

Abstract
Key points
Na+ conducting hypertonicity‐induced cation channels (HICCs) are key players in the volume restoration of osmotically shrunken cells and, under isotonic conditions, considered as mediators of proliferation – thereby opposing apoptosis.
In an siRNA screen of ion channels and transporters in HepG2 cells, with the regulatory volume increase (RVI) as read‐out, δENaC, TRPM2 and TRPM5 were identified as HICCs.
Subsequently, all permutations of these channels were tested in RVI and patch‐clamp recordings and, at first sight, HICCs were found to operate in an independent mode. However, there was synergy in the siRNA perturbations of HICC currents.
Accordingly, proximity ligation assays showed that δENaC was located in proximity to TRPM2 and TRPM5 suggesting a physical interaction.
Furthermore, δENaC, TRPM2 and TRPM5 were identified as mediators of HepG2 proliferation – their silencing enhanced apoptosis. Our study defines the architecture of HICCs in human hepatocytes as well as their molecular functions.
Abstract
Hypertonicity‐induced cation channels (HICCs) are a substantial element in the regulatory volume increase (RVI) of osmotically shrunken cells. Under isotonic conditions, they are key effectors in the volume gain preceding proliferation; HICC repression, in turn, significantly increases apoptosis rates. Despite these fundamental roles of HICCs in cell physiology, very little is known concerning the actual molecular architecture of these channels. Here, an siRNA screening of putative ion channels and transporters was performed, in HepG2 cells, with the velocity of RVI as the read‐out; in this first run, δENaC, TRPM2 and TRPM5 could be identified as HICCs. In the second run, all permutations of these channels were tested in RVI and patch‐clamp recordings, with special emphasis on the non‐additivity and additivity of siRNAs – which would indicate molecular interactions or independent ways of channel functioning. At first sight, the HICCs in HepG2 cells appeared to operate rather independently. However, a proximity ligation assay revealed that δENaC was located in proximity to both TRPM2 and TRPM5. Furthermore, a clear synergy of HICC current knock‐downs (KDs) was observed. δENaC, TRPM2 and TRPM5 were defined as mediators of HepG2 cell proliferation and their silencing increased the rates of apoptosis. This study provides a molecular characterization of the HICCs in human hepatocytes and of their role in RVI, cell proliferation and apoptosis.
Keywords: volume regulation, cation channels, proliferation
Key points
Na+ conducting hypertonicity‐induced cation channels (HICCs) are key players in the volume restoration of osmotically shrunken cells and, under isotonic conditions, considered as mediators of proliferation – thereby opposing apoptosis.
In an siRNA screen of ion channels and transporters in HepG2 cells, with the regulatory volume increase (RVI) as read‐out, δENaC, TRPM2 and TRPM5 were identified as HICCs.
Subsequently, all permutations of these channels were tested in RVI and patch‐clamp recordings and, at first sight, HICCs were found to operate in an independent mode. However, there was synergy in the siRNA perturbations of HICC currents.
Accordingly, proximity ligation assays showed that δENaC was located in proximity to TRPM2 and TRPM5 suggesting a physical interaction.
Furthermore, δENaC, TRPM2 and TRPM5 were identified as mediators of HepG2 proliferation – their silencing enhanced apoptosis. Our study defines the architecture of HICCs in human hepatocytes as well as their molecular functions.
Introduction
Cell volume and its regulation are employed in a multitude of physiological tasks such as cell locomotion, control of hepatocyte metabolism and, most notably, the guidance of proliferation and apoptosis (Wehner et al. 2003a; Hoffmann et al. 2009; Dubois & Rouzaire‐Dubois, 2012). The primary event in the regenerating liver is an increase of parenchymal cell volume that clearly precedes mitotic tissue repair (Furchtgott et al. 2009; Miyaoka et al. 2012; Miyaoka & Miyajima, 2013), suggesting that cell volume regulation may also participate in hepatic regeneration. In the nervous system, cell volume sensing contributes to the detection of systemic tonicity (Liedtke & Kim, 2005; Bourque, 2008) and to the perception of pain (Liu et al. 2007).
Whenever analysed quantitatively, the regulatory volume increase (RVI) of a shrunken cell has proven to critically depend on the activity of hypertonicity‐induced cation channels (HICCs). Likewise, under isotonic conditions, HICCs were identified as essential mediators of the volume increase that occurs in the proliferation of cells. Inhibition of these channels, on the other hand, led to the induction of apoptosis (Shimizu et al. 2006; Numata et al. 2008; Bondarava et al. 2009), a process that is initiated by the so‐called apoptotic volume decrease (Okada et al. 2001; Wehner et al. 2003a; Hoffmann et al. 2009). The tight coupling of apoptosis and HICCs was explicitly shown in a recent study on HeLa cells where staurosporine‐induced cell death could be countered by (osmotic) channel activation, even at 2 h after intoxication with the compound (Numata et al. 2008).
Despite the paramount importance of HICCs in all these processes, information about their molecular correlate has remained rather sparse – in particular with respect to the liver. In primary human hepatocytes, HICC activity is inhibited by amiloride (Li et al. 2005), which is the classic blocker of the epithelial Na+ channel ENaC (Kellenberger & Schild, 2002; Wehner & Olsen, 2004), as well as by flufemamate. In contrast, the HICC in the HeLa human cervix carcinoma cell line is only sensitive to flufenamate and not blocked by amiloride (Wehner et al. 2003b). In these cells the ΔC splice variant of transient receptor potential cation channel, subfamily M, member 2 (TRPM2) has recently been identified as the molecular correlate of the HICC (Numata et al. 2012). Furthermore, the HICC in rat hepatocytes is related to the ENaC (i.e. responsive to amiloride) (Plettenberg et al. 2008), but is clearly insensitive to flufenamate (F. Wehner, unpublished). Are we actually dealing with just two types of HICCs (belonging to subunits and isoforms of ENaC and TRPM2) working separately in rat liver and HeLa cells, but in a synergistic mode in hepatocytes from humans?
In a recent study from this lab, a role of αENaC in the RVI and HICC activity of human hepatoma HepG2 cells was reported. Furthermore, small interfering RNA (siRNA) silencing of αENaC reduced proliferation whereas apoptosis rates were elevated (Bondarava et al. 2009). Of note, however, in that study just one of the three siRNAs tested led to an actual knock‐down (KD) of αENaC expression – with some 40% of the transcript remaining unaffected. It is also worth mentioning that, in the former study, scanning acoustic microscopy (SAM) was employed to determine absolute cell volumes and, given the rather flat appearance of HepG2 monolayers of some 2 μm, this brings acoustic microscopy to its limits (Plettenberg et al. 2008). Clearly, with flat monolayers like HepG2 the determination of relative cell volume changes by measuring acoustic impedance is the better choice (Christmann et al. 2016). So, one of our aims was to revisit the actual contribution of αENaC to the HICC in HepG2 cells by means of this state‐of‐the‐art approach.
And what about β‐, γ‐ and δENaC in setting up HICCs, and what roles do TRPM2 and its congeners play? TRP channels are known specialists in the sensation of all kinds of physicochemical stresses and stimuli (Nilius & Owsianik, 2011), but given their architecture of 4 × 6 transmembrane (TM) regions, is it conceivable that they actually combine with ENaC subunits (n × 2 TM helices) thereby forming functional HICCs?
We report here that δENaC, TRPM2 and TRPM5 are the mediators of HICC currents in HepG2 cells. Currents were closely linked to the actual process of RVI and, with a gradual knockdown, HICC activity and volume recovery were both fading out. There was a clear additivity of the siRNA effects on δENaC, TRPM2 and TRPM5 currents. While independent signalling of each of these proteins may be the most straightforward interpretation of the results, we provide a different conclusion which is based on alternating interactions. All modules of HICC current and RVI turned out to be mediators of cell proliferation and their silencing enforced apoptosis rates. Modulation of both processes appeared in a coordinated fashion – but with no detectable change in cell cycle distribution. These results characterize the molecular architecture of HICCs in human hepatocytes and their actual contribution to RVI, cell proliferation as well as apoptosis.
Methods
Cell culture
Human hepatoma HepG2 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 μg ml−1 streptomycin, 100 U ml−1 penicillin, 5 ml l‐glutamine (200 mm) and 1% non‐essential amino acids. The osmolarity was adjusted to 300 mosmol l−1 by the addition of mannitol. Cells were incubated at 37°C with 95% air and 5% CO2.
Our HepG2 cells were authenticated by short tandem repeat (STR) analysis and did not contain DNA sequences from mouse, rat or hamster (determined by the Leibniz Institute DSMZ, Braunschweig, Germany). Moreover, STR analysis verified our cell profile to be consistent with the one archived for HepG2 cells. Cells were also regularly tested negative for mycoplasma contamination using the MycoAlert Mycoplasma detection kit (from Lonza, Basel, Switzerland).
Spotting of siRNAs and transfection procedures
For the first siRNA screen with the SAM, Lab‐TekTM chambers (Nunc, Thermo Fisher Scientific, Waltham, MA, USA) were coated with 0.1 mg collagen‐R in 2 ml PBS for 20 min, washed 4 times and air‐dried. For reverse transfections, smartPool siRNAs (Dharmacon, Lafayette, CO, USA) were dissolved in siRNA buffer to give stocks of 20 μm; 3 μl OptiMEM (containing 0.2 m sucrose, O+G, Thermo Fisher Scientific), 3.5 μl Lipofectamine 2000 (Thermo Fisher Scientific) and 1.4 μl of the respective siRNA solution were added to the wells of a 384 well low‐volume plate. After 20 min incubation at room temperature, probes were supplemented with 7.5 μl of a 0.2% (w/v) gelatin solution containing 0.01% (v/v) fibronectin and mixed thoroughly (Erfle et al. 2007). Spotting was performed on a QArray2 robot (Genetix) with a pin diameter of 165 μm, and the centre of the spots was 350 μm apart. Cells were seeded at a density of 0.5 × 106 ml−1 and the SAM measurements were performed at 3 days thereafter (see below). Transfection rates were routinely checked (using Alexa‐488 constructs plus 4′,6‐diamidino‐2‐phenylindole (DAPI) as the nucleus reference point; see ‘Cell profiling’) and equalled 70.8 ± 9.5% (mean ± SD). The SAM recordings were always done in the very centre of each spot and we never observed any locomotion of cells that may interfere with our analysis, possibly because of the use of fibronectin in the spotting solution (Fengler et al. 2012). To minimize diffusional artefacts, the position of (typically) seven different siRNAs (in quadruplicate on a single run) was randomly permutated.
For the second RVI screen with the SAM, standard transfection protocols were employed. Single siRNA constructs directed against δENaC, TRPM2 and/or TRPM5 (plus non‐targeting control siRNA) were used as such or in duplicate keeping a total concentration of 25 nm constant. For the triple transfection, however, siRNA concentration had to be elevated to 37.5 nm. Lipofectamine 2000 (1 : 150) served as transfection reagent. Transfection rates (see ‘Cell profiling’) were 79.1 ± 12.0% (mean ± SD). Target sequences were 5′‐CACACTTGGGCTGCTCTGAAA‐3′ (δENaC), 5′‐AAAGACGGAGTTCCTGATCTA‐3′ (TRPM2), 5′‐CTGATCCATATCTTTGCCATA‐3′ (TRPM5). All target sequences were blasted for specificity. No off‐target effects on other ion channels were predicted.
As was determined by qRT‐PCR (see below), siRNA constructs reduced the amount of δENaC, TRPM2 and TRPM5 expression to 21.5 ± 0.1%, 36.9 ± 0.1% and virtually zero, respectively (n = 3).
For the patch‐clamp recordings, total siRNA concentrations were 10 nm except for the triple transfection (15 nm). Constructs were labelled with Alexa 488 and/or Alexa 546, except for the triple siRNAs where labelling and non‐labelling were randomly permutated. HiPerFect (1 : 200, Qiagen, Hilden, Germany) was used as the transfection reagent here yielding clearly higher rates of seal formation than with Lipofectamine. HiPerFect was also instrumental in the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT) assay, significantly improving basal survival rates.
Cell profiling
Cell transfection rates were determined by using the freeware CellProfiler 2.0 (Kamentsky et al. 2011) with DAPI staining of nuclei and with siRNAs identified by their respective fluorophores.
Quantitative RT‐PCR
One microgram of isolated RNA was reverse transcribed using the HighCapacity Reverse Transcription kit (Thermo Fisher Scientific) according to manufacturer's instructions. Commercially available TaqMan assays (Thermo Fisher Scientific) were used for the detection of δENaC (Hs00161595_m1), TRPM2 (Hs01066091_m1), TRPM5 (Hs00175822_m1) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (Hs02786624_g1) as a housekeeping gene. Because expression levels of δENaC, TRPM2 and TRPM5 were too low to be detectable using the standard qPCR protocol, the cDNA was preamplified using the TaqMan PreAmp Kit (Thermo Fisher Scientific) according to manufacturer's instructions. Briefly 250 ng of cDNA was mixed with a pool of 0.2× TaqMan assays and PreAmp Mastermix and preamplified for 14 cycles. The product was diluted 1 : 20 and 5 μl of the diluted product was subjected to the qPCR reaction. The resulting data were analysed using the ΔΔC t method.
RT‐PCR
Isolated RNA was reverse transcribed using the ProtoScript II First Strand cDNA Synthesis kit (New England Biolabs, Ipswich, MA, USA) according to manufacturer's recommendations. The cDNA was subjected to PCR using the Q5 PCR kit (New England Biolabs) following the manufacturer's instructions. The specific primers listed in Table 1 were used. The PCR products were run on agarose gels and visualized using RedSafe DNA dye (iNtRON Biotechnology, Seongnam, South Korea).
Table 1.
Primers used for RT‐PCR
| Name | Sequence |
|---|---|
| hαENaC fwd | CTGCACCTGTCAGGGGAACA |
| hαENaC rev | GTCTTCATGCGGTTGTGCTG |
| hβENaC fwd | CCTGGAACTGAATTCGGCCT |
| hβENaC rev | CTTGGAAGCAGGAGCGAAGA |
| hγENaC fwd | GGTTTCGGAGAAGTGGTTGC |
| hγENaC rev | TACGGGGAGCTTCTGGACAT |
| hδENaC fwd | GGCATCAGGGTCATGGTTCA |
| hδENaC rev | GTAGAAGCAGTGTCCCCAGG |
| hTRPM2 fwd | GGCAGTGGAAGCCTTCAGAT |
| hTRPM2 rev | GATAAAGCGGCTGCGTGAAG |
| hTRPM5 fwd | GAGCACCTGGAGAGAGACCT |
| hTRPM5 rev | AAACCACCTCTGTGGTCAGC |
Scanning acoustic microscopy
Changes of HepG2 cell volumes were determined with the acoustic impedance (Z a) as reciprocal readout, with cell shrinkage causing an increase and cell swelling a decrease of Z a. This was done by means of a SAM unit (Fraunhofer IBMT, St. Ingbert, Germany) mounted on the stage of an inverted microscope (IX81, Olympus, Hamburg, Germany). One‐gigahertz sound waves were focused with a sapphire lens to the cell monolayer and reflected from the glass bottom of the experimental chamber. The same device was used for detection of the echo, which directly depends on Z a. Measurements were performed in a scanning mode of 15 × 15 pixels set to be 4 μm apart and covering a total area of 60 × 60 μm2. A single scan was complete in less than 5 s.
It is noteworthy that the determination of absolute cell volumes with SAM is only feasible if cell layers exhibit a minimum height of some 3–4 μm as is true for primary cultures of rat hepatocytes (Plettenberg et al. 2008). With cells growing at significantly lower heights, as for instance HepG2 cells, determination of absolute cell volumes comes to its limits (Bondarava et al. 2009). A way out is the measurement of the acoustic impedance of cell layers yielding relative cell volumes at high spatial and temporal resolution (Christmann et al. 2016).
Cells were grown in collagen‐coated Lab‐TekTM chambers (Thermo Fisher Scientific) and continuously superfused at 5.0 ml min−1 with piezo‐driven micro‐pumps (Bartels Microtechnique, Dortmund, Germany). Temperature control (37°C) was achieved with a custom‐made Perspex box which enclosed the complete set‐up and was air‐fed by a heating device; in addition, superfusion lines were heated with Peltier elements.
Experimental solutions (of pH 7.4) contained (in mm): NaCl, 144.0; KCl, 2.7; MgCl2, 1.0; CaCl2, 1.8; Hepes, 2.5; Na‐Hepes, 2.5; NaH2PO4, 0.4; glucose, 5.5. Osmolarity was adjusted to 300 (isotonic) and 400 mosmol l−1 (hypertonic) by addition of mannitol.
Patch‐clamp recordings
Patch‐clamp experiments were performed on HepG2 cells that were mechanically detached from culture dishes with a jet‐stream of culture solution, kept in suspension for maximally 2 h thereafter, and finally transferred to the experimental chamber where they seeded for a minimum of 30 min before the actual experiment started.
Membrane currents were recorded in the fast whole‐cell mode of the patch‐clamp technique as described previously (Bondarava et al. 2009). Briefly, 3.5–4.5 MΩ borosilicate pipettes were manufactured from 1.5 mm o.d. × 1.17 mm i.d. capillaries (Harvard Apparatus, Edenbridge, UK) on a programmable multi‐stage puller (DMZ‐Universal Puller; Zeitz‐Instrumente, Munich, Germany). Currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) and the pCLAMP 10.3 software (Molecular Devices) was used for control of the voltage‐protocol, data acquisition and analysis. Series resistances were < 5 MΩ and compensated for. The holding voltage was −30 mV; voltage ramps from −80 to +20 mV of 1 s duration were applied every 10 s. HICC currents (at −30 mV) were referred to membrane capacitance (as determined on the amplifier) yielding current densities in pA pF−1.
The pipette solution (pH 7.3, 300 mosmol l−1) contained (in mm): NaCl, 25.0; NaHCO3 −, 8.0; sodium gluconate, 62.0; MgCl2, 1.0; TEA‐Cl, 2.0; Na2‐ATP, 2.0; Na2‐GTP, 0.5; EGTA, 1.0. The bath solution (pH 7.5) contained (in mm): NaCl, 75.0; NaHCO3 −, 25.0; MgCl2, 1.0; CaCl2, 2.0; TEA‐Cl, 2.0. Osmolarity was adjusted to 320 (isotonic) and 400 mosmol l−1 (hypertonic). With the above ion gradients, the reversal potentials of Na+ and Cl− (plus HCO3 −, as the only permeant ions) are set to 0 and −30 mV, respectively. Solutions were gassed with mixtures of CO2 and O2 to obtain the appropriate HCO3 − buffering of pH values.
Superfusion of cells was achieved by gravity. A thermostat (Intelligent Chiller Control; Huber, Offenburg, Germany) was used for the temperature control of the storage vessels and perfusion lines and a Peltier device (Badcontroller V; Luigs & Neumann, Ratingen, Germany) for heating of the experimental chamber. Recordings were performed at 35°C.
Pull‐downs and mass spectrometry
Pull‐down experiments (for subsequent mass spectrometry (MS) analysis) were prepared as follows. Cells were stably transfected with TRPM2‐, TRPM5‐, αENaC‐, or δENaC‐mCitrine fusion proteins. Transfected cells (1 × 106) were plated on 10 cm dishes and cultured for 2 days. For a better discovery of interacting proteins, cells were incubated with both a cell‐permeant (bis(sulfosuccinimidyl)suberate; BS3) and a cell‐impermeant (disuccinimidyl suberate; DSS) protein cross‐linker (both Thermo Fisher Scientific) at a final concentration of 5 mm (for 30 min at room temperature). Thereafter, cells were harvested and lysed using the recommended buffers for the subsequent green fluorescent protein (GFP)‐Trap usage (ChromoTek, Planegg‐Martinsried, Germany). Cells were resuspended in RIPA lysis‐buffer containing protease inhibitors; lysates were then sonicated and cleared by centrifugation. After determination of protein concentration, lysates were incubated with GFP‐Trap beads for 1 h with end‐over‐end mixing at 4°C. After three washing steps, the proteins bound to beads were subjected to tryptic digestion: they were first incubated for 1 h at room temperature with 5 μg ml−1 trypsin and then iodoacetamide was added for carbamidomethylation. Supernatants were collected and incubated overnight at 37°C under shaking. Peptides were then desalted and purified using StageTips (according to Rappsilber et al. 2007).
For protein identification, tryptic peptides were separated and analysed by nano‐HPLC–tandem mass spectrometry (MS/MS) (Vendrell‐Navarro et al. 2015) using an UltiMateTM 3000 RSLCnano system and a Q Exactive™ Hybrid Quadrupole‐Orbitrap mass spectrometer equipped with a nano‐spray flex ion source (Thermo Fisher Scientific). All solvents were of LC‐MS grade. The lyophilized tryptic peptides were dissolved in 20 μl 0.1% trifluoroacetic acid in water. 3 μl of sample were injected onto a pre‐column cartridge (5 μm, 100 Å, 300 μm i.d. × 5 mm; Dionex, Germany) using 0.1% trifluoroacetic acid in water as eluent with a flow rate of 30 μl min−1. Desalting was performed for 5 min with eluent flow to waste followed by back‐flushing of the sample during the whole analysis from the pre‐column to a PepMap100 RSLC C18 nano‐HPLC column (2 μm, 100 Å, 75 μm i.d. × 25 cm; nanoViper, Dionex) using a linear gradient starting with 95% solvent A (0.1% formic acid in water) vs. 5% solvent B (0.1% formic acid in acetonitrile) and changing to 70% solvent A vs. 30% solvent B after 95 min; the flow rate was 300 nl min−1.
The nano‐HPLC was online coupled to the Quadrupole‐Orbitrap Mass Spectrometer using a standard coated SilicaTip (i.d. 20 μm, tip i.d. 10 μm; New Objective, Woburn, MA, USA). A mass range of m/z 300–1650 was acquired with a resolution of 70,000 for full scan, followed by up to 10 high energy collision dissociation (HCD) MS/MS scans of the most intense at least double charged ions.
Data evaluation was performed using the MaxQuant software (v.1.5.2.8; Cox & Mann, 2008) including the Andromeda search algorithm and searching in the human reference proteome of the UniProt database. Briefly, the search was performed for full enzymatic trypsin cleavages allowing two miscleavages. For protein modifications, carbamidomethylation was chosen as the fixed and oxidation of methionine and acetylation of the N‐terminus as the variable modification. The mass accuracy for full mass spectra was set to 5 ppm and for MS/MS spectra to 20 ppm. The false discovery rates for peptide and protein identification were set to 1%. Only proteins for which at least two peptides were quantified were chosen for further validation. Relative quantification of proteins was carried out using the label‐free algorithm implemented in the MaxQuant software. Samples resulting from affinity enrichments with the active molecule bound to the solid support were grouped together, and also those resulting from a similar enrichment using control molecules. All experiments were done in technical triplicates. Label‐free quantification intensities were logarithmized (log2) and proteins which were not three times quantified in at least one of the groups were filtered off. Missing values were imputed using small normally distributed values. Proteins which were statistically significant outliers were considered as hits.
Proximity ligation assay
Proximity ligation assays (PLAs) using the Duolink‐kit (Sigma‐Aldrich, St. Louis, MO, USA) were prepared as instructed by the manufacturer. Briefly, HepG2 cells were cultured on 8‐well Lab‐Tek chamber slides and left to adhere for 48 h before they were fixed using 4% paraformaldehyde in PBS. Cells were washed and then permeabilized with 0.1% Triton X‐100 in PBS. Subsequent to the blocking with the Duolink blocking buffer, primary antibodies were incubated at 4°C overnight. After washing away unbound primary antibodies, proximity probes were incubated at 37°C for 1 h. Ligation and amplification followed and images were taken using a Zeiss 510 laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
MTT and caspase 3/7 assays
For the MTT assay, HepG2 cells were cultured in plastic 96‐well plates in 110 μl of growth medium and at an initial density of 15,000 cells per well. Four hours later, 0.3 μl of HiPerFect (1 : 400) was added plus the respective siRNAs set to 10 nm in total (except 15 nm, for the triple transfection); 48 h thereafter, 24 μl of 5 mg ml−1 MTT was added, which is quantitatively transformed by viable cells into the green‐fluorescent formazan. After 2 h, cells were lysed with isopropanol–HCl (180 μl of 0.05 M HCl in 100% isopropanol) and optical densities determined at 570 nm on a Multiskan Ascent plate‐reader (Thermo Fisher Scientific); four replicate wells were recorded for each condition. Transfection rates were 97.3 ± 1.1% (mean ± SD).
After 48 h of incubation, culture media (plus siRNAs and transfection reagent) were removed and the Apo‐ONETM Homogeneous Caspase 3/7 Assay (Promega, Madison, WI, USA) conducted following the manufacturer's protocol. Fluorescence was determined with an excitation/emission of 485/538 on a Fluoroskan Ascent plate reader (Thermo Fisher Scientific; Bondarava et al. 2009).
Cell culture impedance recordings
Further evidence for the role of HICCs in cell proliferation was obtained by real time cell analysis (RTCA) measurements on a Dual Plate xCELLigence Instrument (Roche Applied Science, Indianapolis, IN, USA) in which the electric impedance of monolayers is monitored quasi continuously over time (cf. Papke et al. 2016; Martin‐Gago et al. 2017). The readout is a dimensionless parameter referred to as the cell index which is proportional to the number of cells cultured (in so‐called 16‐well E‐plates). Transfection of cells was 4 h after seeding and the linear range between 30 and 50 h was taken as the actual readout of proliferation (Fig. 6 A). E‐plates were arranged in humidified incubators at 37°C and with 5% CO2.
Figure 6. Effects of HICC silencing on HepG2 cell growth and apoptosis.
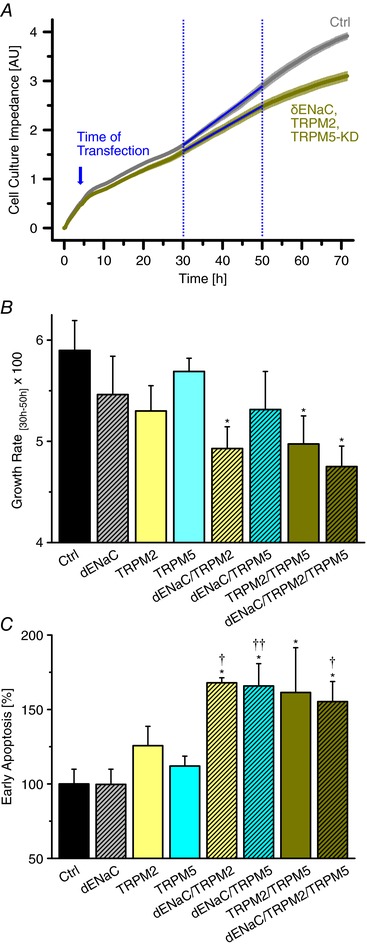
A, typical recordings of cell culture impedance under control conditions and for the triple KD. For quantification of cell proliferation, the quasi linear range of 30–50 h was employed. B, channel silencing inhibits HepG2 cell proliferation as determined with the impedance recordings. * refers to the control siRNA (n = 6). C, effects of HICC silencing on HepG2 early apoptosis determined by FACS (n = 4–6). * and ** refer to the control siRNA, † and †† to δENaC.
Fluorescence‐activated cell sorting analysis of early apoptosis
All fluorescence‐activated cell sorting (FACS) experiments were done on a LSR II flow cytometer (BD Biosciences, San Jose, CA, USA). HepG2 cells were grown in six‐well plates at a density of 2 × 105 per well and transfected at 4 h after seeding; experiments were performed at day 2 thereafter. Annexin V–fluorescein isothiocyanate (FITC; apoptosis marker) and 7‐aminoactinomycin D (7‐AAD; a DNA stain) were used to differentiate between early and late apoptosis as well as necrosis. The excitation wavelength was 488 nm and emission filter sets of 530/30 nm (Annexin V–FITC) and 695/40 nm (7‐AAD) were used for measurements; recordings were gated by the FACSDivaTM software.
FACS analysis of cell cycle distribution
For the cell cycle assay, cells were fixed and permeabilized for 2 h with 70% ethanol at 4°C. DNA staining was done with propidium iodide solution (PI; 12 μg ml−1) also containing 0.1% Triton X‐100. Excitation wavelength was 488 nm and PI fluorescence was determined with a 576/26 nm filter. Analysis was performed with the program FlowJo® (FloJo LLC, Ashland, OR, USA) fitting Gaussians to the G1/G0 and the G2/M phases of the cell cycle.
Statistics
Data are presented as mean values ± SEMs (except for the first siRNA screen where mean values ± SDs are given). n denotes the number of observations and P values ˂ 0.05 were considered significant. For the first siRNA screen, a one‐way ANOVA with Bonferoni post hoc test was used. For all other comparisons, a least significant difference (LSD) post hoc test was applied. One, two and three symbols stand for P ≤ 0.05, 0.01 and 0.001, respectively. All error bars were derived from independent biological experiments.
Results
siRNA screening of various channels and transporters as potential motors of the RVI in HepG2 cells
In a first screen, 34 siRNA pools directed against α‐, β‐, γ‐ and δENaC, NHE1, NKCC1, Piezo1 and ‐2, as well as most TRPs were used to test the contribution of the corresponding gene products to the RVI process. Changes in cell volume were determined by means of SAM (Weiss et al. 2007; Bondarava et al. 2009; Christmann et al. 2016) and, in a typical measurement, four repeats of seven different siRNAs were spotted onto the glass bottom of the experimental chamber (together with the transfection reagent; see ‘Methods’) and, 3 days after reverse transfection, tested in a single run (Fig. 1 A). Osmolarity was step‐wise increased from 300 to 400 mosmol l−1 and the resultant changes in HepG2 cell volumes were monitored with the acoustic impedance Z a as the (reciprocal) read‐out (Fig. 1 Ba). As shown in Fig. 1 Bb and c, hypertonic stress led to a rapid increase of Z a (cell shrinkage) followed by a slow return towards control values (representing the RVI). We considered the time course of the latter process as the most reliable measure of channel and/or transporter activities because it does not depend on RVI amplitudes (and thus on the actual expression levels of a putative volume regulator) and it will not be limited by experimental time frames, even in the case of a strong inhibition of a volume effector. In all instances, RVIs followed single exponentials (as exemplified in Fig. 1 Bb and c) and the τ values obtained from data fitting are referenced to the respective siRNA pools in Fig. 1 Ba. Silencing of Na+/H+ exchange (NHE1) and Na+–K+–2Cl− symport (NKCC1) had no effect on RVI at all, which is in accordance with an earlier transport study from our laboratory (Wehner et al. 2002). Similar results were obtained for Piezo1 and ‐2, both of which are ubiquitously expressed ion channels with a clear mechano‐sensitivity (Bagriantsev et al. 2014). From the series of TRPs, only TRPM2 and TRPM5 exhibited a significant increase of τ values (and both channels were in fact detectable in HepG2 cells; Fig. 2). Notably, the ΔC splice‐variant of TRPM2 was recently identified as the HICC in the human cervix carcinoma cell line HeLa (Numata et al. 2012). TRPM5, on the other hand, is a Ca2+‐activated non‐selective cation channel that per se is Ca2+ impermeable and that is employed in taste reception, insulin release and nasal chemoreception (Nilius & Owsianik, 2011). There has been no report so far on a role of TRPM5 in cell volume regulation or mechano‐reception although its possible contribution to the latter has recently been proposed (Liu & Montell, 2015). Concerning the ENaC group, only subunits α and δ are expressed in HepG2 cells (Fig. 2). Accordingly, β‐ and γENaC siRNAs did not show any effect on the velocity of RVI (Fig. 1 Ba) at all. The siRNA pool designed for δENaC, however, increased the τ value of RVI significantly and to a similar extent as those for TRPM2 and TRPM5. There was also a trend towards higher τ values for αENaC, which, however, remained non‐significant. We reported a role of this ENaC subunit in HepG2 RVI, HICC currents and cell proliferation earlier (Bondarava et al. 2009) – but there only one out of three siRNAs did actually lead to a significant reduction of αENaC expression. So, we did not include αENaC in our analysis here (see ‘Discussion’).
Figure 1. Screening of siRNAs with scanning acoustic microscopy.
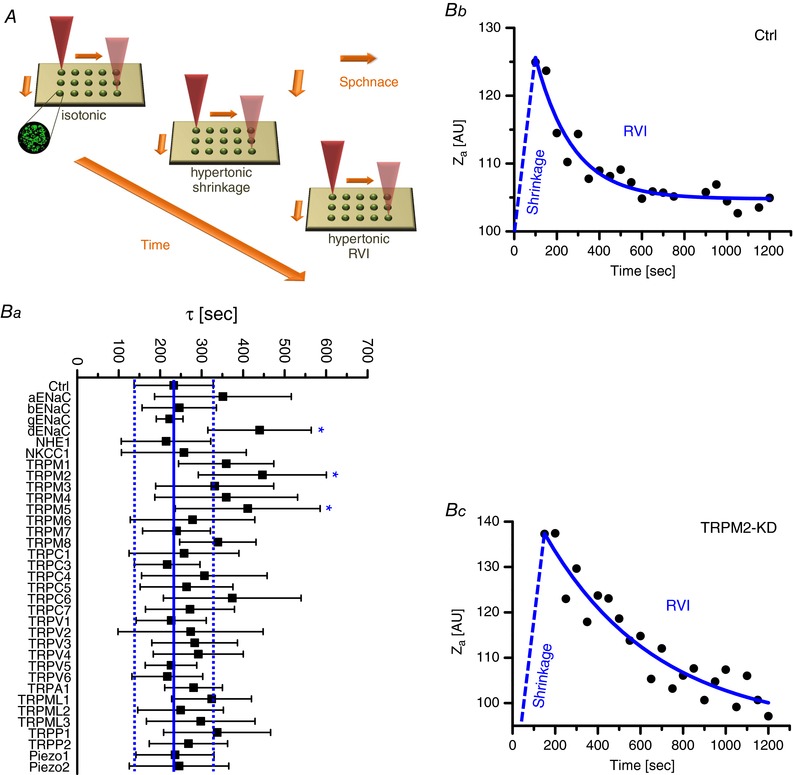
A, HepG2 cells were grown on spots of various (fluorescence labelled) siRNAs plus transfection reagent so that, in the confluent monolayers formed, 70.8 ± 9.5% (mean ± SD) incorporated the respective constructs. Typically, cell volume recordings on 28 spots of siRNAs were complete within 30 s; hence, the effects of hypertonic stress could be determined with a resolution of 40 s or better. Ba, the velocity of RVI was visualized with the acoustic impedance Z a as the (reciprocal) readout of cell volumes; as exemplified in Bb and c, RVIs occurred as single exponentials with defined time constants (τ). Dashed lines in Ba represent the range of ±1.0 σ from the mean τ value of 232.4 s, as determined for control siRNA. Note that only δENaC, TRPM2 and TRPM5 yielded τ values that were significantly different from control, in this first screen (n = 6–21).
Figure 2. RT‐PCR on the proposed HICC elements.
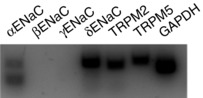
The expression of αENaC, βENaC, γENaC, δENaC, TRPM2 and TRPM5 were tested in untransfected HepG2 cells, with GAPDH as the internal reference.
In Fig. 2, a double band for αENaC was obtained. Size analysis suggests that the upper band is the main αENaC transcript. The identity of the lower band is unknown. According to in silico analysis, however, it is unlikely to be an unspecific amplification product. Primer Blast revealed no PCR product remotely similar in size. A possible explanation could be the existence of an alternatively spliced mRNA. To experimentally exclude unspecific primer binding we performed a RT‐PCR in an unrelated cell line (HeLa) and indeed could only observe one single band for αENaC there (data not shown).
δENaC, TRPM2 and TRPM5: their actual contribution to RVI and HICCs
In the second screen, single siRNAs were employed to silence δENaC, TRPM2 and TRPM5. Again τ‐values were computed from the SAM data to quantify RVI velocity; single, double and triple KDs were performed and all possible permutations were tested. The rationale behind this approach was to check for additivity vs. non‐additivity or, in other words, whether the proposed elements of the HICC(s) act independently (i.e. as individual channels) or if they are actually functioning in a coordinated mode setting up (one or more) ion channels, in a joint fashion. As shown in Fig. 3 A, KD of δENaC, TRPM2 and TRPM5 decreased RVI velocity about 1.4‐fold. Furthermore, the silencing of δENaC, TRPM2 and TRPM5 was clearly additive, in the double KDs. For the triple KD, τ‐values saturated in the range of 550–600 s (Fig. 3 A).
Figure 3. Silencing of ion channels inhibits RVI velocity and HICC currents with a similar pattern.
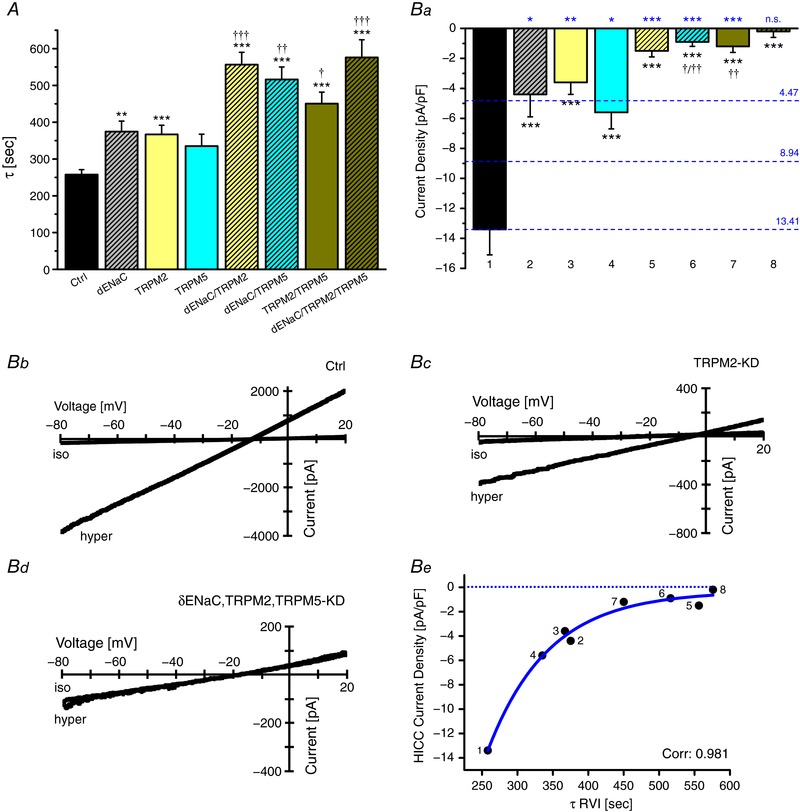
A, time constants of RVI as they were determined by SAM (n = 15–27 for each experimental condition). *, ** and *** refer to the control siRNA, †, †† and ††† to the respective single KDs. Note that the silencing of δENaC, TRPM2 and TRPM5 were additive for the double KDs. Ba, the siRNA inhibition of HICC currents did not differ much on the single KD level, but were again additive for the double KDs of δENaC, TRPM2 and TRPM5. *** refers to the control siRNA, † and †† to the single KDs (n = 6–13 for each condition). Numbers 1–8 at the bottom correspond to the numbers shown in panel Be, for orientation. Dashed blue lines and numbers on the right give the hypothetical current levels of no knockdown (3 active channels), single knockdown (2 active channels) and double knockdown (1 active channel) assuming equal contributions to the overall current. Statistics at the top of the plot (again in blue) refer to these current levels. Note that for the single and double KDs, silencing exceeded simple additivity whereas with the triple KD, currents were reduced to virtually null as expected. Bb–d exemplify current–voltage relations under control conditions, with the TRPM2 KD and with the triple KD, respectively. Be correlates current densities with the τ values of RVI; data were fitted to a single exponential.
Since we are trying to define the integral parts of HICCs as they are employed in RVI (and cell proliferation), a quantification of membrane currents was mandatory. As shown in Fig. 3 B, KD of δENaC, TRPM2 or TRPM5 reduced HICC current density by two‐thirds of the control level. In the double KDs, the effects on δENaC, TRPM2 and TRPM5 silencing were clearly additive, again implying an independent mode of action. With the triple KD, currents were then virtually zero. Accordingly, no further element of conductive Na+ import needs to be assumed for the RVI of HepG2 cells. This also becomes obvious from a plot of HICC current densities vs. the τ of RVIs yielding a correlation coefficient as high as 0.981 (Fig. 3 Be). Together with the SAM recordings outlined above, the patch‐clamp data prove that δENaC, TRPM2 and TRPM5 form (or contribute to) at least three independent HICCs in HepG2 cells that are working in a parallel fashion. Of note, however, in the single and double KDs currents were significantly smaller than expected from a mere one‐to‐one ratio of additivity (Fig. 3 B and ‘Discussion’).
Pull‐downs, mass spectrometry and a proximity ligation assay on HICC architecture
While the additivity observed in the RVI and current density experiments implies that these proteins function independently of each other, an alternative explanation might be that they contribute to different molecular complexes. In this explanation, δENaC would communicate with TRPM2 as well as TRPM5 forming two independent HICCs. Furthermore, a fourth protein would have to interact with TRPM2 and TRPM5 to form two additional channel complexes in order to explain the functional data. Although we did not obtain functional evidence here that αENaC is involved in HICC organization, we proposed that this channel might be the protein contributing to the two putative additional complexes.
In order to test this hypothesis we performed pull‐down experiments with subsequent mass spectrometry analysis on αENaC, δENaC, TRPM2 and TRPM5. A combination of intra‐ and extracellular protein cross‐linkers was employed – so that even weak protein–protein interactions may not be overlooked. As a matter of fact, however, no direct physical correlation between the four HICC elements became detectable with this assay (see Supporting information, Table S1). Nevertheless, αENaC, δENaC, TRPM2 and TRPM5 exhibited a distinct interrelation with one particular membrane protein, namely the voltage‐dependent anion channel VDAC1. Moreover, VDAC2 appeared as a putative partner of αENaC, δENaC and TRPM5 (Table S1). In principle, a coupling of cation to anion transport could facilitate a channel‐mediated import of osmolytes considerably. Furthermore, VDAC1 and VDAC2 may actually function as the core structure of a multi‐channel ion transport complex.
Silencing of VDAC1 and VDAC2, however, had no effect on HICC currents at all, which were −12.4 ± 0.8 pA pF−1 (n = 9) and −13.1 ± 1.1 pA pF−1 (n = 6). This is virtually identical to the control conditions, namely −13.4 ± 1.7 pA pF−1 (n = 10; see Fig. 3 B).
The mass spectrometry experiment supports the hypothesis that all four proteins act independently of each other. However, since membrane proteins are exceptionally difficult to detect with the technique and the fact that these channels are expressed at low level in plasma membranes (see Discussion), we turned to a different method. Given its high specificity and its remarkable amplification rates (Soderberg et al. 2006), the proximity ligation assay (PLA) may be a better choice for a reliable detection of putative HICC channel interactions. And indeed, as the alternative theory suggests, we saw proximity between δENaC and TRPM2 as well as TRPM5. For the sake of integrity, we included αENaC in the analysis and found a similar proximity to TRPM2 and TRPM5. Of note in this respect, a proximity between αENaC and δENaC or TRPM2 and TRPM5 could not be observed (Fig. 4). Proximity detected by PLA means that the proteins cannot be further apart than 40 nm (Koos et al. 2014) indicative of an interaction, which does not necessarily have to be functional. But together with the additivity observed in the HICC and RVI results (Fig. 3 A and B) one could conclude that δENaC–TRPM2 and δENaC–TRPM5 in fact form two independent HICC complexes in HepG2 cells. For αENaC–TRPM2 and αENaC–TRPM5, on the other hand, functional data are not accessible yet.
Figure 4. A proximity ligation assay (PLA) reveals four types of HICC complexes in HepG2 cells.
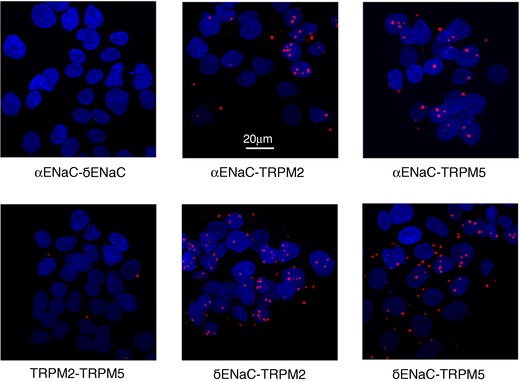
PLA signals shown as red dots were obtained for the various combinations of HICC elements as indicated. The blue areas are cell nuclei stained with DAPI. Data shown are representative of a total of 5 each. Note the close correlation of αENaC or δENaC with either TRPM2 or TRPM5 represented by the red dots of the PLA signal. In contrast, no proximity was detected for αENaC/δENaC and TRPM2/TRPM5.
The role of HICC elements in HepG2 cell proliferation and apoptosis
The contribution of HICCs to cell proliferation is well documented and generally interpreted in terms of their being effectors of the volume gain as it is supposed to precede cell division (Wehner et al. 2003a; Shimizu et al. 2006; Bondarava et al. 2009; Hoffmann et al. 2009; Dubois & Rouzaire‐Dubois, 2012; Numata et al. 2012). We propose a more sophisticated input of ion channels here, so that the availability of HICCs actually sets the checkpoint for a proper mitosis to occur. Following our hypothesis, knocking down one element of the different HICCs (single KD) should result in the shutting down of multiple channel complexes, which should translate in a stronger than expected reduction of current. This hypothesis is supported by the patch‐clamp experiments (see above) where single and double KDs decreased currents in a much higher than just additive way. So, disturbing the system with a single KD led to a current reduction down to 35%, on average; in the double KDs, an inhibition by some 90% was achieved (Fig. 3 B; also see below).
Figure 5 A shows the effects of δENaC, TRPM2 and TRPM5 silencing on the proliferation of HepG2 cells as determined with the MTT assay. There was a progressive decline of HepG2 cell mitosis rates – from 434 ± 70% (mean ± SD) per 48 h (Bondarava et al. 2009), with the control siRNA set at 100.0 ± 1.1% – when going from single to triple KDs, with a maximal inhibition down to 65.8 ± 2.5% achieved for the latter condition. Transfection rates were 97.3 ± 1.1% (mean ± SD) in these experiments.
Figure 5. Silencing of HICC elements inhibits proliferation and induces apoptosis in a cooperative mode.
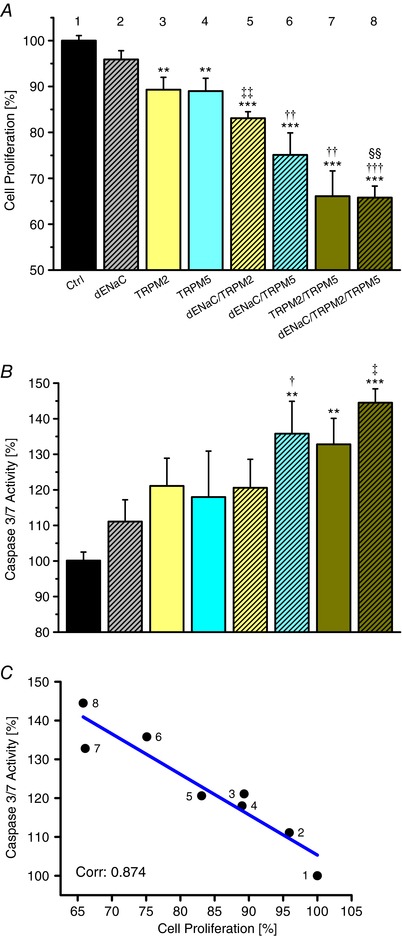
A, effects of channel silencing on cell proliferation determined with the MTT assay. ** and *** refer to the control siRNA, ‡‡ to the single KD of δENaC, †† and ††† to the respective single KDs and §§ to δENaC/TRPM2 (n = 10 to 15). B, effects of HICC silencing on HepG2 apoptosis determined with the caspase 3/7 assay. ** and *** refer to the control conditions, † to δENaC, ‡ to the respective single KDs; there was also a trend to increased caspase activity for the TRPM2 KD (P = 0.06 with reference to control; n = 9–14). As shown in C, there is a clear negative correlation between apoptosis and cell proliferation (with a slope of −1.04 for the linear regression line). Numbering of points in C corresponds to the numbers shown above the bars in A.
The effect of HICC channel silencing on the apoptosis rates in HepG2 cells is depicted in Fig. 5 B. Starting from 100.0 ± 2.4% with the control siRNA, caspase 3/7 activity steadily increased to a final value of 144.5 ± 3.9% for the triple KD. As shown in Fig. 5 C, there was a significant negative correlation between HepG2 apoptosis and cell proliferation over the entire experimental frame with a correlation coefficient as high as 0.874. This proves the distinct interrelation of both processes as well as the key function that the identified HICC elements actually do play in them.
To obtain further evidence for this interrelation, HepG2 cell proliferation was determined with the electrical impedance of monolayers as a read‐out (Fig. 6 A and B) and the amount of early apoptotic cells was visualized by means of a FACS analysis (Fig. 6 C). These approaches strongly supported the data obtained with the MTT and caspase 3/7 assays, with a clear dependence on the actual degree of HICC element silencing.
Given the obvious effects of HICC channel silencing on both proliferation and apoptosis, changes in HepG2 cell‐cycle distribution as the underlying source would not be unexpected. As determined by FACS analysis, however, there was no impact of siRNA silencing detectable for any of the single, double or triple KD protocols used in the present study (Fig. 7 A and B; Table 2).
Figure 7. Silencing of HICCs has no effects on HepG2 cell‐cycle distribution.
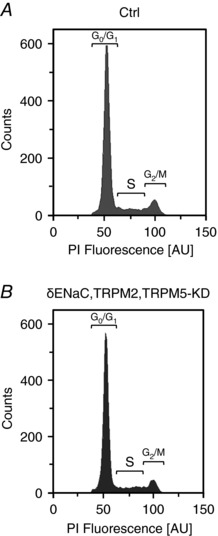
Typical data from FACS under control conditions (A) and for the triple KD (B) are shown (representative of a total of 3). The complete series of siRNA experiments on cell cycles is summarized in Table 2.
Table 2.
HepG2 cell‐cycle analysis
| Percentage of cells | |||
|---|---|---|---|
| siRNA transfection | G0/G1 phase | S phase | G2/M phase |
| Ctrl | 59.58 ± 0.63 | 7.88 ± 0.30 | 9.03 ± 0.39 |
| δENaC | 59.20 ± 0.90 | 7.80 ± 0.05 | 9.95 ± 0.35 |
| TRPM2 | 59.20 ± 0.30 | 7.95 ± 0.15 | 9.60 ± 0.10 |
| TRPM5 | 62.20 ± 0.50 | 7.15 ± 0.15 | 8.05 ± 0.05 |
| δENaC/TRPM2 | 57.90 ± 1.00 | 7.45 ± 0.65 | 9.10 ± 0.10 |
| δENaC/TRPM5 | 60.30 ± 0.30 | 7.20 ± 0.10 | 7.15 ± 0.05 |
| TRPM2/TRPM5 | 60.45 ± 0.25 | 7.35 ± 0.05 | 8.80 ± 0.05 |
| δENaC/TRPM2/TRPM5 | 60.40 ± 0.25 | 7.53 ± 0.34 | 7.73 ± 0.39 |
The silencing of HICCs has no effects on the cell‐cycle distribution of HepG2 cells. Shown are the respective percentages of cells in the G0/G1, S and G2/M phase determined by FACS (n = 3). Apparent differences from 100% are mainly due to the number of pre‐apoptotic cells (not shown) averaging 18.60 ± 1.39% (mean ± SD) throughout the analysis.
Discussion
Why so many channels?
As one of the major findings of the present study, HepG2 cells employ δENaC, TRPM2 and TRPM5 as HICCs. These channels may either act in an independent mode or as molecular complexes, namely as δENaC–TRPM2 and δENaC–TRPM5. The synergy observed in the current and RVI results as well as the interactions found with PLA support the latter hypothesis. But what advantages would such a modularity offer?
First of all, the recruitment of δENaC provides a higher ratio of Na+ over K+ influx to the system than could actually be achieved by mere non‐selective TRPs (Kellenberger & Schild, 2002; Nilius & Owsianik, 2011). This will directly translate into a higher driving force for overall cation entry and thus RVI efficiency. Second, because of the known voltage dependence of TRPM5 (Nilius & Owsianik, 2011), any membrane depolarization, e.g. via δENaC, is expected to stimulate cation uptake through this channel further. Third, with TRPM2 on board, the RVI machinery becomes sensitive to both ROS and CD38 as further amplification and modulation factors (Numata et al. 2012). Finally, a modulatory architecture of HICC currents renders the system flexible so that different tasks can actually be fulfilled – namely compensating for acute volume perturbations, on the one hand, and modulating proliferation/apoptosis, on the other.
Cell volume regulation in the interplay of proliferation and apoptosis
In the present study, the role of three different HICC components in the negative correlation of proliferation and apoptosis could be defined at the molecular level. This strongly supports the actual role of the cell volume regulatory machinery in these processes. As it turns out from the patch‐clamp recordings, however, the sensitivity of proliferation to HICC silencing is not a mere mechanistic one – in a way that channel‐mediated cation and water gain just serves as a prerequisite for mitosis to occur. Rather, the system appears to perceive perturbations of its HICC activities and, consequently, to modulate the checkpoint of its volume set point – best seen in the synergy of HICC currents in response to siRNA silencing (Fig. 3 B).
Given the clear influence of HICC activities on cell proliferation and apoptosis, quite surprisingly, siRNA silencing did not have any effect on cell cycle distribution at all. In our mind, this can only mean that the homeostatic system in HepG2 cells that is triggered via HICC abundance is located upstream of the mitotic mill. A definitive analysis of the signalling network employed in HICC activation, however, was not the aim of the present study.
The inefficiency of αENaC silencing with siRNAs
In an earlier report from this lab, just one of the three siRNAs tested led to an actual KD of αENaC expression – with some 40% of the transcript remaining unaffected (Bondarava et al. 2009). In the first screen employed here, ‘smartPool’ siRNAs with four different sequences against targets (Dharmacon) were used for silencing of the putative Na+ transporters, but no effects of the supposed αENaC siRNAs on RVI became evident (Fig. 1 B). The simplest explanation for this would be that, in HepG2 cells, αENaC simply is not a HICC. Interestingly, however, PLA shows proximities between αENaC and TRPM2 as well as TRPM5 that are very similar to the patterns observed for δENaC (see Fig. 4). The reason for these discrepancies is not clear. Any preselection among cells in response to αENaC siRNA transfection can be excluded since cell numbers were not changing as was frequently checked by automatized cell counting.
Pulldowns/mass spectrometry vs. proximity ligation assays
The HICC complexes implied by the PLA could not be detected with a traditional pulldown plus MS survey. Why is this so? From the patch‐clamp experiments, average whole‐cell HICC currents of −13 nS can be computed for −30 mV. Assuming theoretical single‐channel conductances of 10 and 40 pS, this would translate into the activation of some 1300 and 325 channels or channel complexes per cell. This is a rather limited number that is likely to confine most biochemical approaches. Furthermore, for the pull‐down plus MS survey, cells had to be transfected with the respective bait construct. This overexpression of a particular HICC channel may well interfere with the expression of its congeners explaining the absence of any detectable HICC channel coupling with these techniques. Given that PLA detects endogenous complexes, any effects of overexpression can be ruled out here. Furthermore, while efficiency of PLA is generally low (only a few complexes are detected) the resulting signal is amplified up to a thousand times, which makes any complex detected easily visible. So, whereas some 1300 complexes per cell may by far not be enough to show up in MS surveys they readily do with PLA.
Conclusions
In essence, our cell volume and patch‐clamp recordings as well as the PLA interaction studies suggest that δENaC–TRPM2 and δENaC–TRPM5 form functionally independent HICCs in HepG2 cells. Just based on the PLA results, αENaC–TRPM2 and αENaC–TRPM5 could make up further HICCs which, however, remains to be proven functionally. By combining molecular and physiological data we were able to shed light on the modular organization of HICCs in human hepatocytes and on their role in RVI, cell proliferation and apoptosis.
Additional information
Competing interests
The authors have no conflicts of interest to declare.
Author contributions
B.K. performed the PLA and qPCR and contributed to the manuscript; J.C. optimized the pulldowns and prepared the mass‐spec; S.P. performed the first siRNA screening with the SAM; D.K. and J.B. contributed to the PLA and the pulldowns, respectively; C.K. did the FACS analyses on early apoptosis and cell‐cycle analysis and M.K. contributed to these measurements; S.I. did part of the qPCR. P.J. performed the mass‐spec; P.I.H.B. contributed to the data‐analysis of RVI and to the outline of the study; F.W. did the patch‐clamp recordings, most of the SAM measurements, and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
There was no third party funding of the project.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Table S1. Potential binding partners of αENaC, δENaC, TRPM2 and TRPM5 as they were determined by pulldowns and mass spectroscopy.
Acknowledgements
We wish to thank Holger Vogel for introducing us to the RTCA technique and Klaus C. Schürmann for help with the cell profiling. The invaluable technical assistance of Gabi Beetz and Michael Reichl is also gratefully acknowledged.
B. Koos, J. Christmann and S. Plettenberg contributed equally to this work.
Edited by: Peying Fong & Ruth Murrell‐Lagnado
References
- Bagriantsev SN, Gracheva EO & Gallaghe PG (2014). Piezo proteins: regulators of mechanosensation and other cellular processes. J Biol Chem 289, 31673–31681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarava M, Li T, Endl E & Wehner F (2009). α‐ENaC is a functional element of the hypertonicity‐induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Arch 458, 675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW (2008). Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 9, 519–531. [DOI] [PubMed] [Google Scholar]
- Christmann J, Azer L, Dörr D, Fuhr GR, Bastiaens PIH & Wehner F (2016). Adaptive responses of cell hydration to a low temperature arrest. J Physiol 594, 1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J & Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dubois JM & Rouzaire‐Dubois B (2012). Roles of cell volume in molecular and cellular biology. Prog Biophys Mol Biol 108, 93–97. [DOI] [PubMed] [Google Scholar]
- Erfle H, Neumann B, Liebel U, Rogers P, Held M, Walter T, Ellenberg J & Pepperkok R (2007). Reverse transfection on cell arrays for high content screening microscopy. Nat Protoc 2, 392–399. [DOI] [PubMed] [Google Scholar]
- Fengler S, Bastiaens PIH, Grecco HE & Roda‐Navarro P (2012). Optimizing cell arrays for accurate functional genomics. BMC Res Notes 5, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchtgott LA, Chow CC & Periwal V (2009). A model of liver regeneration. Biophys J 96, 3926–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH & Pedersen SF (2009). Physiology of cell volume regulation in vertebrates. Physiol Rev 89, 193–277. [DOI] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW & Carpenter AE (2011). Improved structure, function and compatibility for CellProfiler: modular high‐throughput image analysis software. Bioinformatics 27, 1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S & Schild L (2002). Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82, 735–767. [DOI] [PubMed] [Google Scholar]
- Koos B, Andersson L, Clausson CM, Grannas K, Klaesson A, Cane G & Soderberg O (2014). Analysis of protein interactions in situ by proximity ligation assays. Curr Top Microbiol Immunol 377, 111–126. [DOI] [PubMed] [Google Scholar]
- Li T, ter Veld F, Nürnberger HR & Wehner F (2005). A novel hypertonicity‐induced cation channel in primary cultures of human hepatocytes. FEBS Lett 579, 2087–2091. [DOI] [PubMed] [Google Scholar]
- Liedtke W & Kim C (2005). Functionality of the TRPV subfamily of TRP ion channels: add mechano‐TRP and osmo‐TRP to the lexicon! Cell Mol Life Sci 62, 2985–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C & Montell C (2015). Forcing open TRP channels: Mechanical gating as a unifying activation mechanism. Biochem Biophys Res Comm 460, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen L, Liedtke W & Simon SA (2007). Changes in osmolality sensitize the response to capsaicin in trigeminal sensory neurons. J Neurophysiol 97, 2001–2015. [DOI] [PubMed] [Google Scholar]
- Martin‐Gago P, Fansa EK, Klein CH, Murarka S, Janning P, Schurmann M, Metz M, Ismail S, Schultz‐Fademrecht C, Baumann M, Bastiaens PIH, Wittinghofer A & Waldmann H (2017). A PDE6δ‐KRas inhibitor chemotype with up to seven H‐bonds and picomolar affinity that prevents efficient inhibitor release by Arl2. Angew Chem Int Ed Engl 56, 2423–2428. [DOI] [PubMed] [Google Scholar]
- Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S & Miyajima A (2012). Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol 22, 1166–1175. [DOI] [PubMed] [Google Scholar]
- Miyaoka Y & Miyajima A (2013). To divide or not to divide: revisiting liver regeneration. Cell Div 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B & Owsianik G (2011). The transient receptor potential family of ion channels. Genome Biol 12, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata T, Sato K, Christmann C, Marx R, Mori Y, Okada Y & Wehner F (2012). The ΔC splice‐variant of TRPM2 is the hypertonicity‐induced cation channel (HICC) in HeLa cells and the ecto‐enzyme CD38 mediates its activation. J Physiol 590, 1121–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata T, Sato K, Okada Y & Wehner F (2008). Hypertonicity‐induced cation channels rescue cells from staurosporine‐elicited apoptosis. Apoptosis 13, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J & Morishima S (2001). Receptor‐mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 532, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke B, Murarka S, Vogel HA, Martin‐Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmerman G, Heinelt K, Schultz‐Fademrecht C, Al Saabi A, Baumann M, Nussbaumer P, Wittinghofer A, Waldmann H & Bastiaens PIH (2016). Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat Comm 7, 11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plettenberg S, Weiss EC, Lemor R & Wehner F (2008). Subunits α, β and γ of the epithelial Na+ channel (ENaC) are functionally related to the hypertonicity‐induced cation channel (HICC) in rat hepatocytes. Pflugers Arch 455, 1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M & Ishihama Y (2007). Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Wehner F & Okada Y (2006). Inhibition of hypertonicity‐induced cation channels sensitizes HeLa cells to shrinkage‐induced apoptosis. Cell Physiol Biochem 18, 295–302. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L & Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Vendrell‐Navarro G, Rua F, Bujons J, Brockmeyer A, Janning P, Ziegler S, Messeguer A & Waldmann H (2015). Positional scanning synthesis of a peptoid library yields new inducers of apoptosis that target karyopherins and tubulin. Chembiochem 16, 1580–1587. [DOI] [PubMed] [Google Scholar]
- Wehner F, Lawonn P & Tinel H (2002). Ionic mechanisms of regulatory volume increase (RVI) in the human hepatoma cell‐line HepG2. Pflugers Arch 443, 779–790. [DOI] [PubMed] [Google Scholar]
- Wehner F & Olsen H (2004). Hypertonicity‐induced cation channels in rat hepatocytes and their intracellular regulation. Adv Exp Med Biol 559, 253–361. [DOI] [PubMed] [Google Scholar]
- Wehner F, Olsen H, Tinel H, Kinne‐Saffran E & Kinne RKH (2003a). Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 148, 1–80. [DOI] [PubMed] [Google Scholar]
- Wehner F, Shimizu T, Sabirov R & Okada Y (2003b). Hypertonic activation of a non‐selective cation conductance in HeLa cells and its contribution to cell volume regulation. FEBS Lett, 551, 20–24. [DOI] [PubMed] [Google Scholar]
- Weiss EC, Wehner F & Lemor RM (2007). Measuring cell volume regulation with time resolved acoustic microscopy. Acoust Imag 28, 73–80. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Table S1. Potential binding partners of αENaC, δENaC, TRPM2 and TRPM5 as they were determined by pulldowns and mass spectroscopy.