

Abstract
Key points
Shank3 increases the HCN channel surface expression in heterologous expression systems.
Shank3 Δ13–16 deficiency causes significant reduction in HCN2 expression and I h current amplitude in thalamocortical (TC) neurons.
Shank3 Δ13–16‐ but not Shank3 Δ4–9‐deficient TC neurons share changes in basic electrical properties which are comparable to those of HCN2−/− TC neurons.
HCN channelopathy may critically mediate events downstream from Shank3 deficiency.
Abstract
SHANK3 is a scaffolding protein that is highly enriched in excitatory synapses. Mutations in the SHANK3 gene have been linked to neuropsychiatric disorders especially the autism spectrum disorders. SHANK3 deficiency is known to cause impairments in synaptic transmission, but its effects on basic neuronal electrical properties that are more localized to the soma and proximal dendrites remain unclear. Here we confirmed that in heterologous expression systems two different mouse Shank3 isoforms, Shank3A and Shank3C, significantly increase the surface expression of the mouse hyperpolarization‐activated, cyclic‐nucleotide‐gated (HCN) channel. In Shank3 Δ13–16 knockout mice, which lack exons 13–16 in the Shank3 gene (both Shank3A and Shank3C are removed) and display a severe behavioural phenotype, the expression of HCN2 is reduced to an undetectable level. The thalamocortical (TC) neurons from the ventrobasal (VB) complex of Shank3 Δ13–16 mice demonstrate reduced I h current amplitude and correspondingly increased input resistance, negatively shifted resting membrane potential, and abnormal spike firing in both tonic and burst modes. Impressively, these changes closely resemble those of HCN2−/− TC neurons but not of the TC neurons from Shank3 Δ4–9 mice, which lack exons 4–9 in the Shank3 gene (Shank3C still exists) and demonstrate moderate behavioural phenotypes. Additionally, Shank3 deficiency increases the ratio of excitatory/inhibitory balance in VB neurons but has a limited impact on the electrical properties of connected thalamic reticular (RTN) neurons. These results provide new understanding about the role of HCN channelopathy in mediating detrimental effects downstream from Shank3 deficiency.
Keywords: HCN channel, Shank3, thalamus, thalamocortical neuron, action potential
Key points
Shank3 increases the HCN channel surface expression in heterologous expression systems.
Shank3 Δ13–16 deficiency causes significant reduction in HCN2 expression and I h current amplitude in thalamocortical (TC) neurons.
Shank3 Δ13–16‐ but not Shank3 Δ4–9‐deficient TC neurons share changes in basic electrical properties which are comparable to those of HCN2−/− TC neurons.
HCN channelopathy may critically mediate events downstream from Shank3 deficiency.
Introduction
SHANK3 encodes a scaffolding protein that is highly enriched in the post‐synaptic density (PSD) of excitatory synapses (Naisbitt et al. 1999; Durand et al. 2007; Monteiro & Feng, 2017). The full‐length mouse Shank3 protein contains 1731 amino acids that fold into highly conserved domains that mediate protein–protein interactions: an ankyrin repeat domain, an SH3 domain, a PDZ domain, a proline‐rich domain and a sterile alpha motif (SAM) domain (Wang et al. 2014). Due to multiple intragenic promoters and alternative splicing, a complex array of Shank3 isoforms exists. These isoforms are often differentially affected by mutations and deletions in the SHANK3 gene discovered from human patients, which have been modelled by over a dozen knockout or transgenic mouse lines (Bozdagi et al. 2010; Peca et al. 2011; Yang et al. 2012; Han et al. 2013; Jiang & Ehlers, 2013; Shcheglovitov et al. 2013; Speed et al. 2015; Hulbert & Jiang, 2016; Jaramillo et al. 2016; Wang et al. 2016; Zhou et al. 2016; Jaramillo et al. 2017). Most of these knockout mice reproduced key features of ASD, including repetitive actions or body movements, deficits in social interaction, and increased levels of anxiety (Jiang & Ehlers, 2013). At the synapse level, the affected neurons show morphological defects including thinner and shorter PSD, sparse spine density, impaired synaptic transmission and other abnormalities in cell physiology (Peca et al. 2011; Speed et al. 2015; Arons et al. 2016; Bariselli et al. 2016; Jaramillo et al. 2016; Peixoto et al. 2016; Wang et al. 2016). However, crucial gaps remain in understanding the impact of Shank3 deficiency on cellular physiology, especially the basic electrical properties of affected neurons.
A unique feature reported earlier for SHANK3‐deficient neurons is the increase in input resistance that leads to an increased response to synaptic inputs and action potential (AP) firing rate (Shcheglovitov et al. 2013). This observation is consistent with a recent report that SHANK3 deficiency leads to HCN channelopathy (Yi et al. 2016). All four HCN channel isoforms are expressed in the brain and they contribute to the integration of synaptic transmission, the maintenance of membrane potential, and the initiation and propagation of APs (Magee, 1999; Biel et al. 2009; Huang & Trussell, 2014). In this study, we focus on the neurons from the ventrobasal (VB) complex in the thalamus where Shank proteins and HCN channels, especially Shank3 and HCN2, are expressed at high levels (Bockers et al. 2004; Notomi & Shigemoto, 2004; Ying et al. 2007, 2012; Han et al. 2013; Wang et al. 2014; Lee et al. 2015). The VB thalamocortical relay neurons have two distinct firing modes, tonic and burst, of which the switch depends on the resting membrane potential and the synergistic interaction between HCN and T‐type calcium channels (Fig. 1; Sherman, 2001). Therefore, the network containing TC neurons and the connected thalamic reticular (RTN) neurons and corticothalamic neurons provides an excellent platform for studying HCN channels and the interaction with Shank proteins.
Figure 1. Location and typical tonic and burst firing modes of a VB neuron in the thalamus.
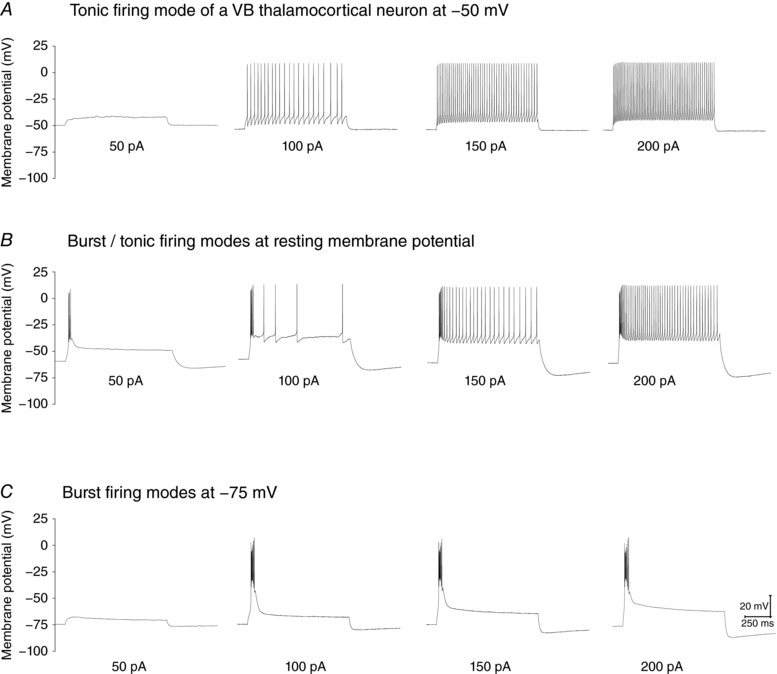
A, tonic firing modes adopted by a VB neuron with the resting membrane potential adjusted to −50 mV by current injection. B, mixture of burst/tonic firing modes for the same VB neuron at resting membrane potential. C, burst firing mode with the resting membrane potential adjust to −75 mV by current injection.
Methods
Ethical approval
All animal experiments were approved by the IACUC Committee of Virginia Commonwealth University (AD20112 for Xenopus laevis frogs and AD20224 for rats and mice). The number of animal as well as procedures introducing pain to the animal have been minimized in compliance with the regulations. The animals were subjected to regular veterinary care on a routine basis. The services of the Centre for Research Animal Resources were available if any veterinary care or consultation was required, and they were available on‐call 24 h per day.
Functional expression in Xenopus oocytes and electrophysiological characterization
The cDNA sequence encoding mouse Shank3A and 3C were generously provided by Dr Yong‐Hui Jiang from Duke University and inserted into the pGHEM vector for expression in Xenopus oocytes. mMessage mMachine (Thermo Fisher Scientific, Waltham, MA, USA) was used for cRNA synthesis. Twenty‐five to thirty nanograms of cRNA encoding Shank3 was first injected into each oocyte at stage VI. After 24 h of incubation at 18°C, injected oocytes were injected again with cRNA of HCN2 channels. For two‐electrode voltage‐clamp experiments, the bath solution contained (in mm): 80 N‐methyl‐d‐glucamine, 2 KCl, 10 KOH, 10 NaOH, 10 HEPES and 1 EGTA (pH 7.4 adjusted by methanesulfonic acid). For patch‐clamp recording of HCN channel activities in the inside‐out configuration, the electrode solution (in contact with the extracellular side of the membrane) and bath solution (intracellular side) were symmetrical and contained (in mm): 110 KCl, 2 NaCl, 10 HEPES and 1 EDTA (pH 7.4 adjusted by KOH). All experiments were carried out at room temperature.
Patch‐clamp fluorometry
To enable simultaneous recording of ionic currents and fluorescence from the membrane patch held within the glass recording pipette, the patch‐clamp fluorometry set‐up was constructed based on an Olympus (Tokyo, Japan) BX50WI microscope equipped with a ×60 water immersion lens (LUMPlanFL, NA 1.0). A 473‐nm diode‐pumped solid‐state laser (Ultralasers Inc., Newmarket, ON, Canada) was used as the excitation light source. The following filter set was used for collecting the enhanced green fluorescent protein (EGFP) fluorescence signal: exciter, D480/30; dichroic mirror, DC505LP; emitter, D510LP (Chroma Technology Corp., Bellows Falls, VT, USA). Optical signals were detected by a 16‐bit EMCCD camera (Cascade 1K by Photometrics Inc., Tucson, AZ, USA). An 18‐bit data acquisition board (PCI‐6289, National Instruments, Austin, TX, USA) was used for analog and digital I/O. WinWCP was used for data acquisition (http://spider.science.strath.ac.uk/sipbs/software_ses.htm). The laser light source, the CCD camera exposure, and the amplifier for patch‐clamp recording were synchronized by transistor‐transistor logic (TTL) signals.
Macroscopic current traces were collected in the presence of a saturating concentration of cAMP and with a hyperpolarizing voltage step based on the Boltzmann equation that was enough to produce maximal channel opening. Fluorescence signals were collected with four increasing exposure times: 25, 50, 100 and 200 ms. No obvious bleaching of the EGFP molecules was detected (Liu et al. 2016). The optical signals were collected within the linear range of the CCD camera. The ImageJ program was used to analyse the fluorescence images (Schneider et al. 2012). To specifically measure the fluorescence signal near the excised membrane, a region of interest (ROI) was selected around the arc of the membrane patch. ΔF was defined as mean fluorescence intensity in the ROI with background fluorescence subtracted. The background fluorescence was collected by moving the tip of the recording pipette out of the field of view.
Shank3 and HCN2 knockout mice
Shank3 knockout mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA; Shank3 Δ13–16, stock no: 017688; Shank3 Δ4–9, stock no: 017890). The brain‐specific HCN2 knockout mice were generated by crossing nestin‐Cre mice (B6.Cg‐Tg(Nes‐Cre)1Kln/J; stock no: 003771; The Jackson Laboratory) with Floxed‐HCN2 mice (exons 3–4 floxed; kindly provided by Dr Peter McNaughton from King's College, London). Mice were housed in a standard 12 h light–dark cycle, with free access to food and water. All experimental procedures we performed were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Acute slice preparation
Horizontal thalamocortical brain slices were collected from young mice (3–4 weeks old) of either sex. In brief, mice were deeply anaesthetized with isoflurane and tribromoethanol (250 mg kg−1, i.p.), and then perfused through the ascending aorta with preoxygenated (95% O2 and 5% CO2) and ice‐cold sucrose‐based artificial cerebrospinal fluid (sucrose‐ACSF) containing (in mm): 240 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3.5 MgCl2, 25 NaHCO3, 0.4 ascorbic acid and 2 sodium pyruvate. After decapitation, brains were rapidly removed and horizontal slices with 300 μm thickness were prepared using a vibrating blade microtome (7550 PSDS, Campden Instruments, Loughborough, UK). Slices were then incubated at 32°C for 30 min in standard ACSF consisting of (in mm): 124 NaCl, 3.6 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 11 glucose, 0.4 ascorbic acid and 2 sodium pyruvate; they were subsequently kept at room temperature for at least another 30 min prior to recording.
In vitro whole‐cell patch‐clamp recording
Brain slices were transferred into the recording chamber maintained at 32°C, and perfused continuously with carbonated standard ACSF at a rate of 2–4 ml min−1. Whole‐cell patch‐clamp recordings were made from visually identified neurons localized in VB or RTN in the thalamus. Unless indicated otherwise, patch pipettes (3–6 MΩ) were filled with internal solution containing (in mm): 130 potassium gluconate, 5 KCl, 10 phosphocreatine, 10 HEPES, 0.5 EGTA, 2 Na2‐ATP, 0.3 Na‐GTP and 2 MgSO4 (pH 7.20–7.30, 290 mosmol l−1). Liquid junction potential was not corrected on‐line or off‐line in this study. Only neurons that showed a resting membrane potential (RMP) negative to −50 mV and had overshoot were selected for study. The series resistance was typically 10–30 MΩ in this study, and neurons were excluded if their series resistance changed by more than 20%. Most recordings were collected at 32°C, unless stated otherwise (room temperature).
RMP of VB thalamocortical or RTN neurons was recorded within 20 s after break‐in, and input resistance (R in) was measured at RMP from the voltage response elicited by a current pulse (+10 pA; 500 ms). Action potentials were elicited with a series of 1 s depolarizing current pulses at different holding potentials (−50 mV, RMP, −75 mV). In voltage‐clamp mode, I h currents were evoked by a series of hyperpolarizing voltage steps from −50 mV to −140 mV in 10 mV decrements at a −40 mV holding potential. To isolate I h currents, a cocktail of blockers (1 mm BaCl2, 1 mm 4‐AP, 0.1 mm NiCl2 and 1 μm TTX) was added to the standard ACSF.
Spontaneous excitatory postsynaptic currents (sEPSC) and inhibitory postsynaptic currents (sIPSC) were recorded at −70 mV and 0 mV, respectively. The caesium‐based pipette solution used for postsynaptic currents recording contained (in mm): 130 CsMeSO4, 5 TEA‐Cl, 10 phosphocreatine, 10 HEPES, 0.5 EGTA, 2 Na2‐ATP, 0.3 Na‐GTP, 2 MgSO4, 5 QX‐314 (pH 7.20–7.30, 290 mosmol l−1). sEPSCs were recorded in the presence of 10 μm bicuculline, and sIPSCs were recorded in the presence of (2R)‐amino‐5‐phosphonovaleric acid (APV; 50 μm) and 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX; 20 μm).
All recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) and acquired with WinWCP (V5.2.6, by Dr. John Dempster, University of Strathclyde). Measurements of I h amplitude, I h activation time constants, and tonic and burst firing properties were performed using Clampfit 10.7 (Molecular Devices). Amplitudes and frequencies of sEPSCs and sIPSCs were analysed with the MiniAnalysis software (Synaptosoft, Fort Lee, NJ, USA).
Western blot
Crude whole cell lysates were prepared from mice of comparable age group. Briefly, the whole brain were dissected out and immersed in ice‐cold lysis buffer which includes (in mm): 150 NaCl, 1% Triton X‐100, 20 Tris–HCl pH 7.6, 0.1% SDS, 1 EDTA, 1 EGTA and 2 PMSF. Then the chilled brain tissue was transferred to a glass grinder and homogenized with 15–20 strokes. After incubation at 4°C for 30 min, the lysate was transferred to an Eppendorf tube and subject to centrifugation for 10 min. The supernatant was collected and mixed with sample buffer. The following antibodies were used during western blot analysis: anti‐HCN2, Antibody Inc., Davis, CA, USA; anti‐beta‐actin, Thermo Fisher Scientific, Waltham, MA, USA.
Statistics
All statistical tests and curve fittings were performed using Pad Prism 5.0 (GraphPad Software, La Jolla, CA, USA) and OriginPro (OriginLab Corp., Northampton, MA, USA). Data were presented as the mean ± SEM, and statistical significance was assessed with Student's unpaired t test. P > 0.05 was considered as statistically non‐significant (n.s.). *** P < 0.001; ** P < 0.01; * P < 0.05.
Results
To study the impact of Shank3 on the expression of the HCN2 channel, we started from the heterologous expression system of Xenopus oocyte and co‐injected the cRNAs encoding HCN2 and Shank3. We separately tested two isoforms, Shank3A and Shank3C (Fig. 2 A), of which the related knockout mice, Shank3 Δ4–9 and Shank3 Δ13–16, respectively, are characterized later in this study (Bozdagi et al. 2010; Peca et al. 2011). The nomenclature of Shank3 isoforms was adopted from previous publications (Wang et al. 2014). Shank3A refers to the full‐length Shank3 protein whereas Shank3C does not contain the ankyrin and SH3 domains. Two‐electrode voltage clamp (TEVC) measurements were used to measure the whole‐cell HCN current. As shown in Fig. 2 B and C, co‐expressing either Shank3A or Shank3C significantly increased the HCN current amplitude (20.4 ± 5.8% for Shank3C and 30.1 ± 6.5% for Sahnk3A) (Fig. 2 D, left). Shank3C or Shank3A had minimal effects on the gating kinetics but shifted the V 1/2 value – the voltage step that leads to half‐maximal opening – towards hyperpolarization by ∼4 mV (Fig. 2 D, right). To check the surface expression of HCN2, we co‐expressed EGFP‐tagged HCN2 channel with Shank3 and subjected the injected cells to confocal microscopy (Fig. 2 E). The presence of Shank3A or Shank3C clearly increased the fluorescence intensity of HCN2–EGFP. However, under the whole‐cell configuration, the arc of fluorescence signals did not exclusively correspond to the signal from the membrane but also that from the HCN2–EGFP molecules in the vicinity. To further clarify the membrane expression, we applied patch‐clamp fluorometry on isolated membrane patches pulled from the cell surface and obtained direct support for the enhancement of HCN channel surface expression upon co‐expression with Shank3 (Fig. 2 F).
Figure 2. Shank3 isoforms improve the surface expression of recombinant HCN2 channel in Xenopus oocyte.
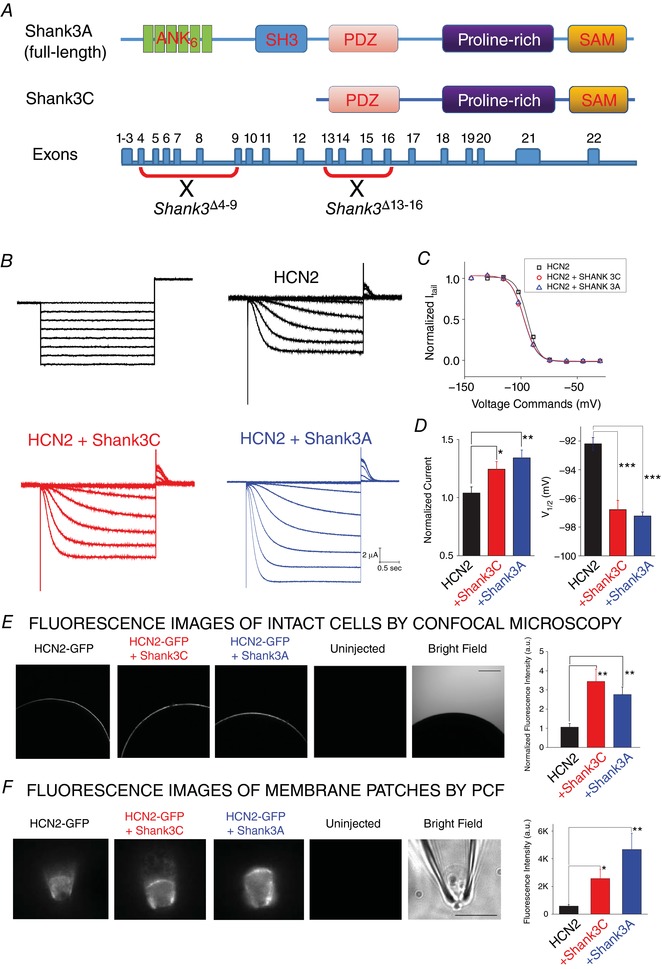
A, schematic representation of the genomic structure of Shank3 gene and two splicing isoforms, Shank3A (full‐length) and Shank3C (missing ankyrin and SH3 domains in the N‐terminus). B, representative HCN2 current (top; no Shank3) recording from whole oocytes by TEVC. A series of hyperpolarizing voltage steps (bottom) were used to activate the channel. C, HCN2 current recorded from oocytes co‐injected with Shank3A (top) or Shank3C (bottom). D, summary of Shank3 effects on the function of HCN2 channel. Values of V 1/2 (mV): HCN2, −92.2 ± 1.1; HCN2 + Shank3C, −96.0 ± 1.5; HCN2 + Shank3A, −97.2 ± 0.6. n = 6 for each group. E, confocal images of whole oocyte expressing (from left to right) HCN2 alone, HCN2 + Shank3A, HCN2 + Shank3C, and uninjected cell. F, fluorescence images of membrane fractions pulled from oocyte surface and held within the glass patch‐clamp pipette in the inside‐out configuration. Statistics are shown on the right. Unpaired t test: * P < 0.05; ** P < 0.01; *** P < 0.001. [Color figure can be viewed at http://wileyonlinelibrary.com]
To examine the effect of Shank3 on the expression of native HCN channels, we isolated the encoded I h current from VB neurons of the WT and Shank3 Δ13–16 mice (Peca et al. 2011). ZD7288 (10 μm), a relatively specific blocker of the HCN channel, was applied later to help identify the I h component (Fig. 3 A). Indeed, both the amplitude and the density of the macroscopic I h current were significantly reduced in Shank3 Δ13–16 VB neurons (32°C, Fig. 3 B and C). The impact on the voltage‐dependent channel activation curve and the shift in the value of V 1/2 were minimal (Fig. 3 D). Finally, to corroborate the electrophysiology recordings, we compared the levels of HCN2 protein in brain extracts from WT and Shank3 Δ13–16 mice using western blot analysis. For negative control, we generated a brain‐specific HCN2 knockout mouse model by crossing floxed‐HCN2 with Nestin‐Cre mice (Emery et al. 2011; Giusti et al. 2014). The HCN2 bands corresponding to the molecular mass of ∼95 kDa were not seen in the HCN2−/− lane (Fig. 3 E). A quantification of the HCN2 bands revealed a 41 ± 6% reduction in the Shank3Δ13–16 brain compared to WT.
Figure 3. Decrease in I h current amplitude in VB neurons of Shank3 Δ13–16 mice.
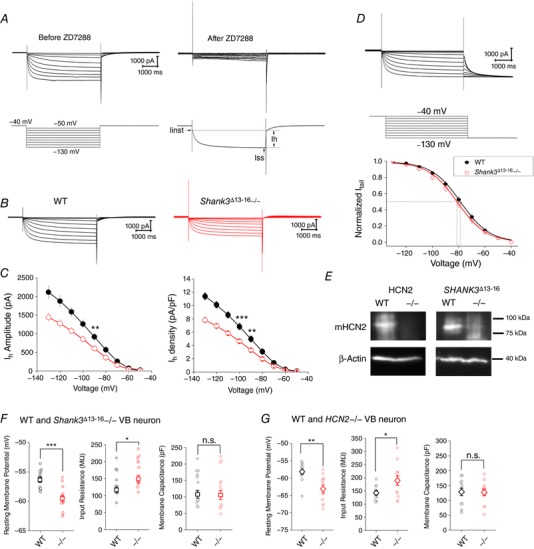
A, representative whole‐cell current traces recorded from a WT VB neuron in response to a series of hyperpolarizing voltage steps. The I h component conducted by HCN channels verified by the later application of ZD7288 (10 μm). B, representative macroscopic I h current of the WT and the Shank3 mutant VB neurons. I–V curve and current density curve are shown at bottom. C, I h current amplitude (left) and current density (right) vs. voltage command. WT, n = 10; Shank3 Δ13–16−/−, n = 12. D, a slightly different voltage protocol with the tail current measured at −130 mV was used to construct the G–V relationship. V 1/2 values (mV): WT, −78.6 ± 1.0 (n = 9); Shank3 Δ13–16−/−, −81.0 ± 1.0 (n = 12). E, immunoblotting of HCN2 from samples of WT, HCN2−/− and Shank3 Δ13–16. F, resting membrane potential (left), neuronal input resistance (middle), and membrane capacitance (right) of WT (n = 13) and Shank3 Δ13–16−/− (n = 13) VB neurons. G, results for WT (n = 12) and HCN2−/− (n = 12) VB neurons. [Color figure can be viewed at http://wileyonlinelibrary.com]
We next examined the impact of Shank3 deficiency on basic electrical properties of VB neurons and compared the results with those of the HCN2−/− neurons. Since Shank3 broadly interacts with many neuronal receptors and channels, similarities between Shank3 Δ13–16 and HCN2−/− neurons should showcase the importance of the HCN channel and the I h current they conduct as a major mechanism in manifesting the effects of Shank3 deficiency. Activities of VB neurons were recorded with physiological solutions (pipette and bath) under current‐clamp recording mode. Compared to the WT neurons, Shank3 Δ13–16−/− VB neurons showed a negative shift in the resting membrane potential (from −56.4 ± 0.5 to −59.6 ± 0.6 mV for the Shank3 Δ13–16−/− neuron) and increases in the cellular input resistance (32°C, Fig. 3 F, Table 1). The size of the neurons as approximated by the membrane capacitance remained unchanged. Indeed, the decrease in RMP and the increase in input resistance are hallmarks of impairment of HCN channel function and reduction in I h amplitude. To directly confirm this, we characterized the VB neuron from the HCN2−/− mice, which demonstrated a ∼5 mV hyperpolarizing shift in RMP and +32.8% increase in input resistance (32°C, Fig. 3 G, Table 2).
Table 1.
Summary of properties of VB thalamocortical neurons from WT and Shank3 Δ13–16−/− mice
| RMP (mV) | R in (MΩ) | Cm (pF) | AP threshold (mV) | AP amplitude (mV) | AP half‐width (μs) | AHP amplitude (mV) | Sag ratio | First spike latency (ms) | No. of rebound spikes | |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | −56.38 ± 0.46 | 138.46 ± 10.15 | 128.97 ± 13.34 | −33.44 ± 1.48 | 41.48 ± 1.43 | 1.11 ± 0.06 | 9.25 ± 0.77 | 0.59 ± 0.02 | 23.00 ± 1.07 | 7.69 ± 0.35 |
| Shank3 Δ13–16−/− | −59.57 ± 0.57 | 177.15 ± 12.98 | 126.70 ± 15.91 | −29.95 ± 1.79 | 39.03 ± 2.37 | 1.19 ± 0.06 | 9.22 ± 0.80 | 0.69 ± 0.03 | 33.438 ± 3.95 | 6.54 ± 0.35 |
Table 2.
Summary of properties of VB thalamocortical neurons from WT and HCN2−/− mice
| RMP (mV) | R in (MΩ) | Cm (pF) | AP Threshold (mV) | AP Amplitude (mV) | AP Half‐width (μs) | AHP Amplitude (mV) | Sag Ratio | First Spike Latency (ms) | No. of Rebound Spikes | |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | −58.17 ± 0.79 | 142.58 ± 7.78 | 128.85 ± 12.47 | −31.48 ± 1.75 | 47.40 ± 1.75 | 1.08 ± 0.05 | 8.28 ± 0.69 | 0.63 ± 0.03 | 29.36 ± 2.28 | 6.58 ± 0.45 |
| HCN2−/− | −63.15 ± 1.12 | 189.33 ± 18.40 | 126.87 ± 11.26 | −27.91 ± 1.37 | 43.05 ± 1.37 | 1.23 ± 0.07 | 7.10 ± 0.79 | 0.95 ± 0.00 | 93.25 ± 10.36 | 3.6 ± 0.4 |
We also repeated the measurements of I h at room temperature and confirmed the significant reduction in I h current amplitude in Shank3 Δ13–16 neurons (Fig. 4 A – C). But in neurons from Shank3 Δ4–9 mice, which still express the Shank3C isoform, no apparent reduction in the I h current amplitude was detected (Fig. 4 D and E; 32°C). This observation indicates that residual Shank3 isoforms – most likely Shank3C – help maintain the normal expression of HCN channels. Impressively, the profile of changes in I h current matched well with the changes in RMP and R in (Fig. 5 A and B). In contrast to Shank3 Δ13−16 neurons, the VB neurons from Shank3 Δ4–9−/− and heterozygous Shank3 Δ4–9+/− mice displayed normal RMP and input resistance (Fig. 5 C and D, Table 3), highly consistent with the normal expression of I h current.
Figure 4. The I h current from Shank3 Δ13–16, but not from Shank3 Δ4–9 VB neurons, exhibits decreased current amplitude.
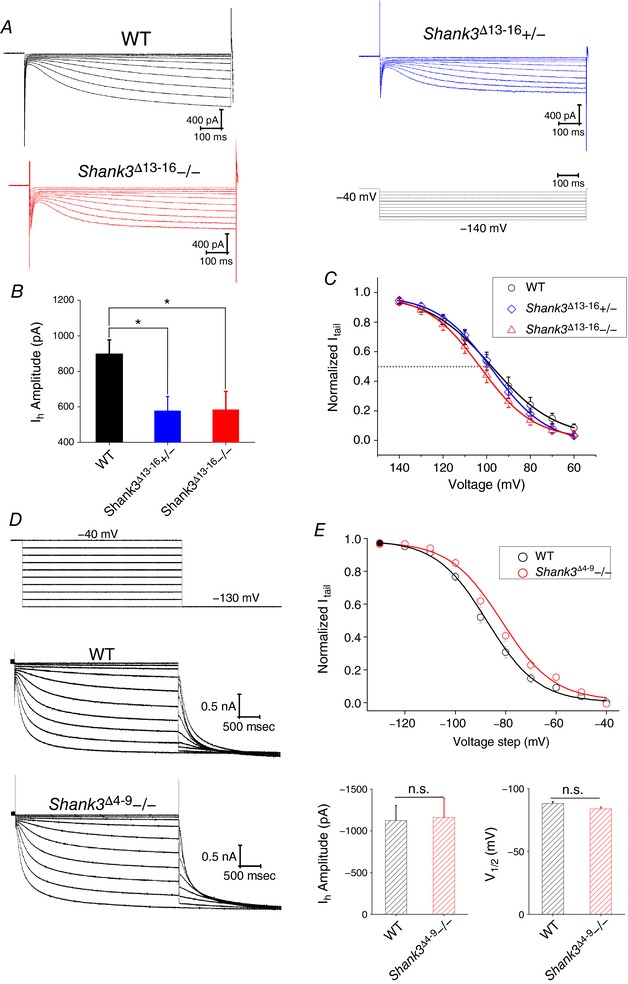
A, representative I h current traces recorded at room temperature from VB neurons of WT, Shank3 Δ13–16+/− and Shank3 Δ13–16−/− mice. I h was activated by a series of hyperpolarizing voltage steps (bottom, right). B, summary graphs of the I h current amplitude measured at −120 mV at room temperature of VB neurons from WT, Shank3 Δ13–16+/− and Shank3 Δ13–16−/− mice (left) or WT, Shank3 Δ4–9+/− and Shank3 Δ4–9−/− mice (right). C, normalized tail current amplitudes (I tail) were plotted against voltage steps and fit with the Boltzmann equation. V 1/2 values (mV): WT, −97.8 ± 0.3; Shank3 Δ13–16+/−, −97.6 ± 0.5; Shank3 Δ13–16−/−, −103.0 ± 0.3. Results were collected at room temperature. D, representative I h current traces recorded at 32°C from VB neurons of WT and Shank3 Δ4–9−/− mice. I h was activated by a series of hyperpolarizing voltage steps. E, top, normalized tail current amplitudes (I tail) were plotted against voltage steps and fit with the Boltzmann equation. V 1/2 values (mV): WT, −88.1 ± 1.5; Shank3 Δ4–9−/−, −83.8 ± 1.5. Bottom, summary graphs of the I h current amplitude and value of V 1/2. All recordings were at 32°C. WT, n = 10; Shank3 Δ4–9−/−, n = 8. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 5. Both Shank3 Δ13–16 and HCN2 knockout neurons show hyperpolarizing shift in resting membrane potential and increase in input resistance, in contrast to Shank3 Δ4–9 neurons.
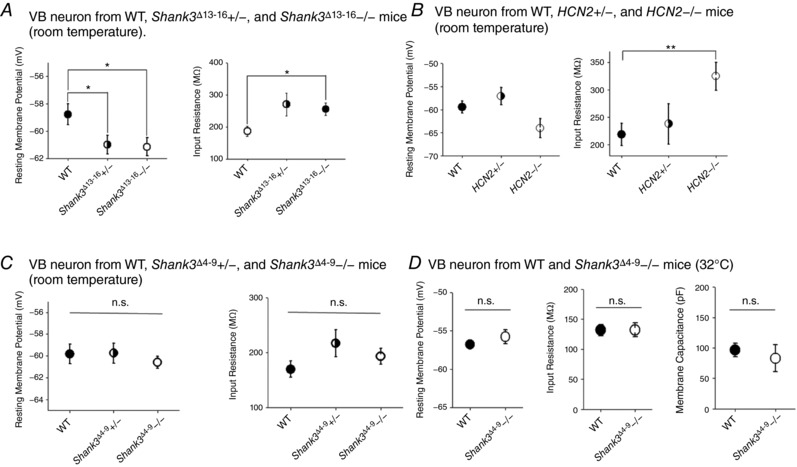
A, VB neurons from both heterozygous and homozygous Shank3 Δ13–16 mice show significantly more negative RMP (left) and increased R in (right). WT, n = 9; Shank3 Δ13–16+/−, n = 11; Shank3 Δ13–16−/−, n = 12. B, VB neurons from HCN2 heterozygous and homozygous mice show almost the same trend of changes compared to Shank3 Δ13–16 neurons. Left, RMP; right, R in. WT, n = 8; HCN2+/−, n = 8; HCN2−/−, n = 7. C, the RMP (left) and R in (right) of VB neurons from Shank3 Δ4–9 heterozygous and homozygous mice remained largely normal. All results were collected at room temperature. WT, n = 6; Shank3 Δ4–9+/−, n = 13; Shank3 Δ4–9−/−, n = 9. All n.s. D, the RMP (left), R in (middle), and the membrane capacitance (right) of VB neurons from Shank3 Δ4–9 homozygous mice remained largely normal compared to WT mice. Results were collected at 32°C. All n.s. WT, n = 9; Shank3 Δ4–9−/−, n = 8.
Table 3.
Summary of properties of VB thalamocortical neurons from WT and Shank3 Δ4−9−/− mice
| RMP (mV) | R in (MΩ) | Cm (pF) | AP threshold (mV) | AP amplitude (mV) | AP half‐width (μs) | AHP amplitude (mV) | Sag ratio | First spike latency (ms) | No. of rebound spikes | |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | −56.78 ± 0.53 | 131.67 ± 9.26 | 96.50 ± 10.85 | −30.99 ± 1.36 | 41.70 ± 1.36 | 1.03 ± 0.03 | 8.30 ± 0.63 | 0.58 ± 0.03 | 24.36 ± 1.76 | 8.00 ± 0.47 |
| Shank3 Δ4–9−/− | −55.76 ± 0.91 | 132.00 ± 11.63 | 83.16 ± 22.05 | −28.58 ± 1.17 | 39.39 ± 1.41 | 1.03 ± 0.01 | 9.51 ± 0.72 | 0.60 ± 0.02 | 22.69 ± 2.07 | 7.00 ± 0.46 |
Since Shank3 Δ13–16 and HCN2−/− neurons showed the same trend of changes in resting membrane potential and input resistance – two very basic properties of neuronal physiology – we asked whether neurons carrying different molecular defects share similar alterations in the firing of action potentials (AP). To separately study the firing modes adopted by VB neuron, the tonic and the burst modes, we manipulated the membrane potential by injecting holding current before applying a series of command currents. For the tonic firing mode, we found that, at both resting and −50 mV membrane potentials, Shank3 Δ13–16 neurons tended to fire fewer APs (32°C, Fig. 6 A–C). Correspondingly, the AP threshold was slightly increased from −33.4 ± 1.5 mV (WT) to −30.0 ± 1.8 mV (Shank3 Δ13–16−/−), accompanied by a slight decrease in AP amplitude and a slight increase in AP half‐width (Fig. 6 D and Table 1). The AHP remained unchanged.
Figure 6. Similarities in VB neuronal physiology between Shank3 Δ13–16 mice and HCN2−/− mice.
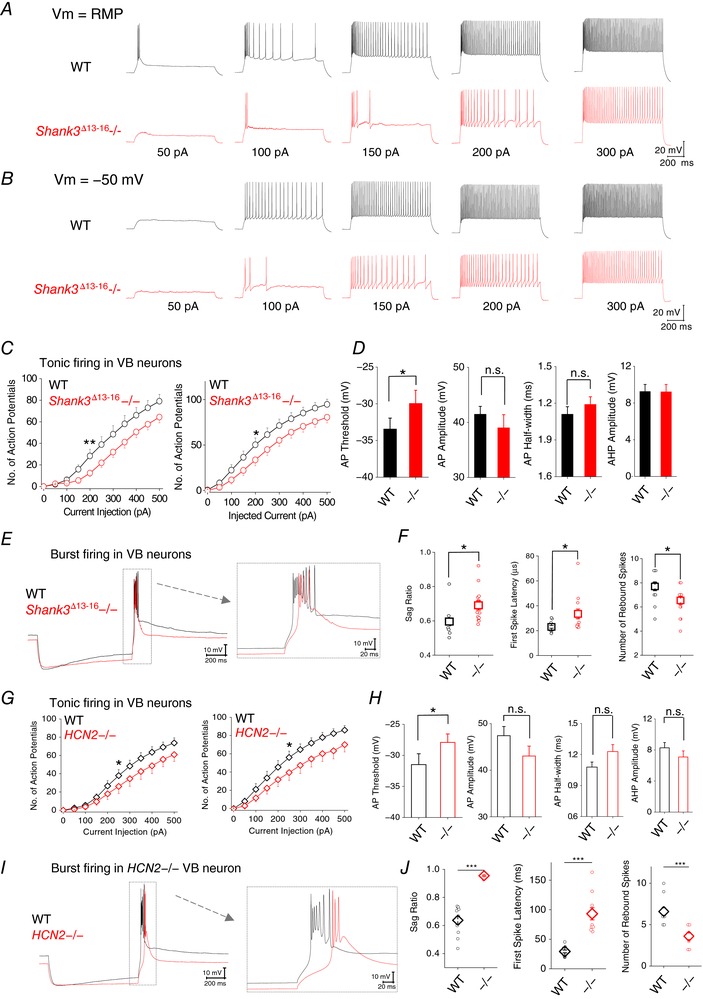
A, representative current traces in response to a series of inward current injection were recorded from neurons at their RMP. B, current traces recorded at −50 mV. C, Shank3 Δ13–16 VB neurons show reduction in the maximal number of spikes generated during tonic firing. For both WT and Shank3 Δ13–16−/−, n = 12. D, characteristics of Shank3 Δ13–16 AP. E, representative membrane responses of a HCN2−/− VB neuron to an episode of hyperpolarizing current injection (−150 pA). F, characteristics of the membrane response of Shank3 Δ13–16 VB neurons: sag ratio, first spike latency and number of rebound spikes. G, HCN2−/− VB neurons also show reduction in the maximal number of spikes generated during tonic firing. n = 11 for both WT and HCN2−/−. H, characteristics of HCN2−/− AP. I, representative membrane responses of a Shank3 Δ13–16 VB neuron to an episode of hyperpolarizing current injection (−150 pA). J, characteristics of the membrane response of HCN2−/− VB neurons: sag ratio, first spike latency and number of rebound spikes. [Color figure can be viewed at http://wileyonlinelibrary.com]
We next examined the burst mode by recording the membrane potential rebound after an episode of membrane hyperpolarization elicited by current injection at resting membrane potential (Fig. 6 E). A typical profile of membrane potential during burst mode is composed of a slow Ca2+ wave crowned with fast Na+ spikes. We monitored the sag in membrane potential, the latency of the first Na+ spike, and the number of rebound Na+ spikes. Consistent with the reduction in I h current, the sag of Shank3 Δ13–16 neurons was significantly reduced (indicated by an increase in the sag ratio; Fig. 6 F, Table 1). Shank3 Δ13–16 neurons showed a delay in firing of the first sodium spike and a slight reduction in the total number of spikes generated. Importantly, Shank3 Δ13–16 neurons showed the same trend of changes compared to HCN2−/− neurons in both tonic and burst firing modes (Fig. 6 G–J). Thus, these results revealed striking similarities in the electrical properties between VB neurons from Shank3 Δ13–16 mice and those from HCN2−/− mice, strongly suggesting the underlying reduction in I h current amplitude shared by both types of neurons might be the direct cause. Impressively, no apparent changes were observed with the VB neurons from the Shank3 Δ4–9 mice for either the tonic or burst firing modes, which are highly consistent with the close to WT I h current, RMP and R in in these neurons (Fig. 7; Tables 2, 3).
Figure 7. VB neurons from Shank3 Δ4–9 homozygous knockout mice show almost normal tonic and burst AP firing.
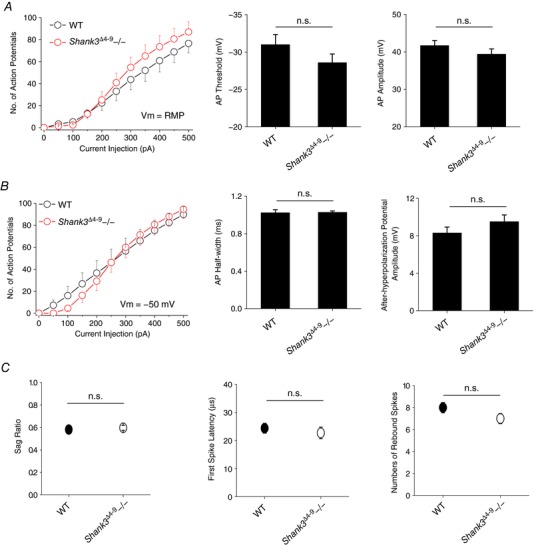
A, tonic action potential firing at resting membrane potential. Left, number of APs vs. amplitude of injected current. Right, summary of AP threshold and AP number. All n.s. B, tonic action potential firing with membrane potential adjusted to −50 mV. Left, number of APs vs. amplitude of injected current. Right, summary of AP half‐width and AHP amplitude. All n.s. C, summary of sag ratio (left), first spike latency (right), and number of rebound spikes (left). WT, n = 9; Shank3 Δ4–9−/−, n = 8. All n.s. [Color figure can be viewed at http://wileyonlinelibrary.com]
Next, we examined whether Shank3 deficiency leads to any changes in the synaptic communication between VB and other types of neurons. VB neurons mainly receive excitatory inputs from corticothalamic neurons and spinothalamic neurons and inhibitory inputs from RTN neurons within the thalamus. Imbalance in excitatory/inhibitory synaptic transmission – at both broad and local space scale – has been observed in many neuropsychiatric disorders including ASD (Nelson & Valakh, 2015). We recorded spontaneous EPSC and IPSC from VB neurons under the voltage‐clamp mode (Fig. 8 A, B, E and F). For Shank3 Δ13–16−/− VB neurons, the averaged amplitude of sEPSC was reduced from 14.2 pA (WT) to 11.9 pA, with no significant changes in sEPSC frequency (Fig. 8 C and D). Recordings of sIPSC were confirmed by a later application of bicuculline (Fig. 8 E and F). Surprisingly, we discovered that the changes in sIPSC were the opposite of those of sEPSC: Shank3 Δ13–16−/− VB neurons display significant increases in sIPSC amplitude but without significant changes in sIPSC frequency (Fig. 8 G and H). These observations confirm that Shank3 causes excitatory/inhibitory imbalance in VB thalamocortical neurons.
Figure 8. Spontaneous EPSC and IPSC recorded from Shank3 Δ13–16 VB neurons and neuronal activities of the connected RTN neuron.
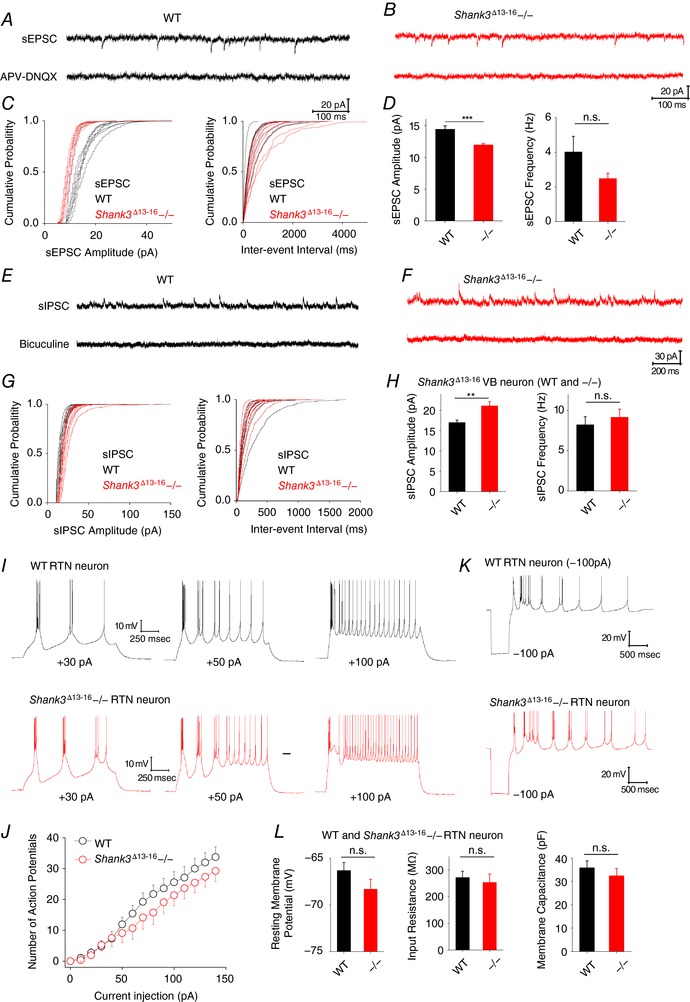
A, representative sEPSC traces recording from WT VB neurons. APV and application of 50 μm APV and 20 μm DNQX confirmed the identify of glutaminergic transmission. B, representative sEPSC traces recording from Shank3 Δ13–16 VB neurons. C, cumulative probabilities of sEPSC amplitude (left) and inter‐event interval (right). D, statistics of sEPC amplitude and frequency. E, representative sIPSC traces recording from WT VB neurons. Bicuculline (10 μm) confirmed the identify of GABAergic transmission. F, representative sIPSC trace recording from Shank3 Δ13–16 VB neurons. G, cumulative probabilities of sIPSC amplitude (left) and inter‐event interval (right). H, statistics of sIPSC amplitude and frequency. I, AP firing in WT (top) and Shank3 Δ13–16 (bottom) neurons elicited by injection of 30, 50 and 100 pA depolarizing current. J, statistics of AP firing vs. amplitude of injected current. All n.s. K, rebound in membrane potential and AP firing in response to a hyperpolarizing current injection. L, resting membrane potential, input resistance and membrane capacitance of WT and Shank3 Δ13–16 RTN neurons. All n.s. WT, n = 12; Shank3 Δ13–16−/−, n = 12. [Color figure can be viewed at http://wileyonlinelibrary.com]
VB neurons function under constant modulation of RTN neurons. We also examined the RTN neurons from Shank3 Δ13–16 mice. The AP firing was examined by a series of positive current injections (Fig. 8 I). The Shank3 Δ13–16−/− and WT RTN neurons exhibited a comparable number of APs with the same amount of current injection, and displayed no other obvious differences from the WT neurons (Fig. 8 J). Consistent with the notion that HCN channels are mainly expressed in distal dendrites, RTN neurons of both WT and Shank3−/− RTN neurons showed no obvious sag upon membrane hyperpolarization (Fig. 8 K). Shank3 Δ13–16−/− neurons presented a slight hyperpolarizing shift in membrane potential (from −66.3 ± 0.8 to −68.3 ± 1.1 mV), but approximately normal input resistance and membrane capacitance (Fig. 8 L). These observations are consistent with the observation that HCN channels are distributed more toward distal dendrites rather than the soma of RTN neurons.
Discussion
Here we investigated HCN channelopathy and alternations in intrinsic electrical properties in Shank3‐deficient neurons. Defective Shank3 has been shown to impair the expression and function of diverse channels and receptors, including TRP channels (Han et al. 2016) and ionotropic and metabotropic glutamate receptors (Wang et al. 2016), of which the effects are largely limited to synapses. In contrast, we found that Shank3 deficiency causes profound changes in basic neuronal properties, including resting membrane potential, input resistance and firing of APs, which are all essential somatic properties. These results expand our understanding of the function of the Shank3 protein expressed outside the synapses. In the soma and nearby proximal dendrites, there could be an important contribution by Shank3 to help regulate basic electrical properties through maintaining the normal expression of HCN and other types of channel and receptors.
Our understanding about the thalamus, compared to that of other brain regions such as hippocampus and striatum, remains relatively underdeveloped. The physiological function of Shank proteins in the thalamus and the relationship to psychiatric disorders remain to be clarified. In the thalamus, the expression levels of Shank3 and HCN2 are higher than those of Shank1 and 2 and HCN1, 3 and 4, respectively (Moosmang et al. 1999; Santoro et al. 2000; Sheng & Kim, 2000; Ludwig et al. 2003; Kanyshkova et al. 2009; Lee et al. 2015; Monteiro & Feng, 2017). We studied two lines of Shank3 knockout mice with deletions of different clusters of exons, Shank3 Δ4–9 and Shank3 Δ13–16, and compared the results with those of neuron‐specific HCN2−/− mice. Consistent with the biophysical result that Shank3 upregulates the expression of HCN channels, we found a significant reduction in HCN current amplitude in the VB neurons of Shank3 Δ13–16 but not of Shank3 Δ4–9 mice. The close correlation between I h reduction and changes in the AP firing suggests that HCN channels might be an important player in affecting the electrical properties of the Shank3‐deficient neurons. Furthermore, the mechanism remains to be determined for the increase in the excitation/inhibition ratio of the Shank3‐deficient VB neuron but the close to normal behaviour of RTN neuron, which should provide new insights at circuitry level into the Shank3 mutation‐related diseases.
The connection between Shank3 deficiency and HCN channelopathy was first reported by Yi et al. (2016). To generate Shank3 mutant human neurons, Yi et al. targeted exon 13, which encodes the N‐terminus of the PDZ domain. This strategy leads to a downstream frameshift from exon 13, a decreased expression of Shank3 in heterozygous neurons, and complete removal of high molecular mass Shank3 isoforms in homozygous neurons. From the aspects of cellular physiology, the results of Yi et al. are largely comparable to our characterization of the VB neurons from Shank3 Δ13–16 mice, including the increase in input resistance and negative shift in resting membrane potential. Notably, Yi et al. reported increases in AP firing when the hippocampal pyramidal neuron was subject to a current injection (−70 mV) but decreases in spontaneous AP firing measured at RMP (−40 mV). Our recording of thalamocortical neuron showed reduced AP firing at RMP (∼ −57 mV), −50 mV (burst firing mode) and −75 mV (tonic firing mode). We needed to inject about 10 time more currents (up to 500 pA) to elicit AP in VB neurons in contrast to the less than 50 pA currents used by Yi et al. to activate hippocampal neurons. These differences could be due to the species of neuron and their reliance on different pools of ion channels.
A number of mutations have been discovered in the SHANK3 gene from autistic patients. An insertion of guanine in exon 21 of the SHANK3 gene (InsG) introduces a premature stop codon and disappearance of SHANK3 isoforms with higher molecular mass (Durand et al. 2007; Speed et al. 2015; Zhou et al. 2016). Thus, it is possible that both InsG and complete SHANK3 deletion have a severe impact on the expression of the HCN channel, similar to the detrimental effects on I h current by exon 13–16 deletion. Since the I h current plays important physiological roles in the heart and the brain and shows unique biophysical properties, HCN channels are appealing targets for drug development. The drugs that specifically interfere with the function of HCN channels could be effective treatments for certain patients carrying Shank3 mutations.
Our results fill in the gaps in our understanding of the SHANK3–HCN interaction and shed light on the pathophysiological mechanisms underlying SHANK3‐related neuropsychiatric disorders. Shank3 is an important scaffolding protein that is critically involved in the expression and membrane trafficking and targeting of receptors and ion channels, which are not necessarily limited to the synapses but also proximal dendrites and somas. The fact that changes in RMP and R in in Shank3 Δ13–16+/− neurons are comparable to those of the Shank3 Δ13–16−/− and even the HCN2−/− neurons illustrate the sensitivity of the system to the expression level of Shank3. On the other hand, since the expression of I h remains largely normal in Shank3 Δ4–9−/− neurons, the Shank3 isoforms that still exist in Shank3 Δ4–9−/− but not Shank3 Δ13–16−/− such as Shank3C might be critical. Moreover, we suspect that Shank3 does not simply help the trafficking and stabilization of the HCN2 channel on the membrane but also may function as a chaperone during the biogenesis of the HCN2 channel. Other than HCN channelopathy, Shank3 deficiency might have impacts on the expression and function of other channels and receptors, which together form the pathological basis for related diseases like ASD at the molecular level. To fully understand the defects caused by SHANK3 mutations, an integrated view that encompasses not only different aspects of cellular physiology but also the coordination of neurons from different regions in the brain is still needed.
Additional information
Competing interest
The authors declare no competing financial interest.
Author contributions
M.Z., V.I., J.W., F.W., V.K., N.S. and C.W. performed the experiments and analysed the data. Q.L. and L.Z. analysed the data and supervised the project. All authors wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health (NIH) grants R01GM098592 and R01GM109193, and the startup funds from Virginia Commonwealth University to Q.L. and L.Z.
Acknowledgements
We acknowledge the technical support and the help during the pilot study from Dr. X. Fang, Dr. R. McQuiston, K. Grant, C. Waite and T. Zhao. We thank Dr Yong‐Hui Jiang from Duke University and Dr Guoping Feng from MIT for cDNA clones of Shank3 and Dr Peter McNaughton from King's College London for floxed HCN2 mice. We thank Dr Tao Liu from Nanchang University for helpful comments on the manuscript.
Biography
Mengye Zhu is a pain physician in the First Affiliated Hospital of Nanchang University in the Jiangxi Province in China. She obtained her Bachelor (2012) and Master of Anaesthesiology (2015) from the same university and has been working on the mechanisms of chronic pain under the supervision of Prof. Daying Zhang and Tao Liu. Between June 2016 and July 2017, she was a visiting scholar in the Department of Physiology and Biophysics, School of Medicine of Virginia Commonwealth University, where in Dr Lei Zhou's lab she studied neurons in the thalamus from HCN and Shank3 knockout mice.

Linked articles This article is highlighted by a Perspective by Monteiro. To read this Perspective, visit https://doi.org/10.1113/JP275828.
Edited by: Jaideep Bains & Katalin Toth
M. Zhu, V. Idikuda1 and J. Wang contribute equally.
References
- Arons MH, Lee K, Thynne CJ, Kim SA, Schob C, Kindler S, Montgomery JM & Garner CC (2016). Shank3 is part of a zinc‐sensitive signaling system that regulates excitatory synaptic strength. J Neurosci 36, 9124–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariselli S, Tzanoulinou S, Glangetas C, Prevost‐Solie C, Pucci L, Viguie J, Bezzi P, O'Connor EC, Georges F, Luscher C & Bellone C (2016). SHANK3 controls maturation of social reward circuits in the VTA. Nat Neurosci 19, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl‐Schott C, Michalakis S & Zong X (2009). Hyperpolarization‐activated cation channels: from genes to function. Physiol Rev 89, 847–885. [DOI] [PubMed] [Google Scholar]
- Bockers TM, Segger‐Junius M, Iglauer P, Bockmann J, Gundelfinger ED, Kreutz MR, Richter D, Kindler S & Kreienkamp HJ (2004). Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol Cell Neurosci 26, 182–190. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR & Buxbaum JD (2010). Haploinsufficiency of the autism‐associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, Sponheim E, Goubran‐Botros H, Delorme R, Chabane N, Mouren‐Simeoni MC, de Mas P, Bieth E, Roge B, Heron D, Burglen L, Gillberg C, Leboyer M & Bourgeron T (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 39, 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L & McNaughton PA (2011). HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333, 1462–1466. [DOI] [PubMed] [Google Scholar]
- Giusti SA, Vercelli CA, Vogl AM, Kolarz AW, Pino NS, Deussing JM & Refojo D (2014). Behavioral phenotyping of Nestin‐Cre mice: implications for genetic mouse models of psychiatric disorders. J Psychiatr Res 55, 87–95. [DOI] [PubMed] [Google Scholar]
- Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu HC & Zoghbi HY (2013). SHANK3 overexpression causes manic‐like behaviour with unique pharmacogenetic properties. Nature 503, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Kim YH, Wang X, Liu D, Zhang ZJ, Bey AL, Lay M, Chang W, Berta T, Zhang Y, Jiang YH & Ji RR (2016). SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron 92, 1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H & Trussell LO (2014). Presynaptic HCN channels regulate vesicular glutamate transport. Neuron 84, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulbert SW & Jiang YH (2016). Monogenic mouse models of autism spectrum disorders: Common mechanisms and missing links. Neuroscience 321, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Escamilla CO, Weaver TP, Liu S, Filonova I & Powell CM (2017). Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res 10, 42–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S & Powell CM (2016). Altered striatal synaptic function and abnormal behaviour in Shank3 exon4‐9 deletion mouse model of autism. Autism Res 9, 350–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH & Ehlers MD (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanyshkova T, Pawlowski M, Meuth P, Dube C, Bender RA, Brewster AL, Baumann A, Baram TZ, Pape HC & Budde T (2009). Postnatal expression pattern of HCN channel isoforms in thalamic neurons: relationship to maturation of thalamocortical oscillations. J Neurosci 29, 8847–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Chung C, Ha S, Lee D, Kim DY, Kim H & Kim E (2015). Shank3‐mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front Cell Neurosci 9, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Xie C, Grant K, Su Z, Gao W, Liu Q & Zhou L (2016). Patch‐clamp fluorometry‐based channel counting to determine HCN channel conductance. J Gen Physiol 148, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M & Hofmann F (2003). Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC ( 1999). Dendritic I h normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2, 848. [DOI] [PubMed] [Google Scholar]
- Monteiro P & Feng G (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci 18, 147–157. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F & Ludwig A (1999). Differential distribution of four hyperpolarization‐activated cation channels in mouse brain. Biol Chem 380, 975–980. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF & Sheng M (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD‐95/GKAP complex and cortactin. Neuron 23, 569–582. [DOI] [PubMed] [Google Scholar]
- Nelson SB & Valakh V (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T & Shigemoto R (2004). Immunohistochemical localization of Ih channel subunits, HCN1‐4, in the rat brain. J Comp Neurol 471, 241–276. [DOI] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z & Feng G (2011). Shank3 mutant mice display autistic‐like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang WG, Croney DM, Kozorovitskiy Y & Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B − / − mice. Nat Neurosci 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR & Siegelbaum SA (2000). Molecular and functional heterogeneity of hyperpolarization‐activated pacemaker channels in the mouse CNS. J Neurosci 20, 5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF & Dolmetsch RE (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M & Kim E (2000). The Shank family of scaffold proteins. J Cell Sci 113, 1851–1856. [DOI] [PubMed] [Google Scholar]
- Sherman SM (2001). Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci 24, 122–126. [DOI] [PubMed] [Google Scholar]
- Speed HE, Kouser M, Xuan Z, Reimers JM, Ochoa CF, Gupta N, Liu S & Powell CM (2015). Autism‐associated insertion mutation (InsG) of Shank3 exon 21 causes impaired synaptic transmission and behavioral deficits. J Neurosci 35, 9648–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, Dutta N, Hayrapetyan V, Yu C, Gaidis E, Zhao S, Ding JD, Xu Q, Chung L, Rodriguiz RM, Wang F, Weinberg RJ, Wetsel WC, Dzirasa K, Yin H & Jiang YH (2016). Altered mGluR5‐Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun 7, 11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu Q, Bey AL, Lee Y & Jiang YH (2014). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol Autism 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wohr M, Roullet FI, Katz AM, Abrams DN, Kalikhman D, Simon H, Woldeyohannes L, Zhang JY, Harris MJ, Saxena R, Silverman JL, Buxbaum JD & Crawley JN (2012). Reduced excitatory neurotransmission and mild autism‐relevant phenotypes in adolescent Shank3 null mutant mice. J Neurosci 32, 6525–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M & Sudhof TC (2016). Autism‐associated SHANK3 haploinsufficiency causes I h channelopathy in human neurons. Science 352, aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Jia F, Abbas SY, Hofmann F, Ludwig A & Goldstein PA (2007). Dendritic HCN2 channels constrain glutamate‐driven excitability in reticular thalamic neurons. J Neurosci 27, 8719–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Kanda VA, Hu Z, Purtell K, King EC, Abbott GW & Goldstein PA (2012). Targeted deletion of Kcne2 impairs HCN channel function in mouse thalamocortical circuits. PLoS One 7, e42756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C, Wells M, Amaya A, Nguyen S, Lewis M, Sanjana N, Zhou Y, Zhang M, Zhang F, Fu Z & Feng G (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]