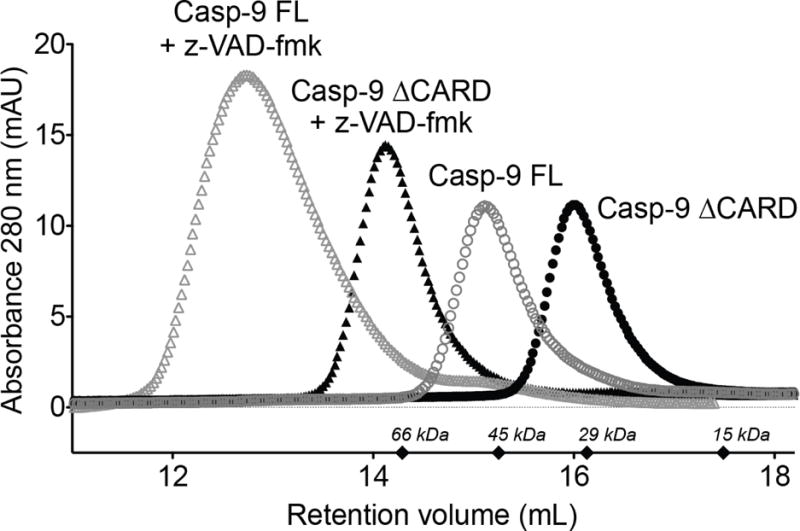
Size exclusion chromatography of full-length caspase-9 (Casp-9 FL) and caspase-9 ΔCARD (Casp-9 ΔCARD) in the presence and absence of active-site ligand z-VAD-fmk. Both Casp-9 FL and Casp-9 ΔCARD are capable of dimerization induced by z-VAD-fmk. The molecular weights for the standards are marked as diamonds.
Table 1.
Molecular weights of caspase-9 variants from Size Exclusion Chomatography.
| Caspase-9 variant | Molecular weight (kDa)
|
|
|---|---|---|
| Observed | Expected3 | |
| Casp-9 ΔCARD | 30 | 29.0 |
| Casp-9 ΔCARD + zVAD-fmk | 71 | 58.0 |
| Casp-9 Full-length (FL) | 45 | 47.2 |
| Casp-9 Full-length + z-VAD-fmk | 136 | 94.4 |
Expected/Theoretical molecular weights were calculated from the protein sequence of caspase-9 variants using ExPASy ProtParam tool (61).
The presence of CARD influences stability of caspase-9
Another potential reason for the increased activity of full-length caspase-9 could be that the presence of the CARD affects the protein’s stability. To assess this, both full-length and ΔCARD caspase-9 in monomeric and dimeric forms were analyzed for changes in thermal stability by circular dichroism (CD) spectroscopy. Monomeric, full-length caspase-9, which was cleaved at the intersubunit linker between the large and small subunits, showed a three-state unfolding curve (Figure 4A). The first melting transition occurred at 48 ± 2°C while a second occurred at 62 ± 2°C. To determine the domain of full-length caspase-9 that unfolds at each melting transition, the catalytic core (ΔCARD) and CARD domains were expressed independently and interrogated in a similar fashion. The unfolding of the catalytic core of the enzyme corresponded to the first melting transition at 49 ± 1°C (Figure 4B) while the second transition corresponded to that of the CARD with a melting temperature of 61 ± 2°C (Figure 4C). These results suggest that when caspase-9 is in its cleaved monomeric state, the presence of CARD does not affect the overall thermal stability of the caspase-9 catalytic core and the two domains (CARD and core) of the enzyme unfold independently.
Figure 4. Monomeric and dimeric states of caspase-9 have different unfolding properties.
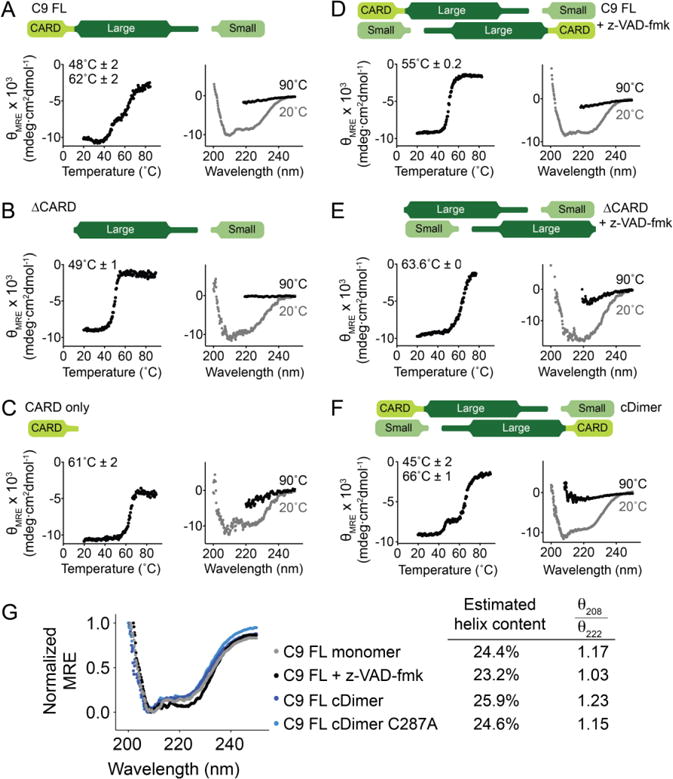
Thermal denaturation profile monitored by circular dichroism (left) with the thermal melting temperatures (Tm) for each transition listed and circular dichroism spectra (right) of various forms of caspase-9 (top schematics).
(A) Two melting transitions are observed in the cleaved full-length, monomeric, caspase-9.
(B) and (C) Thermal denaturation profile of cleaved caspase-9 core and the CARD, respectively, showing that the first melting transition in full-length, monomeric caspase-9 (A) is due to the unfolding of the core, while the second is due to that of the CARD.
(D) Full-length, cleaved caspase-9 is dimeric in the presence of an active-site ligand z-VAD-fmk. Upon thermal denaturation, caspase-9 in this state exhibits a single melting transition, likely due either to dimerization, or the presence of an ordered active site, or both.
(E) Dimeric, cleaved caspase-9 ΔCARD with bound z-VAD-fmk is stabilized by 14°C compared to monomeric, cleaved ΔCARD (B).
(F) Constitutive dimer (cDimer) full-length caspase-9 cleaved at the intersubunit linker exhibits two melting transitions, suggesting independent unfolding of CARD and core domains.
(G) The secondary structure of caspase-9 with (+ z-VAD-fmk) and without (FL monomer, cDimer and cDimer C287A) an active-site ligand bound was assessed by circular dichroism spectroscopy. C9 FL cDimer is a constitutive dimer variant of caspase-9. There is no significant change in the helical content of caspase-9 upon dimerization and substrate binding. Estimation of helix content was performed using BeStSel structure prediction and fold recognition software (http://bestsel.elte.hu/index.php) (62).
Plots are representative of three trials. Tm values shown are means ± SEM of three trials done on three separated days.
A physical interaction exists between the CARD and catalytic core
Although caspase-9 exists in equilibrium between monomer and dimer, it is predominantly monomeric in solution (Figure 3). To assess whether the oligomeric state of caspase-9 influences its stability, thermal denaturation studies were similarly performed on the cleaved full-length and ΔCARD versions of caspase-9 when the enzyme is in a dimeric state due to binding of active-site ligand (z-VAD-fmk), which is known to promote dimerization (Figures 4D and 4E). Both versions of caspase-9 showed an increase in thermal stability. Caspase-9 ΔCARD had a 14°C increase in thermal stability in the presence of active-site ligand (Figure 4E), which is similar to the increase in stability observed when caspase-7 binds active-site ligand (27). Full-length caspase-9 (Figure 4D) showed only a 6°C increase in thermal stability in the presence of active-site ligand, more similar to the 3°C increase in stability observed for caspase-6 upon ligand binding (30). Strikingly, in the presence of substrate, the three-state unfolding (two melting transitions) of the full-length caspase-9 was no longer observed (Figure 4A vs. 4D). It appears that binding a ligand to the active site of caspase-9, which induces dimerization and ordering of the active-site loop bundle, also transitions caspase-9 to a two-state unfolding mechanism (single melting transition). In addition, full-length caspase-9 is completely unfolded at 90°C as observed in the circular dichroism spectrum (Figure 4D, 4E). These data suggest that in the cleaved, dimeric and active-site bound state, the catalytic core and the CARD of caspase-9 unfold cooperatively. To discriminate the influence of substrate-binding from dimerization on the observed cooperative unfolding, thermal denaturation was performed on a caspase-9 variant that exists as a constitutive dimer (cDimer) (Figure 4F). This dimeric version of caspase-9 (10) was constructed by substituting residues in the dimer interface with those present in caspase-3, which is constitutively dimeric. We observed that cDimer also exhibits LEHDase activity, albeit less active (Figure S2). Full-length, unbound dimeric caspase-9, which was cleaved at the intersubunit linker exhibited three-state unfolding (Figure 4F), suggesting that substrate binding was responsible for the observed changes in unfolding properties. Thus, the single melting transition observed when cleaved, full-length caspase-9 in the dimeric, active-site bound state (Figure 4D) indicates that substrate binding-induced dimerization either results in the complete unfolding of CARD, or causes the catalytic core and CARD to unfold as one cooperative unit. Comparison of the circular dichroism spectra of the full-length, cleaved caspase-9 in monomeric and dimeric states (Figure 4G) shows that there is no significant change overall in the secondary structural content. The CARD is composed of six helices (31); if the CARD became unfolded upon substrate binding we would expect to see a significant loss in the CD signal. Secondary structure content analyses from the CD spectra revealed that there is ~5 % loss in helical content when full-length caspase-9 binds an active-site ligand to induce dimerization (Figure 4G, FL monomer vs FL + z-VAD-fmk). The similarity in the CD spectra with and without active-site ligand suggests that the CARD remains folded and that the caspase-9 catalytic core and CARD must be unfolding as a single cooperative unit. This observation suggests that a physical interaction between the CARD and core domains occurs, which causes the two domains to unfold as a single unit.
An ordered active site supports CARD:core interactions
Caspase-9 has been shown to possess catalytic function even as an uncleaved zymogen (5) possibly due to its increased intersubunit linker length. Caspase-9 possesses a longer L2 loop, allowing L2′ some flexibility to assume a productive conformation which enables caspase-9 to support a properly formed active site even without linker cleavage (32). Therefore, the uncleaved/zymogen form of caspase-9 can be utilized to interrogate whether the interaction observed between the CARD and the catalytic core of caspase-9 is due to changes in the active site conformation. Analysis of full-length monomeric caspase-9 in the uncleaved state (catalytic site-inactivated variant C287A) resulted in a two-state unfolding mechanism (Figure 5A) similar to that observed for the cleaved, dimeric and active-site-bound state (Figure 4D). The monomeric, uncleaved caspase-9 zymogen appears to support the interaction of the core and CARD domains because they unfold as a single unit. To further test this mechanism, we cleaved the same caspase-9 zymogen construct with caspase-3. Cleavage of the intersubunit linker by caspase-3 disrupted the interaction of the CARD and catalytic core as observed by the independent three-state unfolding (Figure 5B), which is similar to that of the cleaved wild-type monomeric caspase-9 (Figure 4A). The presence of an intact linker also appears to support CARD:core interactions in the constitutively dimeric (cDimer) full-length caspase-9 zymogen (C287A). This version of caspase-9 had a two-state unfolding mechanism (Figure 5C), similar to that of a full-length, dimeric caspase-9 with a bound active-site ligand (Figure 4D). Together, these data suggest that the CARD and the catalytic core of caspase-9 do not physically interact and unfold independently in monomeric or in dimeric caspase-9 that has a disordered active site resulting from cleavage of the intersubunit linker. In contrast, these domains physically interact and unfold cooperatively, as a single unit, when the active-site region assumes an ordered conformation as supported by either an intact linker in both monomeric and dimeric states, or by binding of a substrate to the active site.
Figure 5. The CARD and core of caspase-9 unfold as a single unit when the intersubunit linker is intact.
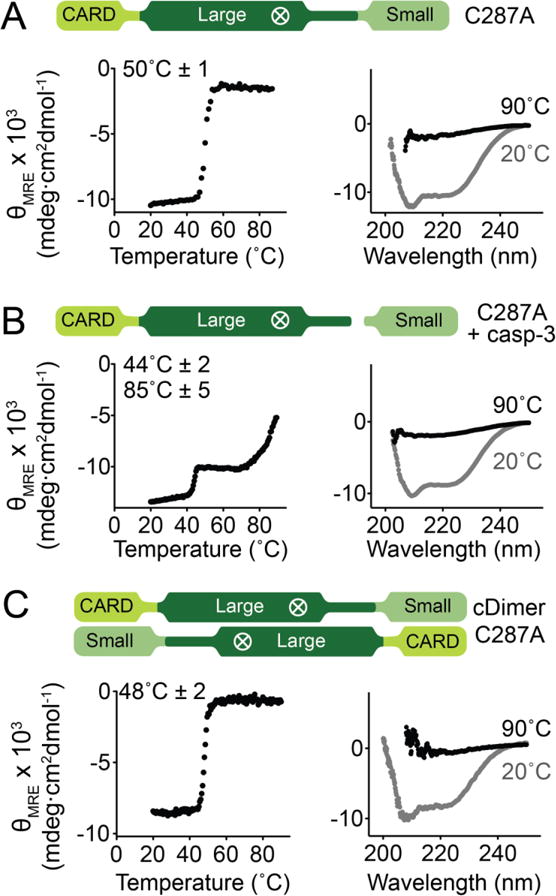
Thermal denaturation profiles monitored by circular dichroism (left) with the thermal melting temperatures (Tm) for each transition listed and circular dichroism spectra (right) of various forms of caspase-9.
(A) Monomeric, zymogen (uncleaved) caspase-9 (catalytic site-inactivated C287A) exhibited a single melting transition, which suggests cooperative unfolding of CARD and core of caspase-9.
(B) Cleavage of the linker of (A) by caspase-3 results in independent unfolding, as manifested by two separate melting transitions.
(C) Zymogen (catalytic site-inactivated C287A), uncleaved caspase-9 in a constitutively dimeric state (cDimer) shows a single melting transition.
Plots are representative of three trials. Tm values shown are means ± SEM of three trials done on three separated days.
Characterizing the interaction between caspase-9 catalytic core and CARD
The ΔCARD caspase-9 variant is less active than full-length caspase-9 (Figure 2A), suggesting that the presence of the CARD could increase the catalytic activity of the caspase-9 core. Indeed, an increase of caspase-9 activity was observed when the CARD was incubated with ΔCARD caspase-9 (Figure 6A). This increase in activity was not simply due to molecular crowding since adding BSA in place of CARD did not cause any significant change in activity. However, this enhancement did not reflect the full activity of full-length caspase-9, suggesting that the tether between the CARD and the catalytic core of caspase-9 is necessary for CARD’s impact on enzymatic activity. Since the linker between the CARD and core domains is essential to mediate the increase in the activity of caspase-9, we reasoned that perhaps there were either specific interactions with the tether and the adjacent domains or a length-dependence to the interaction. To test this, a five amino acid Ser-Gly extension was inserted within the linker between CARD and the large subunit of caspase-9′s catalytic core (Figure 6B). This variant behaves like the native full-length form of caspase-9 in both thermal stability (Figure 6B) and catalytic efficiency (Figure 6C), suggesting that a longer and potentially more flexible linker does not negatively impact the function of caspase-9.
Figure 6. Linker between CARD and core supports CARD-core interaction.
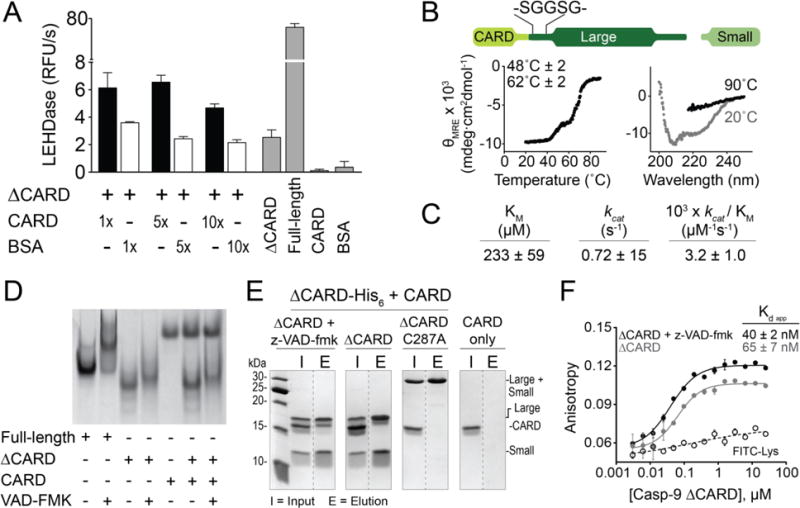
(A) In trans activity assay of ΔCARD caspase-9 with CARD. The presence of CARD enhances caspase-9 activity, but does not recapitulate the full activity of a full-length caspase-9. Error bars are SD from three trials done on three separate days.
(B) Thermal denaturation analysis (left) and circular dichroism spectra (right) of the Ser-Gly linker extension variant of caspase-9 showing the same melting transitions as that of full-length, cleaved caspase-9. Plots are representative of three trials. Tm values shown are means ± SEM of three trials done on three separated days.
(C) Caspase-9 Ser-Gly linker extension variant exhibits similar kinetic behaviors to wild-type full-length, cleaved caspase-9.
(D) Native gel electrophoresis showed no visible mobility shift, which indicate interaction between CARD and caspase-9 core (unbound/monomeric and z-VAD-fmk-bound/dimeric).
(E) His-tagged, dimeric, z-VAD-fmk-bound ΔCARD was able to pull down untagged CARD by Ni-NTA affinity assay, suggesting interaction between CARD and catalytic core of caspase-9.
(F) Fluorescence anisotropy shows binding of FITC-labeled CARD to both monomeric and dimeric (+ z-VAD-fmk) ΔCARD. Error bars are SD from three trials done on three separate days.
The cooperative unfolding observed between the CARD domain and the catalytic core of caspase-9 implies a physical interaction between the two domains. To characterize this, the interaction between the isolated CARD and catalytic core domains in trans was interrogated. The catalytic core (ΔCARD) in its monomeric or dimeric form (ΔCARD + z-VAD-fmk) was incubated with the CARD and analyzed for an interaction between the two domains by native gel electrophoresis (Figure 6D). An interaction between the two domains would result in a band migrating with the molecular weight of the full-length enzyme during native gel analysis. However, no visible shift in band migration was observed that would correspond to complex formation between CARD and ΔCARD, which suggests either no interaction between the domains, or very weak and transient interactions, or that the conditions for native gel electrophoresis did not promote CARD:core interactions. Ni-affinity isolation assay was then performed (Figure 6E) in which ΔCARD-His6 in different states (cleaved with z-VAD-fmk-bound, cleaved with no ligand bound, or uncleaved with no ligand bound (C287A)) was incubated with CARD (no His6). Only ΔCARD with bound z-VAD-fmk was able to isolate the CARD after elution out of the Ni beads, suggesting complex formation between the catalytic core and the CARD. In addition, fluorescent polarization/anisotropy binding experiments showed that while FITC-labeled CARD also binds to monomeric, cleaved ΔCARD caspase-9, tighter binding was observed with dimeric ΔCARD caspase-9 bound with an active-site ligand (+ z-VAD-fmk) (Figure 6F). The magnitude of the difference in the fluorescence anisotropy assessment was not as significant as might be predicted by the affinity isolation assessment (Figure 6E), perhaps indicating that fluorophore labeling impacted the interaction of the two domains. Nevertheless, together these data are consistent with those observed from thermal denaturation studies, where an ordered active site appears to promote CARD:core interactions.
Given the observation that the CARD and core domain physically interact, we undertook a program designed to uncover sites where substitution of the native amino acids by alternatively charged amino acids might disrupt interactions between the CARD and core domain. To generate candidate sites for interaction and mutations, we performed docking studies between reported crystal structures of the CARD (PDB ID: 3YGS) (31) and the dimeric form of the caspase-9 catalytic core (PDB ID: 1JXQ) (16) using the RosettaDock server (33). The top docking models were those that avoided interactions of CARD residues involved in the caspase-9 CARD/Apaf-1 CARD complex and avoiding unfavorable (e.g. steric) interactions between caspase-9 CARD and core. Two models fit these criteria (Supplemental Figure S3). Single and combination charge swapping variations were constructed and analyzed for decrease in catalytic efficiency similar to what was observed for ΔCARD (Table S1). However, none of the charge-swapped variants resulted in a significant change in caspase-9 activity as all of them showed kcat/KM parameters that were within 3-fold of wild type.
Phosphomimetic S183E breaks CARD:core interactions
The propensity of the CARD:core interactions to exist when caspase-9 is in a conformation with an ordered active site suggests that any modification in caspase-9 that would lead to a disordered active-site loop bundle would disrupt these interactions. One particular caspase-9 variant that we have shown to inactivate caspase-9 by that precise mechanism is the phosphomimetic S183E (34). S183E was observed to have a profound effect on caspase-9 function. After overexpression S183E remains uncleaved and exhibits no LEHDase activity. Since S183E is in its monomeric and uncleaved form, we expected its stability to be comparable to that of the zymogen, catalytic-site inactivated caspase-9 C287A, and predicted it would exhibit only one melting transition upon thermal denaturation (Figure 4A). However, S183E underwent two melting transitions (Figure 7A), corresponding to the unfolding of the core (43°C) and the CARD domain (64°C). The S183E thermal denaturation profile suggests that the interaction between the CARD and core domain has been disrupted by the S183E substitution, leading the two domains to unfold independently. Importantly, the melting temperature of the core is unchanged from WT caspase-9 suggesting that the core is intact when the S183E variant is in the uncleaved zymogen conformation. Unfortunately, S183E does not bind to z-VAD-fmk under any conditions, so it is not possible to interrogate the direct interaction of the free CARD with the S183E core as was done for WT caspase-9 (Figure 6). Nevertheless, the striking observation that the CARD:core interactions were eliminated upon modification of the Ser183 site suggests that Ser183 must sit at the binding interface between the caspase-9 core and its CARD.
Figure 7. Phosphomimetic S183E disrupts CARD:core interactions.
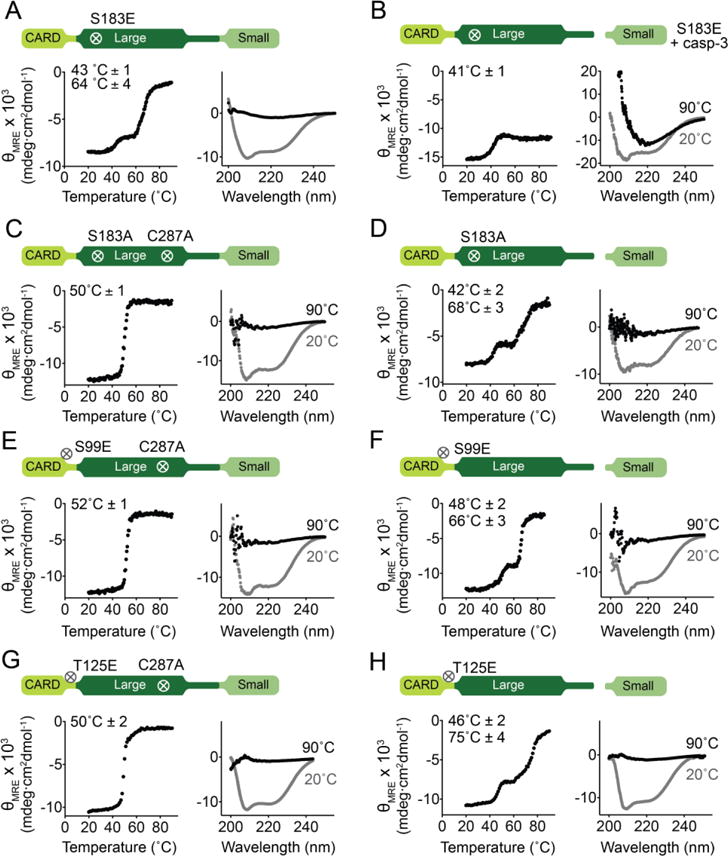
Thermal denaturation curves monitored by circular dichroism with the thermal melting temperatures (Tm) for each transition listed (left) and circular dichroism spectra (right) of caspase-9 variants.
(A) Full-length, monomeric, uncleaved S183E exhibits three-state unfolding (two melting transitions), unlike other full-length, uncleaved caspase-9 variants, suggesting that S183E breaks CARD:core interactions because the two domains unfold independently.
(B) Cleavage of S183E by caspase-3 leads to destabilization and formation of aggregates.
(C) Full-length, monomeric, uncleaved S183A (catalytic-site inactivated C287A) shows a single melting transition, indicating an intact CARD:core interaction.
(D) Full-length, monomeric, cleaved S183A behaves similarly to full-length, monomeric, cleaved caspase-9 that exhibits three-state unfolding.
(E) and (G) Full-length, monomeric, uncleaved (constructed in the background of C287A) S99E (E) and T125E (G) exhibit two-state unfolding (single melting transition), suggesting intact CARD:core interactions.
(F) and (H) Full-length, monomeric, cleaved S99E and T125E behaves similarly to full-length, monomeric, cleaved caspase-9 that exhibits three-state unfolding.
Thermal denaturation curves (left) and CD spectra (right) of caspase-9 variants. Plots are representative of three trials. Tm values shown are means ± SEM of three trials done on three separated days.
However, our results from interrogating the effect of S183 phosphorylation on caspase-9 structure point to a different mechanism in which S183E imparts conformational instability to caspase-9 in the cleaved state. We found that S183E prevents substrate binding by displacing a specific arginine residue (Arg180) in the S1 specificity pocket, resulting in the active-site loop bundle becoming disoriented (34). This most likely contributes to the unusual three-state unfolding of S183E upon thermal denaturation – its unstable conformation cannot fully support CARD:core interactions. Moreover, cleaving S183E with caspase-3 to generate a fully mature caspase-9 led to its aggregation, which was evident in both its thermal denaturation curve, which showed a decrease in thermal stability of the core (41°C in cleaved S183E vs 48°C in WT caspase-9), and a negative signal in its CD spectrum at 90°C (Figure 7B), which was typical of an unfolded protein. These results are consistent with our observations that the core of S183E is becomes extremely unstable upon linker cleavage, and ultimately leads to the formation of ordered aggregates (34). In addition, the CARD:core interactions appear to remain intact in the alanine variant S183A. Full-length, monomeric S183A showed a single melting transition in its uncleaved state (Figure 7C), suggesting cooperative unfolding of domains, and two melting transitions in its cleaved state (Figure 7D), indicating independent unfolding of CARD and core domains. Thus, although Ser183 did not emerge as a critical site of CARD:core interactions, our model of caspase-9 inactivation by S183E by disorienting the active-site loops is in agreement with our hypothesis that a properly formed active site is crucial for the interaction between caspase-9 core and its CARD domain.
Phosphorylation has been shown to be a robust mechanism to disrupt binding interfaces (35–37). Three reported phosphorylation sites - S99 (38), T107 and T125 (39,40) - reside in the potentially highly flexible linker region between the CARD and the large subunit (Figure 1). Given that the linker which tethers the CARD to the catalytic core seems to be required for increased catalytic activity (Figure 6A, 6B), it is conceivable that phosphorylation at this region could impact interactions between the CARD and core. We examined whether the phosphomimetic versions of these residues would break the CARD:core interactions by conducting the same thermal denaturation studies on both cleaved and zymogen/uncleaved forms. Both S99E and T125E in monomeric, uncleaved form showed cooperative unfolding of domains, exhibiting a single melting transition (Figure 7E, 7F), suggesting that the interaction between CARD and core is still present and was not interrupted by these mutations. Cleavage at the intersubunit linker of S99E and T125E resulted in independent unfolding of the CARD and core domains (Figure 7G, 7H). These results suggest that S99 and T125 sites are not within the binding interface of the CARD and catalytic core.
DISCUSSION
Full control of the caspases involved in apoptosis, inflammation and neurodegeneration requires detailed understanding of the functions and regulatory mechanisms for each individual caspase. Caspase-9 has a particularly unique activation mechanism including changes in its conformational and oligomeric states and association with the apoptosome activation platform. Furthermore, the presence of individual domains such as the caspase-9 CARD (in cis) or the Apaf-1 CARD (in trans) have the ability to increase caspase-9 basal activity (9). Altering enzymatic activities by additional domains have been observed in other proteins including PAS (Per-Arnt-Sim) Kinase (41), Dnmt1 DNA methyltransferase (42), and ADAMTS-4 (A disintegrin and metalloproteinase with thrombospondin motifs 4) (43), suggesting that this property may be of widespread significance. Therefore studying the individual activation effects of a particular domain provides further insights towards how caspase-9 becomes activated on the apoptosome.
Here we investigated the mechanism by which the caspase-9 CARD influences caspase-9 activity. We have demonstrated that the oligomeric states of both full-length and CARD-deleted (ΔCARD) caspase-9 are similar, thus the increase in caspase-9 activity in the presence of the CARD is not due to a shift the oligomeric state as had been previously suggested (16). We also observed that the mere presence of CARD is not responsible for the increased enzymatic activity but requires specific interactions between the CARD and core domain, particularly with the active site. Furthermore, our results suggest that the caspase-9 CARD:core interaction is controlled by the ordered state of the active site (Figure 8). Specifically, in procaspase-9, the length of the intersubunit linker is sufficient to support ordering of the active site (32), and an interaction with the CARD. The CARD and catalytic core domains of caspase-9 unfold independently and do not physically interact in either monomeric or dimeric states of the cleaved enzyme because the cleaved loops that form the active site are unable to form a properly ordered substrate-binding groove. The disordered active site is therefore unable to support the interactions between the CARD and caspase-9 core. In the dimeric state with a ligand bound to the active site, these domains unfold cooperatively, as a single folding unit, indicating a physical interaction between the CARD and core domain when the cleaved enzyme dimerizes and has a properly ordered active site, which is capable of binding CARD.
Figure 8. Model for caspase-9 conformational states in the presence of CARD domain.
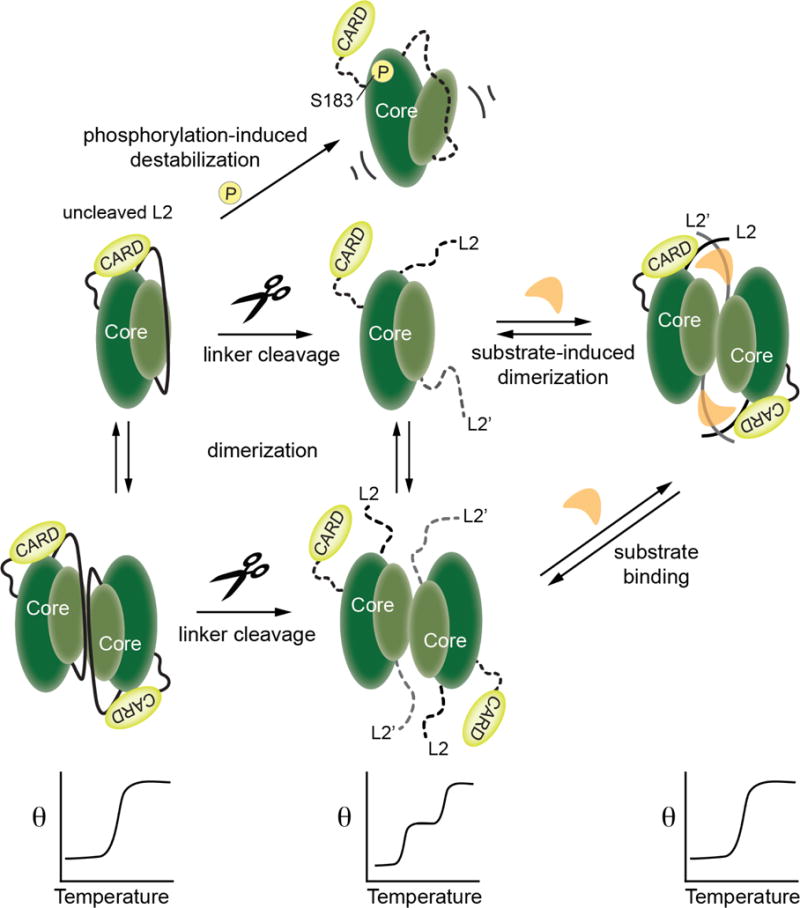
Relevant conformations of caspase-9 are shown as cartoons in the upper panels. When caspase-9 is in the uncleaved state the length of the intact, uncleaved intersubunit linker promotes the ordered conformation of the active site loops and thus facilitates interactions of the CARD with the caspase-9 core. A similar conformation is attained in a cleaved, dimeric caspase-9 when substrate binding promotes an ordered active site and thus supports the interaction of CARD with the core of the caspase-9. These CARD-core interactions result in cooperative unfolding of the two domains, as depicted in the lower panels. Caspase-9 assumes a disordered active-site conformation which abolishes the CARD-core interaction, either by transitioning to a cleaved monomeric or dimeric state in the absence of substrate, or by introducing a mutation in the core (as in S183E) that leads to its destabilization and disorder of active-site loops.
The substrate-binding groove is ordered in dimeric, cleaved caspases (-1, -3, -6, -7, -8 and -9) only when a substrate is bound. Notably a similar ordered conformation can be formed in uncleaved zymogen of caspase-9, due to linkage effects which allow the intersubunit linker to buttress the L3 and L4 loops in an ordered conformation (16,44) (Figures 4, 7). This manifests in the cooperative unfolding of CARD and the catalytic core of caspase-9 observed in the zymogen/uncleaved form of full-length caspase-9, whether monomeric or dimeric, because the intact intersubunit linker can properly order the active site even prior to cleavage. Intriguingly, this CARD:core domain interaction is disrupted either by cleavage of the intersubunit linker by self-processing or by caspase-3, or by a mutation in the core such as S183E, all of which prevent the active site from assuming an ordered conformation. Thus, the CARD appears to be interacting with the caspase-9 core in any version of caspase-9 presenting a properly formed substrate-binding groove. From these studies, the biochemical interactions between the CARD and core are clear. Future intracellular investigations should shed light on when each of these states of caspase-9 are populated.
Our attempt to pinpoint the binding interface between the CARD and catalytic core of caspase-9 showed that single charge swap mutations on the surface of the protein distal from the active site were not strong enough or properly positioned to disrupt the activating affect of CARD. A more extensive alanine scanning mutagenesis study or charge repulsion analysis around the substrate-binding groove may further define the region of interaction between the CARD and core domains mediated by the active-site region of the enzyme. Once the binding interface is defined, its role in the caspase-9 activation cascade can be further interrogated, and may serve as a potential junction to control caspase-9’s intrinsic activity.
The primary known role of the CARD is to facilitate recruitment and subsequent activation of caspase-9 in the apoptosome. Prior to our work there has been no data to suggest that there are existing interactions between the CARD and the catalytic core of caspase-9. However, there have been reports that point to an inhibitory role of CARD in caspase-9 activity prior to apoptosome binding, either by potentially interfering with caspase-9 homodimerization (11), or by binding to the active site and/or other regions of caspase-9 (13) to keep the enzyme in its latent state, and that Apaf-1 CARD binding relieves this inhibition. Here we observed that the CARD directly participates in stabilizing interactions with the core of caspase-9. The role of these interactions in the context of the caspase-9 activation via the apoptosome remains to be explored, but seems to be consistent with the induced conformational changes model (17). The CARD:core interactions that we observed here could resemble the interactions that are hypothesized to support the active conformation of caspase-9 in the apoptosome. Recent cryo-EM structures of caspase-9-bound apoptosome show that the catalytic core of caspase-9 is bound to the apoptosome hub (18,19), and mechanistic studies have also shown that the catalytic core is able to interact with the nucleotide binding domain of Apaf-1 in the apoptosome (12). Our results complement these observations in the sense that there are regions in the catalytic core that engage in stabilizing interactions with the apoptosome and with the CARD, possibly influencing capase-9 activity. Given our observations that CARD:core interactions influence caspase-9 stability, it is possible that these interactions exist to stabilize caspase-9 prior to its recruitment to the apoptosome, which is in line with the notion that another role for the CARD is to keep free caspase-9 in its latent state. One can envision caspase-9 utilizing the same binding interface in the CARD to interact with its core in its zymogen state, free from the apoptosome. Once the apoptosome is formed, the caspase-9 CARD:core interaction gives way to caspase-9 CARD:Apaf-1 CARD binding, allowing caspase-9 to be finally be recruited and activated on the apoptosome. Moreover, in light of observations that caspase-9 is activated independent of the apoptosome to facilitate alternative pathways (both apoptotic and non-apoptotic) (22,23,45), the presence of CARD:core interactions could also serve as a mechanism to retain, modulate or even enhance caspase-9 activity as it functions outside the apoptosome. Other human caspases (caspase-1, -2, -4/-5, -12) also possess a CARD (review (46)). Among these caspases, caspase-2 is most similar to caspase-9. It would be interesting to examine whether caspase-2 CARD is also able to form these interactions with the catalytic core, which would suggest natural prevalence of these interdomain interactions in caspases and not limited to caspase-9. This could also be a relevant theme in caspase-2 activation. Although caspase-2 has been shown to be activated via proximity-induced oligomerization via the PIDDosome (47), genetic experiments have challenged this mode of activation, since caspase-2 was observed to be activated in the absence of this activating scaffold (48,49). Alternative modes of caspase-2 activation have since been proposed depending on the type of cellular death signals (review (50)); in these cases it is tempting to speculate that CARD:core interactions may play a role in regulating caspase-2 function, should they be present. Interdomain interactions have been demonstrated to be critical in controlling the different conformational states and in regulating the catalytic activity of several proteins including the deubiquitinating enzyme USP4 (51), phenylalanine hydroxylase (52) and ERAP-1 (endoplasmic reticulum aminopeptidase-1) (53).
Prior work on other caspases has suggested that regulation may be dependent on the most unique regions within the caspase structure, the prodomain and intersubunit linker. It is well established that the cleavage of the intersubunit linker primarily acts as an activation switch in executioner caspases (and in some initiator caspases such as caspase-8). However, it seems that there is less consensus as to the function of prodomain in executioner caspases. For example, while the prodomains of caspase-3 and caspase-7 are dispensable for activity in vitro, it appears that in vivo, having an intact prodomain keeps the enzyme in its inactive state until cleaved by another downstream caspase (54,55). The caspase-3 prodomain has been also shown to bind Hsp27 in monocytes, leading to inhibition of its proteolytic activation (56). In caspase-6, an intact prodomain was reported to inhibit self-cleavage at the linker region in vivo (57) and both the prodomain and linker are predicted to be highly disordered protein-binding regions (58) that dramatically affect the stability of caspase-6 (29). In caspase-7 the region just adjacent to the short prodomain contains an exosite for substrate recognition (59,60), so it may be possible that the casp-9 CARD could play similar roles. In the case of caspase-9 it appears that the cleaved state of the intersubunit linker and the interactions between the CARD (prodomain) and the catalytic core is essential for the appropriate function, which is a unique feature of caspase-9. Perhaps this interaction between the CARD and a substrate-bound active site could serve as a valuable therapeutic target for uniquely controlling caspase-9 function.
Supplementary Material
Acknowledgments
We thank Scott Eron for providing the caspase-9 constitutive dimer protein.
FUNDING INFORMATION
This work was supported by the National Institutes of Health (GM 080532) to JH. KH and BS were supported in part by the UMass Chemistry-Biology Interface Training Program (National Research Service Award T32 GM 08515 from the National Institutes of Health).
ABBREVIATIONS
- CARD
Caspase Activation and Recruitment Domain
- Apaf-1
Apoptotic Protease Activation Factor-1
- z-VAD-fmk
carbobenzoxy-Val-ALa-Asp-[O-methyl]fluoromethylketone
- Ac-LEHD-afc
N-acetyl-Leu-Glu-His-Asp-7-aminotrifluoromethylcoumarin
Footnotes
AUTHOR CONTRIBUTIONS
KH and BS designed and performed the experiments, analyzed and interpreted data, prepared the figures and wrote the manuscript. JH conceptualized and directed the research project, secured funding, analyzed and interpreted data, wrote and edited the manuscript.
CONFLICT OF INTEREST
The authors declare no competing interest associated with the manuscript.
References
- 1.Benchoua A, Gué C, Couriaud C, Hosseini H, Sampao N, Morin D, et al. Specific Caspase Pathways Are Activated in the Two Stages of Cerebral Infarction. J Neurosci. 2001;21(18):7127–34. doi: 10.1523/JNEUROSCI.21-18-07127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander RM, et al. Cytochrome C and caspase-9 expression in Huntington’s disease. NeuroMolecular Med. 2002;1(3):183–95. doi: 10.1385/NMM:1:3:183. [DOI] [PubMed] [Google Scholar]
- 3.Sträter J, Herter I, Merkel G, Hinz U, Weitz J, Möller P. Expression and prognostic significance of APAF-1, caspase-8 and caspase-9 in stage II/III colon carcinoma: caspase-8 and caspase-9 is associated with poor prognosis. Int J Cancer. 2010;127(4):873–80. doi: 10.1002/ijc.25111. [DOI] [PubMed] [Google Scholar]
- 4.Inoue H, Tsukita K, Iwasato T, Suzuki Y, Tomioka M, Tateno M, et al. The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 2003;22(24):6665–74. doi: 10.1093/emboj/cdg634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274(13):8359–62. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez J, Lazebnik Y. Caspase-9 and Apaf-1 form an active holoenzyme. Genes Dev. 1999;13:3179–84. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou H, Li Y, Liu X, Wang X. An Apaf-1-cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 8.Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22(2):269–75. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Shiozaki EN, Chai J, Shi Y. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci U S A. 2002;99(7):4197–202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao Y, Shiozaki EN, Srinivasula SM, Rigotti DJ, Fairman R, Shi Y. Engineering a dimeric caspase-9: a re-evaluation of the induced proximity model for caspase activation. PLoS Biol. 2005;3(6):e183. doi: 10.1371/journal.pbio.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A Unified Model for Apical Caspase Activation. Mol Cell. 2003;11(2):529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 12.Wu C-C, Lee S, Malladi S, Chen M-D, Mastrandrea NJ, Zhang Z, et al. The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nat Commun. 2016;7:13565. doi: 10.1038/ncomms13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Zhou M, Hu Q, Bai X-C, Huang W, Scheres SHW, et al. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A. 2017;114(7):1542–7. doi: 10.1073/pnas.1620626114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-Dimensional Structure of the Apoptosome : Implications for Assembly, Procaspase-9 Binding, and Activation. Mol Cell. 2002;9:423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 15.Salvesen G, Dixit V. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A. 1999 Sep;96:10964–7. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase-9. Proc Natl Acad Sci U S A. 2001;98(25):14250–5. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117(7):855–8. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Yuan S, Yu X, Asara JMJ, Heuser JJE, Ludtke SJ, Akey CW. The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure. 2011;19(8):1084–96. doi: 10.1016/j.str.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng TC, Hong C, Akey IV, Yuan S, Akey CW. A near atomic structure of the active human apoptosome. Elife. 2016 Oct;5:1–28. doi: 10.7554/eLife.17755. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB. The Apaf-1-procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer. EMBO J. 2009;28(13):1916–25. doi: 10.1038/emboj.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manns J, Daubrawa M, Driessen S, Paasch F, Hoffmann N, Löffler A, et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: activation of caspase-9 in the absence of Apaf-1. FASEB J. 2011;25(9):3250–61. doi: 10.1096/fj.10-177527. [DOI] [PubMed] [Google Scholar]
- 22.Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Hoyer-Hansen M, Elling F, et al. Apoptosome-Independent Activation of the Lysosomal Cell Death Pathway by Caspase-9. Mol Cell Biol. 2006;26(21):7880–91. doi: 10.1128/MCB.00716-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mille F, Thibert C, Fombonne J, Rama N, Guix C, Hayashi H, et al. The Patched dependence receptor triggers apoptosis through a DRAL–caspase-9 complex. Nat Cell Biol. 2009;11(6):739–46. doi: 10.1038/ncb1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber KL, Hardy JA. Mechanism of zinc-mediated inhibition of caspase-9. Protein Sci. 2012;21(7):1056–65. doi: 10.1002/pro.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target Protease Specificity of the Viral Serpin CrmA: ANALYSIS OF FIVE CASPASES. J Biol Chem. 1997;272(12):7797–800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 26.Stennicke HR, Salvesen GS. Caspases: Preparation and Characterization. Methods. 1999;17(4):313–9. doi: 10.1006/meth.1999.0745. [DOI] [PubMed] [Google Scholar]
- 27.Witkowski WA, Hardy JA. L2′ loop is critical for caspase-7 active site formation. Protein Sci. 2009;18(7):1459–68. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steuber H, Siszler G, Nisa S, Schwarz F, Blasche S, Mo M, et al. The E. coli Effector Protein NleF Is a Caspase Inhibitor. PLoS One. 2013;8(3):e58937. doi: 10.1371/journal.pone.0058937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaidya S, Velázquez-Delgado EM, Abbruzzese G, Hardy JA. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J Mol Biol. 2011;406(1):75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaidya S, Hardy JA. Caspase-6 Latent State Stability Relies on Helical Propensity. Biochemistry. 2011;50(16):3282–7. doi: 10.1021/bi2001664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399(6736):549–57. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y. Mechanisms of Caspase Activation and Inhibition during Apoptosis Caspases are central components of the machinery. Mol Cell. 2002;9:459–70. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 33.Lyskov S, Gray JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36:W233–8. doi: 10.1093/nar/gkn216. Web Server. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serrano BP, Hardy JA. Phosphorylation by Protein Kinase A Disassembles the Caspase-9 Core. Cell Death Differ. 2017;1 doi: 10.1038/s41418-017-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishi H, Fong JH, Chang C, Teichmann SA, Panchenko AR. Regulation of protein–protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol Biosyst. 2013;9(7):1620. doi: 10.1039/c3mb25514j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure. 2011;19(12):1807–15. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishi H, Shaytan A, Panchenko AR. Physicochemical mechanisms of protein regulation by phosphorylation. Front Genet. 2014;5:270. doi: 10.3389/fgene.2014.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin MC, Allan La, Lickrish M, Sampson C, Morrice N, Clarke PR. Protein kinase A regulates caspase-9 activation by Apaf-1 downstream of cytochrome c. J Biol Chem. 2005;280(15):15449–55. doi: 10.1074/jbc.M414325200. [DOI] [PubMed] [Google Scholar]
- 39.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5(7):647–54. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 40.Seifert A, Allan LA, Clarke PR. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008;275(24):6268–80. doi: 10.1111/j.1742-4658.2008.06751.x. [DOI] [PubMed] [Google Scholar]
- 41.Rutter J, Michnoff CH, Harper SM, Gardner KH, McKnight SL. PAS kinase: an evolutionarily conserved PAS domain-regulated serine/threonine kinase. Proc Natl Acad Sci U S A. 2001;98(16):8991–6. doi: 10.1073/pnas.161284798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fatemi M, Hermann A, Pradhan S, Jeltsch A. The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J Mol Biol. 2001;309(5):1189–99. doi: 10.1006/jmbi.2001.4709. [DOI] [PubMed] [Google Scholar]
- 43.Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279(11):10109–19. doi: 10.1074/jbc.M312123200. [DOI] [PubMed] [Google Scholar]
- 44.Twiddy D, Cain K. Caspase-9 cleavage, do you need it? Biochem J. 2007;405(1):e1–2. doi: 10.1042/BJ20070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murray TVA, McMahon JM, Howley BA, Stanley A, Ritter T, Mohr A, et al. A non-apoptotic role for caspase-9 in muscle differentiation. J Cell Sci. 2008;121(Pt 22):3786–93. doi: 10.1242/jcs.024547. [DOI] [PubMed] [Google Scholar]
- 46.Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tinel A, Tschopp J. The PIDDosome, a Protein Complex Implicated in Activation of Caspase-2 in Response to Genotoxic Stress. Science (80-) 2004;304(5672) doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 48.Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M, et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 2012;19(10):1722–32. doi: 10.1038/cdd.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim IR, Murakami K, Chen N-J, Saibil SD, Matysiak-Zablocki E, Elford AR, et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis. 2009;14(9):1039–49. doi: 10.1007/s10495-009-0375-1. [DOI] [PubMed] [Google Scholar]
- 50.Fava LL, Bock FJ, Geley S, Villunger A. Caspase-2 at a glance. J Cell Sci. 2013;125(24) doi: 10.1242/jcs.115105. [DOI] [PubMed] [Google Scholar]
- 51.Clerici M, Luna-Vargas MPA, Faesen AC, Sixma TK. The DUSP–Ubl domain of USP4 enhances its catalytic efficiency by promoting ubiquitin exchange. Nat Commun. 2014;5 doi: 10.1038/ncomms6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, et al. Structural basis of autoregulation of phenylalanine hydroxylase. Nat Struct Biol. 1999;6(5):442–8. doi: 10.1038/8247. [DOI] [PubMed] [Google Scholar]
- 53.Stamogiannos A, Maben Z, Papakyriakou A, Mpakali A, Kokkala P, Georgiadis D, et al. Critical Role of Interdomain Interactions in the Conformational Change and Catalytic Mechanism of Endoplasmic Reticulum Aminopeptidase 1. Biochemistry. 2017;56(10):1546–58. doi: 10.1021/acs.biochem.6b01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meergans T, Hildebrandt A-K, Horak D, Haenisch C, Wendel A. The short prodomain influences caspase-3 activation in HeLa cells. Biochem J. 2000;349:135–40. doi: 10.1042/0264-6021:3490135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denault J-B, Salvesen GS. Human caspase-7 activity and regulation by its N-terminal peptide. J Biol Chem. 2003;278(36):34042–50. doi: 10.1074/jbc.M305110200. [DOI] [PubMed] [Google Scholar]
- 56.Voss OH, Batra S, Kolattukudy SJ, Gonzalez-Mejia ME, Smith JB, Doseff AI. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J Biol Chem. 2007;282(34):25088–99. doi: 10.1074/jbc.M701740200. [DOI] [PubMed] [Google Scholar]
- 57.Klaiman G, Champagne N, LeBlanc AC. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim Biophys Acta - Mol Cell Res. 2009;1793(3):592–601. doi: 10.1016/j.bbamcr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 58.Dagbay KB, Hardy JA. Multiple proteolytic events in caspase-6 self-activation impact conformations of discrete structural regions. Proc Natl Acad Sci U S A. 2017;201704640 doi: 10.1073/pnas.1704640114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boucher D, Blais V, Denault J-B. Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc Natl Acad Sci U S A. 2012;109(15):5669–74. doi: 10.1073/pnas.1200934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martini C, Bédard M, Lavigne P, Denault JB. Characterization of Hsp90 Co-Chaperone p23 Cleavage by Caspase-7 Uncovers a Peptidase-Substrate Interaction Involving Intrinsically Disordered Regions. Biochemistry. 2017;56(38):5099–111. doi: 10.1021/acs.biochem.7b00298. [DOI] [PubMed] [Google Scholar]
- 61.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker John M., editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press Inc; 2005. pp. 571–607. [Google Scholar]
- 62.Micsonai A, Wien F, Kernya L, Lee Y-H, Goto Y, Réfrégiers M, et al. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci U S A. 2015;112(24):E3095–103. doi: 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.