

Abstract
Key points
Pulmonary arterial hypertension (PAH) triggers right ventricle (RV) hypertrophy and left ventricle (LV) atrophy, which progressively leads to heart failure.
We designed experiments under conditions mimicking those encountered by the heart in vivo that allowed us to investigate whether consequent structural and functional remodelling of the ventricles affects their respective energy efficiencies.
We found that peak work output was lower in RV trabeculae from PAH rats due to reduced extent and velocity of shortening. However, their suprabasal enthalpy was unaffected due to increased activation heat, resulting in reduced suprabasal efficiency. There was no effect of PAH on LV suprabasal efficiency.
We conclude that the mechanism underlying the reduced energy efficiency of hypertrophied RV tissues is attributable to the increased energy cost of Ca2+ cycling, whereas atrophied LV tissues still maintain normal mechano‐energetic performance.
Abstract
Pulmonary arterial hypertension (PAH) greatly increases the afterload on the right ventricle (RV), triggering RV hypertrophy, which progressively leads to RV failure. In contrast, the disease reduces the passive filling pressure of the left ventricle (LV), resulting in LV atrophy. We investigated whether these distinct structural and functional consequences to the ventricles affect their respective energy efficiencies. We studied trabeculae isolated from both ventricles of Wistar rats with monocrotaline‐induced PAH and their respective Control groups. Trabeculae were mounted in a calorimeter at 37°C. While contracting at 5 Hz, they were subjected to stress–length work‐loops over a wide range of afterloads. They were subsequently required to undergo a series of isometric contractions at various muscle lengths. In both protocols, stress production, length change and suprabasal heat output were simultaneously measured. We found that RV trabeculae from PAH rats generated higher activation heat, but developed normal active stress. Their peak external work output was lower due to reduced extent and velocity of shortening. Despite lower peak work output, suprabasal enthalpy was unaffected, thereby rendering suprabasal efficiency lower. Crossbridge efficiency, however, was unaffected. In contrast, LV trabeculae from PAH rats maintained normal mechano‐energetic performance. Pulmonary arterial hypertension reduces the suprabasal energy efficiency of hypertrophied right ventricular tissues as a consequence of the increased energy cost of Ca2+ cycling.
Keywords: Right heart failure, cardiac efficiency, cardiac activation heat
Key points
Pulmonary arterial hypertension (PAH) triggers right ventricle (RV) hypertrophy and left ventricle (LV) atrophy, which progressively leads to heart failure.
We designed experiments under conditions mimicking those encountered by the heart in vivo that allowed us to investigate whether consequent structural and functional remodelling of the ventricles affects their respective energy efficiencies.
We found that peak work output was lower in RV trabeculae from PAH rats due to reduced extent and velocity of shortening. However, their suprabasal enthalpy was unaffected due to increased activation heat, resulting in reduced suprabasal efficiency. There was no effect of PAH on LV suprabasal efficiency.
We conclude that the mechanism underlying the reduced energy efficiency of hypertrophied RV tissues is attributable to the increased energy cost of Ca2+ cycling, whereas atrophied LV tissues still maintain normal mechano‐energetic performance.
Introduction
Pulmonary arterial hypertension (PAH) is an incapacitating disease. If untreated, its mortality rate of 40% (Sitbon et al. 2002; Humbert et al. 2010) 3 years post‐diagnosis remains extreme. Death results primarily from right heart failure (van Wolferen et al. 2007; Campo et al. 2010). This is because the increased pulmonary vascular resistance and resulting elevated pulmonary artery pressure impose a high afterload on the right ventricle (RV), inducing sustained pathological RV hypertrophy, which ultimately leads to RV failure (Vonk‐Noordegraaf et al. 2013). In addition to RV structural remodelling, PAH patients suffer a constellation of RV systolic abnormalities: lower ejection fraction (Gan et al. 2006; Rain et al. 2013), lower stroke volume (Marcus et al. 2001; Gan et al. 2006; Schafer et al. 2009), and reduced peak systolic strain and strain rate (Li et al. 2013). A comprehensive study by Wong et al. (2011) comparing two cohorts of PAH patients (New York Heart Association (NYHA) Class II and Class III) showed markedly reduced myocardial energy efficiency of the RV owing to increased myocardial oxygen consumption.
A widely studied rat model induces PAH by a single injection of monocrotaline (MCT). This agent selectively damages the vascular endothelium of the lung causing a series of RV pathological changes that are consistent with those seen in PAH patients (Hardziyenka et al. 2011, 2012). Using isolated RV papillary muscles obtained from such a PAH rat model, Wong et al. (2010) showed increased oxygen consumption and hence reduced efficiency – results that are in striking accord with those from their study of human patients (Wong et al. 2011). The consistency of these findings between the animal and patient studies clearly implicates disturbance of energy utilisation in the aetiology of PAH.
Nevertheless, the mechanism underlying RV energy disturbance in PAH remains unknown. This uncertainty provides the primary aim of the present study. We hypothesise that the observed energy disturbance in RV myocardium in PAH arises from a shift towards high energy costs of both crossbridge cycling and Ca2+ cycling. Our hypothesis stems from various lines of evidence: (i) disruption of the transverse tubular network (Xie et al. 2012), (ii) prolongation of the action potential (Lee et al. 1997; Piao et al. 2010; Hardziyenka et al. 2012), (iii) prolongation of the Ca2+ transient (Lamberts et al. 2007; Miura et al. 2011; Lookin et al. 2015), and (iv) decreased expression of the sarcoplasmic reticulum Ca2+‐ATPase (Kogler et al. 2003; Hardziyenka et al. 2011; Hadri et al. 2013). Experimental testing of our hypothesis is designed to provide an explanation for the reported decreased RV mitochondrial energy‐producing ability (Redout et al. 2007; Daicho et al. 2009; Wust et al. 2016) and the resulting myocardial contractile dysfunction observed in PAH RV tissue preparations (Korstjens et al. 2002; Miura et al. 2011; Lookin et al. 2015).
A secondary aim of our study was to investigate whether energy disturbance also presents in the myocardium of the left ventricle (LV) as a consequence of PAH. Over the past decade, the importance of ‘ventricular interdependence’ (Hsia & Haddad, 2012; Naeije & Badagliacca, 2017) has been demonstrated in the PAH disease setting. This high degree of ventricular interdependency means that, as the RV undergoes hypertrophy, the interventricular septum constantly bows leftward. Thus LV diastolic filling is greatly reduced, subsequently leading to LV atrophic remodelling (Vonk‐Noordegraaf et al. 2005; Gan et al. 2006; Marcus et al. 2008). A number of studies have reported several anomalies in the LV myocardium during PAH. These include reduction of peak LV systolic pressure (Correia‐Pinto et al. 2009), prolongation of the action potential (Hardziyenka et al. 2012), lower longitudinal conduction velocity (Hardziyenka et al. 2012), and a shift of myosin heavy chain (MHC) profile from the fast to the slow isoform (Lowes et al. 1997; Correia‐Pinto et al. 2009). Nevertheless, whether these contractile and ionic dysfunctions are associated with disturbed LV energy utilisation is unknown. We hypothesise that PAH disturbs LV energy utilisation such that LV efficiency is also reduced.
To test these two hypotheses, we have investigated energy efficiency using isolated trabeculae from both ventricles of MCT‐treated and Control rats. Different energy expenditure sources (crossbridge‐ and Ca2+ cycling‐related metabolism) were partitioned using two protocols: isometric pre‐shortened and afterloaded isotonic work‐loops. We measured heat as a proxy of oxygen consumption, and experiments were performed over a wide range of afterloads. We chose isolated cardiac trabeculae because of the linear arrangement of their cardiomyocytes along the longitudinal axis, unlike the multi‐orientated fibre directions of the whole‐heart. Unlike papillary muscles, trabeculae are sufficiently thin to obviate the risk of hypoxia during high metabolic demand (Han et al. 2011), which is an important factor to consider since we challenge them at high afterloads to mimic the hypertensive condition.
Methods
Ethical approval
Experiments were conducted in accordance with protocols approved by the University of Auckland Animal Ethics Committee (R1403) and conform to the principles and regulations described in a Journal of Physiology Editorial (Grundy, 2015).
Animal model
Male Wistar rats (300–325 g) received either a single subcutaneous injection of monocrotaline (MCT, Sigma Aldrich, Auckland, NZ, 60 mg kg−1) as the PAH group (n = 26) or an equivalent volume of saline as the Control group (n = 20). Treated animals had ad libitum access to standard rat chow and water, and experienced a 12 h light/dark environment. They were weighed three times a week for the first 4 weeks, but daily thereafter for another 2 weeks. The latter frequency was necessary as we observed that the MCT‐treated rats began to show several phenotypical signs of RV failure, including laboured respiration, lethargy, ruffled fur and lack of inquisitiveness, but most importantly, they started to experience consecutive days of body weight loss (>15%, a criterion for end‐stage RV failure). A previous study from our laboratory has reported that PAH rats, commencing at Week 4 post‐injection, show pronounced cardiac haemodynamic changes in vivo. These include decreased heart rate, decreased mean arterial blood pressure and, most importantly, a 3‐fold increase in RV systolic pressure (Han et al. 2018).
Muscle preparation
Not later than post‐injection Week 6, each rat was deeply anaesthetised with isoflurane and weighed before injecting with heparin (1000 IU kg−1). Following cervical dislocation, the heart was rapidly excised and plunged into cold Tyrode solution and immediately Langendorff‐perfused with oxygenated Tyrode solution (at room temperature), which contained (in mmol L−1): 130 NaCl, 6 KCl, 1 MgCl2, 0.5 NaH2PO4, 0.3 CaCl2, 10 HEPES, 10 glucose and 20 2,3‐butanedione monoxime (BDM), with pH adjusted to 7.4 using Tris.
Under a dissecting microscope, intact trabeculae were dissected from the endocardial surfaces of both left and right ventricles. A suitable trabecula, in terms of its geometrical uniformity and small diameter to enhance diffusive oxygen supply, was transferred to a work‐loop calorimeter (Taberner et al. 2015) and mounted between two platinum hooks connected to a custom‐built force transducer at the downstream end and a length motor at the upstream end. In the calorimeter, the muscle was superfused with the same oxygenated Tyrode solution as used during dissection, except at a higher concentration of CaCl2 (1.5 mmol L−1) and in the absence of BDM. The pH of the superfusate was adjusted to 7.4 with Tris at body temperature (37°C). The rate of flow of Tyrode superfusate over the muscle in the measurement chamber was electronically maintained at 0.55 μL s−1 (Taberner et al. 2017). This flow rate provides adequate oxygenation to the muscle (Han et al. 2011) while maximising the thermal signal‐to‐noise ratio (Johnston et al. 2015).
Once mounted and superfused, the trabecula was electrically stimulated to contract at 3 Hz (via platinum electrodes in the measurement chamber) for at least 1 h before it was gradually stretched to optimal length (L o; the length that maximises developed force). The length of the trabecula was measured at L o in the calorimeter, as was its diameter in two orthogonal views via a 45° mirror, using a microscope graticule. Since each studied trabecula resembled an ellipse in cross‐section (Goo et al. 2009), cross‐sectional areas were calculated from the two orthogonal measurements of major and minor diameters.
The force developed by the trabecula was determined by a laser interferometer system, using a custom‐made, low‐compliance transducer, while its length was controlled using a motor under software control. The rate of muscle suprabasal heat production was measured and quantified from the difference in temperature between downstream and upstream thermopile arrays and the flow rate‐dependent temperature sensitivity (Taberner et al. 2011). The entire calorimeter system was then optically isolated and thermally insulated in an enclosure to ensure that the environmental temperature was maintained at 37°C.
Experimental protocols
A period of about 1 h was required to gradually increase the temperature of the calorimeter to 37°C, during which time muscle force and rate of suprabasal heat production both reached steady states. Experiments were performed with the trabecula contracting at 5 Hz to mimic the physiological heart rate of the rat. Each trabecula was first required to contract isometrically, where its length was servo‐maintained using the upstream length motor. The trabecula was subsequently required to undergo force–length work‐loop contractions, preloaded at L o, at six different afterloads. Each afterloaded work‐loop reached a steady state of force and heat output after 2–3 min. The muscle was returned to isometric contractions between each bout of work‐loops to allow comparison of the baselines of the rates of suprabasal heat production at different afterloads. Isotonic work‐loop contractions approximate the pressure–volume loops of the heart, by allowing measurements of stress–length work output, suprabasal enthalpy and suprabasal efficiency, as well as both the extent and velocity of contraction.
Upon completion of the work‐loop protocol experiments, the trabecula was then required to undergo a series of isometric contractions at different preloads, in which muscle length was progressively reduced, in five steps, from L o to L min (the length at which negligible macroscropic active force was observed). Electrical stimulation was halted for 3 min between each length step transition to provide baselines for zero force and zero active heat.
Post‐experiment quantifications
The heat artifact resulting from the small cyclic movement (up to about 0.3 mm) of the upstream hook (required to change muscle length during work‐loop contractions) was quantified by oscillating the hook at a frequency of 5 Hz at the extent of muscle shortening achieved at the lowest afterload while the muscle was quiescent. A second heat artefact, resulting from electrical stimulation at 5 Hz, was quantified in the absence of a muscle. Net muscle heat output was calculated by subtracting these two artefactual sources of heat, which typically averaged at most 10% of maximal active heat.
Upon completion of an experiment, the thickness of the ventricular free wall was measured using a dissecting microscope graticule. Tibial length of the rat was measured, and the heart (including the dissected trabeculae) and lungs dried in an oven at 60°C for at least 24 h prior to weighing.
Definitions
Measurements of force, length and rate of heat output were acquired using LabVIEW software (National Instruments, Austin, TX, USA) and analysed offline using a custom‐written MATLAB (MathWorks, Natick, MA, USA) program. Force was converted to stress (kPa) by normalising to muscle cross‐sectional area. Twitch duration was quantified at 5% and 50% of peak active stress. Twitch heat (kJ m−3) was calculated by dividing the steady‐state rate of heat production by the stimulus frequency (5 Hz) and normalising it to muscle volume. Relative active stress is the ratio of active stress, at which the muscle transitioned from the isometric phase to isotonic shortening, to the peak isometric active stress at L o. External work output (kJ m−3) was calculated by integrating stress as a function of relative muscle length over the period of the twitch. Suprabasal enthalpy (kJ m−3) was calculated as the sum of work and suprabasal heat. Suprabasal efficiency was defined as the ratio of work to suprabasal enthalpy. Crossbridge efficiency was defined as the ratio of work to crossbridge enthalpy (following subtraction of the activation heat from the suprabasal enthalpy). Relative extent of shortening (%) was given by the distance that the muscle shortened during the isotonic phase at each afterloaded work‐loop) normalised to L o. Velocity of shortening (s−1) was calculated as the maximal slope of the relative length–time trace during the isotonic phase of the work‐loop. Heat–stress relations were fitted using linear regression for each muscle. Activation heat was defined as the averaged heat intercept of the heat–stress relations arising from the isometric contractions. Peak ‘shortening heat’ was calculated as the difference between the heat intercepts from the work‐loop (shortening) and isometric (non‐shortening) protocols.
Statistical analyses
Measured variables were plotted as functions of either relative muscle length or stress (afterload). Data were fitted using polynomial regression (up to third order) and the regression lines were averaged within groups using the ‘random coefficient model’ (a ‘Maximum Likelihood’ curve‐fitting technique advocated by Feldman, 1988) within PROC MIXED of the SAS software package (SAS Institute Inc., Cary, NC, USA). For clarification, as illustrated in Fig. 7 A, there are 16 fitted regression lines (thin lines) representing 16 Control LV trabeculae; the resulting average line (thick line) is superimposed.
Figure 7. Variability of the data underlying Fig. 5 .

Data were fitted using second‐order polynomials for enthalpy panels and third‐order polynomials for the other panels. Each panel shows individual data points, trabecula‐specific regression lines (LV‐CON (n = 16), LV‐PAH (n = 22), RV‐CON (n = 14), RV‐PAH (n = 21)), and the resulting averaged regression lines. Means ± SEMs of peak values were subsequently superimposed.
Suitable trabeculae could not always be found in both ventricles from the same heart. In Control rats, six hearts each provided one LV and one RV trabecula, 12 hearts each provided one trabecula from either ventricle, and three hearts each provided two RV trabeculae. In MCT‐treated rats, nine hearts each provided one LV and one RV trabecula, three hearts each provided one LV and two RV trabeculae, 12 hearts each provided one trabecula from either ventricle, and two hearts each provided two RV trabeculae. In consequence, a nested design in which trabeculae were nested within hearts was adopted.
The significance of differences among regression lines, or peak mean values, of the four groups was tested for the effect of PAH both between ventricles (LV versus RV) and within ventricles (Control versus PAH) using a set of mutually orthogonal contrast vectors. For each variable, peak values arising from each regression line were averaged (expressed as means ± standard errors) and superimposed on the resulting plot. Statistical significance of differences was declared when P < 0.05.
Results
Morphological characteristics of the rats after humane killing
PAH rats had lower body mass and tibial length than those of Control rats (Table 1). Lung dry mass and heart dry mass were significantly higher in PAH rats. The latter cohort developed RV hypertrophy and LV atrophy, as evident by the relative change of ventricular free wall thickness when normalised to heart mass. As shown in Table 2, the average lengths, cross‐sectional areas and volumes of the LV and RV trabeculae were not different among groups.
Table 1.
General characteristics of Control and MCT‐treated PAH rats
| Parameter | Control (n = 20) | PAH (n = 26) |
|---|---|---|
| Body mass (g) | 471.2 ± 6.0 | 395.5 ± 7.1* |
| Tibial length (mm) | 43.4 ± 0.04 | 40.0 ± 0.03* |
| Lung dry mass (g) | 0.27 ± 0.01 | 0.39 ± 0.03* |
| Lung dry mass/body mass (%) | 0.057 ± 0.001 | 0.101 ± 0.008* |
| Lung dry mass/tibial length (g mm−1) | 6.22 ± 0.15 | 9.15 ± 0.59* |
| Heart dry mass (g) | 0.29 ± 0.01 | 0.32 ± 0.01* |
| Heart dry mass/body mass (%) | 0.061 ± 0.001 | 0.081 ± 0.002* |
| Heart dry mass/tibial length (g mm−1) | 6.62 ± 0.15 | 7.45 ± 0.19* |
| LV wall thickness (mm) | 3.58 ± 0.07 | 3.39 ± 0.07* |
| LV thickness/heart dry mass (mm g−1) | 12.59 ± 0.34 | 10.74 ± 0.30* |
| RV wall thickness (mm) | 1.52 ± 0.03 | 2.05 ± 0.06* |
| RV thickness/heart dry mass (mm g−1) | 5.36 ± 0.18 | 6.48 ± 0.21* |
Values are means ± SEM. * P < 0.05.
Table 2.
Trabeculae dimensions of Control and MCT‐treated PAH rats
| LV trabeculae | RV trabeculae | |||
|---|---|---|---|---|
| Parameter | CON (n = 16) | PAH (n = 22) | CON (n = 14) | PAH (n = 21) |
| Length (mm) | 3.44 ± 0.15 | 3.50 ± 0.12 | 3.38 ± 0.12 | 3.03 ± 0.15 |
| Cross‐sectional area (mm2) | 0.070 ± 0.015 | 0.080 ± 0.009 | 0.072 ± 0.009 | 0.075 ± 0.007 |
| Volume (mm3) | 0.24 ± 0.05 | 0.27 ± 0.03 | 0.25 ± 0.03 | 0.22 ± 0.02 |
Values are means ± SEM; n, number of trabeculae.
Isometric contractions
Reduction of muscle length resulted in decreases of both steady‐state active stress production and suprabasal heat output (Fig. 1). The average relations of active stress–length and heat–length from either the LV (Fig. 1 D and G) or the RV (Fig. 1 E and H) trabeculae were not different between PAH and Control groups. As shown in Fig. 2, RV trabeculae from PAH animals showed prolonged twitch duration and lower maximal rates of rise and fall of twitch stress, but no difference was detected in the area under the twitch‐time profile (stress–time integral, STI). In contrast, normal isometric twitch kinetics were obtained in the LV trabeculae.
Figure 1. Results from the isometric pre‐shortened protocol at 5 Hz.
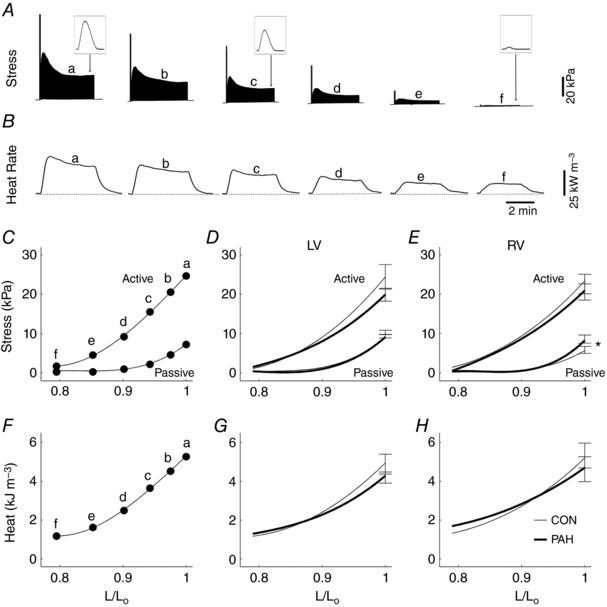
Typical experimental records of twitch stress (A) and rate of heat production (B) of a representative trabecula at various lengths (b–f) below its optimal length (a), where the insets depict single twitch, and where the dotted line segments were drawn to indicate the heat‐rate baseline. The first force twitch at each muscle length depicts the rested‐state contraction after the pause of stimulation, indicating the maximal post‐rest potentiation of stress. Passive stress, active stress and active heat at steady state as functions of relative muscle length (L/L o) for a single trabecula (C and F, respectively) and for average relations of LV (D and G) and RV trabeculae (E and H) from Control (thin lines) and PAH (thick lines) rats. Data were fitted using third‐order polynomials (in panels C–H). Data from panels C and F were obtained from an RV trabecula (length, 3.4 mm; cross‐sectional area, 0.087 mm2). Mean ± SEM values at optimal length (L o). * P < 0.05 comparing the regression lines of PAH and Control groups.
Figure 2. Temporal and kinetic characteristics of steady‐state isometric twitches.

Twitch duration at 5% (t 5) and at 50% (t 50) of peak stress, maximal rate of twitch stress development (+dS/dt, labelled ‘rise’) and relaxation (−dS/dt, labelled ‘fall’), and stress–time integral (STI) as functions of active stress for a single trabecula (A, D and G), and for average LV (B, E and H) and RV trabeculae (C, F and I) from Control (thin lines) and PAH (thick lines) rats. Data were fitted using linear regressions in A–F and second‐order polynomials in G–I. The data in A, D and G were obtained from an RV trabecula (length, 3.8 mm; cross‐sectional area, 0.073 mm2). * P < 0.05 comparing the regression lines of PAH and Control groups.
Figure 3 A shows steady‐state twitch heat as a function of active stress arising from the two distinct protocols: ‘work‐loop’ and ‘pre‐shortened isometric’ contractions. The slopes of both heat–stress relations were lower for the PAH RV trabeculae (Fig. 3 D and E). The averaged heat intercept of the isometric heat–stress relation (an index of activation heat) of RV trabeculae from PAH rats was significantly greater than that of their Control group, whereas no difference was detected in the LV trabeculae (Fig. 3 F). There was no difference in the averaged peak shortening heat between either groups or ventricles (Fig. 3 G).
Figure 3. Twitch heat as functions of active stress for quantifying activation heat and shortening heat.

A, linear regression of heat–stress relations arising from work‐loop (open circles, dashed lines) and isometric (filled circles, continuous lines) protocols for a representative trabecula and for the averages of LV trabeculae (B) and of RV trabeculae (C) from PAH (thick lines) and Control (thin lines) groups. The averaged values of the slopes were estimated from the heat–stress relations arising from both the isometric protocol (D) and the work‐loop protocol (E). The averaged activation heat (Q a, heat intercepts of relations from the isometric protocol) was greater in the RV PAH group compared with their respective Control group (F), as indicated by *. Peak shortening heat (peak Q s) was calculated as the difference between heat intercepts from work‐loop and isometric protocols (G). Representative data (A) were from an RV trabecula (length, 3.8 mm; cross‐sectional area, 0.073 mm2).
Work‐loop contractions
In Fig. 5, work output, suprabasal enthalpy (work plus suprabasal heat) and suprabasal efficiency, quantified from Fig. 4, are plotted as functions of relative afterload. Peak work output was significantly lower in RV trabeculae from PAH rats, despite there being no difference in suprabasal enthalpy; hence, suprabasal efficiency was lower compared to that of the Control group. However, when account was taken of the difference in activation heat (Fig. 3 E), crossbridge efficiency of RV trabeculae was revealed to be not significantly different between groups. In contrast, the LV trabeculae showed no difference in work, or in either suprabasal or crossbridge efficiency between the two groups. Both extent and velocity of shortening as functions of relative active stress (Fig. 6) were significantly lower in the RV trabeculae from PAH rats compared with those of their respective Control groups, whereas no difference between groups was detected in the LV trabeculae for either relation.
Figure 5. Energetics of afterloaded work‐loop contractions at steady state.

Data were fitted using second‐order polynomials for enthalpy panels and third‐order polynomials for the other panels. The left‐most panels (A, D and G) show data from a single representative trabecula (length, 3.4 mm; cross‐sectional area, 0.087 mm2) whereas the middle panels (B, E and H) show averaged relations from the LV trabeculae and the right‐most panels (C, F and I) show averaged relations from the RV trabeculae. The data (A, D and G) were obtained from a RV trabecula. * P < 0.05 comparing the regression lines of PAH and Control groups.
Figure 4. Typical experimental records arising from the work‐loop protocol at 5 Hz.

A, twitch stress of a trabecula undergoing progressively decreasing afterloaded work‐loop contractions (b–g), bounded by return to isometric contractions at L o (a). The first twitch illustrates the rested‐state contraction immediately following the commencement of electrical stimulation. B, the corresponding rates of heat output, where the dotted line is drawn to signify the heat‐rate baseline. C, steady‐state twitch stress profiles at various afterloads. D, the corresponding steady‐state muscle length trajectory during each twitch in C. E, parametric plots of the stress and length profiles shown in C and D, resulting in stress–length work‐loops. In all panels, data were obtained from an RV trabecula (length, 3.4 mm; cross‐sectional area, 0.087 mm2).
Figure 6. Shortening‐related parameters arising from the work‐loop protocol.

Velocity of shortening and extent of shortening as functions of relative active stress for a single representative trabecula (A and D) of length 2.9 mm and cross‐sectional area 0.035 mm2, and for averaged relations of LV (B and E) and RV trabeculae (C and F) from Control and PAH groups. SEM values were superimposed on the plots. The relations were lower for the RV PAH group compared with its Control group (as indicated by *).
Discussion
Overview
We find that energy utilisation by trabeculae isolated from the hypertrophied RV in end‐stage right‐ventricular failure induced by PAH is indeed disturbed. PAH RV trabeculae developed the same active stress, but they have reduced extent and velocity of shortening. Thus their production of peak external work was reduced. Since they released the same amount of suprabasal enthalpy, their suprabasal efficiency was reduced. Whereas the component of myocardial efficiency attributed to crossbridge shortening was maintained, the component apportioned to the performance of stress–length work‐loops was greatly reduced. Hence, their reduced suprabasal efficiency was due to the increased energy cost of the Ca2+‐triggered activation process. Surprisingly, despite the LV having undergone a period of atrophic remodelling, the ability of its trabeculae to utilise energy, and hence their energy efficiency, was unaffected.
Energetics of the right‐ventricular trabeculae in PAH
This is the first study to have examined the mechano‐energetic responses of trabeculae in the PAH setting. We distinguished the energy used for Ca2+ cycling and that consumed by crossbridge cycling. Crossbridge cycling heat was further partitioned into shortening and non‐shortening (isometric) components (Tran et al. 2017; Pham et al. 2017a). Separation of the energy demands of these two energy sinks clarifies the cellular mechanisms underlying disturbed energy utilisation in hypertrophied RV myocardium. Studies of human patients, and of the MCT‐hypertensive rats, have provided ample evidence that the oxygen consumption of the hypertrophied RV myocardium is higher. As work output is unchanged, RV efficiency is thus lower in PAH (Wong et al. 2010, 2011). We extended these findings by showing that the PAH RV trabeculae require greater thermal expenditure for the Ca2+‐triggered activation of contractile events (Fig. 3). This source of energy cost, coined ‘activation heat’ or ‘Ca2+ cycling heat’, is indexed by the heat intercept of the isometric heat–stress relation (Hill, 1949; Gibbs et al. 1967; Loiselle & Gibbs, 1979; Pham et al. 2017b). It comprises the thermal accompaniment of the hydrolysis of ATP by the sarcoplasmic reticulum Ca2+‐ATPase (SERCA) and the sarcolemmal Na+–K+‐ATPase, as well as the Na+/Ca2+ exchanger and the sarcolemmal Ca2+‐ATPase (Schramm et al. 1994). We have recently shown that activation heat is independent of muscle length and hence muscle stress (Pham et al. 2017b). The latter does not differ between PAH and Control trabeculae in either ventricle (Fig. 1 B and C). Nevertheless, PAH RV trabeculae demonstrated greater thermal expenditure associated with Ca2+‐triggered activation (Fig. 3 F). Our results align with literature findings on studies of RV failure showing impaired sarcoplasmic reticulum Ca2+ uptake (Endo et al. 2006) with higher diastolic [Ca2+]i and larger Ca2+ waves (Miura et al. 2011).
Suprabasal enthalpy consists of activation heat and crossbridge enthalpy. Our finding of similar suprabasal enthalpy between RV groups (Fig. 5 F) cannot be directly compared with that of Wong et al. because their measurements of oxygen consumption of the whole hearts (Wong et al. 2011) as well as papillary muscles (Wong et al. 2010) necessarily included a basal component. These somewhat disparate results could be reconciled if future measurements were to show a higher rate of basal metabolism in the hypertrophic RV myocardium, possibly as a result of a higher rate of protein turnover (Everett et al. 1977) in response to hypertrophy.
The lower peak work output of the PAH RV trabeculae does not stem from their inability to produce contractile stress, as they show the same development of isometric active stress as the Control group (Fig. 1). Despite developing the same active stress, they reveal an increased passive stress–length relation, consistent with increased fibrosis in the RV myocardium in PAH (Daicho et al. 2009; Handoko et al. 2009). The PAH RV trabeculae also show prolonged twitch duration (Fig. 2 C), which has been attributed to prolongation of the action potential (Lee et al. 1997; Piao et al. 2010; Hardziyenka et al. 2012), down‐regulation of voltage‐gated K+ channels (Piao et al. 2010) and prolongation of the Ca2+ transient (Kuramochi et al. 1994; Xie et al. 2012; Lookin et al. 2015). We note that comparable results of normal active stress production have been reported in isolated RV trabeculae (Kogler et al. 2003) and papillary muscles (Wong et al. 2010) from the same animal model under similar experimental conditions. However, previous studies have reported lower active stress in RV trabeculae from PAH tissues (Korstjens et al. 2002; Miura et al. 2011; Lookin et al. 2015), and higher active stress in isolated permeabilised cardiomyocytes of RV tissue from PAH patients (Rain et al. 2013). Whether such discrepancies can be attributed to differences of protocol or of experimental or environmental conditions awaits further exploration.
The failure of the PAH RV trabeculae to develop stress–length work as great as that of the Control group can be attributed to their inability to shorten to the same extent. Their lower extent of shortening arises from their reduced speed of shortening (Fig. 6), consistent with the reported shift in the myosin heavy chain isoform from predominantly V1 to V3 in the PAH model (Korstjens et al. 2002; Kogler et al. 2003; Hardziyenka et al. 2011). Our findings of reduced extent and velocity of shortening are likewise consistent with data from PAH patients, obtained using echocardiography, showing reduced peak RV wall systolic strain and strain rate (Li et al. 2013). Thus, the above comprehensive results extend previous observations by Wong et al. (2010), and imply that reduced work output, and increased thermal costs of the Ca2+‐triggered activation of contractile events are the main mechanisms underlying the reduction of suprabasal efficiency in PAH RV tissue.
Energetics of the left‐ventricular trabeculae in PAH
Our study has extended the investigation of the consequences of PAH‐induced RV hypertrophy to include the atrophied LV myocardium. The LV myocardium in PAH, like the RV, also demonstrates a shift of MHC profile from the fast to the slow isoforms (Lowes et al. 1997; Correia‐Pinto et al. 2009). Despite these reported MHC shifts, we find that the PAH LV trabeculae maintain both their extents and velocities of shortening. These results are consistent with those seen in human patients where the maximal velocities of contractile element shortening and circumferential fibre shortening are unaffected by chronic PAH (Krayenbuehl et al. 1978).
We also find that PAH LV trabeculae are capable of developing normal stress (Fig. 1), a result which is in agreement with that reported by Kogler et al. (2003). Their normal stress as a function of muscle length is associated with normal twitch kinetics (Fig. 2). Hence, their work output is normal, and concomitantly, their heat output is also normal. Hence, both their suprabasal and crossbridge efficiencies remain normal (Fig. 5).
Our null results of mechano‐energetics changes in PAH LV trabeculae are somewhat unexpected. Our MCT‐treated rats clearly suffered LV atrophy, as confirmed by a decrease in their normalised LV wall thickness (Table 1). Furthermore, in the same rat model (Kogler et al. 2003; Umar et al. 2012), as well as in patients (Hardziyenka et al. 2011, 2012), LV atrophic remodelling is manifested as impaired diastolic filling due to leftward interventricular septal bowing during contraction (Gan et al. 2006). Electrophysiological alteration has also been shown to occur in the atrophic LV of both PAH patients and rats (Hardziyenka et al. 2012). Despite these well‐documented changes in both animal models and human patients, we saw negligible effects of PAH on LV mechano‐energetics.
Summary
The null effect of PAH on crossbridge efficiency underscores our contention that the reduction of suprabasal efficiency in RV trabeculae arises largely from increased energy costs of Ca2+ cycling. The mechano‐energetics of the LV myocardium remain preserved in PAH rats.
Additional information
Competing interests
None declared.
Author contributions
All authors contributed to the conception and design of the work; acquisition, analysis and interpretation of data; and drafting the work. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, while all those who qualify for authorship are listed. All experimental work was performed at Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand.
Funding
This work was supported by the Heart Foundation of New Zealand: Project Grant 1601 (D.L.), Research Fellowship 1611 (J.‐C.H.) and Postgraduate Scholarship 1572 (T.P.), Marsden Fast‐Start grant (15‐UOA‐209) from the Royal Society of New Zealand (J.‐C.H., D.L. and A.T.), and Emerging Researcher First Grant (16/510) from the Health Research Council of New Zealand (J.‐C.H.).
Biography
Toan Pham received his MSc degree in Biological Science from the University of Auckland, New Zealand in 2013. He is currently pursuing a PhD degree in Physiology at the same university. His research interest is in cardiac muscle physiology, particularly focusing on cardiac energetic efficiency at the tissue and subcellular (mitochondria) levels in health and disease.

Edited by: Don Bers & Jolanda Van der Velden
References
- Campo A, Mathai SC, Le Pavec J, Zaiman AL, Hummers LK, Boyce D, Housten T, Champion HC, Lechtzin N, Wigley FM, Girgis RE & Hassoun PM (2010). Hemodynamic predictors of survival in scleroderma‐related pulmonary arterial hypertension. Am J Respir Crit Care Med 182, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia‐Pinto J, Henriques‐Coelho T, Roncon‐Albuquerque R Jr, Lourenco AP, Melo‐Rocha G, Vasques‐Novoa F, Gillebert TC & Leite‐Moreira AF (2009). Time course and mechanisms of left ventricular systolic and diastolic dysfunction in monocrotaline‐induced pulmonary hypertension. Basic Res Cardiol 104, 535–545. [DOI] [PubMed] [Google Scholar]
- Daicho T, Yagi T, Abe Y, Ohara M, Marunouchi T, Takeo S & Tanonaka K (2009). Possible involvement of mitochondrial energy‐producing ability in the development of right ventricular failure in monocrotaline‐induced pulmonary hypertensive rats. J Pharmacol Sci 111, 33–43. [DOI] [PubMed] [Google Scholar]
- Endo H, Miura M, Hirose M, Takahashi J, Nakano M, Wakayama Y, Sugai Y, Kagaya Y, Watanabe J, Shirato K & Shimokawa H (2006). Reduced inotropic effect of nifekalant in failing hearts in rats. J Pharmacol Exp Ther 318, 1102–1107. [DOI] [PubMed] [Google Scholar]
- Everett AW, Taylor RR & Sparrow MP (1977). Protein synthesis during right‐ventricular hypertrophy after pulmonary‐artery stenosis in the dog. Biochem J 166, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HA (1988). Families of lines: random effects in linear regression analysis. J Appl Physiol (1985) 64, 1721–1732. [DOI] [PubMed] [Google Scholar]
- Gan CT, Lankhaar JW, Marcus JT, Westerhof N, Marques KM, Bronzwaer JG, Boonstra A, Postmus PE & Vonk‐Noordegraaf A (2006). Impaired left ventricular filling due to right‐to‐left ventricular interaction in patients with pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 290, H1528–H1533. [DOI] [PubMed] [Google Scholar]
- Gibbs CL, Mommaerts M & Ricchiuti NV (1967). Energetics of cardiac contractions. J Physiol 191, 25–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo S, Joshi P, Sands G, Gerneke D, Taberner A, Dollie Q, LeGrice I & Loiselle D (2009). Trabeculae carneae as models of the ventricular walls: implications for the delivery of oxygen. J Gen Physiol 134, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmuller P, Ladage D, Guignabert C, Ishikawa K, Aguero J, Ibanez B, Turnbull IC, Kohlbrenner E, Liang L, Zsebo K, Humbert M, Hulot JS, Kawase Y, Hajjar RJ & Leopold JA (2013). Therapeutic efficacy of AAV1.SERCA2a in monocrotaline‐induced pulmonary arterial hypertension. Circulation 128, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Guild SJ, Pham T, Nisbet L, Tran K, Taberner A & Loiselle D (2018). Left‐ventricular energetics in pulmonary arterial hypertension‐induced right‐ventricular hypertrophic failure. Front Physiol 8, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Taberner A, Kirton RS, Nielsen P, Archer R, Kim N & Loiselle D (2011). Radius‐dependent decline of performance in isolated cardiac muscle does not reflect inadequacy of diffusive oxygen supply. Am J Physiol Heart Circ Physiol 300, H1222–H1236. [DOI] [PubMed] [Google Scholar]
- Handoko ML, de Man FS, Happe CM, Schalij I, Musters RJ, Westerhof N, Postmus PE, Paulus WJ, van der Laarse WJ & Vonk‐Noordegraaf A (2009). Opposite effects of training in rats with stable and progressive pulmonary hypertension. Circulation 120, 42–49. [DOI] [PubMed] [Google Scholar]
- Hardziyenka M, Campian ME, Reesink HJ, Surie S, Bouma BJ, Groenink M, Klemens CA, Beekman L, Remme CA, Bresser P & Tan HL (2011). Right ventricular failure following chronic pressure overload is associated with reduction in left ventricular mass: evidence for atrophic remodeling. J Am Coll Cardiol 57, 921–928. [DOI] [PubMed] [Google Scholar]
- Hardziyenka M, Campian ME, Verkerk AO, Surie S, van Ginneken ACG, Hakim S, Linnenbank AC, de Bruin‐Bon HACMR, Beekman L, van der Plas MN, Remme CA, van Veen TAB, Bresser P, de Bakker JMT & Tan HL (2012). Electrophysiologic remodeling of the left ventricle in pressure overload‐induced right ventricular failure. J Am Coll Cardiol 59, 2193–2202. [DOI] [PubMed] [Google Scholar]
- Hill AV (1949). The heat of activation and the heat of shortening in a muscle twitch. Proc R Soc Lond B Biol Sci 136, 195–211. [DOI] [PubMed] [Google Scholar]
- Hsia HH & Haddad F (2012). Pulmonary hypertension: a stage for ventricular interdependence? J Am Coll Cardiol 59, 2203–2205. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier J‐F, Chabot F, Dromer C, Pison C, Reynaud‐Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jaïs X, Montani D, Souza R & Simonneau G (2010). Survival in patients with idiopathic, familial, and anorexigen‐associated pulmonary arterial hypertension in the modern management era. Circulation 122, 156–163. [DOI] [PubMed] [Google Scholar]
- Johnston CM, Han JC, Ruddy BP, Nielsen PMF & Taberner AJ (2015). A high‐resolution thermoelectric module‐based calorimeter for measuring the energetics of isolated ventricular trabeculae at body temperature. Am J Physiol Heart Circ Physiol 309, H318–H324. [DOI] [PubMed] [Google Scholar]
- Kogler H, Hartmann O, Leineweber K, Nguyen van P, Schott P, Brodde OE & Hasenfuss G (2003). Mechanical load‐dependent regulation of gene expression in monocrotaline‐induced right ventricular hypertrophy in the rat. Circ Res 93, 230–237. [DOI] [PubMed] [Google Scholar]
- Korstjens IJ, Rouws CH, van der Laarse WJ, Van der Zee L & Stienen GJ (2002). Myocardial force development and structural changes associated with monocrotaline induced cardiac hypertrophy and heart failure. J Muscle Res Cell Motil 23, 93–102. [DOI] [PubMed] [Google Scholar]
- Krayenbuehl HP, Turina J & Hess O (1978). Left ventricular function in chronic pulmonary hypertension. Am J Cardiol 41, 1150–1158. [DOI] [PubMed] [Google Scholar]
- Kuramochi T, Honda M, Tanaka K, Enomoto K, Hashimoto M & Morioka S (1994). Calcium transients in single myocytes and membranous ultrastructures during the development of cardiac hypertrophy and heart failure in rats. Clin Exp Pharmacol Physiol 21, 1009–1018. [DOI] [PubMed] [Google Scholar]
- Lamberts RR, Hamdani N, Soekhoe TW, Boontje NM, Zaremba R, Walker LA, de Tombe PP, van der Velden J & Stienen GJ (2007). Frequency‐dependent myofilament Ca2+ desensitization in failing rat myocardium. J Physiol 582, 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Kodama I, Honjo H, Anno T, Kamiya K & Toyama J (1997). Stage‐dependent changes in membrane currents in rats with monocrotaline‐induced right ventricular hypertrophy. Am J Physiol 272, H2833–H2842. [DOI] [PubMed] [Google Scholar]
- Li Y, Xie M, Wang X, Lu Q & Fu M (2013). Right ventricular regional and global systolic function is diminished in patients with pulmonary arterial hypertension: a 2‐dimensional ultrasound speckle tracking echocardiography study. Int J Cardiovasc Imaging 29, 545–551. [DOI] [PubMed] [Google Scholar]
- Loiselle D & Gibbs CL (1979). Species differences in cardiac energetics. Am J Physiol 237, H90–H98. [DOI] [PubMed] [Google Scholar]
- Lookin O, Balakin A, Kuznetsov D & Protsenko Y (2015). The length‐dependent activation of contraction is equally impaired in impuberal male and female rats in monocrotaline‐induced right ventricular failure. Clin Exp Pharmacol Physiol 42, 1198–1206. [DOI] [PubMed] [Google Scholar]
- Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM & Bristow MR (1997). Changes in gene expression in the intact human heart. Downregulation of alpha‐myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 100, 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JT, Gan C, Zwanenburg JJ, Boonstra A, Allaart CP, Götte MJ & Vonk‐Noordegraaf A (2008). Interventricular mechanical asynchrony in pulmonary arterial hypertension: left‐to‐right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 51, 750–757. [DOI] [PubMed] [Google Scholar]
- Marcus JT, Vonk Noordegraaf A, Roeleveld RJ, Postmus PE, Heethaar RM, Van Rossum AC & Boonstra A (2001). Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension: noninvasive monitoring using MRI. Chest 119, 1761–1765. [DOI] [PubMed] [Google Scholar]
- Miura M, Hirose M, Endoh H, Wakayama Y, Sugai Y, Nakano M, Fukuda K, Shindoh C, Shirato K & Shimokawa H (2011). Acceleration of Ca2+ waves in monocrotaline‐induced right ventricular hypertrophy in the rat. Circ J 75, 1343–1349. [DOI] [PubMed] [Google Scholar]
- Naeije R & Badagliacca R (2017). The overloaded right heart and ventricular interdependence. Cardiovasc Res 113, 1474–1485. [DOI] [PubMed] [Google Scholar]
- Pham T, Han JC, Taberner A & Loiselle D (2017a). Do right‐ventricular trabeculae gain energetic advantage from having a greater velocity of shortening? J Physiol 595, 6477–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T, Tran K, Mellor KM, Hickey A, Power A, Ward ML, Taberner A, Han JC & Loiselle D (2017b). Does the intercept of the heat‐stress relation provide an accurate estimate of cardiac activation heat? J Physiol 595, 4725–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD & Archer SL (2010). The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 88, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmuller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk‐Noordegraaf A, van der Velden J & de Man FS (2013). Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 128, 2016–2025. [DOI] [PubMed] [Google Scholar]
- Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ & Simonides WS (2007). Right‐ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res 75, 770–781. [DOI] [PubMed] [Google Scholar]
- Schafer S, Ellinghaus P, Janssen W, Kramer F, Lustig K, Milting H, Kast R & Klein M (2009). Chronic inhibition of phosphodiesterase 5 does not prevent pressure‐overload‐induced right‐ventricular remodelling. Cardiovasc Res 82, 30–39. [DOI] [PubMed] [Google Scholar]
- Schramm M, Klieber HG & Daut J (1994). The energy expenditure of actomyosin‐ATPase, Ca2+‐ATPase and Na+,K+‐ATPase in guinea‐pig cardiac ventricular muscle. J Physiol 481, 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M & Simonneau Gé (2002). Long‐term intravenous epoprostenol infusion in primary pulmonary hypertension. J Am Coll Cardiol 40, 780–788. [DOI] [PubMed] [Google Scholar]
- Taberner A, Pham T, Han JC, Uddin R & Loiselle D (2017). A flow‐through infusion calorimeter for measuring muscle energetics during pharmacological interventions 2017 IEEE International Instrumentation and Measurement Technology Conference (I2MTC), Turin, 2017, pp. 1–5. https://doi.org/10.1109/I2MTC.2017.7969675. [Google Scholar]
- Taberner AJ, Han JC, Loiselle D & Nielsen PM (2011). An innovative work‐loop calorimeter for in vitro measurement of the mechanics and energetics of working cardiac trabeculae. J Appl Physiol (1985) 111, 1798–1803. [DOI] [PubMed] [Google Scholar]
- Taberner AJ, Johnston CM, Pham T, June‐Chiew H, Ruddy BP, Loiselle DS & Nielsen PM (2015). Measuring the mechanical efficiency of a working cardiac muscle sample at body temperature using a flow‐through calorimeter. Conf Proc IEEE Eng Med Biol Soc 2015, 7966–7969. [DOI] [PubMed] [Google Scholar]
- Tran K, Han JC, Crampin EJ, Taberner AJ & Loiselle DS (2017). Experimental and modelling evidence of shortening heat in cardiac muscle. J Physiol 595, 6313–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Lee JH, de Lange E, Iorga A, Partow‐Navid R, Bapat A, van der Laarse A, Saggar R, Saggar R, Ypey DL, Karagueuzian HS & Eghbali M (2012). Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhythm Electrophysiol 5, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE & Vonk‐Noordegraaf A (2007). Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 28, 1250–1257. [DOI] [PubMed] [Google Scholar]
- Vonk‐Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF & Hassoun PM (2013). Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 62, D22–33. [DOI] [PubMed] [Google Scholar]
- Vonk‐Noordegraaf A, Marcus JT, Gan CT, Boonstra A & Postmus PE (2005). Interventricular mechanical asynchrony due to right ventricular pressure overload in pulmonary hypertension plays an important role in impaired left ventricular filling. Chest 128, 628S–630S. [DOI] [PubMed] [Google Scholar]
- Wong YY, Handoko ML, Mouchaers KT, de Man FS, Vonk‐Noordegraaf A & van der Laarse WJ (2010). Reduced mechanical efficiency of rat papillary muscle related to degree of hypertrophy of cardiomyocytes. Am J Physiol Heart Circ Physiol 298, H1190–H1197. [DOI] [PubMed] [Google Scholar]
- Wong YY, Ruiter G, Lubberink M, Raijmakers PG, Knaapen P, Marcus JT, Boonstra A, Lammertsma AA, Westerhof N, van der Laarse WJ & Vonk‐Noordegraaf A (2011). Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ Heart Fail 4, 700–706. [DOI] [PubMed] [Google Scholar]
- Wust RC, de Vries HJ, Wintjes LT, Rodenburg RJ, Niessen HW & Stienen GJ (2016). Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc Res 111, 362–372. [DOI] [PubMed] [Google Scholar]
- Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME & Song LS (2012). Sildenafil prevents and reverses transverse‐tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension 59, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]