

Abstract
The tightly regulated opening and closure of ion channels underlies the electrical signals that are vital for a wide range of physiological processes. Two decades ago the first atomic level view of ion channel structures led to a detailed understanding of ion selectivity and conduction. In recent years, spectacular developments in the field of cryo‐electron microscopy have resulted in cryo‐EM superseding crystallography as the technique of choice for determining near‐atomic resolution structures of ion channels. Here, we will review the recent developments in cryo‐EM and its specific application to the study of ion channel gating. We will highlight the advantages and disadvantages of the current technology and where the field is likely to head in the next few years.

Keywords: ion channels, structure, channel gating
Ion channels are the molecular building blocks of electrical signalling
Ion channels are integral membrane proteins that generate electrical signals in cells (Armstrong & Hille, 1998). Ion channel activity is critical for a wide range of physiological functions including information transfer in the nervous system, the rhythm of the heartbeat, hormone secretion and fluid homeostasis. The importance of ion channels is further underscored by the number of diseases caused by inherited mutations in genes encoding ion channels, the so‐called channelopathies (Ashcroft, 2000). Analysis of protein structures at atomic level resolution, most commonly using X‐ray crystallography, has provided unparalleled insights into how proteins work. In the case of ion channels, crystallographic studies of the bacterial KcsA potassium channel underpin our understanding of the chemical basis of ion selectivity and conduction (Doyle et al. 1998; Zhou et al. 2001). The structural analysis of ion channel proteins, however, has lagged behind the study of soluble proteins. This is due to multiple factors, not least of which is the inherently dynamic nature of ion channel proteins which makes them hard to crystallize. However, as with all proteins whose function is mediated by rapid conformational changes, it is their inherent dynamics that makes them so critical for physiological processes as well as fascinating to study.
The latest exciting development in the quest to understand the molecular basis of how these exquisite nano‐machines work is the ‘resolution revolution’ in cryo‐electron microscopy (Kuhlbrandt, 2014). Cryo‐EM not only removes the need for crystallization (see below) but can also capture the full range of a protein's conformational states. For these reasons, the spectacular improvements in the resolution of cryo‐EM have been of particular interest for the study of dynamic proteins such as ion channels. Indeed, the important contribution that cryo‐EM has made to modern biomedical research is reflected in the recent awarding of the Nobel Prize in Chemistry to Joachim Frank, Richard Henderson and Jacques Dubochet “for developing cryo‐electron microscopy for the high‐resolution structure determination of biomolecules in solution” (https://www.nobelprize.org/nobel_prizes/chemistry/).
A brief overview of cryo‐EM
It is not our intention here to give an extensive overview of the methods for solving protein structures using cryo‐EM as there are many excellent reviews that have covered this topic in recent years (Bai et al. 2015; Cheng, 2015; Vinothkumar & Henderson, 2016; Liu et al. 2017). However, it is worth giving a brief overview for non‐structural biologists to highlight why there has been so much excitement caused by the recent improvements in cryo‐EM.
In contrast to X‐ray crystallography, which relies on crystals containing a highly ordered array of protein molecules, cryo‐EM (single particle analysis) utilizes thousands of randomly orientated particles to determine the structure. Purified protein is snap frozen in a thin layer of vitrified ice, preserving its in‐solution structure. In the case of dynamic proteins such as ion channels, the steady‐state distribution of the population of conformations is also captured. These images are then computationally clustered based on protein orientation and conformation before being combined into 3D density maps. However, extracting this information can be challenging as micrographs are typically noisy with low contrast due to the low electron doses that are used to reduce radiation damage and minimize motion‐induced artefacts caused when electrons interact with the sample.
The recent advancements in electron detection hardware and image processing algorithms have played an important role in tackling these issues. Old generation charge‐coupled device (CCD) electron cameras detect electrons indirectly (Faruqi & Henderson, 2007) by converting electrons to photons with a scintillator for subsequent detection by semiconductors. These could therefore produce multiple readouts (i.e. noise) from a single incident electron due to the scattering of light across pixels. The new generation of cameras, called direct electron detector (DED) cameras, detect electrons using electronics that are fabricated on a thin semiconductor membrane, thereby easily detecting individual incident electrons. Moreover, the detective quantum efficiency (DQE) of DEDs when operated in electron counting mode is much higher than CCD cameras, especially at high spatial frequency, allowing more atomic information to be captured (Kuijper et al. 2015). The fast readout speed of DED cameras provides yet another advantage as it allows a single micrograph to be recorded as a movie with separated frames. Together with newly developed algorithms, beam‐induced motion and sample drift (stage movement) can be compensated computationally (Campbell et al. 2012). In addition, advances in image processing algorithms based on maximum likelihood methods has enabled data processing with a minimum amount of user input thereby reducing the chance of overfitting (Scheres, 2012). Together, these advances have improved the resolution of cryo‐EM and unveiled the structures of many ion channels in the past 5 years.
The workflow for a typical cryo‐EM experiment is summarized in Fig. 1 and for a more detailed review on the process involved in single particle cryo‐EM and image processing readers are referred to the excellent recent reviews by Cheng et al. (2015) and White et al. (2017).
Figure 1. Workflow for cryo‐EM structure determination.
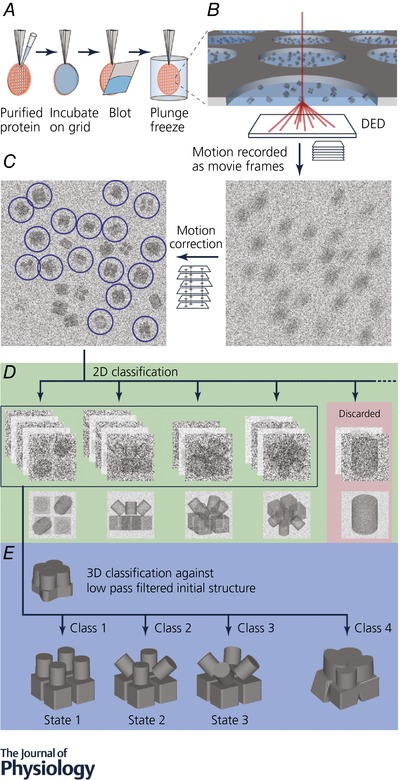
Proteins are overexpressed recombinantly in the expression system of choice, but purification from natural source is also possible as cryo‐EM requires less sample than X‐ray crystallography (Yan et al. 2015, 2017). A, purified protein is applied on a holey carbon grid. Excess protein is removed by blotting with a filter paper immediately before being plunge frozen in liquid ethane. B, protein vitrified in a thin layer of ice in the holey carbon grid is transferred to an electron microscope and imaged. Typically, thousands of micrographs are collected. Micrographs are dose fractionated into subframes. C, computational processing after image acquisition to recover information lost in the imaging process (e.g. motion correction). Particles are selected and extracted for processing. D, extracted particles are classified and averaged in two dimensions to enhance signal. Noise is significantly reduced in averaged particles and therefore ‘bad’ particles can be identified and removed. E, ‘good’ particles (green background in D) are classified in three dimensions against an initial model to generate subsets of particles with homogeneous conformation. These homogeneous subsets are used for further processing to generate the final high resolution EM map in different conformational states.
Cryo‐EM studies of ion channels
For over 50 years, electron microscopy has been used to study the structure of biomolecules (De Rosier & Klug, 1968; Dubochet et al. 1971, 1981) and in the case of large and highly symmetrical particles, such as viruses, it has been possible to achieve near‐atomic resolution (3.8 Å) structures for as many as 10 years (Zhang et al. 2008). Cryo‐EM has also been used to study structures of ion channels for over 20 years (Radermacher et al. 1994), albeit at limited resolution until recently. A major advantage of using cryo‐EM to study protein structure is that protein particles are frozen directly from solution making it possible to capture different conformational states of a channel in proportion to their steady‐state distribution (see Fig. 2). Although, in practice most investigators still try to generate samples under conditions where occupation of a single state is heavily favored as this simplifies the subsequent data analysis. Nigel Unwin exploited this to determine structures of both the open and closed state of the nAChR by freeze‐trapping the open state following rapid application of the activating ligand (Unwin, 1995; Miyazawa et al. 2003; Unwin & Fujiyoshi, 2012). In contrast, when using X‐ray crystallography to compare apo‐ and ligand bound states it is often necessary to purify the two states separately and use different crystalisation conditions to obtain high‐resolution structures for the different states (Armstrong et al. 2003; Song et al. 2017).
Figure 2. Ion channels exist in multiple conformational states.
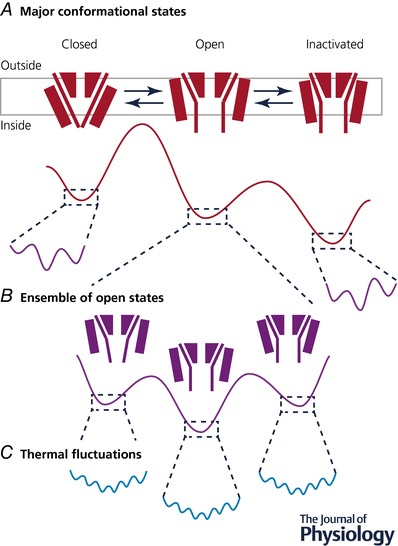
A, ion channels are an example of highly dynamic proteins. They undergo major conformational changes associated with transitions between open (ion conducting), closed (non‐conducting) and inactivated (another non‐conducting state that occurs in response to a prolonged activating stimulus) states. These states are separated by energy barriers, which can be altered, e.g. by binding of ligands or changes in voltage, to bias transitions in one direction or another. B, in addition, they are likely to exist in multiple substates within each of these major gating states that presumably reflect local minima in the energetic profiles of these states. C, finally, all proteins are in a state of thermal fluctuation. When a sample is plunge frozen, particles in each of the different states will be captured in proportion to the probability of occupying that state.
The last 5 years have seen spectacular improvements in the resolution of cryo‐EM and ion channel structural biologists have been at the forefront of utilizing these technical improvements to determine multiple structures from every 6 transmembrane domain 1 pore domain (6TM1P) ion channel subfamily (see Table 1). Due to space constraints, we will focus in this review on a few examples that demonstrate the power of cryo‐EM rather than provide an exhaustive overview.
Table 1.
Table of ion channel structures that have been determined
| Particles | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | Channel | Species | Detergent1 | Grid2 | (mg/ml) | Size (kDa) | Picked | Final | Resolution | PDB | EMD | Notes | Reference |
| 2013 | TRPV1 | Rat | A8‐35 | Q;R1.2/–;400;–/– | 0.3 | 270 | 97k | 90k | 3.3 | 3J5P | 5778 | (Cao et al. 2013) | |
| TRPV1 | Rat | A8‐35 | Q;R1.2/–;400;–/– | 0.3 | 270 | 149k | 36k | 3.8 | 3J5Q | 5776 | RTX/DkTx | (Liao et al. 2013) | |
| 96k | 33k | 4.2 | 3J5R | 5777 | Capsaicin | ||||||||
| 2015 | Insp3R | Rat | CHAPS | Q;–/–;–;–/– | 0.5 | 1250 | 157k | 96k | 4.7 | 3JAV | 6369 | (Fan et al. 2015) | |
| Piezo1 | Mouse | C12E10 | Q;R2/2;–/–(AmC) | 0.2 | 900 | 180k | 30k | 4.8 | 3JAC | 6343 | (Ge et al. 2015) | ||
| RYR1 | Rabbit | MSP1E3D1 | C;R1.2/1.3;–;–/– | 2 | 2260 | 75k | 25k | 6.1 | 4UWA | 2751 | Closed | (Efremov et al. 2015) | |
| 101k | 94k | 8.5 | 4UWE | 2752 | Open | ||||||||
| RYR1 | Rabbit | Tween20 | Q;R2/2;–;Cu/C(AmC) | 0.1 | 2260 | 246k | 66k | 3.8 | 3J8H | 2807 | (Yan et al. 2015) | ||
| RYR1 | Rabbit | CHAPS/DOPC | C;R1.2/1.3;200;Cu/C | 10 | 2260 | 230k | 47k | 4.8 | 3J8E | 6106 | (Zalk et al. 2015) | ||
| 225k | 48k | 5.0 | N/A | 6107 | CIP‐treated | ||||||||
| Slo2.2 | Chicken | DDM/Lipids | Q;R1.2/1.3;–;Cu/C | 5 | 550 | 41k | 24k | 4.5 | 5A6E | 3062 | (Hite et al. 2015) | ||
| TRPA1 | Human | PMAL‐C8 | Q;R1.2/–;400;Cu/C | N/A | 690 | 233k | 44k | 4.2 | 3J9P | 6267 | AITC | (Paulsen et al. 2015) | |
| 106k | 21k | 3.9 | N/A | 6268 | HC030031/A967079 | ||||||||
| 123k | 22k | 4.7 | N/A | 6269 | HC030031 | ||||||||
| Cav1.1 | Rabbit | Digitonin | N/A | 0.1 | 450 | 1482k | 353k | 4.2 | 3JBR | 6475 | (Wu et al. 2015) | ||
| 2016 | CaV1.1 | Rabbit | Digitonin | Q;R1.2/1.3;300;Cu/C | 2 | 500 | 1630k | 528k | 3.6 | 5GJV | 9513 | Class I | (Wu et al. 2016) |
| 123k | 3.9 | 5GJW | 9515 | Class II | |||||||||
| CFTR | Zebrafish | Digitonin | Q;R1.2/1.3;400;Au/C | 5.5 | 170 | 803k | 803k | 3.7 | 5UAR | 8461 | (Zhang & Chen, 2016) | ||
| Eag1 | Rat | DDM/CHS/Lipids | Q;R1.2/1.3;–;Cu/C | 4 | 450 | 240k | 145k | 3.8 | 5K7L | 8215 | (Whicher & MacKinnon, 2016) | ||
| PKD2 | Human | MSP2N2:soy PC | Q;R1.2/1.3;400;Cu/C | 3.5 | 240 | 368k | 94k | 3.0 | 5T4D | 8354 | 198‐703 | (Shen et al. 2016) | |
| 1 | 156k | 35k | 4.0 | N/A | 8355 | 198‐792 | |||||||
| A8‐35 | 3.5 | 290k | 66k | 4.0 | N/A | 8356 | 53‐792 | ||||||
| RYR1 | Rabbit | CHAPS/DOPC | C;R1.2/1.3;400;Cu/C | 10 | 2260 | 511k | 360k | 4.4 | 5TB0 | 8391 | EGTA | (des Georges et al. 2016) | |
| 1639k | 263k | 4.4 | 5T9V | 8376 | Ca2+/ATP/Caf. | ||||||||
| Q;R1.2/1.3;400;Cu/C | 123k | 67k | 4.6 | 5TAP | 8381 | ATP/Caf. | |||||||
| 123k | 65k | 3.8 | 5T15 | 8342 | Ca2+ | ||||||||
| 200k | 45k | 4.4 | 5TAW | 8387 | ryanodine | ||||||||
| RYR2 | Pig | Digitonin | –;–/–;–;Cu/C(AmC) | 0.1 | 2200 | 214k | 48k | 4.4 | 5GO9 | 9528 | Closed | (Peng et al. 2016) | |
| 589k | 133k | 4.2 | 5GOA | 9259 | Open | ||||||||
| TRPV1 | Rat | MSP2N2/soy lipid | Q;R1.2/1.3;400;Cu/C | 0.5 | 270 | 219k | 74k | 3.0 | 5IRX | 8117 | RTX/DkTx | (Gao et al. 2016) | |
| 159k | 31k | 3.3 | 5IRZ | 8118 | Apo | ||||||||
| 199k | 81k | 3.4 | 5IS0 | 8119 | Capsazepine | ||||||||
| TRPV2 | Rabbit | A8‐35 | C;R1.2/1.3;400;Cu/C | 1 | 280 | 427k | 44k | 3.8 | 5AN8 | 6455 | (Zubcevic et al. 2016) | ||
| 2017 | CFTR | Zebrafish | Digitonin | Q;R1.2/1.3;400;Au/C | 5.5 | 170 | 804k | 804k | 3.7 | 5TSI | 8461 | (Zhang et al. 2017) | |
| CLC‐K | Bovine | DDM/CHS | Q;R1.2/1.3;–;Cu/C | 3 | 190 | 442k | 82k | 3.8 | 5TQQ | 8435 | Class 1 | (Park et al. 2017) | |
| 67k | 4.0 | 5TR1 | 8454 | Class 2 | |||||||||
| CNG | C. elegans | A8‐35 | Q;R1.2/1.3;–;–/– | 0.6 | 330 | 231k | 100k | 3.5 | 5H3O | 6656 | (Li et al. 2017a) | ||
| HCN1 | Human | Digitonin | Q;R1.2/1.3;400;Au/C | 5 | 300 | N/A | 56k | 3.5 | 5U6O | 8511 | Apo | (Lee & MacKinnon, 2017) | |
| 125k | 3.5 | 5U6P | 8512 | cAMP | |||||||||
| hERG | Human | DDM/CHS/lipids | Q;R1.2/1.3;400;Au/C | 6 | 350 | 555k | 144k | 3.7 | 5VA1 | 8650 | T | (Wang & MacKinnon, 2017) | |
| 830k | 213k | 3.8 | 5VA2 | 8651 | TS | ||||||||
| 333k | 206k | 4.0 | 5VA3 | 8652 | TS S631A | ||||||||
| KCNQ1/Cam | Frog | DDM/CHS | Q;R1.2/1.3;400;Cu/C | 5 | 320 | 382k | 148k | 3.7 | 5VMS | 8712 | (Sun & MacKinnon, 2017) | ||
| Kir6.2/SUR1 | Mouse | Digitonin | L;R1/1;300;Au/C | 5 | 1000 | 599k | 35k | 5.6 | 5WUA | 6689 | (Li et al. 2017b) | ||
| Nav1.4β1 | Electric eels | Digitonin | Q;R1.2/1.3;300;Au/C | 1 | 230 | 2187k | 123k | 4.0 | 5XSY | 6770 | (Yan et al. 2017) | ||
| Slo1 | Sea hare | DDM/CHS | C;–/–;–;Cu/C | 7 | 480 | 638k | 230k | 3.8 | 5TJI | 8414 | (Hite et al. 2017) | ||
| Slo1.1 | Sea hare | DDM/CHS | Q;R1.2/1.3;–;Cu/C | 7 | 480 | 233k | 133k | 3.5 | 5TJ6 | 8410 | (Tao et al. 2017) | ||
| Slo2.2 | Chicken | DDM/lipids | Q;R1.2/1.3;–;Cu/C | 6 | 550 | 162k | 130k | 3.8 | 5U70 | 8515 | Open | (Hite & MacKinnon, 2017) | |
| 32k | 4.3 | 5U76 | 8517 | Closed | |||||||||
| TRPN (nompC) | Drosophila | MSP2N2:soy PC | C:R1.2/1.3;–;–/C | 3 | 750 | 338k | 175k | 3.6 | 5VKQ | 8702 | (Jin et al. 2017) | ||
| TRPML1 | Human | Digitonin | Q:R1.2/1.3;400;Au/C | 7 | 260 | 111k | 60k | 3.7 | 5WJ5 | 8840 | Apo | (Schmiege et al. 2017) | |
| 201k | 60k | 3.5 | 5WJ9 | 8841 | ML‐SA1 | ||||||||
| TRPML1 | Mouse | MSP1:POPC:POPG:POPE | Q;R1.2/1.3;200;Cu/C | 1.3 | 260 | 519k | 20k | 3.6 | 5WPQ | 8881 | Overall | (Chen et al. 2017) | |
| 9k | 3.6 | 5WPT | 8882 | Closed I | |||||||||
| 11k | 3.8 | 5WPV | 8883 | Closed II | |||||||||
| TRPML3 | Marmoset | PMAL‐C8 | Q;R1.2/1.3;300;Au/Au | 0.5 | 260 | 839k | 104k | 2.9 | 5W3S | 8764 | (Hirschi et al. 2017) | ||
| TRPM4 | Human | DDM/CHS | Q:R1.2/1.3;300;Au/C | 4 | 530 | N/A | N/A | 3.8 | 5WP6 | 8871 | (Winkler et al. 2017) | ||
| TRPM4 | Mouse | MSP1:POPC:POPG:POPE | Q;R1.2/1.3;200;Cu/C | 1.8 | 560 | 438k | 140k | 3.1 | 6BCJ | 7081 | Apo | (Guo et al. 2017) | |
| 565k | 197k | 2.9 | 6BCO | 7083 | ATP bound | ||||||||
| TRPM4 | Human | MSP2N2:Soy lipid | Q:R1.2/1.3;‐;Cu/C | 2.2 | 530 | 529k | 47k | 3.0 | N/A | N/A | EDTA | (Autzen et al. 2018) | |
| 829k | 45k | 2.9 | 6BQV | N/A | CaCl2 | ||||||||
| TRPM8 | Collared flycatcher | Digitonin | Q;R1.2/1.3;300;Au/Au | 0.5 | 400 | 3143k | 20k | 4.1 | 6BPQ | 7127 | (Yin et al. 2017) | ||
| NaVPaS | Cockroach | Digitonin | Q;R1.2/1.3;–;Cu/C | 1 | 180 | 4739k | 1373k | 3.8 | 5X0M | 6698 | (Shen et al. 2017) | ||
1A8‐35: amphipols; PMAL‐C8: amphipols; MSP2N2: nanodiscs; MSP1: nanodiscs. 2Brand; shape hole size (μm)/hole spacing(μm); mesh size (mesh); gird material/film material (modification). Q: Quantifoil; C: C‐flat; L: Lantuo Biotechnology; R: round; Cu: copper; Au: gold; AmC: continuous amorphous carbon film.
TRP channels
The resolution revolution began with the first ever near‐atomic resolution (3.4 Å) structure of a transient receptor potential (TRP) channel (Cao et al. 2013; Liao et al. 2013), which belongs to an ion channel family that has been particularly resistant to crystallization. Using cryo‐EM, the Cheng and Julius lab has determined structures of the TRPV1 channel in the unliganded closed state (Liao et al. 2013), as well as the open state (toxin bound or capsaicin bound; Cao et al. 2013). They were able to determine these structures by simply incubating purified proteins with ligand (capsaicin) or activating toxins (DkTx and RTx) before sample vitrification. These studies together revealed for the first time the structural basis of gating in this important channel family as well as revealing both similarities (e.g. the domain swap architecture within the transmembrane regions) and differences (extensive intra‐subunit interactions in the cytoplasmic domains) between TRP and the architecturally similar voltage‐gated ion channels.
These were the first structures of any ion channel that enabled identification of side‐chains using cryo‐EM (Fig. 3). It was achieved by utilizing many of the technical developments that ushered in the resolution revolution including the use of direct electron detector and movie mode recording to correct for beam‐induced motion. They also demonstrated that amphipols‐stabilized membrane proteins can be used for cryo‐EM structural determination if the use of detergents is undesirable. These papers were also noteworthy for the very detailed methodology and extensive supplementary material included with the manuscript that provides a detailed blueprint for how to follow in their footsteps.
Figure 3. The first high resolution cryo‐EM structure of an ion channel (Liao et al. 2013).
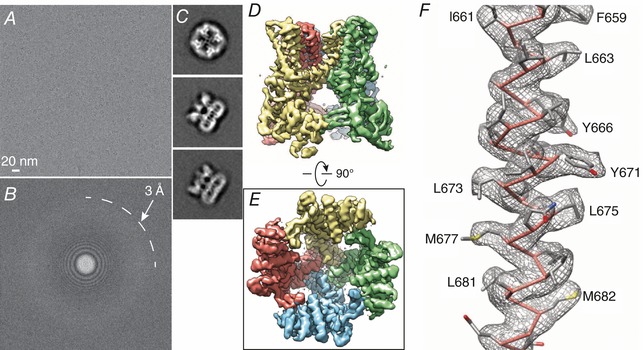
A, cryo‐electron micrograph of TRPV1. Micrographs typically have low contrast. B, Fourier transform of micrograph shown in A showing high resolution information approaching 3 Å. C, representative 2D class averages of particles. D and E, 3D density map of TRPV1 with each subunit colour‐coded, viewed from side (D) and bottom (E). F, cryo‐EM density map of the TRPV1 S6 domain superimposed on the atomic model. The clearly defined geometry of α‐helices and side‐chain densities allowed de novo model building. Reproduced with permission from Nature Publishing.
Since 2013, more structures of the TRP family have been determined using cryo‐EM, including TRPA1 (Paulsen et al. 2015), TRPN (Jin et al. 2017), and TRPV1 purified in the presence of lipid nanodiscs (Gao et al. 2016), which identified the role of lipids in influencing toxin binding. Recently, structures of TRPML determined in amphipol (Hirschi et al. 2017), nanodisc (Chen et al. 2017), and detergent (Schmiege et al. 2017) have been published back‐to‐back by three different laboratories highlighting the potential of cryo‐EM for structural determination of difficult‐to‐crystallize proteins.
Ryanodine receptor
The ryanodine receptor type 1 (RyR1) is the largest mammalian ion channel, with a mass of ∼2.2 MDa for the functional tetramer, which has made it an attractive target for cryo‐EM studies for over 25 years (Radermacher et al. 1992, 1994). It is predominantly expressed in skeletal muscle and regulated by a wide range of small molecules (most notably Ca2+ and ATP), as well as interacting protein partners (e.g. voltage‐gated calcium channel, immunophilins FKBP12). Functional studies of the RyR1 channel have also shown inconsistent results due to the different conditions used to purify and reconstitute the protein from the inner membrane. These subtle differences in purification methods were suggested to result in the presence or absence of different binding partners. Direct visualisation of proteins prepared under different conditions has therefore been a long‐term goal in the field.
In January 2015 three groups published cryo‐EM structures of the type 1 ryanodine receptor (Efremov et al. 2015; Yan et al. 2015; Zalk et al. 2015). The resolutions of these structures ranged from 3.8 Å to 6.1 Å. At this resolution, it was possible to obtain a detailed analysis of the overall architecture and in some parts model side‐chain orientations. There were a number of notable features in these structures. First, the architecture of the transmembrane region was broadly similar to that of the voltage‐gated ion channels, including the characteristic domain swap architecture whereby the ‘voltage‐sensor’ domain of one subunit swaps over to interact with the pore domain of a neighbouring subunit. Second, both the intracellular and extracellular entrances to the pore domain contain numerous acidic residues that would attract cations to the mouth of the channel, thereby contributing to its high conductance (Yan et al. 2015; Zalk et al. 2015). The two higher resolution studies also identified a novel cytosolic linker between the second and third transmembrane domains that interacts with the Ca2+ binding EF‐hand motifs in the cytosolic assembly, thereby suggesting a plausible mechanism by which Ca2+ binding to the cytosolic domains could allosterically modulate pore gating.
Efermov and colleagues exploited the power of cryo‐EM to capture different conformational states by solving structures of an open conformation (in the presence of 10 mm Ca2+) and a closed conformation (in the presence of EGTA) (Efremov et al. 2015). Whilst the resolution of these structures was lower (8.5 Å and 6.1 Å for the open and closed conformations, respectively) they confirmed the basic principles of gating suggested by the higher resolution closed state structures. Interestingly, they also noted an additional density in the cytosolic domains of the open state structure, which the authors suggested may represent endogenous FKBP that was bound to some but not all individual particles in the sample. Efremov and colleagues also noted considerable conformational heterogeneity with at least three different conformations for the closed state and four different conformations for the open state and suggested that the major contributor to this heterogeneity was the inherent flexibility of the cytosolic domain assembly.
In follow‐up studies, des Georges and colleagues determined the structures of structures of the Ryanodine receptor bound to different combinations of EGTA, Ca2+, ATP, caffeine and ryanodine to explore the full range of conformational changes in the cytoplasmic domain and pore region, as well as the pseudo‐voltage sensor (des Georges et al. 2016). Whilst the resolution of these structures remains lower than ideally we would like to see, a clearer picture of the structural basis of channel gating in this complex channel has started to emerge and we refer the interested reader to an excellent recent review by Zalk & Marks, (2017) for a more detailed description of these findings.
Slo2.2 channels and the dynamics of ion channel gating
Cryo‐EM is undoubtedly the technique of choice to use for determining multiple conformation states of ion channels by imaging the protein in different conditions, as discussed above for the TRP and RyR channels. However, it should be possible to capture distinct conformational states from a single sample provided there are enough particles from each conformational state.
The best demonstration of the possibility of this approach is a recent study of the Slo2.2 Na+‐activated K+ channel (Hite & MacKinnon, 2017). Similar to the nAChR channels, Slo2.2 is a ligand‐gated ion channel. Hite and Mackinnon exploited this property to determine structures of the open and closed conformations of the channel. They prepared samples with different concentrations of sodium ions (the ligand, ranging from 20 to 300 mm) prior to plunge‐freezing to titrate occupancy of the different conformations. Particles from all samples were then combined and subjected to 3D classification as a single data set. The whole dataset with 461k particles was analysed and one 3D class was found to be sensitive to changes in the sodium ion concentration and represents the open conformation. Surprisingly, the conformational open probability in saturating ligand concentrations was projected to reach 100% and appeared to be much higher than the functional open probability of ∼70%. The authors suggested this discrepancy is due to the fact that although the high ligand concentration pushed the global conformation of the channel to an open state, functionally around ∼30% of these channels have subtle changes which render them non‐conductive. Unfortunately, the resolution of these structures is still not sufficient to definitively determine such subtle structural or chemical factors, which are contributing to this non‐conducting open state. This study has demonstrated that the structure of different conformational states can be determined from a single data set if large enough numbers of each of the conformations are present in the sample.
The Hite and Mackinon (2017) study has also highlighted the dynamic nature of ion channels. The nine closed state 3D classes were similar to each other, with the main differences being in the rotation of the gating ring. These classes appeared to represent a continuous distribution of conformation. The distribution of these classes didn't correlate with Na+ concentration as demonstrated with the open state class. This study also highlights the power of numbers, i.e. if you can capture images of enough particles (hundreds of thousands in this case) then it should be possible to capture multiple substates as well as the major classes of conformational states (see Fig. 2).
EAG channels and novel domain sensing architecture
In classical voltage‐gated ion channels the ‘voltage‐sensor’ domain of one subunit swaps over to interact with the pore domain of a neighbouring subunit (Long et al. 2005, 2007). An intriguing finding to emerge from the recent cryo‐EM studies on other members of the 6TM1P superfamily of ion channels was the appearance of a non‐domain swapped voltage‐sensor/pore domain architecture which was first clearly delineated in the ether‐a‐go‐go gene (EAG) K+ channel (Whicher & MacKinnon, 2016). Furthermore, in the EAG family of ion channels it is possible to sever the link between the voltage‐sensor and pore domains and yet still retain normal voltage‐dependent gating (Lorinczi et al. 2015), suggesting that the EAG channels may have a distinct mechanism for transducing voltage signalling to opening of the activation gate. Subsequently, this non‐domain swapped architecture was also found to be present in the cyclic nucleotide‐gated channel TAX‐4 (Li et al. 2017a), hyperpolarization‐activated channel HCN1 (Lee & MacKinnon, 2017), human ether‐a‐go‐go‐related gene (hERG) K+ channel (Wang & MacKinnon, 2017) and the Na+‐activated Slo2.2 K+ channel (Hite & Mackinnon, 2017). In retrospect, it is clear that the structure reported for the Slo2.2 channels in 2015 (i.e. prior to publication of the EAG channel structure) must have a non‐domain‐swapped architecture. However, this was not reported at the time, as it was not possible to determine the location of the S4S5 linker (Hite et al. 2015). Presumably this was due to there being insufficient resolution in this region of the channel (∼5.2 Å), and so it was not possible to definitively assign the voltage‐sensor and pore domains to the same or different subunits. Conversely, the TRP and RyR receptors share the domain swapped architecture seen in the canonical voltage‐gated ion channels. Within the non‐domain swapped channels one common feature appears to be that gating is regulated by the binding of ligands to an intracellular domain that interacts with the voltage‐sensor domain, thereby modulating voltage‐sensor and gate movement. The hERG K+ channels is one potential exception to this rule, as gating in these channels is not known to be regulated by any exogenous ligands. However, an intrinsic ligand, i.e. a component of the hERG K+ channel itself, regulates gating by binding to the cyclic nucleotide homology domain (Ng et al. 2014).
The most intriguing finding of all with regard to domain swapping is that from Sobolevsky & coworkers (Singh et al. 2017), who showed that a point mutation in the TRPV6 pore domain could convert the channel from a voltage‐sensor domain swapped architecture (WT) to a non‐domain swapped architecture (mutant). This suggests that the energetic difference between these two architectures is probably very small and it is likely that during channel biogenesis and domain assembly there is an intermediate structure that determines whether channels adopt a voltage‐sensor domain swapped architecture or not.
CFTR
The ability to control channel conformation using specific ligands is a powerful approach for deconvolving the ‘what state is it?’ question that confronts the interpretation of every ion channel structure (as described above for nAChR, RyR and Slo2.2). For most channels, however, we have poor or no pharmacological and/or toxinological tools that can be used to trap states. Hence, there is still considerable need to develop these tools to help unlock the power of cryo‐EM to trap and assign states.
In the meantime, for channels where the steady‐state distribution of conformational states cannot be biased by titration of ligand concentrations, an alternative approach to capturing different states is to study mutants that bias distributions. The cystic fibrosis transmembrane conductance regulator is an anion channel evolved from the ATP‐binding cassette (ABC) transporter family (essentially a broken transporter; Gadsby, 2009). Electrophysiology studies have shown that a glutamic acid to glutamine mutation could slow the pore‐closing rate by ∼1000‐fold therefore greatly biasing channel occupancy in the open state (Vergani et al. 2005). Zhang et al. used this mutation to try to capture the structure of the zebrafish CFTR in the open conformation (Zhang et al. 2017). Whilst the structure has most of the characteristics expected of an open channel, the pore remained closed at the extracellular end. The authors suggest that the observed closed gated is due to the intraburst closure observed in electrophysiological recordings (Vergani et al. 2003). The structure represents the average of the open and closed states and this is consistent with the less defined EM densities at the gate region. However, in electrophysiology records, the dwell time for the open state is much longer than the intraburst close state (Vergani et al. 2003). The authors suggested that one possible explanation could be the existence of fast gating events that could not be resolved in electrophysiology recordings. Again, this illustrates the phenomenon of new experimental methods revealing new insights that forces us to think more carefully about old data and suggests new sets of experiments that can be done combining both the old and new methods.
Cryo‐EM limitations
Resolution
Beam‐induced motion will continue to affect resolution unless it can be completely avoided. Although sample movement can now be compensated using motion‐correction algorithms, beam‐induced motion is still problematic, as the movement is most severe when the beam first hits the sample. This is also when the structural information is still intact. High resolution information is lost when the motion is too strong to be corrected. Development of new algorithms and faster cameras has also improved motion correction by accounting for non‐uniform localized movement (Zheng et al. 2017). Recently, it has been shown that sample supports made entirely of gold can reduce the initial movement to below 2 Å (Russo & Passmore, 2014). A related issue is that signal to noise is a major hurdle to overcome in the chase to atomic level resolution. High defocus improves contrast but at the cost of reducing the maximum resolution of the final model. Improvements in phase plates (see below) will reduce the need for high defocus levels.
Structural basis of drug binding
Identifying drug interactions with ion channels is one of the most important areas of structural study, as many of these ion channels are important drug targets. At present, the resolution of cryo‐EM structures is generally not as high as X‐ray crystallography. In order to model drug binding, resolution below 2 Å, at least in the area of the drug binding cavity, is highly desirable in order to precisely position the small molecules (Subramaniam et al. 2016). Whilst there are structures that have been determined to 1.8 Å resolution using cryo‐EM (Merk et al. 2016), for most ion channels the resolution has been in the range of 3–4 Å (Table 1). This is partly due to the highly dynamic nature of ion channels. The conformational heterogeneity becomes problematic when the differences are too small to be identified causing information to be averaged out in flexible regions (White et al. 2017).
Ion channels pose another unique problem when there is a symmetry mismatch between drugs and channels. Many ion channels have rotational symmetry, which can facilitate structure determination. When a drug symmetrically binds to the channel and saturates all binding sites, it is possible to identify interacting residues on the channel (Cao et al. 2013). However, when the drug cannot bind in a symmetrical manner (e.g. the pore cavity of most ion channels where the drug can only occupy one of the four overlapping binding sites), symmetry cannot be applied during reconstruction. This effectively lowers the resolution for a given dataset and reduces the feasibility of obtaining detailed information on the chemical basis of drug binding. Furthermore, the ability to orient the particles based on such a small comparative asymmetry of just a few atoms would likely be impossible given the current equipment and techniques. One possibility to overcome this hurdle will be to combine cryo‐EM studies at modest resolution with molecular dynamics simulations to gain significant insights into how drugs bind to ion channels (Vandenberg et al. 2017).
Assessing model quality
The lack of adequate validation tests for assessing cryo‐EM structure quality has been well recognized for many years (Henderson et al. 2012). Prior to 2013 there were relatively few laboratories working with cryo‐EM and they were very well aware of the limitations of the technology. For example, it is possible to reconstruct a desired high‐resolution model simply from noise in the micrographs if data is not properly processed. This issue is eloquently described in a short review, ‘Avoiding the pitfalls of single particle cryo‐electron microscopy: Einstein from noise’, written by Richard Henderson (Henderson, 2013).
As the cryo‐EM technology becomes more robust and more ‘non‐experts’ start to use it there is always a risk that simple errors can lead to flaws in interpretation of the quality of a structure. The current gold standard relies on the Fourier shell correlation (Rosenthal & Henderson, 2003; Rosenthal & Rubinstein, 2015), which gives an indication of the correlation between two independently refined half‐maps. This is, however, more akin to a consistency score for the selected particles rather than an absolute measure of the resolution (Herzik et al. 2017) and does not represent the quality of the fitted model. Recently, the Lander lab devised an alternative multi‐model approach to assess the quality of EM models on a residue‐by‐residue basis. Multiple structures are modelled using the EM map as constraint and the structures are scored using structural analysis programs. The top 10 scored structures are then used for RMSD calculation. Regions with low RMSD score represent well‐ordered density while less well‐defined regions will have high RMSD scores. This provides information on the quality of the fitted model and care can then be taken when interpreting structures in the less defined region. In addition, regions with high RMSD could also represent high flexibility that may be very important for protein function. The Lander lab have applied this approach to most of the cryo‐EM structures determined to date and the results are posted on their lab website: http://www.lander-lab.com/convergence/.
What does the future hold?
Single particle cryo‐EM requires proteins to be purified from their native environment. Ion channels have to be extracted from the lipid bilayer (most commonly using detergents), which may affect their functions. Exciting developments in cryo‐electron tomography (cryo‐ET) will enable us to determine sub‐nanometre resolution structures of proteins, including ion channels, in situ by averaging molecules in tomograms to improve signal‐to‐noise ratio (subtomogram averaging). In addition to improvements in camera technology and computing algorithms, cryo‐ET is further improved by the development of the volta phase plates (VPP) and the use of cryo focus ion beam (cryo‐FIB) milling for sample preparation.
Cryo‐ET has already started to be employed for larger protein complexes such as determination of ATP synthase structures in intact mitochondria (Davies et al. 2012), investigation of nuclear pore complexes (Beck et al. 2007; Maimon et al. 2012), ribosomes (Mahamid et al. 2016) and the 26S proteasome (Asano et al. 2015). Some of these complexes are as small as 25 nm (Asano et al. 2015), which is comparable to the size of RyR1 (Yan et al. 2015) and RyR2 (Peng et al. 2016). Typically, these experiments have a resolution of ∼2 nm but sub‐nanometre resolution is achievable for larger complexes (Pfeffer et al. 2015). Global architectures of the protein can be studied and high resolution structure from crystal or single particle EM can be fitted into the 3D model to identify changes in subunit interactions (Stewart et al. 2012). In the case of ion channels, one can immediately imagine how powerful this technique could be for studying RyR–I CaL interactions during excitation–contraction coupling. One could also start to manipulate phosphorylation status in intact cells to start addressing how signalling pathways modulate channel structure (and function) in situ.
Advancement in sample preparation for single particle cryo‐EM will enable us to dissect the molecular rearrangement during gating movements on the millisecond timescale. Time‐resolved cryo‐EM was originally applied by Unwin to study the opening of acetylcholine receptor by spraying droplets of acetylcholine on the receptor immediately (5 ms) before vitrification to capture the open conformation (Unwin, 1995; Unwin & Fujiyoshi, 2012). More recently similar techniques have been used to study the formation of the ribosome providing structural information on the millisecond scale (Shaikh et al. 2014). Recent developments in fast‐mixing microfluidic devices (Lu et al. 2014) and microsprayers (Feng et al. 2017) for grid preparation have shown some promising results. Perhaps in the near future we will be able to dissect not only the steady‐state conformations but also the channel gating motions on the millisecond timescale and better understand these nano‐machines that are never at rest.
Conclusions
Technological advances in cryo‐electron microscopy over the last 5 years represents yet another significant step forward in our quest to understand the structural basis of the extraordinary feats of molecular gymnastics that underlie ion channel gating. Undoubtedly, many more structures will be worked out in the next few years, and these efforts are likely to focus on the use of ligands and/or mutations to elucidate the structures of different conformational states. In the not too distant future we should also start to see structures of channels in liposomes or intact cells determined using cryo‐electron tomography.
Additional information
Competing interests
None.
Author contributions
C.L. co‐wrote the first draft of the manuscript and prepared figures, M.H. co‐wrote the first draft of the manuscript, A.S. helped draft the outline of the manuscript and revised the manuscript, E.P. helped draft the outline of the manuscript and revised the manuscript, J.V. helped draft the outline of the manuscript, prepared figures and revised the manuscript.
Funding
National Health and Medical Research Council of Australia (App1116948, J.V.; App1141974, J.V., A.S. and E.P.; App1090408, A.S.), National Institutes of Health (R01‐GM057846, E.P.; U54‐GM087519, E.P.).
Acknowledgements
We than the members of the Mark Cowley Lidwill Research Programme in Cardiac Electrophysiology for helpful discussions.
Biographies
Carus Lau gained his PhD degree from the University of Queensland studying the interaction of spider toxins and voltage‐gated ion channels. He is currently a postdoctoral scientist at the Victor Chang Cardiac Research Institute undertaking cryo‐EM studies of ion channels.

Mark Hunter is a science graduate of the University of Western Sydney. He has a long‐standing interest in cardiac ion channels and is currently a Senior Research Officer at the Victor Chang Cardiac Research Institute undertaking cryo‐EM studies of ion channels.
Edited by: Ole Petersen & Jian Yang
This is an Editor's Choice article from the 1 April 2018 issue.
References
- Armstrong CM & Hille B (1998). Voltage‐gated ion channels and electrical excitability. Neuron 20, 371–380. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M & Gouaux E (2003). Tuning activation of the AMPA‐sensitive GluR2 ion channel by genetic adjustment of agonist‐induced conformational changes. Proc Natl Acad Sci USA 100, 5736–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, Danev R & Baumeister W (2015). Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 347, 439–442. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM (2000). Ion Channels and Disease: Channelopathies. Academic Press, San Diego. [Google Scholar]
- Autzen HE, Myasnikov AG, Campbell MG, Asarnow D, Julius D & Cheng Y (2018). Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, McMullan G & Scheres SH (2015). How cryo‐EM is revolutionizing structural biology. Trends Biochem Sci 40, 49–57. [DOI] [PubMed] [Google Scholar]
- Beck M, Lucic V, Forster F, Baumeister W & Medalia O (2007). Snapshots of nuclear pore complexes in action captured by cryo‐electron tomography. Nature 449, 611–615. [DOI] [PubMed] [Google Scholar]
- Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B & Grigorieff N (2012). Movies of ice‐embedded particles enhance resolution in electron cryo‐microscopy. Structure 20, 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao E, Liao M, Cheng Y & Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, She J, Zeng W, Guo J, Xu H, Bai XC & Jiang Y (2017). Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 550, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y ( 2015). Single‐particle Cryo‐EM at crystallographic resolution. Cell 161, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Grigorieff N, Penczek PA & Walz T (2015). A primer to single‐particle cryo‐electron microscopy. Cell 161, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Anselmi C, Wittig I, Faraldo‐Gomez JD & Kuhlbrandt W (2012). Structure of the yeast F1Fo‐ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosier DJ & Klug A (1968). Reconstruction of three dimensional structures from electron micrographs. Nature 217, 130–134. [DOI] [PubMed] [Google Scholar]
- des Georges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, Hendrickson WA, Marks AR & Frank J (2016). Structural basis for gating and activation of RyR1. Cell 167, 145–157.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT & MacKinnon R (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Booy FP, Freeman R, Jones AV & Walter CA (1981). Low temperature electron microscopy. Annu Rev Biophys Bioeng 10, 133–149. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Ducommun M, Zollinger M & Kellenberger E (1971). A new preparation method for dark‐field electron microscopy of biomacromolecules. J Ultrastruct Res 35, 147–167. [DOI] [PubMed] [Google Scholar]
- Efremov RG, Leitner A, Aebersold R & Raunser S (2015). Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43. [DOI] [PubMed] [Google Scholar]
- Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ & Serysheva II (2015). Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqi AR & Henderson R (2007). Electronic detectors for electron microscopy. Curr Opin Struct Biol 17, 549–555. [DOI] [PubMed] [Google Scholar]
- Feng X, Fu Z, Kaledhonkar S, Jia Y, Shah B, Jin A, Liu Z, Sun M, Chen B, Grassucci RA, Ren Y, Jiang H, Frank J & Lin Q (2017). A fast and effective microfluidic spraying‐plunging method for high‐resolution single‐particle cryo‐EM. Structure 25, 663–670.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC (2009). Ion channels versus ion pumps: the principal difference, in principle. Nat Rev Mol Cell Biol 10, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D & Cheng Y (2016). TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, Li R, Gao N, Xiao B & Yang M (2015). Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527, 64–69. [DOI] [PubMed] [Google Scholar]
- Guo J, She J, Zeng W, Chen Q, Bai XC & Jiang Y (2017). Structures of the calcium‐activated, non‐selective cation channel TRPM4. Nature. 552, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R ( 2013). Avoiding the pitfalls of single particle cryo‐electron microscopy: Einstein from noise. Proc Natl Acad Sci USA 110, 18037–18041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, Jiang W, Ludtke SJ, Medalia O, Penczek PA, Rosenthal PB, Rossmann MG, Schmid MF, Schroder GF, Steven AC, Stokes DL, Westbrook JD, Wriggers W, Yang H, Young J, Berman HM, Chiu W, Kleywegt GJ & Lawson CL (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzik MA Jr, Fraser JS & Lander GC (2017). A multi‐model approach to assessing local and global cryo‐EM map quality. bioRxiv https://doi.org/10.1101/128561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi M, Herzik MA Jr, Wie J, Suo Y, Borschel WF, Ren D, Lander GC & Lee SY (2017). Cryo‐electron microscopy structure of the lysosomal calcium‐permeable channel TRPML3. Nature 550, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite RK & MacKinnon R (2017). Structural titration of Slo2.2, a Na+‐dependent K+ channel. Cell 168, 390–399.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite RK, Tao X & MacKinnon R (2017). Structural basis for gating the high‐conductance Ca2+‐activated K+ channel. Nature 541, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite RK, Yuan P, Li Z, Hsuing Y, Walz T & MacKinnon R (2015). Cryo‐electron microscopy structure of the Slo2.2 Na+‐activated K+ channel. Nature 527, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Bulkley D, Guo Y, Zhang W, Guo Z, Huynh W, Wu S, Meltzer S, Cheng T, Jan LY, Jan YN & Cheng Y (2017). Electron cryo‐microscopy structure of the mechanotransduction channel NOMPC. Nature 547, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrandt W (2014). Biochemistry. The resolution revolution. Science 343, 1443–1444. [DOI] [PubMed] [Google Scholar]
- Kuijper M, van Hoften G, Janssen B, Geurink R, De Carlo S, Vos M, van Duinen G, van Haeringen B & Storms M (2015). FEI's direct electron detector developments: Embarking on a revolution in cryo‐TEM. J Struct Biol 192, 179–187. [DOI] [PubMed] [Google Scholar]
- Lee CH & MacKinnon R (2017). Structures of the human HCN1 hyperpolarization‐activated channel. Cell 168, 111–120.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou X, Wang S, Michailidis I, Gong Y, Su D, Li H, Li X & Yang J (2017a). Structure of a eukaryotic cyclic‐nucleotide‐gated channel. Nature 542, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wu JX, Ding D, Cheng J, Gao N & Chen L (2017b). Structure of a pancreatic ATP‐sensitive potassium channel. Cell 168, 101–110.e10. [DOI] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D & Cheng Y (2013). Structure of the TRPV1 ion channel determined by electron cryo‐microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gutierrez‐Vargas C, Wei J, Grassucci RA, Sun M, Espina N, Madison‐Antenucci S, Tong L & Frank J (2017). Determination of the ribosome structure to a resolution of 2.5 A by single‐particle cryo‐EM. Protein Sci 26, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB & Mackinnon R (2005). Crystal structure of a mammalian voltage‐dependent Shaker family K+ channel. Science 309, 897–903. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB & MacKinnon R (2007). Atomic structure of a voltage‐dependent K+ channel in a lipid membrane‐like environment. Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- Lorinczi E, Gomez‐Posada JC, de la Pena P, Tomczak AP, Fernandez‐Trillo J, Leipscher U, Stuhmer W, Barros F & Pardo LA (2015). Voltage‐dependent gating of KCNH potassium channels lacking a covalent link between voltage‐sensing and pore domains. Nat Commun 6, 6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Barnard D, Shaikh TR, Meng X, Mannella CA, Yassin A, Agrawal R, Wagenknecht T & Lu TM (2014). Gas‐assisted annular microsprayer for sample preparation for time‐resolved cryo‐electron microscopy. J Micromech Microeng 24, 115001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Cuellar LK, Forster F, Hyman AA, Plitzko JM & Baumeister W (2016). Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351, 969–972. [DOI] [PubMed] [Google Scholar]
- Maimon T, Elad N, Dahan I & Medalia O (2012). The human nuclear pore complex as revealed by cryo‐electron tomography. Structure 20, 998–1006. [DOI] [PubMed] [Google Scholar]
- Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS & Subramaniam S (2016). Breaking cryo‐EM resolution barriers to facilitate drug discovery. Cell 165, 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y & Unwin N (2003). Structure and gating mechanism of the acetylcholine receptor pore. Nature 423, 949–955. [DOI] [PubMed] [Google Scholar]
- Ng CA, Phan K, Hill AP, Vandenberg JI & Perry MD (2014). Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J Biol Chem 289, 25822–25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Campbell EB & MacKinnon R (2017). Structure of a CLC chloride ion channel by cryo‐electron microscopy. Nature 541, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Armache JP, Gao Y, Cheng Y & Julius D (2015). Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR & Yan N (2016). Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354, aah5324. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Burbaum L, Unverdorben P, Pech M, Chen Y, Zimmermann R, Beckmann R & Forster F (2015). Structure of the native Sec61 protein‐conducting channel. Nat Commun 6, 8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S & Wagenknecht T (1994). Cryo‐electron microscopy and three‐dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J Cell Biol 127, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Grassucci R, Frank J, Inui M, Chadwick C & Fleischer S (1992). Cryo‐EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys J 61, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB & Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J Mol Biol 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Rosenthal PB & Rubinstein JL (2015). Validating maps from single particle electron cryomicroscopy. Curr Opin Struct Biol 34, 135–144. [DOI] [PubMed] [Google Scholar]
- Russo CJ & Passmore LA (2014). Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). A Bayesian view on cryo‐EM structure determination. J Mol Biol 415, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiege P, Fine M, Blobel G & Li X (2017). Human TRPML1 channel structures in open and closed conformations. Nature 550, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TR, Yassin AS, Lu Z, Barnard D, Meng X, Lu TM, Wagenknecht T & Agrawal RK (2014). Initial bridges between two ribosomal subunits are formed within 9.4 milliseconds, as studied by time‐resolved cryo‐EM. Proc Natl Acad Sci USA 111, 9822–9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J & Yan N (2017). Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 355, eaal4326. [DOI] [PubMed] [Google Scholar]
- Shen PS, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE & Cao E (2016). The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167, 763–773.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Saotome K & Sobolevsky AI (2017). Swapping of transmembrane domains in the epithelial calcium channel TRPV6. Sci Rep 7, 10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Yang D, Wang Y, de Graaf C, Zhou Q, Jiang S, Liu K, Cai X, Dai A, Lin G, Liu D, Wu F, Wu Y, Zhao S, Ye L, Han GW, Lau J, Wu B, Hanson MA, Liu ZJ, Wang MW & Stevens RC (2017). Human GLP‐1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315. [DOI] [PubMed] [Google Scholar]
- Stewart AG, Lee LK, Donohoe M, Chaston JJ & Stock D (2012). The dynamic stator stalk of rotary ATPases. Nat Commun 3, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Earl LA, Falconieri V, Milne JL & Egelman EH (2016). Resolution advances in cryo‐EM enable application to drug discovery. Curr Opin Struct Biol 41, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J & MacKinnon R (2017). Cryo‐EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Hite RK & MacKinnon R (2017). Cryo‐EM structure of the open high‐conductance Ca2+‐activated K+ channel. Nature 541, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N (1995). Acetylcholine receptor channel imaged in the open state. Nature 373, 37–43. [DOI] [PubMed] [Google Scholar]
- Unwin N & Fujiyoshi Y (2012). Gating movement of acetylcholine receptor caught by plunge‐freezing. J Mol Biol 422, 617–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Perozo E & Allen TW (2017). Towards a structural view of drug binding to hERG K+ channels. Trends Pharmacol Sci 38, 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC & Gadsby DC (2005). CFTR channel opening by ATP‐driven tight dimerization of its nucleotide‐binding domains. Nature 433, 876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC & Gadsby DC (2003). On the mechanism of MgATP‐dependent gating of CFTR Cl− channels. J Gen Physiol 121, 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR & Henderson R (2016). Single particle electron cryomicroscopy: trends, issues and future perspective. Q Rev Biophys 49, e13. [DOI] [PubMed] [Google Scholar]
- Wang W & MacKinnon R (2017). Cryo‐EM structure of the open human ether‐a‐go‐go‐related K+ channel hERG. Cell 169, 422–430.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher JR & MacKinnon R (2016). Structure of the voltage‐gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HE, Ignatiou A, Clare DK & Orlova EV (2017). Structural study of heterogeneous biological samples by cryoelectron microscopy and image processing. Biomed Res Int 2017, 1032432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler PA, Huang Y, Sun W, Du J & Lu W (2017). Electron cryo‐microscopy structure of a human TRPM4 channel. Nature 552, 200–204. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q & Yan N (2016). Structure of the voltage‐gated calcium channel Cav1.1 at 3.6 A resolution. Nature 537, 191–196. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M & Yan N (2015). Structure of the voltage‐gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- Yan Z, Bai X, Yan C, Wu J, Li Z, Xie T, Peng W, Yin C, Li X, Scheres SHW, Shi Y & Yan N (2015). Structure of the rabbit ryanodine receptor RyR1 at near‐atomic resolution. Nature 517, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J & Yan N (2017). Structure of the Nav1.4‐beta1 complex from electric eel. Cell 170, 470–482.e11. [DOI] [PubMed] [Google Scholar]
- Yin Y, Wu M, Zubcevic L, Borschel WF, Lander GC & Lee SY (2017). Structure of the cold‐ and menthol‐sensing ion channel TRPM8. Science 359, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J & Marks AR (2015). Structure of a mammalian ryanodine receptor. Nature 517, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R & Marks AR (2017). Ca2+ release channels join the ‘resolution revolution’. Trends Biochem Sci 42, 543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC & Grigorieff N (2008). Near‐atomic resolution using electron cryomicroscopy and single‐particle reconstruction. Proc Natl Acad Sci USA 105, 1867–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z & Chen J (2016). Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 167, 1586–1597.e9. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu F & Chen J (2017). Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 170, 483–491.e8. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y & Agard DA (2017). MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Morais‐Cabral JH, Kaufman A & MacKinnon R (2001). Chemistry of ion coordination and hydration revealed by a K+ channel‐Fab complex at 2.0 A resolution. Nature 414, 43–48. [DOI] [PubMed] [Google Scholar]
- Zubcevic L, Herzik MA Jr, Chung BC, Liu Z, Lander GC & Lee SY (2016). Cryo‐electron microscopy structure of the TRPV2 ion channel. Nat Struct Mol Biol 23, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]