

Abstract
Key points
Traumatic brain injury (TBI) in children remains a leading cause of death and disability and it remains poorly understood why children have worse outcomes and longer recover times.
TBI has shown to alter cortical excitability and inhibitory drive onto excitatory neurons, yet few studies have directly examined changes to cortical interneurons.
This is addressed in the present study using a clinically relevant model of severe TBI (controlled cortical impact) in interneuron cell type specific Cre‐dependent mice.
Mice subjected to controlled cortical impact exhibit specific loss of parvalbumin (PV) but not somatostatin immunoreactivity and cell density in the peri‐injury zone.
PV interneurons are primarily of a fast‐spiking (FS) phenotype that persisted in the peri‐injury zone but received less frequent inhibitory and stronger excitatory post‐synaptic currents.
The targeted loss of PV‐FS interneurons appears to be distinct from previous reports in adult mice suggesting that TBI‐induced pathophysiology is dependent on the age at time of impact.
Abstract
Paediatric traumatic brain injury (TBI) is a leading cause of death and disability in children. Traditionally, ongoing neurodevelopment and neuroplasticity have been considered to confer children with an advantage following TBI. However, recent findings indicate that the paediatric brain may be more sensitive to brain injury. Inhibitory interneurons are essential for proper cortical function and are implicated in the pathophysiology of TBI, yet few studies have directly investigated TBI‐induced changes to interneurons themselves. Accordingly, in the present study, we examine how inhibitory neurons are altered following controlled cortical impact (CCI) in juvenile mice with targeted Cre‐dependent fluorescence labelling of interneurons (Vgat:Cre/Ai9 and PV:Cre/Ai6). Although CCI failed to alter the number of excitatory neurons or somatostatin‐expressing interneurons in the peri‐injury zone, it significantly decreased the density of parvalbumin (PV) immunoreactive cells by 71%. However, PV:Cre/Ai6 mice subjected to CCI showed a lower extent of fluorescence labelled cell loss. PV interneurons are predominantly of a fast‐spiking (FS) phenotype and, when recorded electrophysiologically from the peri‐injury zone, exhibited intrinsic properties similar to those of control neurons. Synaptically, CCI induced a decrease in inhibitory drive onto FS interneurons combined with an increase in the strength of excitatory events. The results of the present study indicate that CCI induced both a loss of PV interneurons and an even greater loss of PV expression. This suggests caution is required when interpreting changes in PV immunoreactivity alone as direct evidence of interneuronal loss. Furthermore, in contrast to reports in adults, TBI in the paediatric brain selectively alters PV‐FS interneurons, primarily resulting in a loss of interneuronal inhibition.
Keywords: traumatic brain injury, cortex, pediatric, parvalbumin, interneuron
Key points
Traumatic brain injury (TBI) in children remains a leading cause of death and disability and it remains poorly understood why children have worse outcomes and longer recover times.
TBI has shown to alter cortical excitability and inhibitory drive onto excitatory neurons, yet few studies have directly examined changes to cortical interneurons.
This is addressed in the present study using a clinically relevant model of severe TBI (controlled cortical impact) in interneuron cell type specific Cre‐dependent mice.
Mice subjected to controlled cortical impact exhibit specific loss of parvalbumin (PV) but not somatostatin immunoreactivity and cell density in the peri‐injury zone.
PV interneurons are primarily of a fast‐spiking (FS) phenotype that persisted in the peri‐injury zone but received less frequent inhibitory and stronger excitatory post‐synaptic currents.
The targeted loss of PV‐FS interneurons appears to be distinct from previous reports in adult mice suggesting that TBI‐induced pathophysiology is dependent on the age at time of impact.
Introduction
Children are one of the most at risk groups with respect to suffering a traumatic brain injury (TBI), with more than one in five children experiencing a TBI before reaching adulthood (Kraus et al. 1990; Bruns & Hauser, 2003). A TBI may range from mild to severe, although it will often lead to the development of lifelong cognitive and behavioural deficits (Caveness et al. 1979; Annegers et al. 1998; Barlow et al. 2000). Emerging evidence indicates that children have worse outcomes and take longer to recover then adults (Anderson & Moore, 1995; Schmidt et al. 2012; Cook et al. 2014). How TBI disrupts paediatric brain function and leads to the development of post‐traumatic neurological disorders, such as post‐traumatic epilepsy, remains unknown.
Disruption to cortical inhibition has been repeatedly implicated in the pathophysiology of numerous neurological disorders, including migraine (Palmer et al. 2000; Aurora et al. 2005), schizophrenia (Molloy et al. 2011; Lewis et al. 2012), epilepsy and seizures (Bromfield et al. 2006; Trevelyan & Schevon, 2013; Trevelyan et al. 2015), as well as the sequela that follow TBI. In adult animals, TBI has been shown to induce enhanced cortical excitability (Carron et al. 2016), as well as a loss of synaptic inhibition onto excitatory neurons and a decrease in expression of known markers for multiple subtypes of interneurons (Cantu et al. 2015). However, we have previously found that juvenile mice respond uniquely after TBI with a lack of generalized hyperexcitability (Goddeyne et al. 2015; Nichols et al. 2015) and the presence of unique synaptic bursts following more severe TBI (Nichols et al. 2015). This suggests the pathophysiology of TBI in juvenile animals is distinct, at least in part, from adults. In children, TBI has been shown to disrupt brain function and maturation via mechanisms distinct from adult TBI (Anderson & Moore, 1995; Schmidt et al. 2012; Cook et al. 2014). GABAergic neurons play a critical role in cortical development and disruption of the immature inhibitory circuit can have severe effects long into adulthood (Wang & Kriegstein, 2009; Judson et al. 2016). However, the precise nature of the changes to interneurons and the inhibitory circuit induced by paediatric TBI is poorly understood. Even in the larger body of studies performed in adult animals, there is a paucity of direct examination of synaptic and cellular changes to cortical interneurons. In the present study, we directly examine how inhibitory interneurons in the cortex are altered in an animal model of paediatric TBI.
Cortical interneurons are a diverse population of cells with unique intrinsic and synaptic properties (Hensch, 2005; Yuste, 2005; Wonders & Anderson, 2006; Rudy et al. 2011) To examine for changes in the interneuron population, conventional approaches have often used cell type specific immunohistochemical markers. Parvalbumin (PV) and somatostatin (SST) immunohistochemical positive interneurons account for >70% of the interneuron population (Rudy et al. 2011) and have been implicated in the pathophysiology of TBI (Hunt et al. 2011; Cantu et al. 2015) and epilepsy (Caveness et al. 1979; Cossart et al. 2001; Jin et al. 2014). However, examination of immunohistochemical markers is unable to differentiate between a loss of immunoreactivity and overt cell death. Distinguishing between these two possibilities in interneurons is of particular importance to understanding the pathophysiology of TBI and potential interneuron targeted therapeutic interventions. Therefore, in the present study, we took advantage of the fact that almost all GABAergic interneurons selectively express the vesicular GABA transporter (VGAT) (McIntire et al. 1997; Chaudhry et al. 1998; Wojcik et al. 2006). We examined the outcome of TBI in transgenic animals that express Cre recombinase under the control of the Vgat promoter (Vgat:Cre) (Vong et al. 2011) and a Cre‐dependent red fluorescent protein reporter (CAG‐loxp‐STOP‐loxp‐tdTomato; hereafter referred to as Ai9) (Madisen et al. 2010) to ubiquitously label GABAergic neurons. Furthermore, we similarly investigated interneuron subtype specific changes using transgenic mice with PV:Cre‐dependent activation of a green fluorescent protein reporter (CAG‐loxp‐STOP‐loxp‐ZsGreen1). Consequently, because the fluorescent proteins are expressed under the control of the strong and ubiquitous CAG promoter, we were able to directly determine whether TBI induces an actual loss of interneurons and also examine for functional changes to cortical inhibition.
Juvenile mice were subjected to controlled cortical impact (CCI), which has previously been shown to induce a clinical relevant injury that effectively models a severe TBI (Brody et al. 2007; Hunt et al. 2009; Nichols et al. 2015). Combined with whole‐cell patch clamp and fluorescence immunohistochemical (IHC) approaches, we quantified CCI‐induced changes to cortical interneurons. Although CCI did not induce a general interneuron loss, in the peri‐injury zone, a significant reduction in the expression of PV immunoreactivity was observed in fluorescence labelled interneurons. By contrast to similar experiments conducted in adults, no accompanying loss of SST expression was observed (Cantu et al. 2015). PV positive interneurons are predominantly of a fast‐spiking (FS) phenotype (Hu et al. 2014). Despite TBI‐induced loss of PV interneurons, electrophysiological recordings confirmed the presence of FS interneurons with intrinsic properties similar to those of control neurons. However, there was a marked decrease in inhibitory synaptic drive onto FS interneurons with an accompanying increase in the strength of excitatory synaptic events. Our data indicate that, in contrast to previous reports in adult animals, CCI in juvenile mice preferentially alters PV‐FS cortical interneurons.
Methods
Ethical approval
All laboratory and animal experiments were conducted in accordance with NIH guidelines, were approved by the University of Arizona Institutional Animal Care and Use Committee and conform to the principles and guidelines of The Journal of Physiology (Grundy, 2015). Genetically modified mouse lines were bred in‐house from commercially available strain (Jackson Laboratories, Bar Harbor, ME, USA). Specifically, Vgat:Cre mice (B6J.129S6(FVB)‐Slc32a1tm2(cre)Lowl/MwarJ , #028862) have been characterized previously (Vong et al. 2011) and have Cre‐recombinase directed to inhibitory GABAergic neurons. Vgat:Cre mice were bred with Ai9 mice (Gt(ROSA)26Sortm9(CAG‐tdTomato)Hze , #007909) allowing for the Cre‐dependent expression of red‐fluorescent protein (tdTomato) under the ubiquitous CAG promoter selectively in inhibitory GABAergic neurons. Similarly, PV:Cre mice (B6;129P2‐Pvalbtm1(cre)Arbr/J, #008069) selectively express Cre recombinase in PV‐expressing neurons (Hippenmeyer et al. 2005) and were bred with Ai6 mice (B6.Cg‐GT(ROSA) 26Sortm6(CAG‐ZSGreen1)Hze, #007906) allowing for Cre‐dependent expression of green‐fluorescent protein (ZsGreen1) under the CAG promoter selectively in PV neurons.
CCI
Juvenile mice, at post‐natal day (P)22, were subjected to a severe CCI as described previously (Hunt et al. 2009; Nichols et al. 2015). Animal temperature was maintained for the duration of surgery and until mice were ambulatory post‐surgery. The skull was exposed with a midline incision and a 5 mm craniotomy was made over the right somatosensory region (lateral to the sagittal suture between bregma and lambda). Precaution was taken during the craniotomy not to disturb the underlying dura. A frontoparietal CCI (3 mm diameter tip, 3.0 m s−1, 2 mm depth, 500 ms in duration) was performed using an electromagnetic cortical impactor (Hatteras Instruments, Cary, NC, USA). After the impact, the removed bone flap was placed over the site of injury and sealed with dental cement. Control animals were naïve because previous reports have found that a craniotomy alone may be sufficient to cause an inflammatory response and/or induce brain injury (Olesen, 1987; Cole et al. 2010).
Preparation of brain slices
We have previously shown that all animals subjected to CCI develop in vivo epileptic activity by 14 days after injury and that this is an effective time‐point for examining pathophysiological network changes after TBI (Nichols et al. 2015). Accordingly, to examine for CCI‐induced changes to the inhibitory network, mice were deeply anaesthetized by inhalation of isoflurane and decapitated on post‐injury day (PID) 14–19. The brain was rapidly removed and coronal slices (350 μm thick) prepared on a vibratome (VT 1200; Leica, Nussloch, Germany) as described previously (Anderson et al. 2010; Nichols et al. 2015). Slices were obtained from the somatosensory cortex at the site of injury in CCI animals or from the corresponding control cortex in control animals. The site of CCI was readily identifiable in whole brain and slices as indicated by significant cavitation and tissue loss (Fig. 1). Initial harvesting of brain slices was performed in an ice‐cooled (4°C) carboxygenated (95% O2, 5% CO2) high sucrose solution containing (in mm): 234 sucrose, 11 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4H2O, 10 MgSO47H2O and 0.5 CaCl22H2O. Slices were then incubated for 1 h at 32°C in carboxygenated artificial CSF (aCSF) containing (in mm): 126 NaCl, 26 NaHCO3, 2.5 KCl, 10 Glucose, 1.25 Na2H2PO4H2O, 1 MgSO47H2O and 2 CaCl2H2O (pH 7.4). Slices were then returned to room temperature before being individually moved to the recording chamber for whole‐cell patch clamp recording.
Figure 1. CCI induces marked tissue loss and cavitation at site of injury.
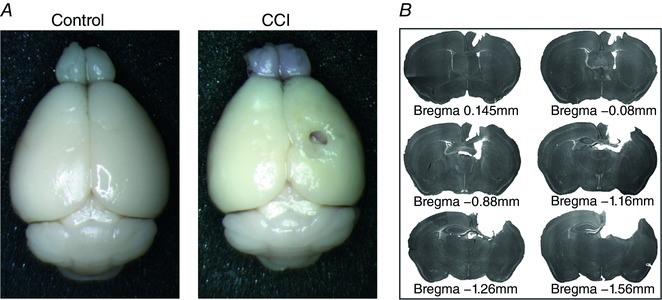
A, example images of perfused control and CCI VGAT cre; Ai9 P36 mouse brains 14 days after injury. B, serial images of 60 μm slices showing extent of CCI induced injury. [Color figure can be viewed at http://wileyonlinelibrary.com]
In vitro electrophysiological recording
Coronal slices were prepared from CCI or control animals, placed in the recording chamber and submerged in flowing carboxygenated aCSF heated to 32°C. Submerged slices were first visualized under 4× brightfield for identification of layer V cortex. For slices from impacted mice, recordings were made in the peri‐injury zone within 700 μm of the injury‐induced cavitation. Recordings from control slices were made in the corresponding cortex to the peri‐injury zone of CCI animals. Whole‐cell recordings were obtained from FS interneurons using an upright microscope (Axioexaminer; Carl Zeiss, Thornwood, NY, USA) fitted with infrared differential interference contrast (DIC) optics. Fast‐spiking neurons were distinguished based on their current clamp firing behaviour (Kawaguchi & Kubota, 1997). The electrode capacitance and bridge circuit were appropriately adjusted. The series resistance (R s) of neurons chosen for analysis was <20% of membrane input resistance and monitored for stability. Membrane potential was not corrected for a calculated 10 mV liquid junction potential. A Multiclamp 700B patch clamp amplifier (Axon Instruments, Union City, CA, USA) was used for both current and voltage clamp mode. Recordings were obtained at 32°C using borosilicate glass microelectrodes (tip resistance, 2–5 MΩ). For recording of excitatory events, electrodes were filled with an intracellular solution containing (in mm): 135 KGluconate, 4 KCl, 2 NaCl, 10 Hepes, 4 EGTA, 4 Mg ATP and 0.3 Na Tris. With a calculated E Cl = −80 mV and a holding potential of −70 mV, excitatory events were isolated as inward currents. Pharmacological isolation was avoided to prevent induced hyperexcitability that would interfere with baseline measurements of synaptic activity. For recording of inhibitory events, a high‐chloride intracellular solution containing was used containing (in mm): 70 KGluconate, 70 KCl, 2 NaCl, 10 Hepes, 4 EGTA, 4 Mg ATP and 0.3 GTP. This internal solution has been used previously (Anderson et al. 2010; Nichols et al. 2015) and shown to facilitate the detection of inhibitory events. The calculated E Cl was −16 mV, resulting in inward GABAA currents at a holding potential of −70 mV. Inhibitory events were pharmacologically isolated by bath application of kynurenic acid (2 mm).
Whole‐cell patch clamp analysis
Data analysis was performed using pCLAMP (Axon Instruments, Sunnyvale, CA, USA) Prism (GraphPad, La Jolla, CA, USA) and MiniAnalysis (Synaptosoft, Decatur, GA, USA). Data are reported as the mean ± SEM. For the detection of spontaneous events and parameters, automated threshold detection was employed using MiniAnalysis and detected events were subsequently manually verified. Decay time was calculated as time from peak to reach 37% of the peak amplitude. Input resistance was calculated from the steady‐state voltage response to the input of a 1 s, −50 pA current step. Firing frequency was calculated by the number of spikes elicited per current step (−150 pA to 300 pA steps, 1 s duration). Resting membrane potential was measured prior to induced current steps. Action potentials were induced by intracellular current steps (2 nA, 0.5 ms), repeated 10 times and averaged prior to analysis. Rheobase was measured as the minimum 50 ms long current step that induced an action potential and was assessed with current delivered in +5 pA increments.
Immunohistochemistry
PID 14 mice and age‐matched control animals were deeply anaesthetized and perfused transcardially with 4% paraformaldehyde/PBS essentially as described previously (Xing et al. 2016). Immunohistochemistry was performed using 60 μm free‐floating vibratome sections. Brain sections were collected in PBS, blocked with 5% normal serum in PBS with 0.1% Triton X‐100 and incubated with primary antibodies in blocking solution for 24–48 h at 4°C. The primary antibodies used in the present study were: mouse anti‐NeuN (dilution 1:1000; Chemicon, Temecula, CA, USA), goat anti‐PV (dilution 1:1000; PVG213; Swant, Barron, WI, USA) and rabbit anti‐SST‐14 (dilution 1:1000; T4103; Peninsula Laboratories, Belmont, CA, USA). After rinsing, sections were then incubated with Alexa conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA) and 4′,6‐diamidino‐2‐phenylindole (DAPI) (dilution 1:2000; Roche, Basel, Switzerland) overnight at 4°C, rinsed three times in PBS, and mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA, USA). For biocytin filled neurons, sections were labelled with Alexa‐488 conjugated streptavidin (dilution 1:1000; Jackson Immunoresearch, West Grove, PA, USA) prior to rinsing and mounting.
Image acquisition and data analysis
Images were collected on a model SP5 (Leica) and a model 710 (Carl Zeiss) laser scanning confocal microscope using 10× or 20× objectives. Single fields were tiled together to generate high‐resolution images of whole brain coronal sections. Images were corrected for brightness and contrast in Photoshop (Adobe Inc., San Jose, CA, USA). For the assessment of changes in neuron density, we defined a 350 μm wide region of interest (ROI) that extended across all cortical layers and was oriented perpendicular to the white matter. The ROI was positioned 3.15 mm lateral to midline on the injured hemisphere (i.e. over the lateral peri‐injury site). For semi‐automated assessment of Vgat:Cre/Ai9 + and NeuN+ cell density, single channel images from three separate sections were imported into ImageJ (NIH, Bethesda, MD, USA). Images were manually thresholded to optimize binary masking and the detection of immunofluorescent soma. To separate clearly overlapping soma, a watershed algorithm was applied. The analyse particle plugin was then employed using a minimal particle size of 70 pixels2 (∼50 μm2), which was chosen to enrich the detection of well labelled cell soma and to minimize counting cross‐sectioned neuritis (Ippolito & Eroglu, 2010; Xing et al. 2016) (Ippolito & Eroglu, 2010; Xing et al. 2016). For the analysis of the less abundant PV‐ and SST‐expressing neurons, area and number were manually determined in Photoshop. To determine density, the number of labelled neurons was recorded and divided by the area of tissue within the ROI. Density values were then averaged across three sections per animal. The results from this analysis were averaged using the data from at least three independent mice originating from separate litters per condition. For sholl analysis, 100 μm wide regions, covering all cortical layers, were positioned serially beginning with the edge of the injury site. Data are reported as the mean ± SEM. Statistical significance was tested with a one‐way ANOVA with Tukey's multiple comparison test or Student's t test (paired or unpaired). P < 0.05 was considered statistically significant.
Results
To examine TBI‐induced changes in the cortical inhibitory neuronal population, we performed unilateral CCI injuries on juvenile (P22) Vgat:Cre/Ai9 mice. As previously described, animals were given 14 days to recover before further experimentation because this was the earliest time‐point by which all juvenile animals subjected to CCI reliably showed spontaneous epileptic activity (Nichols et al. 2015). By PID 14, animals subjected to CCI showed marked cavitation and tissue loss at the site of injury (Fig. 1). Despite the significant tissue loss at the site of the CCI injury, no significant change in the thickness of the cortex was observed in the peri‐injury zone (control: 1219.27 ± 22.10 μm; CCI: 1093.83 ± 56.33 μm) (P = 0.13) (n = 3).
Inhibitory interneuron (Ai9) cell density was unchanged after CCI
To quantify CCI‐induced changes in different populations of cortical neurons, we performed IHC staining using the neuron specific marker NeuN or examined tdTomato labelled interneurons in transgenic mice (Vgat:Cre/Ai9). First, utilizing counts of the NeuN positive cells, we observed that CCI induced no significant change in neuronal density in the peri‐injury zone (control: 2321.1 ± 106 cells mm−2; CCI: 2212.7 ± 74 cells mm−2) (Fig. 2) (P = 0.15). Next, we were able to directly examine for CCI‐induced changes in the interneuron population by taking advantage of the select Vgat:Cre‐dependent expression of tdTomato (Ai9) in interneurons. Similar to the NeuN findings, we observed no significant difference in tdTomato+ cell density between control and CCI mice (control: 471.7 ± 29 cells mm−2; CCI: 434.6 ± 39 cells mm−2) (P = 0.49) (Fig. 2). These experiments determined that, 14 days after a CCI in the juvenile brain, there is no significant change in the density of either total excitatory or inhibitory neurons within the peri‐injury zone.
Figure 2. Ai9 cell density is not significantly changed in the peri‐injury zone after CCI.
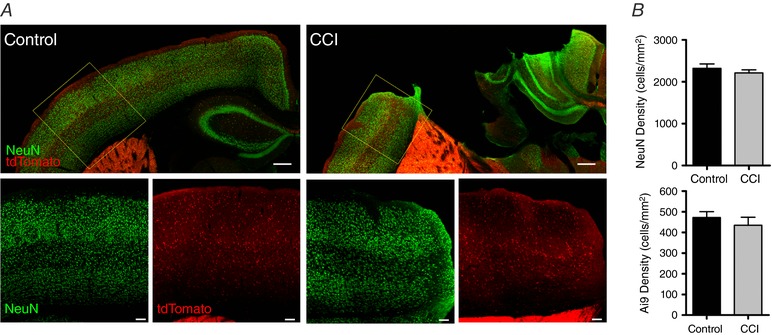
A, top: NeuN (green) is qualitatively similar between control and CCI. Similarly, Ai9 (tdTomato) distribution is not altered after CCI. Bottom: higher magnification images taken from the peri‐injury zone (yellow dotted box). Note the presence of total neurons (NeuN) and GABAergic neurons (Ai9) throughout this region. B, quantitative analysis shows that neither NeuN (top) or Ai9 cell density (bottom) are significantly reduced in CCI animals (n = 3 mice) compared to control (n = 3 mice) (NeuN: Control vs. CCI, P = 0.15; Ai9: Control vs. CCI, P = 0.49, unpaired Student's t test). Top: scale bar = 300 μm. Bottom: scale bar = 100 μm.
VGAT is reported to be expressed in 99% of all cortical interneurons (McIntire et al. 1997; Chaudhry et al. 1998; Vong et al. 2011) and the use of a Vgat:Cre/Ai9 transgenic mouse allowed us to directly examine for changes in the global interneuron population without confounds related to alterations in the expression of typical immunological targets. However, cortical interneurons are a mixed population of neurons with numerous subtypes and unique physiological roles (Gupta et al. 2000; Markram et al. 2004). One effective method for segregating interneurons subtypes is based on the gene expression pattern (Rudy et al. 2011). Specifically, in the cortex, the two most common interneuron subtypes express either PV (40–50%) or SST (∼30%) (Markram et al. 2004, Rudy et al. 2011). Having established a lack of global cortical interneuron loss following CCI, we next investigated whether CCI induced changes in select interneuron subtypes by examining the expression of PV and SST.
Selective loss of PV expression following CCI
To examine for TBI‐induced changes in PV expression, we used an immunohistochemical approach and again performed CCI in juvenile Vgat:Cre/Ai9 mice. By contrast to tdTomato labelled interneurons where we observed no significant change, CCI induced a dramatic reduction in the number of cells that express PV (Fig. 3 A). Specifically, CCI significantly reduced the density of neurons positively labelled by anti‐PV antibody by >71% in the peri‐injury zone (32.2 ± 5 cells mm−2) compared to control (110.4 ± 5 cells mm−2) (P < 0.0001) (Fig. 3 A and B). Finally, to investigate whether the CCI‐induced loss of PV expression was correlated with distance from the site of the injury, we examined the number of PV‐expressing neurons within 100 um ROIs (bins) extending even further to 1 mm lateral from the edge of the injury. The loss of PV expression induced by CCI was significantly greater near (<300 μm) the injury site (P < 0.05) (Fig. 3 B).
Figure 3. PV immunoreactivity is reduced after CCI.
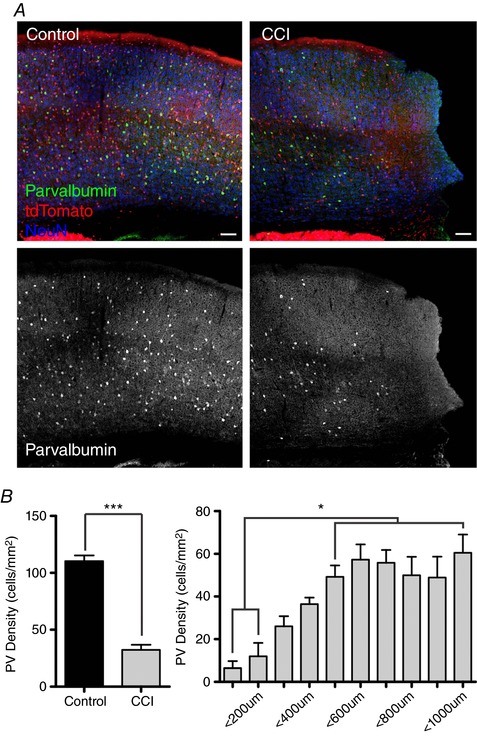
A, peri‐injury regions with PV in green, Ai9 in tdTomato and NeuN in blue. (Bottom) Monochrome image of immunostaining of PV shows loss of expression after injury. Scale bar = 100 μm. B, left: quantification of PV density is significantly reduced after CCI (n = 5 mice per group, *** P < 0.0001, unpaired Student's t test). Right: distal analysis with 100 μm wide columns was used to analyse changes in PV expression in relation to injury. Quantitatively, it shows that the loss of PV expression is greatest closest to the injury site (n = 5 mice, * P < 0.05, one‐way ANOVA with a post hoc Tukey's test).
SST expression is not altered by CCI
Using an IHC approach, we examined for changes in SST expression in the peri‐injury zone after CCI in juvenile Vgat:Cre/Ai9 mice (Fig. 4 A). By contrast to PV, we found that CCI induced no statistically significant difference in the density of SST+ neurons in the peri‐injury zone compared to control animals (control: 108.3 ± 14 cells mm−2; CCI: 111.9 ± 11 cells mm−2) (P = 0.85) (Fig. 4 B). Similarly, no significant loss of SST positive neurons was observed across any 100 μm ROIs extending laterally from the site of injury (P > 0.05, one‐way ANOVA). Taken together, these results show that, in juvenile mice, CCI does not induce a loss of SST‐expression but, instead, selectively reduces PV‐expression in the peri‐injury zone.
Figure 4. CCI does not alter the density of SST neurons in the cortex after CCI.
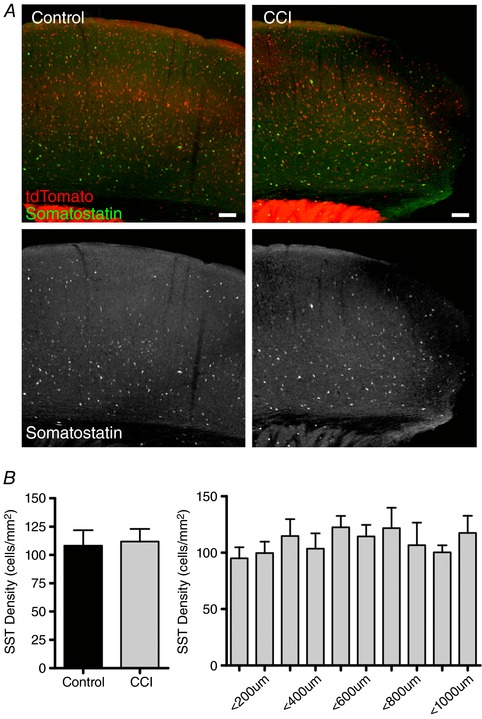
A, top: control co‐labelling of GABAergic neurons (tdTomato) and SST interneurons (green). Bottom: monochrome images of immunostaining of SST interneurons reveals no change in number after injury. Scale bar = 100 μm. B, left: quantification of SST density is not significantly altered after CCI (n = 3 mice per group, P = 0.63, unpaired Student's t test. Right: distal analysis with 100 μm wide columns was used to analyse changes in SST expression in relation to injury. Quantitatively, it shows no loss of SST expression ranging from peri‐injury zone to 1 mm lateral of injury site (n = 5 mice), one‐way ANOVA with a post hoc Tukey's test.
PV interneuron (Ai6) cell density is decreased in the peri‐injury zone
The observed decrease in PV expression may be solely the result of a loss of PV immunoreactivity or may reflect an actual loss of PV interneurons. To distinguish between these possibilities, we employed Cre‐dependent genetic labelling of PV‐expressing neurons with ZsGreen1 (PV:Cre/Ai6). PV expression rises postnally and is well established by the time of impact at P22 (de Lecea et al. 1995; Philpot et al. 1997; Erickson & Lewis, 2002), thereby indelibly labelling PV interneurons with ZsGreen1 (del Río et al. 1994; Huang et al. 1999; Madisen et al. 2010). We were therefore able to fluorescence‐label neurons that expressed PV at some point during development. Ongoing reporter gene expression was therefore independent of subsequent changes in PV promoter activity or immunoreactivity that might occur following CCI. One caveat to this approach is the potential labelling of ‘off‐target’ cells, such as a few layer 5 excitatory neurons and SST neurons (Tanahira et al. 2009; Kobayashi & Hensch, 2013; Fujihara et al. 2015; Nassar et al. 2015) In line with these previous studies, we noted a significant fraction of ZsGreen1+ labelled cortical neurons in PV:Cre/Ai6 mice do not express immunohistochemically significant levels of PV (Fig. 5).
Figure 5. PV expression and cell density are decreased after CCI in the peri‐injury zone.
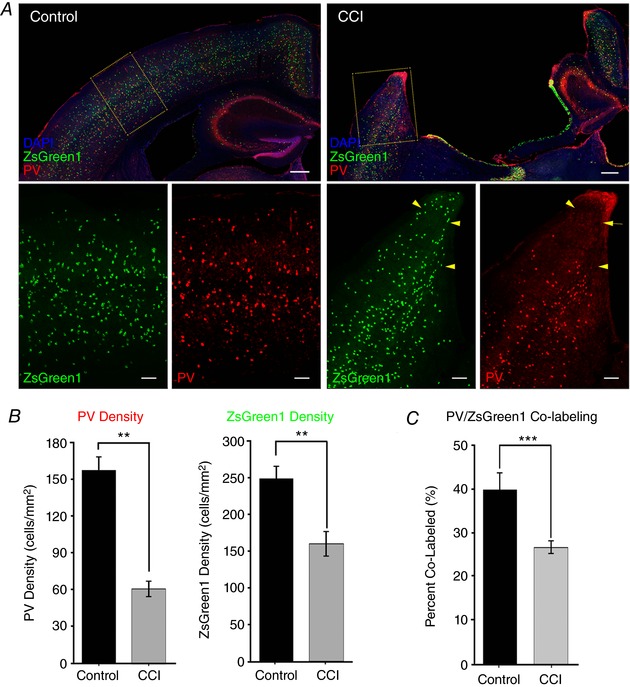
A, representative images of ZsGreen1 expressing cells in PV:cre Ai6 mouse cortex from control or after CCI. Sections were counter‐stained with DAPI (blue) and PV antibody (red). Higher magnification images from the region of the peri‐injury zone (yellow box) are shown below. Note the presence of ZsGreen1 positive neurons in the peri‐injury zone that are not co‐immunoreactive with PV (yellow arrows). B, bar charts quantifying the statistically significant reduction in PV and ZsGreen density in the per‐injury zone (n = 4 mice per group). C, the proportion of PV/ZsGreen1 co‐labelled cells is signficantly reduced in the peri‐injury zone following CCI. ** P < 0.01,*** P < 0.0001 unpaired Student's t test). Top: scale bar = 300 μm. Bottom: scale bar = 100 μm.
In agreement with our IHC findings in Vgat:Cre/Ai9 mice, TBI in PV:Cre/Ai6 mice led to a 61% reduction in the density of cells labelled by PV IHC in the peri‐injury zone (control: 157.14 ± 12.5 cells mm−2; CCI: 60.9 ± 7 cells mm−2, P < 0.001). Interestingly, we found that the relative density of ZsGreen1+ (i.e. PV:Cre dependently labelled) cells was only reduced by 39% in the peri‐injury zone compared to control mice (control: 248.7 ± 17 cells mm−2; CCI: 152.9 ± 21 cells mm−2 CCI, P < 0.01) (Fig. 5 A and B). Consistent with these results, the relative proportion of ZsGreen1+ cells that are also immunoreactive for PV [(PV+ and ZsGreen+)/Total ZsGreen+] was significantly reduced by almost 34% in the peri‐injury zone (control: 39.09 ± 4.1%; CCI: 25.97 ± 1.7%, P < 0.0001) (Fig. 5 C). Overall, these data demonstrate that the global reduction of PV expression in the peri‐injury zone is attributable to both the overt loss of a significant fraction of PV‐expressing interneurons in addition to a reduction in PV expression in a subset of PV:Cre labelled neurons that persist following TBI.
Fast‐spiking interneurons are not significantly altered following CCI
Subtypes of cortical interneurons can also be categorized by their electrophysiological patterns of action potential firing in response to sustained depolarizing current injection (Kawaguchi & Kubota, 1997; Markram et al. 2004; Uematsu et al. 2008; Rudy et al. 2011). PV positive interneurons are predominantly of a FS electrophysiological phenotype (Galarreta & Hestrin, 1999) and are considered to play an important role in the maintenance of spike timing (Kinney et al. 2006; Orduz et al. 2013) and rhythmic firing (Orduz et al. 2013). Consequently, the select loss of PV expression following CCI led us to investigate whether the interneurons that persist in the peri‐injury zone exhibit altered electrophysiological properties. We performed whole‐cell patch clamp experiments in the peri‐injury zone of coronal brain slices taken from Vgat:Cre/Ai9 mice 14 days after CCI or in corresponding regions of un‐injured control cortices. Neurons were first visually identified via DIC bright field imaging and confirmed to be tdTomato positive. Under current clamp, neurons were confirmed to have a FS phenotype based on the firing pattern induced by a series of intracellular current steps (−150 pA to 300 pA, 50 pA steps, 1 s) (Fig. 6 A) (Anderson et al. 2010; Hu et al. 2014). Following CCI, FS interneurons had a more depolarized resting membrane potential (‐65.52 ± 1.1 mV) compared to control (–69.69 ± 1.6 mV) (P < 0.05). However, this was not accompanied by any significant difference in the input resistance (control: 150.42 ± 18.1 MΩ; CCI: 156.46 ± 17.3 MΩ, P = 0.82) or average firing frequency across various current steps (P > 0.05, one‐way ANOVA) (Fig. 6 A). Similarly, compared with the control, no significant differences were observed in action potential height (control: 84.43 ± 4.1 mV; CCI: 83.20 ± 2.9 mV, P = 0.80), threshold (control: −40.49 ± 0.9 mV; CCI: −41.33 ± 0.9 mV, P = 0.76) or half‐width (control: 0.63 ± 0.1 ms; CCI: 0.87 ± 0.1 ms, P = 0.07) (Fig. 6 B). Rheobase appeared decreased following CCI, although the change failed to reach significance (control: 152.5 ± 25 pA; CCI: 107.3 ± 18 pA, P = 0.14) (Fig. 6 C). These findings confirmed that, although CCI induces a decrease in the immunohistochemical expression of PV, the FS interneurons that remain in the peri‐injury zone possess similar intrinsic electrophysiological properties as control FS interneurons.
Figure 6. FS interneurons remain in the peri‐injury zone following CCI.
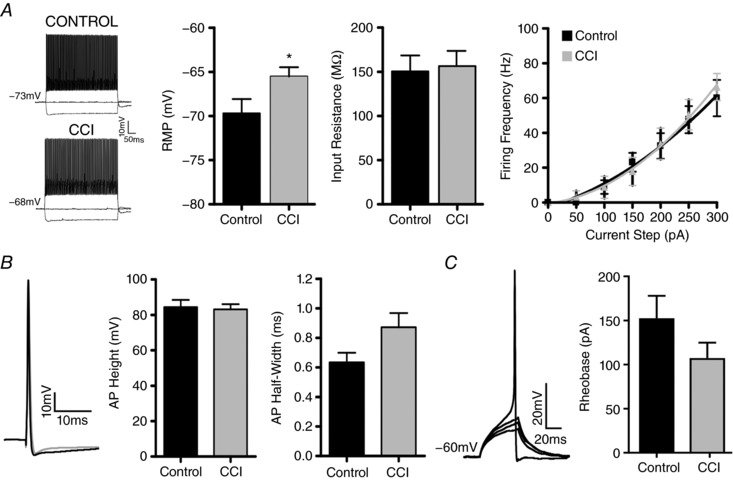
Whole‐cell patch clamp recordings from FS neurons recorded from Vgat:Ai9 positive neurons from control (n = 10) or CCI (n = 15) mice. A, current clamp recordings of FS interneurons in response to intracellular current steps (−150, 0 and 250 pA). Resting membrane potential of FS interneurons is more depolarized following CCI (* P < 0.05, unpaired Student's t test), whereas input resistance (P > 0.05, unpaired Student's t test) and firing frequency remain unchanged (curves fit with second‐order polynomial, R2 = 0.990 control, 0.997 CCI). B, examples of average single action potential (2 nA, 0.5 ms) from control (black) or CCI (grey) (left) and quantification of action potential height and half‐width (right) (P > 0.05, unpaired Student's t test). C, example of a series of rheobase trial traces (left) and quantification (right) indicating no change between control and CCI (P > 0.05, unpaired Student's t test).
CCI reduces inhibitory sIPSC frequency and increases synaptic kinetics
Next, we aimed to investigate whether synaptic neurotransmission onto FS interneurons was altered following CCI. We first examined for changes in inhibitory activity by recording spontaneous IPSCs (sIPSCs) in FS interneurons in the peri‐injury zone 14 days after CCI or from corresponding cortical region in age‐matched control animals. sIPSCs were pharmacologically isolated by bath application of the glutamate receptor antagonist kynurenic acid (2 mm). To increase the detection of inhibitory synaptic events, a modified internal solution (E Cl‐ = −16 mV) was used as described previously (Anderson et al. 2010). No significant change was observed in the amplitude (control = 23.10 ± 2.9 mV, CCI = 21.69 ± 1.9 mV) (P = 0.68) or charge (control = 77.43 ± 9.6 C, CCI = 91.84 ± 7.1 C) (P = 0.23) of sIPSCs. However, CCI significantly reduced the frequency of sIPSCs by >50% (control = 4.34 ± 0.8 Hz; CCI 1.96 ± 0.3 Hz) (P = 0.004) (Fig. 7 A–D). CCI also induced significant changes to the kinetics of sIPSC, as indicated by increases in both the rise (control = 2.56 ± 0.3 ms, CCI = 3.36 ± 0.2 ms) (P < 0.05) and decay (control = 3.32 ± 0.2 ms, CCI = 4.43 ± 0.4 ms) (P < 0.05) time (Fig. 7 E–G). This suggests that CCI primarily induces a loss of inhibitory drive onto FS interneurons.
Figure 7. sIPSC frequency and kinetics are increased onto FS interneurons after CCI.
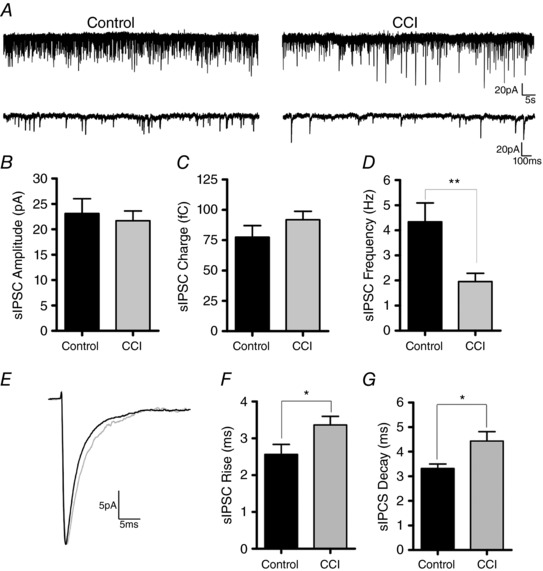
A, gap‐free traces of sIPSCs recorded in voltage clamp from FS interneurons in control (n = 10) or after CCI (n = 15) (Vhold = −70 mV). B–C, bar graphs of average sIPSC properties indicating no significant change in sIPSC amplitude or charge following CCI. D, average sIPSC frequency was significantly reduced after injury (** P < 0.01, unpaired Student's t test). E, overlaid average sIPSC traces recorded in FS interneurons from control (black) or CCI (red). F–G, sIPSC rise and decay times are significantly increased after injury (* P < 0.05, unpaired Student's t test).
Excitatory synaptic strength increases after CCI
Finally, we examined for changes in excitatory input onto FS interneurons by recording spontaneous EPSCs (sEPSCs). For these experiments, a physiological internal was used (E Cl‐ = −80 mV) that allowed the detection of inward glutamatergic events isolated from outward GABAergic synaptic events. Excitatory activity was not pharmacologically isolated because blocking GABAergic activity may disinhibit the network and alter the ability to directly record CCI‐induced changes. Again, whole‐cell recording of sEPSCs in FS interneurons was performed in the peri‐injury zone 14 days after CCI or in corresponding control animals. Analysis revealed a statistically significant increase in the amplitude (control = 14.33 ± 1.7 mV, CCI = 25.55 ± 2.5 mV) (P < 0.001) and charge (control = 32.92 ± 1.9 C, CCI = 61.98 ± 5.8 C) of sEPSCs following CCI compared to control (P < 0.0001) (Fig. 8 A–C). However, in contrast to CCI‐induced changes to sIPSCs, there was no change in sEPSC frequency (control = 8.34 ± 1.1 Hz, CCI = 9.16 ± 1.2 Hz) (P = 0.61), rise time (control = 3.04 ± 0.3 ms, CCI = 2.48 ± 0.2 ms) (P = 0.20) or decay time (control = 2.17 ± 0.2, CCI = 2.47 ± 0.4) (P = 0.50) (Fig. 8 D–G). This suggests that CCI primarily induces an increase in the strength of excitation onto FS interneurons.
Figure 8. The strength of sESPSs onto FS interneurons is increased after CCI.
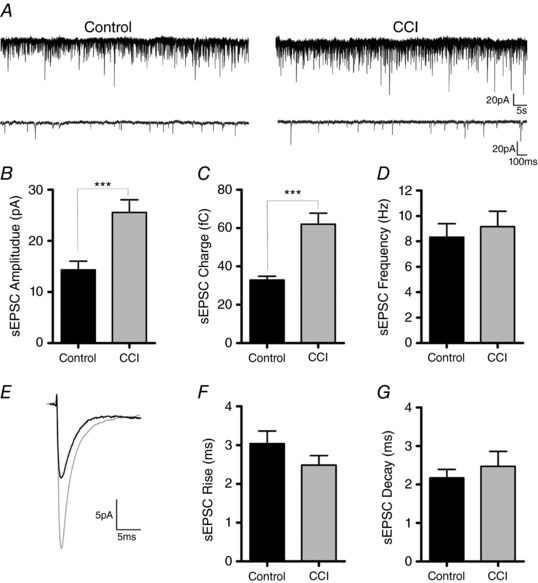
A, gap‐free voltage clamp traces from FS interneurons recorded in control (n = 13) or after CCI (n = 12) (Vhold = −70 mV). B–C, bar graphs of average sEPSC properties indicating a significant increase in amplitude (P < 0.001) and charge (P < 0.0001) following CCI. D, average sEPSC frequency was not significantly reduced after injury. E, overlaid average sEPSC traces recorded in FS interneurons from control (black) or CCI (red). F–G, sEPSC kinetic properties (rise and decay times) are not significantly altered after injury. An unpaired Student's t test was used for all of the statistics.
Discussion
In the present study, we examined how cortical interneurons in juvenile animals are altered following TBI induced by CCI. To our knowledge, this is the first study to examine changes to the cortical interneuron network in a paediatric model of TBI. Using a combined IHC and electrophysiological approach in mice with targeted Cre‐dependent fluorescence labelled interneurons, we identified five key findings. First, as reported previously (Nichols et al. 2015), CCI induced significant tissue loss at the site of impact. However, within the surrounding peri‐injury zone, there was no significant loss in the density of total neurons (NeuN) or inhibitory interneurons (tdTomato). Second, despite the lack of change in overall neuronal density, CCI induced a loss of immunoreactivity for the interneuron subtype specific marker PV but not SST. This is in contrast to TBI in adult mice where more global interneuron changes have been reported (Hunt et al. 2011; Cantu et al. 2015). Third, the loss of PV expression following CCI was accompanied, although to a lesser extent, by a decrease in the density of Cre‐dependent fluorescence labelled PV interneurons (PV:Cre/Ai6). Fourth, despite the loss of PV immunoreactivity, a population of FS interneurons persisted in the peri‐injury zone and had intrinsic electrophysiological parameters similar to control mice. Fifth, at a synaptic level, CCI decreased the frequency of inhibition at the same time as increasing the strength of excitation onto FS interneurons. These studies suggest that, in juvenile animals, PV‐FS interneurons may be selectively vulnerable to the effects of CCI.
The pathogenesis of neurological symptoms associated with TBI remains poorly understood and even less is known about TBI in children and adolescents. A loss of inhibitory interneurons and inhibition has been repeatedly implicated in the pathophysiology of TBI and also in the development of subsequent neurological disorders, including post‐traumatic epilepsy (Huusko & Pitkänen, 2014; Cantu et al. 2015). However, most studies have been conducted in adult animals and focused on the excitatory neuronal population, necessitating a direct examination of cortical interneurons in juvenile animals following TBI. Comparing mouse with human development is fraught with difficulty, although careful consideration was given to choice of the age of the mice at the time of the impact. At P22, the mouse is considered to be well within the ‘paediatric’ age range relevant to the present study (Ben‐Ari, 2002; Vanhooren & Libert, 2013), whereas cortical interneurons are reaching maturity, are outside the period of maximum synaptogenesis, have inhibitory chloride potentials, and NMDA receptor insertion is nearing completion (Sutor & Luhmann, 1995). To investigate changes in the interneuron population, traditional approaches have utilized immunohistochemical labelling of neurochemical markers including GAD65/67, as well as subtype specific markers (e.g. PV, SST, 5‐HT3A, cholecystokinin). However, a major caveat to this approach is the inability to differentiate the reduced expression of these markers from actual neuronal loss. To overcome this, we took advantage of methods that Cre‐dependently fluorescence label interneurons either ubiquitously (Vgat:Cre/Ai9) or subtype specifically (PV:Cre/Ai6). Accordingly, we were able to directly examine for loss of GABAergic neurons independent of changes in their immunoreactivity for select markers.
Traumatic brain injury selectively targets PV interneurons
As reported previously (Nichols et al. 2015), TBI in juvenile animals induced significant tissue loss and cavitation similar to that reported in adult animals (Hunt et al. 2009, 2011; Cantu et al. 2015). The cortical region most adjacent to the site of injury is considered to be a key component in the generation of epileptic activity and, in adult animals, it is a reported site of inhibitory dysfunction (Cantu et al. 2015). Despite the significant tissue loss at the site of injury, we observed no change in the density of excitatory (NeuN) neurons. At an inhibitory level, the Vgat:Cre/Ai9 transgenic mouse approach allowed us to directly examine changes in the interneuron population. Similar to the excitatory population, no loss of inhibitory (tdTomato positive) neurons was observed in the peri‐injury zone following CCI. This suggests that, 14–19 days after CCI, there is no significant cell death in either the excitatory or inhibitory peri‐injury neuronal populations.
Inhibition in the cortex plays an essential role in regulating excitability and is governed by a broad group of interneurons with complex morphological, cellular, molecular and synaptic properties (Gupta et al. 2000; Markram et al. 2004; Rudy et al. 2011). In the present study, we tested how TBI alters the population of interneurons in juvenile mice by examining changes in the two most prominent interneuronal subtypes: PV and SST. PV and SST are independent neurochemical markers of cortical interneurons that display little overlap in expression and account for over 70% of the interneuron population (Kawaguchi & Kubota, 1997; Lee et al. 2010; Petilla Interneuron Nomenclature Group et al. 2008; Xu et al. 2010). We determined that TBI selectively reduces the expression of PV within the peri‐injury zone without altering the expression of SST. This is in contrast to the more widespread changes in multiple interneuron subtypes reported in adult mice after CCI (Cantu et al. 2015). The selective loss of PV may therefore be a unique response to TBI in juvenile animals.
To determine whether the loss of PV expression was indicative of actual PV interneuron loss, we examined the effects of CCI on Cre‐dependently labelled PV interneurons (PV:Cre/Ai6). PV mRNA can be detected starting in the second post‐natal week (P8) and reaches near mature levels by P21 (de Lecea et al. 1995; Dong, 2008). As a result, PV interneurons are well and indelibly labelled by ZsGreen1 prior to the age (P22) that mice were subjected to CCI. The observed loss of density of ZsGreen1 positive neurons following CCI is therefore a clear indication of injury induced PV interneuron loss. Of note, the loss of PV density following CCI was substantially greater as indicated by IHC PV immunoreactivity (61%) then by Cre‐dependent ZsGreen1 (39%) labelling. This suggests that, following TBI, some PV interneurons die, although a greater proportion only undergo phenotypic loss of PV immunoreactivity. This finding was validated by the electrophysiological determined presence of FS interneurons in the peri‐injury (for further discussion, see below). This suggests caution is required with respect to the use and interpretation of IHC measures alone for indicating interneuron loss. Conversely, an important caveat to the use of the PV:Cre‐dependent approach is indicated by the significant number of ZsGreen1+ cells that did not express detectable levels of PV (Fig. 5 C). Off‐target fluorescence labelling has been previously noted in PV:Cre mice (Tanahira et al. 2009; Kobayashi & Hensch, 2013; Fujihara et al. 2015; Nassar et al. 2015) and may be the result of a relatively reduced PV IHC detection efficacy or transient PV expression during development. Other independent IHC markers of PV interneurons (e.g. Kv3.1) (Chow et al. 1999; Yanagi et al. 2014) may yield a higher co‐labelling efficiencies but, again, these are limited by distinguishing changes in expression over interneuron loss. In addition, the decrease in ZsGreen1 labelling following CCI was not detected using a GABAergic neuron‐wide analysis approach (i.e. VGAT). This may reflect the increased detection sensitivity of our subtype specific targeted approach (i.e. PV:Cre/Ai6); however, subtle compensatory changes in other interneuron subtypes cannot be ruled out. The results of the present study clearly demonstrate reduced PV‐Ai6 co‐labelling after CCI, consistent with the loss of PV expression, although they also highlight the need for a combined IHC and genetic labelling approach to begin delineating TBI‐induced changes with respect to the expression of interneuron markers and overt interneuron loss.
Synaptic input onto FS interneurons is disrupted by traumatic brain injury
PV neurons alone represent ∼40% of GABAergic cortical interneurons and are predominantly of an FS phenotype (Kawaguchi & Kondo, 2002; Rudy et al. 2011). FS interneurons have unique properties, including sustained high frequency firing, which allows for electrophysiological identification (Connors & Gutnick, 1990; Cauli et al. 1997; Kawaguchi & Kubota, 1997; Gibson et al. 1999). PV‐FS neurons has been shown to influence neuronal excitability (e.g. firing rate) and spike timing (Caillard et al. 2000; Schwaller et al. 2002; Orduz et al. 2013). However, the loss of PV expression in FS interneurons following CCI did not alter the majority of the intrinsic membrane properties we tested, including those known to be important to the pathophysiology of other neurological disorders (van Zundert et al. 2012; Prinz et al. 2013). The resting membrane potential of FS interneurons was more depolarized following CCI and this may contribute to the trend towards a reduced rheobase. However, this change in membrane potential did not translate into alterations in the input–output relationship of FS interneurons, as indicated by no significant difference in the firing frequency or action potential properties. This suggests that, although FS interneurons may lose their PV immunoreactivity, they are still present in the peri‐injury zone and maintain a near ‘normal’ FS electrophysiological phenotype.
Beyond intrinsic excitability, regulation of calcium homeostasis by PV is considered to play an important role in synaptic neurotransmission (Kinney et al. 2006; Orduz et al. 2013). The loss of PV is predicted to disrupt inhibitory neurotransmission which is accordance with previous work indicating inhibitory network dysfunction after brain injury (Hunt et al. 2011; Jin et al. 2011; Cantu et al. 2015; Nichols et al. 2015). Using an electrophysiological approach, we found that FS interneurons receive less frequent inhibition after CCI, indicative of a decrease in pre‐synaptic release probability. We also observed an increased rise and decay time of inhibitory spontaneous synaptic events. Kinetic properties of synaptic events are often controlled by the specific expression of GABA receptor subunits that have been shown to be altered after TBI (Huusko & Pitkänen, 2014) and will be evaluated in future studies. Although the altered inhibitory synaptic kinetics may influence the synaptic integration window, this was not sufficient to increase synaptic charge and compensate for the >50% loss in inhibitory event frequency suggesting an overall loss of inhibitory drive onto FS interneurons.
On the excitatory side, the amplitude and charge transfer of sEPSCs onto FS interneurons was also increased after CCI, indicating increased excitatory synaptic strength. By contrast to the inhibitory changes, the frequency of sEPSC events was not significantly altered by CCI. The strengthening of excitatory events may also be a result of post‐synaptic receptor changes, potentially driven by homeostatic mechanisms (Pozo & Goda, 2010; Turrigiano, 2012).
PV neurons preferentially synapse onto other PV and excitatory pyramidal neurons (Tamás et al. 1998; Gibson et al. 1999). The loss of PV interneurons makes it tempting to suggest that the reduced inhibitory input onto FS interneurons is the result of a loss of input from other PV interneurons. However, PV interneurons receive synaptic input from a wide array of interneuron subtypes and from various excitatory lamina and regions (Markram et al. 2004; Pfeffer et al. 2013; Pi et al. 2013; Jin et al. 2014). The recorded post‐synaptic currents in FS interneurons in the present study are a compilation of these various inputs, foregoing the ability to discretely determine the subtype and/or source of these synaptic changes. Consequently, it remains possible that the reduced inhibitory synaptic activity following CCI is contributed to by alterations in other unstudied interneuron subtypes (e.g. 5‐HT3A, VIP). In our previous work, we similarly found that excitatory pyramidal neurons in juvenile mice undergo unique changes following a TBI. By contrast to reports in adult mice (Cantu et al., 2015), recordings from pyramidal neurons in juvenile mice revealed a lack of generalized hyperexcitability (Goddeyne et al. 2015; Nichols et al. 2015) and the presence of unique excitatory and inhibitory synaptic bursts following CCI (Nichols et al. 2015). These changes were evident at a time post‐injury where epileptic activity was evident on EEG (i.e. by post‐injury day 14) as examined in the present study. PV interneurons are considered as being important with respect to regulating the output of pyramidal neurons, gain control, and contributing to network oscillations and synchrony (Bartos et al. 2007; Puig et al. 2008; Cardin et al. 2009; Wilson et al. 2012). Determining how the TBI‐induced loss of PV expression and interneurons and associated TBI‐induced synaptic changes contribute, or potentially even resist, synaptic bursting and network level excitability remains to be determined. Overall, the findings of the present study indicate that juvenile PV interneurons are particularly sensitive to brain injury and undergo unique changes following a TBI.
PV interneurons have been shown to play a crucial role in higher cognitive function (Schwaller et al. 2004; Sohal et al. 2009; Lewis et al. 2012; Ma & Prince, 2012). PV loss has been shown to increase seizure susceptibility (Schwaller et al. 2004) and induce behavioural deficits relevant to the core symptoms of autism (Wöhr et al. 2015) and is also implicated in the pathophysiology of schizophrenia (Sohal et al. 2009; Lewis et al. 2012) and epilepsy (Schwaller et al. 2004; Ma & Prince, 2012; Jin et al. 2014). Given that TBI is a known risk factor for both schizophrenia (Molloy et al. 2011), autism spectrum disorders (Wöhr et al. 2015) and epilepsy (Barlow et al. 2000; Hunt et al. 2009), the loss of PV expression following TBI may be a common denominator in the development of numerous neurological disorders. However, at present, it is unknown whether PV expression loss is an adaptive or maladaptive response to injury. Furthermore, it might be the case that PV loss serves in both capacities dependent on where an individual is along the time course of TBI and recovery. What is evident from these studies is that the response to TBI in juvenile brains appears to be distinct from that in adults. The alteration to PV‐FS neurons after TBI may be critical to understanding how TBI in the paediatric brain may alter neurodevelopment and why children have worse outcomes and take longer to recover after brain injury.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JNi, JNe and TA all contributed to the conception and design of experiments. All authors contributed to the data analysis, as well as to its interpretation and the writing of the manuscript. TA and JNe contributed financial support for materials and staffing. JNi performed all of the experimental procedures and collected data. GRB contributed to the immunohistochemical processing, imaging and analysis. All authors approved the final version of the manuscript submitted for publication.
Funding
TA is supported by R01‐NS087031, R01‐NS097537 and R21‐EB020767 from the NIH and IOS‐1353804 from the NSF. JNe is supported by R00‐NS07661 and R01‐NS097537.
Acknowledgements
We thank Dr David Adelson for scientific discussions and providing research equipment, as well as Mario Moreno and Johan Martinez from the Newbern laboratory for their technical assistance.
Biography
Joshua Nichols studied how cortical neurons are altered after traumatic brain injury in the developing brain during his PhD at the University of Arizona and Arizona State University. This work led to the acceptance of a postdoctoral position at the Salk Institute, where he now investigates how cortical microcircuits process visual information. Addressing this phenomenon requires working at both the cellular and system‐wide level to assess how populations of neurons co‐operate to encode information. The combination of in vivo work with an analysis of synaptic connectivity and network dynamics in vitro aims to provide a more complete understanding of how neural circuits process information.

Edited by: Ole Paulsen & Katalin Toth
This is an Editor's Choice article from the 1 April 2018 issue.
References
- Anderson V & Moore C (1995). Age at injury as a predictor of outcome following pediatric head injury: a longitudinal perspective. Child Neuropsychol 1, 187–202. [Google Scholar]
- Anderson TR, Huguenard JR & Prince DA (2010). Differential effects of Na+‐K+ ATPase blockade on cortical layer V neurons. J Physiol 588, 4401–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP & Rocca WA (1998). A population‐based study of seizures after traumatic brain injuries. N Engl J Med 338, 20–24. [DOI] [PubMed] [Google Scholar]
- Aurora SK, Barrodale P, Chronicle EP & Mulleners WM (2005). Cortical inhibition is reduced in chronic and episodic migraine and demonstrates a spectrum of illness. Headache 45, 546–552. [DOI] [PubMed] [Google Scholar]
- Barlow KM, Spowart JJ & Minns RA (2000). Early posttraumatic seizures in non‐accidental head injury: relation to outcome. Dev Med Child Neurol 42, 591–594. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I & Jonas P (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci 8, 45–56. [DOI] [PubMed] [Google Scholar]
- Ben‐Ari Y (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 3, 728–739. [DOI] [PubMed] [Google Scholar]
- Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM & Bayly PV (2007). Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma 24, 657–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromfield EB, Cavazos JE & Sirven JI (2006). An Introduction to Epilepsy. West Hartford (CT): American Epilepsy Society. Available from: https://www.ncbi.nlm.nih.gov/books/NBK2508. [PubMed] [Google Scholar]
- Bruns J & Hauser WA (2003). The epidemiology of traumatic brain injury: a review. Epilepsia 44(Suppl 10), 2–10. [DOI] [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR & Marty A (2000). Role of the calcium‐binding protein parvalbumin in short‐term synaptic plasticity. Proc Natl Acad Sci USA 97, 13372–13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu D, Walker K, Andresen L, Taylor‐Weiner A, Hampton D, Tesco G & Dulla CG (2015). Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb Cortex 25, 2306–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai L‐H & Moore CI (2009). Driving fast‐spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron SF, Alwis DS & Rajan R (2016). Traumatic brain injury and neuronal functionality changes in sensory cortex. Front Syst Neurosci 10, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S & Rossier J (1997). Molecular and physiological diversity of cortical nonpyramidal cells. J. Neurosci Off J Soc Neurosci 17, 3894–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caveness WF, Meirowsky AM, Rish BL, Mohr JP, Kistler JP, Dillon JD & Weiss GH (1979). The nature of posttraumatic epilepsy. J Neurosurg 50, 545–553. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Bellocchio EE, Danbolt NC, Osen KK, Edwards RH & Storm‐Mathisen J (1998). The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J Neurosci Off J Soc Neurosci 18, 9733–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Erisir A, Farb C, Nadal MS, Ozaita A, Lau D, Welker E & Rudy B (1999). K(+) channel expression distinguishes subpopulations of parvalbumin‐ and somatostatin‐containing neocortical interneurons. J Neurosci Off J Soc Neurosci 19, 9332–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JT, Yarnell A, Kean WS, Gold E, Lewis B, Ren M, McMullen DC, Jacobowitz DM, Pollard HB, O'Neill JT, Grunberg NE, Dalgard CL, Frank JA & Watson WD (2010). Craniotomy: true sham for traumatic brain injury, or a sham of a sham? J Neurotrauma 28, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW & Gutnick MJ (1990). Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci 13, 99–104. [DOI] [PubMed] [Google Scholar]
- Cook LG, Chapman SB, Elliott AC, Evenson NN & Vinton K (2014). Cognitive gains from gist reasoning training in adolescents with chronic‐stage traumatic brain injury. Front Neurol. 5, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan‐Perez A, De Felipe J, Ben‐Ari Y, Esclapez M & Bernard C (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4, 52–62. [DOI] [PubMed] [Google Scholar]
- Dong HW (2008). The Allen Reference Atlas: A Digital Color Brain Atlas of the C57BL/6J Male Mouse. Hoboken, NJ: John Wiley & Sons, Inc. [Google Scholar]
- Erickson SL & Lewis DA (2002). Postnatal development of parvalbumin‐ and GABA transporter‐immunoreactive axon terminals in monkey prefrontal cortex. J Comp Neurol 448, 186–202. [DOI] [PubMed] [Google Scholar]
- Fujihara K, Miwa H, Kakizaki T, Kaneko R, Mikuni M, Tanahira C, Tamamaki N & Yanagawa Y (2015). Glutamate decarboxylase 67 deficiency in a subset of GABAergic neurons induces schizophrenia‐related phenotypes. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 40, 2475–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarreta M & Hestrin S (1999). A network of fast‐spiking cells in the neocortex connected by electrical synapses. Nature 402, 72–75. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M & Connors BW (1999). Two networks of electrically coupled inhibitory neurons in neocortex. Nature 402, 75–79. [DOI] [PubMed] [Google Scholar]
- Goddeyne C, Nichols J, Wu C & Anderson T (2015). Repetitive mild traumatic brain injury induces ventriculomegaly and cortical thinning in juvenile rats. J Neurophysiol 113, 3268–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Wang Y & Markram H (2000). Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science 287, 273–278. [DOI] [PubMed] [Google Scholar]
- Hensch TK (2005). Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6, 877–888. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR & Arber S (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J & Jonas P (2014). Fast‐spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345, 1255263. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L & Tonegawa S (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW & Smith BN (2009). Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW & Smith BN (2011). Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci Off J Soc Neurosci 31, 6880–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N & Pitkänen A (2014). Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience 267, 30–45. [DOI] [PubMed] [Google Scholar]
- Ippolito DM & Eroglu C (2010). Quantifying synapses: an immunocytochemistry‐based assay to quantify synapse number. J Vis Exp JoVE 45, 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR & Prince DA (2011). Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex 21, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Jiang K & Prince DA (2014). Excitatory and inhibitory synaptic connectivity to layer V fast‐spiking interneurons in the freeze lesion model of cortical microgyria. J Neurophysiol 112, 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Wallace ML, Sidorov MS, Burette AC, Gu B, van Woerden GM, King IF, Han JE, Zylka MJ, Elgersma Y, et al (2016). GABAergic neuron‐specific loss of Ube3a causes angelman syndrome‐Like EEG abnormalities and enhances seizure susceptibility. Neuron 90, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y & Kondo S (2002). Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J. Neurocytol. 31, 277–287. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y & Kubota Y (1997). GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7, 476–486. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T & Behrens MM (2006). A specific role for NR2A‐containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci Off J Soc Neurosci. 26, 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y & Hensch TK (2013). Germline recombination by conditional gene targeting with Parvalbumin‐Cre lines. Front Neural Circuits 7, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus JF, Rock A & Hemyari P (1990). Brain injuries among infants, children, adolescents, and young adults. Am J Dis Child 1960 144, 684–691. [DOI] [PubMed] [Google Scholar]
- de Lecea L, del Río JA & Soriano E (1995). Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res Mol Brain Res 32, 1–13. [DOI] [PubMed] [Google Scholar]
- Lee S, Hjerling‐Leffler J, Zagha E, Fishell G & Rudy B (2010). The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci 30, 16796–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier J & Volk DW (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y & Prince DA (2012). Functional alterations in GABAergic fast‐spiking interneurons in chronically injured epileptogenic neocortex. Neurobiol Dis 47, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al (2010). A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo‐Rodriguez M, Wang Y, Gupta A, Silberberg G & Wu C (2004). Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5, 793–807. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH & Jorgensen EM (1997). Identification and characterization of the vesicular GABA transporter. Nature 389, 870–876. [DOI] [PubMed] [Google Scholar]
- Molloy C, Conroy RM, Cotter DR & Cannon M (2011). Is traumatic brain injury a risk factor for schizophrenia? A meta‐analysis of case‐controlled population‐based studies. Schizophr Bull 37, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar M, Simonnet J, Lofredi R, Cohen I, Savary E, Yanagawa Y, Miles R & Fricker D (2015). Diversity and overlap of parvalbumin and somatostatin expressing interneurons in mouse presubiculum. Front Neural Circuits 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Perez R, Wu C, Adelson PD & Anderson T (2015). Traumatic brain injury induces rapid enhancement of cortical excitability in juvenile rats. CNS Neurosci Ther 21, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen SP (1987). Leakiness of rat brain microvessels to fluorescent probes following craniotomy. Acta Physiol Scand 130, 63–68. [DOI] [PubMed] [Google Scholar]
- Orduz D, Bischop DP, Schwaller B, Schiffmann SN & Gall D (2013). Parvalbumin tunes spike‐timing and efferent short‐term plasticity in striatal fast spiking interneurons. J Physiol 591, 3215–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JE, Chronicle EP, Rolan P & Mulleners WM (2000). Cortical hyperexcitability is cortical under‐inhibition: evidence from a novel functional test of migraine patients. Cephalalgia Int J Headache 20, 525–532. [DOI] [PubMed] [Google Scholar]
- Petilla Interneuron Nomenclature Group , Ascoli GA, Alonso‐Nanclares L, Anderson SA, Barrionuevo G, Benavides‐Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, et al (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ & Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Lim JH & Brunjes PC (1997). Activity‐dependent regulation of calcium‐binding proteins in the developing rat olfactory bulb. J Comp Neurol 387, 12–26. [PubMed] [Google Scholar]
- Pi H‐J, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ & Kepecs A (2013). Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K & Goda Y (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz A, Selesnew L‐M, Liss B, Roeper J & Carlsson T (2013). Increased excitability in serotonin neurons in the dorsal raphe nucleus in the 6‐OHDA mouse model of Parkinson's disease. Exp Neurol 248, 236–245. [DOI] [PubMed] [Google Scholar]
- Puig MV, Ushimaru M & Kawaguchi Y (2008). Two distinct activity patterns of fast‐spiking interneurons during neocortical UP states. Proc Natl Acad Sci USA 105, 8428–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Río JA, de Lecea L, Ferrer I & Soriano E (1994). The development of parvalbumin‐immunoreactivity in the neocortex of the mouse. Brain Res Dev Brain Res 81, 247–259. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S & Hjerling‐Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AT, Hanten GR, Li X, Vasquez AC, Wilde EA, Chapman SB & Levin HS (2012). Decision making after pediatric traumatic brain injury: trajectory of recovery and relationship to age and gender. Int J Dev Neurosci Off J Int Soc Dev Neurosci 30, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Meyer M & Schiffmann S (2002). “New” functions for “old” proteins: the role of the calcium‐binding proteins calbindin D‐28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum Lond Engl 1, 241–258. [DOI] [PubMed] [Google Scholar]
- Schwaller B, Tetko IV, Tandon P, Silveira DC, Vreugdenhil M, Henzi T, Potier M‐C, Celio MR & Villa AEP (2004). Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol Cell Neurosci 25, 650–663. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O & Deisseroth K (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutor B & Luhmann HJ (1995). Development of excitatory and inhibitory postsynaptic potentials in the rat neocortex. Perspect Dev Neurobiol 2, 409–419. [PubMed] [Google Scholar]
- Tamás G, Somogyi P & Buhl EH (1998). Differentially interconnected networks of GABAergic interneurons in the visual cortex of the cat. J Neurosci Off J Soc Neurosci 18, 4255–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanahira C, Higo S, Watanabe K, Tomioka R, Ebihara S, Kaneko T & Tamamaki N (2009). Parvalbumin neurons in the forebrain as revealed by parvalbumin‐Cre transgenic mice. Neurosci Res 63, 213–223. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ & Schevon CA (2013). How inhibition influences seizure propagation. Neuropharmacology 69, 45–54. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Muldoon SF, Merricks EM, Racca C & Staley KJ (2015). The role of inhibition in epileptic networks. J Clin Neurophysiol Off Publ Am Electroencephalogr Soc 32, 227–234. [DOI] [PubMed] [Google Scholar]
- Turrigiano G (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4, a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu M, Hirai Y, Karube F, Ebihara S, Kato M, Abe K, Obata K, Yoshida S, Hirabayashi M, Yanagawa Y, et al (2008). Quantitative chemical composition of cortical GABAergic neurons revealed in transgenic venus‐expressing rats. Cereb Cortex 18, 315–330. [DOI] [PubMed] [Google Scholar]
- Vanhooren V & Libert C (2013). The mouse as a model organism in aging research: usefulness, pitfalls and possibilities. Ageing Res Rev 12, 8–21. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S & Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD & Kriegstein AR (2009). Defining the role of GABA in cortical development. J Physiol 587, 1873–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Runyan CA, Wang FL & Sur M (2012). Division and subtraction by distinct cortical inhibitory networks in vivo. Nature 488, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhr M, Orduz D, Gregory P, Moreno H, Khan U, Vörckel KJ, Wolfer DP, Welzl H, Gall D, Schiffmann SN & Schwaller B (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5, e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N & Rhee J‐S (2006). A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50, 575–587. [DOI] [PubMed] [Google Scholar]
- Wonders CP & Anderson SA (2006). The origin and specification of cortical interneurons. Nat Rev Neurosci 7, 687–696. [DOI] [PubMed] [Google Scholar]
- Xing L, Larsen RS, Bjorklund GR, Li X, Wu Y, Philpot BD, Snider WD & Newbern JM (2016). Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. ELife 5, e11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Roby KD & Callaway EM (2010). Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. J Comp Neurol 518, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi M, Joho RH, Southcott SA, Shukla AA, Ghose S & Tamminga CA (2014). Kv3.1‐containing K+ channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. Mol Psychiatry 19, 573–579. [DOI] [PubMed] [Google Scholar]
- Yuste R (2005). Origin and classification of neocortical interneurons. Neuron 48, 524–527. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Izaurieta P, Fritz E & Alvarez FJ (2012). Early pathogenesis in the adult‐onset neurodegenerative disease amyotrophic lateral sclerosis. J Cell Biochem 113, 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]