

Abstract
A compartmental blood-brain barrier (BBB) model describing drug transport across the BBB was implemented to evaluate the influence of efflux transporters on the rate and extent of the multikinase inhibitor ponatinib penetration across the BBB. In vivo pharmacokinetic studies in wild-type and transporter knockout mice showed that two major BBB efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp), cooperate to modulate the brain exposure of ponatinib. The total and unbound (free) brain-to-plasma ratios were approximately 15-fold higher in the triple knockout mice lacking both P-gp and Bcrp [Mdr1a/b(−/−)Bcrp1(−/−)] compared with the wild-type mice. The triple knockout mice had a greater than an additive increase in the brain exposure of ponatinib when compared with single knockout mice [Bcrp1(−/−) or Mdr1a/b(−/−)], suggesting functional compensation of transporter-mediated drug efflux. Based on the BBB model characterizing the observed brain and plasma concentration-time profiles, the brain exit rate constant and clearance out of the brain were approximately 15-fold higher in the wild-type compared with Mdr1a/b(−/−)Bcrp1(−/−) mice, resulting in a significant increase in the mean transit time (the average time spent by ponatinib in the brain in a single passage) in the absence of efflux transporters (P-gp and Bcrp). This study characterized transporter-mediated drug efflux from the brain, a process that reduces the duration and extent of ponatinib exposure in the brain and has critical implications for the use of targeted drug delivery for brain tumors.
Introduction
Nearly 12,000 new cases of glioblastoma (GBM), the most common malignant primary brain cancer, are projected for 2017 (Ostrom et al., 2016). Currently, the 2-year survival rate of GBM patient is only approximately 17%, despite aggressive treatment that combines surgery, radiation, and adjuvant chemotherapy (Ostrom et al., 2016). Many potent anticancer agents, targeting known drivers of GBM, have failed to demonstrate efficacy in clinical trials in the past decade (De Witt Hamer, 2010). Our proteomic analysis of patient-derived xenograft (PDX) GBM tumors has identified overexpression of platelet-derived growth factor receptor-α (PDGFR-α) and rearranged during transfection (RET) as a possible mechanism of drug resistance (erlotinib and temozolomide) upon orthotopic implantation of epidermal growth factor receptor–driven GBM tumors (unpublished data) (Laramy et al., 2017). Based on literature review, a multitargeted kinase inhibitor (ponatinib), a Food and Drug Administration–approved leukemia therapy, has activities against (PDGFR-α) and RET, resulting in proteomic-guided drug selection for GBM6, one of the drug-resistant PDX GBM tumors (Laramy et al., 2017). Interestingly, depending on the characteristics of an individual brain tumor, the same drug can lead to differential orthotopic efficacy. Ponatinib represents an interesting case because our previous preclinical study showed negative preclinical efficacy in an adult GBM xenograft model (GBM6) (Laramy et al., 2017), whereas a positive outcome was observed in another brain tumor model, pediatric brain tumor xenograft (D-2159MG) (Keir et al., 2012). The tumor-dependent preclinical efficacy indicates the potential influence of the oncogenic makeup of GBM tumor on the blood-brain barrier (BBB) integrity (i.e., the extent of BBB leakiness). This can also subsequently lead to differential drug distribution and resultant orthotopic efficacy, warranting further in-depth assessment of the brain penetration of ponatinib.
Free tissue drug concentration is often considered to be the therapeutically relevant concentration, not the total drug concentration, based on the free drug hypothesis (Dubey et al., 1989; Hammarlund-Udenaes et al., 2008). At the BBB, the net “clearance” or transport (i.e., influx vs. efflux at the BBB) governs the delivery of free drug concentration from plasma to the brain. Drug distribution across the BBB is often restricted by the physical and biochemical barriers, such as tight junctions and efflux transporters, respectively. Tight junctions located between the BBB epithelial cells often prevent paracellular drug transport, and efflux transporters can actively reduce the brain penetration of a compound (Abbott et al., 2006). Functional compensation by breast cancer resistance protein (Bcrp) and P-glycoprotein (P-gp) has been commonly reported for various compounds (Kodaira et al., 2010), including tyrosine kinase inhibitors (Agarwal et al., 2011a; Mittapalli et al., 2012; Oberoi et al., 2013). The triple knockout genotype [Mdr1a/b(−/−)/Bcrp1(−/−)] often exhibits a brain-to-plasma ratio that is higher than what would be expected from brain-to-plasma ratios observed in the Bcrp1(−/−) and Mdr1a/b(−/−) genotypes individually. Restricted drug distribution across the BBB, and thus subtherapeutic drug exposure at the GBM tumor-bearing brain, can lead to a lack of efficacy even for a potent, highly lipophilic compound that would readily diffuse across the BBB cell layer if efflux transporters were not involved. Utilization of genetic knockout mice has been useful in determining the mechanism by which BBB efflux transporters limit CNS drug delivery and in quantifying the rate and extent of CNS penetration of a compound (Agarwal et al., 2011a; Mittapalli et al., 2012; Oberoi et al., 2013).
A compound exhibiting a greater extent and duration of tissue exposure within the therapeutic window is assumed to elicit a superior therapeutic outcome at the targeted tissue site, such as a tumor (Suzuki et al., 2009). The BBB efflux transporters, however, may reduce both the extent and duration of drug exposure in the brain, which can lead to suboptimal efficacy in the tumor-bearing brain, despite the tumor-inhibitory potency of a compound. The present study aimed to quantitatively assess the influence of BBB efflux transporters on the extent and duration of CNS penetration of ponatinib. Experimental approaches to achieve this included in vivo studies utilizing the transporter knockout and wild-type mice, in conjunction with in vitro brain slices, to determine brain-specific volumes of distribution. Subsequently, quantitative analyses of CNS distribution employed noncompartmental analysis (NCA) and mechanistic modeling to quantify the extent and duration of CNS penetration of ponatinib by estimating several parameters, including the tissue transfer rate constants, unbound (free) brain-to-plasma ratio (Kp,uu), and total brain-to-plasma ratio (Kp), and the mean transit time (MTT; i.e., the mean time spent in the brain [MTT in the brain (MTTbrain)] by ponatinib in a single passage). The results of this study provide insight into the role of efflux transporters on the CNS penetration of ponatinib, and the implication of limited CNS delivery on the efficacy of ponatinib for GBM.
Materials and Methods
Chemicals and Reagents
Ponatinib hydrochloride [3-(2-imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]benzamide hydrochloride] was purchased from Chemietek (Indianapolis, IN), imatinib methanesulfonate (4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide) (>99% purity) from LC Laboratories (Woburn, MA), and [2H8]-ponatinib (>98% purity) from Alsachim SAS (Illkirch, France). Analytical-grade reagents were purchased from Thermo Fisher Scientific (Waltham, MA).
Animals
Pharmacokinetic studies were conducted using Friend leukemia virus strain B (FVB) wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice (Taconic Biosciences, Inc., Germantown, NY). Animals were sourced from Taconic Biosciences, Inc., which has maintained an animal colony following the established procedures of breeding and back-crossing. Mice were bred and maintained in the American Association for the Accreditation of Laboratory Animal Care International–accredited animal housing facility at the Academic Health Center at the University of Minnesota. Animals were housed in a standard 12-hour dark/light cycle with unlimited access to food and water. For in vivo pharmacokinetic studies, we used both female and male mice (50% male and 50% female) ranging in age from 8 to 14 weeks. All animal experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee and conducted in accordance with the Guide for the Care and Use of Laboratory Animals established by the U.S. National Institutes of Health (Bethesda, MD).
Plasma and Brain Concentration-Time Profiles of Ponatinib
The oral dose of 30 mg/kg, the optimal dose used in mice in several studies (Gozgit et al., 2012; De Falco et al., 2013), was selected in the current study to examine the CNS distribution of ponatinib. This was the dose previously used to test the in vivo efficacy of ponatinib in a PDX GBM model (Laramy et al., 2017). Equivalent doses for the distribution and efficacy studies allow assessment of how the extent of brain penetration of ponatinib affects efficacy in the PDX GBM model. The dose was reduced to 3 mg/kg in the intravenous cohort to achieve similar exposures following the various modes of administration. The dosing formulation of ponatinib was prepared in a vehicle of dimethyl sulfoxide/Tween 80 (Sigma-Aldrich, St. Louis, MO)/water (2:1:7) for intravenous administration, and 0.5% methylcellulose (percentage grams per volume) and 0.2% Tween 80 for oral administration on the day of the animal experiment. A single intravenous bolus dose (3 mg/kg) was administered to FVB wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice via tail vein injection, followed by serial sacrifice at 0.25, 0.5, 1, 2, 4, 8, and 16 hours (N = 3 to 4 at each time point). In a separate study, a single oral dose (30 mg/kg) of ponatinib was administered to FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice via oral gavage followed by serial sacrifice at 0.5, 2, 4, 8, 12, 16, and 24 hours (N = 4 at each time point). The mice were euthanized in a carbon dioxide chamber, followed by the collection of blood via cardiac puncture and rapid surgical removal of the brain. The brain was rinsed with water and blotted to remove superficial meninges. Plasma was obtained by centrifuging the blood at 3500 rpm for 15 minutes at 4°C. Plasma and brain samples were stored at −80°C until LC-MS/MS analysis.
Steady-State Brain Distribution of Ponatinib after Intraperitoneal Infusion
A continuous intraperitoneal infusion was achieved by surgical implantation of an Alzet osmotic mini pump (model 1000D; DURECT Corporation, Cupertino, CA) in the intraperitoneal cavity of FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice as described previously (Agarwal et al., 2010). Ponatinib was dissolved in dimethyl sulfoxide at a concentration of 40 µg/µl, and the solution was loaded into the mini pumps. The drug-loaded pumps were primed overnight by soaking them in sterile saline at 37°C. Mice were anesthetized with isoflurane, and, after surgical implantation, the mice were infused with ponatinib at a constant rate of 1 µl/h (40 µg/h) for 48 hours. Plasma and brain samples were collected and stored following the same procedure as described earlier.
Brain Slice Method to Estimate the Volume of Distribution of Free (Unbound) Drug in the Brain
Brain slice experiment was conducted as previously reported (Friden et al., 2009; Loryan et al., 2013) with the following modifications. Briefly, drug-native FVB wild-type animals were sacrificed under isoflurane anesthesia, and the brain was immediately harvested and immersed in ice-cold, oxygenated, HEPES-buffered artificial fluid or artificial extracellular fluid (aECF) (129 mM NaCl, 3 mM KCl, 1.4 mM CaCl2, 1.2 mM MgSO4, 0.4 mM K2HPO4, 25 mM HEPES, 10 mM glucose, and 0.4 mM ascorbic acid). Two consecutive 300-µm coronal sections were cut using a Vibratome at a cutting speed of 5 and an amplitude (vibration) of 10 (Lancer TPI Vibratome Series 1000 Sectioning Microtome System; The Vibratome Company, St. Louis, MO). The slices were transferred into a 50-mm-high, 90-mm-diameter, flat-bottomed glass dish containing 200 nM ponatinib (10 ml) in aECF. A parafilm-covered beaker containing the aECF was gently infused with 100% oxygen. The beaker was gently shaken (UVP Multidizer Hybridization Oven; UVP, LLC, Upland, CA) and incubated for 5 hours at 37°C with a rotation speed of 90 cycles/min. The slices were removed, dried on filter paper, and weighted in an Eppendorf tube. Each slice was individually homogenized with nine volumes (w/v) of aECF buffer by vigorous vortexing. The 200 µl of buffer was collected in an Eppendorf tube containing 200 µl of blank brain homogenate (1:3 volume of aECF buffer). The samples were stored at −80°C until LC-MS/MS analysis. The volume of distribution of unbound (free) drug (Vu,brain) was calculated by the ratio of drug amount in the brain slice (Aslice) to the buffer concentration (Cbuffer):
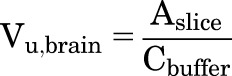 |
(1a) |
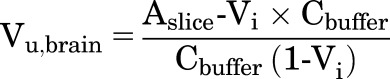 |
(1b) |
The eqs. 1a and 1b, are with or without, respectively, correction for the residual volume of buffer that can remain on the slice surface (Vi). The value of Vi found in the literature is 0.094 ml/g of brain slice (Kakee et al., 1996; Friden et al., 2009; Loryan et al., 2013). The Kp,uu values of ponatinib were calculated using the following equations adapted from Loryan et al. (2013). These equations incorporate the parameters, including the unbound (free) drug fraction in plasma (fu,plasma) of ponatinib (fu,plasma = 0.0023) and unbound (free) drug fraction in brain homogenate (fu,brain = 0.00029), as follows:
 |
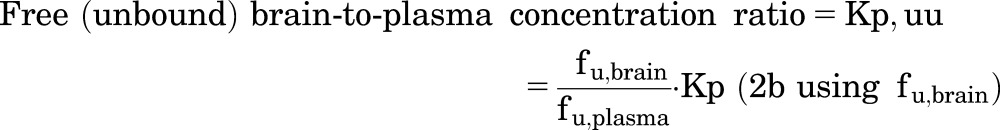 |
LC-MS/MS Assay to Measure Ponatinib Concentration
Total drug concentrations of ponatinib in plasma and brain specimens were measured using the LC-MS/MS method previously reported (Laramy et al., 2017). In summary, brain samples resulting from in vivo animal studies were homogenized with three tissue volumes of 5% bovine serum albumin (grams per volume) solution using a homogenizer (PowerGen 125; Thermo Fisher Scientific). Liquid-liquid extraction was performed for an aliquot of 25 µl of plasma or 50 µl of brain homogenate by adding 75 ng of internal standard (imatinib), 10 volumes of ice-cold ethyl acetate, and five volumes of 0.2 M sodium hydroxide (pH 13). The mixture was vortexed for 5 minutes, followed by centrifugation at 7500 rpm for 5 minutes (4°C). The organic layer was dried under nitrogen and reconstituted with 150 µl of mobile phase (acetonitrile and 20 mM ammonium acetate with 0.05% formic acid), followed by centrifugation at 14,000 rpm for 5 minutes (4°C). Five microliters of the sample was injected into the Zorbax XDB Eclipse C18 column (4.6 × 50 mm, 1.8 µm; Agilent Technologies, Santa Clara, CA) for liquid chromatography. Gradient elution was performed to separate analyte for the samples resulting from all studies except of those from intravenous bolus dosing. For the samples from in vivo pharmacokinetic studies utilizing the intravenous bolus dosing, a new isocratic method with a deuterated internal standard ([2H8]-ponatinib) was used to reduce the total run time to 7 minutes. For each of these LC-MS/MS methods, the calibration curve was sensitive and linear over the range of 0.4–2000 ng/ml (weighting factor of 1/Y2) with a CV of less than 15%. All of the measured concentrations were within the range of the calibration curve.
Pharmacokinetic Data Analysis
Noncompartmental Analysis.
Plasma and brain concentration-time profiles resulting from the administration of an oral or intravenous bolus of ponatinib in different genotypes of mice were analyzed using Phoenix WinNonlin version 6.4 (Certara USA, Inc., Princeton, NJ). NCA was performed by trapezoidal rule integration [i.e., area under the curve (AUC)] to the last time point [AUC(0→t)] and the AUC to the time infinity including the extrapolated area [AUC(0→∞)]. The Phoenix NCA module also reported other parameters/metrics, such as clearance (CL) or apparent CL (CLapparent), volume of distribution (Vd) or apparent volume of distribution, bioavailability (F), half-life, and Cmax. The Kp value was calculated using the following three approaches for comparison: 1) the ratio of AUC(0→∞) of brain concentration-time profile ([AUC(0→∞),brain]) to that of plasma concentration-time profile ([AUC(0→∞),plasma]); 2) the ratio of steady-state brain concentration to steady-state plasma concentration; and 3) the ratio of the Cmax of brain concentration (Cmax,brain) to the corresponding plasma concentration at transient steady state (Cp,tss) at that time point, since a “transient” steady state occurs at the time of Cmax,brain (Oberoi et al., 2013). The free derivative of Kp (Kp,uu) was calculated by multiplying the Kp value by the relative magnitude of the free fraction of ponatinib in brain homogenate to plasma (fu,brain/fu,plasma = 0.00029/0.0023 = approximately 0.1) (Laramy et al., 2017). A distribution advantage (DA) due to the lack of efflux transporters was quantitated by the ratio of Kp,knockout to Kp,wild-type (Oberoi et al., 2013; Parrish et al., 2015a; Vaidhyanathan et al., 2016) or Kp,uu,knockout to Kp,uu,wild-type. Using the calculated AUC(0→∞) values, the oral bioavailability (F) values in the wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice were calculated using the following equation (Rowland and Tozer, 2011):
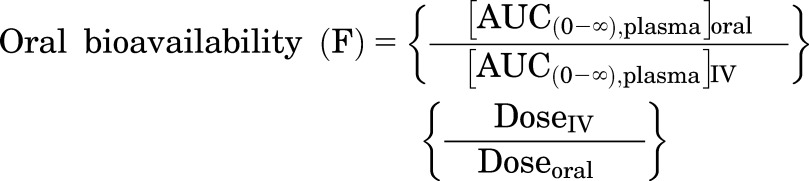 |
(3) |
Compartmental Analysis with a BBB Model.
A compartmental BBB model (Fig. 1) was adapted from the literature (Wang and Welty, 1996; Hammarlund-Udenaes et al., 1997) and was implemented to quantitatively assess CNS distributional kinetics of ponatinib into and out of the brain. The model was fitted to the observed data (total plasma and brain concentration-time profiles) resulting from intravenous bolus and oral administration in the wild-type and genetic knockout mice. The assumptions for this model included the following: 1) instantaneous equilibrium exists between the bound and free (unbound) drug in each compartment; 2) only free drug is available to move into and out of the brain at the BBB; and 3) the extent of drug binding in plasma and brain homogenates does not differ among the four genotypes. Nonlinear regression analysis for the BBB model was performed using SAAM II (version 2.3; The Epsilon Group, Charlottesville, VA).
Fig. 1.

A compartmental BBB model was adapted from the literature (Wang and Welty, 1996; Hammarlund-Udenaes et al., 1997) to describe drug movement between plasma and brain tissue after administration of a single intravenous bolus or oral dose. (A) An open two-compartment model described the total drug concentration-time profile in plasma with the parameters (k12, k21, k10, and/or Ka) that were used to generate a forcing function. The forcing function was subsequently input and used in the BBB model, which simultaneously described the Cplasma and Cbrain values. (B) A BBB model for free (unbound) drug in plasma and brain compartments. Xperipheral, total drug amount in the peripheral compartment.
The implementation of the BBB model was completed in two stages. The first stage involved describing the total plasma concentration with an open two-compartment model with drug elimination from the central compartment (Fig. 1A). For the total plasma concentration resulting from intravenous or oral administration, the intercompartmental rate constants (K12 and K21), the elimination rate constant from the central compartment (K10), the volume of distribution in the central compartment (Vcentral), and the absorption rate constant after oral dosing (Ka) were estimated. The terminal elimination rate constant (Kelim) for drug elimination from the body was calculated by using the following equation (Gibaldi and Perrier, 1998):
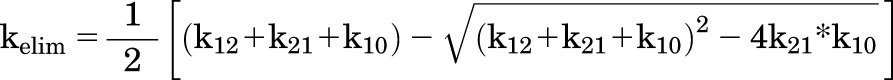 |
(4) |
The half-life and clearance of ponatinib (total drug) from the systemic circulation (body) (CLsystemic) were calculated using kelim.
In the second stage, the forcing function, or the analytical solution to the open two-compartment model describing the total plasma concentration, was input into the compartmental BBB model to estimate model parameters that describe the distribution of drug to the brain (Fig. 1A).
over time was the following:
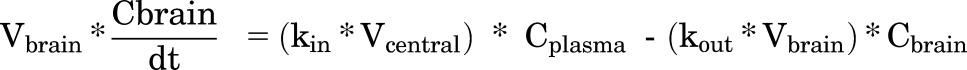 |
(5) |
where Vbrain is the volume of distribution of total drug in the brain, kin is the tissue transfer rate constant into the brain, and kout is the tissue transfer rate constant out of the brain, and Cplasma and Cbrain are the total drug concentrations in plasma and brain, respectively.
After these parameters were estimated, the differential equation for the brain compartment was rewritten to describe the movement of free drug as follows (Fig. 1B):
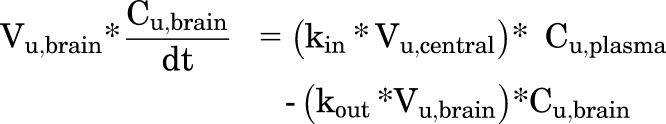 |
(6) |
where Vu,central (milliliters per kilogram) is the volume of distribution of unbound (free) drug in the central compartment, Cu,plasma is the unbound (free) drug concentration in plasma, and Cu,brain is the unbound (free) drug concentration in brain.
Clearance of total drug into the brain (CLin), CL of unbound (free) drug into the brain (CLu,in), CL of total drug out of the brain (CLout), and CL of unbound (free) drug out of the brain (CLu,out) can be expressed as a product of the rate constants and the corresponding volumes of distribution terms as follows:
| (7) |
| (8) |
| (9) |
| (10) |
The clearance terms CLin and CLu,in (milliliters per hour per kilogram) represent the net drug clearance of total and free drug, respectively, from plasma to the brain. The clearance terms, 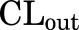 and CLu,out (milliliters per hour per kilogram), represent the sum of all drug clearances out of the brain resulting from efflux transport, drug metabolism, and flow processes.
and CLu,out (milliliters per hour per kilogram), represent the sum of all drug clearances out of the brain resulting from efflux transport, drug metabolism, and flow processes.
During the second stage of the model-fitting procedure (BBB model), the rate constants (kin and kout) were estimated, and other parameters (Vcentral, Vu,central, fu,plasma, Vbrain, Vu,brain, fu,brain) were fixed. The Vcentral (1478.3 ml/kg body weight) was obtained from the first stage of the model-fitting procedure (a two-compartment model describing the plasma concentration-time profiles). Given that the weight of the brain constitutes approximately 1.8% of body weight in mice (Davies and Morris, 1993), the experimental value of Vu,brain (1.62 ml/g of brain slices from the brain slice method) was converted to 29.2 ml/kg body weight and fixed in the BBB model (assuming 20% CV). Using the free fraction values of ponatinib in plasma (fu,plasma = 0.0023) and brain homogenate (fu,brain = 0.00029) that were previously reported (Laramy et al., 2017), the Vu,central (6.458 × 105 ml/kg, or 645.8 l/kg) and Vbrain (0.0086 ml/kg) were calculated according to eqs. 7 and 8.
The duration of brain tissue exposure to ponatinib was quantitated with the MTT using the following equation (Kong and Jusko, 1988):
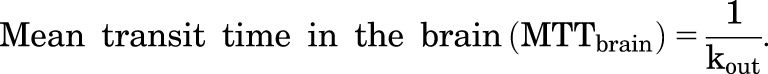 |
(11) |
The BBB model-predicted Kp (Kp,pred) or model-predicted Kp,uu (Kp,uu,pred) value was calculated by the ratio of model-predicted areas under the curve (ratio of AUC for the brain to the AUC for plasma). These model-predicted Kp and Kp,uu values are expected to equal the ratio of the clearance of total or free drug in and out of the brain, as follows:
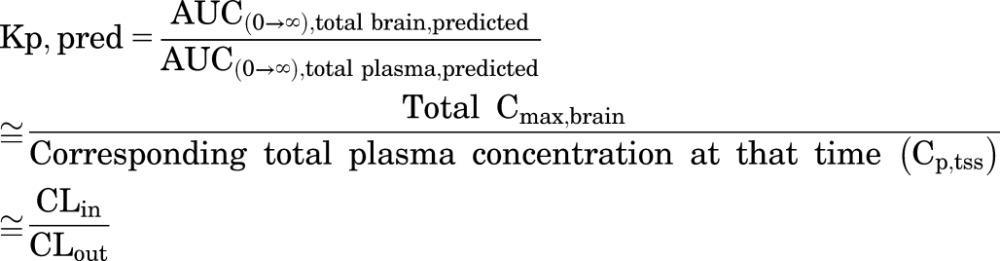 |
(12) |
 |
(13) |
The predicted DA (DA,pred) due to the lack of efflux transporters (the ratio of Kp,pred,knockout and Kp,pred,wild-type, or Kp,uu,pred,knockout and Kp,uu,pred,wild-type) was also calculated using the model-predicted brain-to-plasma ratio values.
Statistical Analysis
All experimental data were presented as the mean ± S.D. or S.E.M. The sample size in this study was determined from a power analysis assuming 20% variance and an alpha (α) value of 0.05, where the power is about 80% to detect a true difference between the anticipated means (about 50%) in drug distribution studies. Two-sample t test or analysis of variance with Bonferroni correction for multiple testing was used to compare the drug concentration or Kp values of wild-type animals with each of the three other genotypes [Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−)]. Visual display of data and statistical tests was completed using GraphPad Prism (version 6; GraphPad Software, La Jolla, CA). A significance level at P < 0.05 was applied to all statistical tests.
Results
Plasma and Brain Concentration-Time Profiles of Ponatinib after a Single Intravenous Bolus Dose.
The total plasma and brain concentration-time profiles and brain-to-plasma ratio profiles after the administration of a single intravenous bolus dose of ponatinib (3 mg/kg) are presented in Fig. 2. The total plasma concentration exhibited a biexponential decline with respect to time for both wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice, suggesting that a two-compartmental model may describe the systemic disposition of ponatinib. The terminal slopes in the total plasma and brain concentrations were similar (Fig. 2A) regardless of the genotype, as reflected in the similar half-life calculated for plasma and brain concentration-time profiles (Table 1). In the wild-type mice, there was no consistent difference between the total plasma and brain concentrations at each time point (P > 0.05), and the AUC(0→∞),plasma and AUC(0→∞),brain values were comparable (Table 1), resulting in the AUC-based Kp value of 1 (unity) for total drug concentrations (bound plus free). On the other hand, the Mdr1a/b(−/−)Bcrp1(−/−) mice had total brain concentrations that were consistently higher than the plasma concentrations at all time points (*P < 0.05; *statistical significance) (Fig. 2B), resulting in an AUC(0→∞),brain value that was 10 times higher than the AUC(0→∞),plasma value (Table 1). The total brain ponatinib concentration showed a relative time delay in reaching the maximum concentration in Mdr1a/b(−/−)Bcrp1(−/−) mice, as the time of Cmax,brain was 2 hours postdose in Mdr1a/b(−/−)Bcrp1(−/−) mice and 0.25 hour (15 minutes) in the wild-type mice. The brain-to-plasma ratio profile reached a plateau at an earlier time point (between 0.5 and 1 hour postdose) in the wild-type mice compared with the Mdr1a/b(−/−)Bcrp1(−/−) mice (approximately 2 hours postdose) (Fig. 2C). At each time point, the brain-to-plasma ratio was consistently higher in the Mdr1a/b(−/−)Bcrp1(−/−) mice than the wild-type mice (*P < 0.05; *statistical significance). The brain-to-plasma ratio (AUC-based Kp) was approximately 10-fold higher in Mdr1a/b(−/−)Bcrp1(−/−) mice than in the wild-type mice, as reflected in the DA value of 10 (Fig. 2C; Table 1). The Kp and DA values estimated using either AUC(0→t) or AUC(0→∞) provided similar results, as the percentage of extrapolated area between AUC(0→t) or AUC(0→∞) was below 10%. The wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice had systemic clearance values of 12.3 and 11.1 ml/min per kilogram, Vd values of 3.2 and 3.4 l/kg, and plasma half-life of 3.0 and 3.5 hours, respectively, indicating no difference in the systemic elimination of ponatinib between the two genotypes. Further supporting that genotype has minimal influence on systemic exposure of ponatinib, the two genotypes had comparable AUC(0→∞),plasma values and similar total plasma concentrations at all time points (P > 0.05). However, unlike the total plasma concentrations, the total brain concentrations statistically differed between the two genotypes at all time points (*P < 0.05; *statistical significance).
Fig. 2.

Total plasma (solid line with square) concentration and brain (dashed line with circle) concentration in wild-type mice (A) and Mdr1a/b(−/−)Bcrp1(−/−) mice (B), and brain-to-plasma ratio time course of ponatinib after administration of a single intravenous bolus (3 mg/kg) in FVB wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 3 to 4 at each time point) (C). Data are presented as the mean ± S.D.
TABLE 1.
Pharmacokinetic/metric parameters estimated from NCA of total brain and plasma concentration-time profiles after administration of a single intravenous bolus of ponatinib (3 mg/kg) in FVB wild-type and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 3 to 4 at each time point)
Data are presented as mean or mean ± S.E.M, unless otherwise indicated.
| Metric/Parameters | Plasma |
Brain |
||
|---|---|---|---|---|
| FVB Wild-Type | Mdr1a/b(−/−)Bcrp1(−/−) | FVB Wild-Type | Mdr1a/b(−/−)Bcrp1(−/−) | |
| Cmax (µg/ml) | 1.2 ± 0.05 | 1.1 ± 0.08 | 1.8 ± 0.1 | 4.7 ± 0.2 |
| Tmax (h) | 0.25 | 0.25 | 0.25 | 2 |
| CL (ml/min per kilogram) | 12.3 | 11.1 | ||
| Vd (l/kg) | 3.2 | 3.4 | ||
| Half-life (h) | 3.0 | 3.5 | 3.3 | 3.6 |
| AUC(0→t) (h*μg/ml) | 4.0 ± 0.3 | 3.96 ± 0.1 | 4.27 ± 0.2 | 42.1 ± 1.2 |
| AUC(0→∞) (h*μg/ml) | 4.1 | 4.5 | 4.1 | 44.8 |
| AUC-based Kpa | 1.0 | 10.0 | ||
| AUC-based Kp,uub | 0.11 | 1.1 | ||
| Transient steady-state Kpc | 1.7 | 11.6 | ||
| Transient steady-state Kp,uud | 0.11 | 1.3 | ||
| AUC based DAe | 9.9 | |||
| Transient steady-state DAe | 6.9 | |||
Calculated by [AUC(0–∞),brain]/[AUC(0–∞),plasma].
Calculated by [AUC(0–∞),brain]/[AUC(0–∞),plasma] × [fu,brain/fu,plasma].
Calculated by Cmax,brain/corresponding plasma concentration at that time (Cp,tss).
Calculated by (Cmax,brain/Cp,tss) × (fu,brain/fu,plasma).
DA due to the lack of efflux transporters, or Kp,knockout/Kp,wild-type or Kp,uu,knockout/Kp,uu,wild-type.
Plasma and Brain Concentration-Time Profiles of Ponatinib after a Single Oral Dose.
The total plasma and brain concentration-time profiles, and brain-to-plasma ratio profiles after administration of a single oral dose of ponatinib (30 mg/kg) in the wild-type mice were previously reported (Laramy et al., 2017). The present study compared the pharmacokinetic data resulting from the three genotypes that lack efflux transporters and those from the wild-type mice that were previously reported. In the wild-type mice, the total plasma and brain concentrations were similar at almost all time points (P > 0.05 except for the 0.5-hour time point) (Fig. 3A), resulting in an AUC(0→∞),brain/AUC(0→∞),plasma ratio of 0.82 (Fig. 4; Table 2). The three other genotypes, including Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−), had the AUC-based Kp values of 1.3, 3.6, and 14.2, respectively (Fig. 4; Table 2), as the total brain concentration differed from the plasma concentration at most of the time points (*P < 0.05; *statistical significance) (Fig. 3, B–D). The brain-to-plasma ratio profile reached a plateau at a relatively earlier time point (approximately 2 hours postdose) in the wild-type and Bcrp1(−/−) genotypes. A later plateau (4–8 hours postdose) was observed in the Mdr1a/b(−/−) and Mdr1a/b(−/−)Bcrp1(−/−) genotypes (Fig. 4). The Kp values did not statistically differ between the wild-type and Bcrp1(−/−) genotypes (P > 0.05). However, Mdr1a/b(−/−) and Mdr1a/b(−/−)Bcrp1(−/−) genotypes, respectively, had a significantly higher Kp value compared with the wild type (*P < 0.05; *statistical significance). The Kp value in the Mdr1a/b(−/−)Bcrp1(−/−) genotype was greater than the additive value of Kp in Bcrp1(−/−) and Mdr1a/b(−/−) genotypes (Fig. 4; Table 2), indicating functional compensation between P-gp and Bcrp in modulating the brain distribution of ponatinib. The corresponding DA values (AUC based) were 1.6, 4.4, and 17.3, respectively (Table 2). Regardless of the methods of calculation (AUC or transient steady state), the estimated values of Kp and DA values were consistent. The use of either AUC(0→t) or AUC(0→∞) also led to consistent values of Kp and DA, as the percentage of extrapolated areas between AUC(0→t) and AUC(0→∞) was below 10%.
Fig. 3.

Total plasma (solid line with square) and brain (dashed line with circle) concentration-time profiles after administration of a single oral dose (30 mg/kg) of ponatinib in FVB wild-type (A), Bcrp1(−/−) (B), Mdr1a/b(−/−) (C), and Mdr1a/b(−/−)Bcrp1(−/) (D) mice (N = 4 at each time point). Data are presented as mean ± S.D. Data for the wild-type mice were previously reported (Laramy et al., 2017) and included in this present study to compare the wild-type genotype with the three other genotypes that lack efflux transporters.
Fig. 4.

Total brain-to-plasma ratio profiles after administration of a single oral dose (30 mg/kg) in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 4 at each time point). Data are presented as the mean ± S.D. Data for the wild-type were previously reported (Laramy et al., 2017) and were included in this present study to compare the wild-type genotype with the three other genotypes that lack efflux transporters.
TABLE 2.
Pharmacokinetic/metric parameters determined by NCA of total brain and plasma concentration-time profiles after a single oral dose (30 mg/kg) of ponatinib in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 4 at each time point)
Data are presented as the mean or mean ± S.E.M, unless otherwise indicated. Data for the wild-type mice were previously reported (Laramy et al., 2017) and are included in this present study to compare with the three other genotypes that lack efflux transporters.
| Metric/Parameters | Plasma |
Brain |
||||||
|---|---|---|---|---|---|---|---|---|
| FVB Wild-Type | Bcrp1(−/−) | Mdr1a/b(−/−) | Mdr1a/b(−/−)Bcrp1(−/−) | FVB Wild-Type | Bcrp1(−/−) | Mdr1a/b(−/−) | Mdr1a/b(−/−)Bcrp1(−/−) | |
| Cmax (µg/ml) | 1.1 ± 0.1 | 0.98 ± 0.2 | 1.5 ± 0.4 | 1.0 ± 0.04 | 0.96 ± 0.07 | 1.0 ± 0.09 | 5.1 ± 0.7 | 13.1 ± 0.8 |
| Tmax (h) | 2 | 4 | 2 | 4 | 2 | 8 | 4 | 4 |
| CL/F (ml/min per kilogram) | 46.6 | 48.2 | 39.1 | 43.3 | ||||
| Vd/F (l/kg) | 17.6 | 25.4 | 19.6 | 20.0 | ||||
| Half-life (h) | 4.4 | 6.1 | 5.8 | 5.3 | 5.7 | 6.6 | 4.3 | 5.5 |
| AUC(0→t) (h*μg/ml) | 10.4 ± 0.6 | 9.5 ± 0.8 | 12.1 ± 0.8 | 10.8 ± 0.6 | 8.3 ± 0.7 | 12.5 ± 0.7 | 45.0 ± 3.1 | 151.1 ± 7.4 |
| AUC(0→∞) (h*μg/ml) | 10.7 | 10.4 | 12.8 | 11.5 | 8.8 | 13.7 | 46.4 | 163.6 |
| AUC-based Kpa | 0.82 | 1.3 | 3.6 | 14.2 | ||||
| AUC-based Kp,uub | 0.11 | 0.14 | 0.40 | 1.6 | ||||
| Transient steady-state Kpc | 0.87 | 1.6 | 4.9 | 12.7 | ||||
| Transient steady-state Kp,uud | 0.11 | 0.18 | 0.54 | 1.4 | ||||
| AUC-based DAe | 1.6 | 4.4 | 17.3 | |||||
| Transient steady-state DAe | 1.9 | 5.7 | 14.6 | |||||
Calculated by [AUC(0–∞),brain]/[AUC(0–∞),plasma].
Calculated by [AUC(0–∞),brain]/[AUC(0–∞),plasma] × [fu,brain/fu,plasma].
Calculated by Cmax,brain/corresponding plasma concentration at that time (Cp,tss).
Calculated by [Cmax,brain/corresponding plasma concentration at that time (Cp,tss)] × (fu,brain/fu,plasma).
DA due to lack of transporters, or Kp,knockout/Kp,wild-type or Kp,uu,knockout/Kp,uu,wild-type.
As observed upon intravenous bolus dosing, the total plasma concentrations at each time point (P > 0.05), and thus the AUC(0→∞),plasma values, were comparable between the genotypes (Fig. 3; Table 2) upon oral dosing, suggesting a lack of influence of P-gp and Bcrp on the systemic exposure of ponatinib. The calculated oral bioavailability of ponatinib (26.5% based on eq. 3) was identical between the wild-type and Mdr1a/b(−/−)Bcrp1(−/−) genotypes. The total brain concentration at each time point significantly differed between the wild-type genotype and each of the three other genotypes (*P < 0.05; *statistical significance). This demonstrates that whereas P-gp and Bcrp dramatically affect the targeted bioavailability of ponatinib to the brain, the efflux transporters do not influence the oral bioavailability of ponatinib at the doses administered.
Steady-State Brain Distribution of Ponatinib after Continuous Intraperitoneal Infusion.
After continuous intraperitoneal infusion of 40 µg/h of ponatinib for 48 hours, the steady-state total plasma and brain concentrations, and the corresponding total brain-to-plasma ratios were compared between the wild-type and each of the three genotypes [Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−)] using analysis of variance with Bonferroni correction for multiple testing. The steady-state total plasma concentration of ponatinib did not differ between the wild-type and each of the three genotypes (P = 0.58), whereas the total brain concentration differed between the wild-type and Mdr1a/b(−/−)Bcrp1(−/−) genotypes (*P = 0.03 *statistical significance) (Fig. 5A). The steady-state total brain-to-plasma ratios were approximately 1.7-, 3.7-, and 15-fold higher in the Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) genotypes, respectively, compared with the wild-type genotype (Fig. 5B; Table 3), as reflected in the calculated DA values (Table 3). The Mdr1a/b(−/−)Bcrp1(−/−) genotype had a Kp value that was greater than the additive value of the Kp values of Bcrp1(−/−) and Mdr1a/b(−/−) genotypes. A data summary table (Table 4) compares the Kp estimates resulting from the different routes of administration (intravenous bolus, oral dosing, or steady-state continuous intraperitoneal infusion) and methods of calculation (based on AUC, transient steady state, or steady state). The Kp estimates were consistent and robust, regardless of the route of administration and the method of calculation.
Fig. 5.

Total steady-state plasma and brain concentrations (A) and corresponding total steady-state brain-to-plasma ratios (B) of ponatinib after continuous intraperitoneal infusion (40 µg/h) for 48 hours in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 4 in each genotype) (*P < 0.05; *statistical significance). Data are presented as the mean ± S.D.
TABLE 3.
Steady-state plasma and brain concentration, brain-to-plasma ratio, and distribution advantage of ponatinib after continuous intraperitoneal infusion (40 µg/h) for 48 h in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice (N = 4 in each genotype)
Data are presented as the mean ± S.D, unless otherwise indicated.
| Mice | Steady-State Total Concentration (μg/ml) |
Kp | Kp,uua | DAb | |
|---|---|---|---|---|---|
| Plasma | Brain | ||||
| FVB wild-type | 0.72 ± 0.66 | 0.53 ± 0.37 | 1.0 ± 0.51 | 0.11 ± 0.056 | |
| Bcrp1(−/−) | 0.42 ± 0.21 | 0.71 ± 0.30 | 1.8 ± 0.26 | 0.20 ± 0.029 | 1.7 |
| Mdr1a/b(−/−) | 0.39 ± 0.14 | 1.3 ± 0.70 | 3.8 ± 2.5 | 0.42 ± 0.28 | 3.7 |
| Mdr1a/b(−/−)Bcrp1(−/−) | 0.40 ± 0.34 | 6.1 ± 5.0* | 15.6 ± 2.6* | 1.7 ± 0.29* | 15.0 |
Calculated by Kp × [fu,brain/fu,plasma].
DA due to lack of transporters, or Kp,knockout/Kp,wild-type or Kp,uu,knockout/Kp,uu,wild-type.
Statistical difference (P < 0.05) compared with the FVB wild-type mice.
TABLE 4.
Data summary: comparison of the Kp estimates among different routes of administration, including a single intravenous bolus (3 mg/kg, i.v.; N = 3 to 4 at each time point), an oral dose (30 mg/kg, PO; N = 4 at each time point), or a continuous steady-state intraperitoneal infusion (40 µg/h, i.p.; N = 4 in each genotype)
Data are presented as the mean. Data for the wild-type genotype after administration of a single oral dose were previously reported (Laramy et al., 2017) and are included in this present study to compare with the three other genotypes that lack efflux transporters.
| Genotype | Kpa |
DAb |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| i.v. |
PO |
i.p. |
i.v. |
PO |
i.p. |
|||||
| A | B | A | B | C | A | B | A | B | C | |
| FVB wild-type | 1.0 | 1.7 | 0.82 | 0.87 | 1.0 | — | — | — | — | — |
| Bcrp1(−/−) | — | — | 1.3 | 1.6 | 1.8 | — | — | 1.6 | 1.9 | 1.7 |
| Mdr1a/b(−/−) | — | — | 3.6 | 4.9 | 3.8 | — | — | 4.4 | 5.7 | 3.7 |
| Mdr1a/b(−/−) | — | — | — | — | — | — | — | — | — | — |
| Bcrp1(−/−) | 10.0 | 11.6 | 14.2 | 12.7 | 15.6 | 9.9 | 6.9 | 17.3 | 14.6 | 15.0 |
Method A, Kp = [AUC(0→∞),brain]/[AUC(0→∞),plasma]; Method B, Kp = Transient steady-state concentration ratio = Cmax,brain/Corresponding plasma concentration at that time point (Cp,tss); Method C, Kp = Steady-state brain-to-plasma concentration ratio.
Kp,knockout/Kp,wild-type.
Ponatinib Distribution in the Brain Based on the Brain Slice Method.
The brain slice method was conducted to determine the volume of distribution of free (unbound) ponatinib in the brain. The Vu,brain value of ponatinib was estimated to be 1.62 ml/g per brain from eq. 1a. The same value of Vu,brain was obtained either with or without correction (eqs. 1a and 1b, respectively) for the buffer film Vi value. The estimated Vu,brain value was greater than the physiologic volume of total brain fluids (0.8 ml/g of brain slice) (Reinoso et al., 1997; Loryan et al., 2013). This indicates that ponatinib extensively binds to the nonfluid components of the brain (i.e., the parenchymal components or lysosomes). Based on the Kp estimate of ponatinib in the wild-type mice (i.e., 1.0) (Tables 1 and 2), the Kp,uu values were calculated to be 268.4 (using the Vu,brain parameter; eq. 2a) and 0.11 (using the fu,brain parameter; eq. 2b). Such a drastic discrepancy in Kp,uu estimation depending on the equations used (eq. 2a vs. eq. 2b) could arise due to the pH partitioning of a basic drug (e.g., ponatinib and other anticancer drugs, such as paclitaxel and mitoxantrone) in its intrabrain distribution (Friden et al., 2011). The calculation of Vu,brain in the present study used the Vi value of 0.094 ml/g of brain slice. However, this Vi value was previously determined in Sprague-Dawley male rats (Kakee et al., 1996), not in the FVB wild-type mice, so that the possible interspecies differences in the Vi value could lead to a misleading value of Vu,brain. Therefore, for the assessment of brain penetration of ponatinib, a Kp,uu value of 0.11 (based on eq. 2b) was assumed.
Equilibration of Total and Free Ponatinib across the BBB: Insight from a BBB Model.
For the implementation of a BBB model, the total plasma concentrations were pooled across all genotypes after either a single intravenous bolus or an oral dose, given that we found essentially the same concentration-time profiles, bioavailability, and systemic exposure (i.e., AUC(0→∞),plasma) of ponatinib among the genotypes. The total plasma concentration-time profiles were best described by an open two-compartment model, and the resultant estimates of pharmacokinetic parameters are shown in Table 5. The predicted plasma concentration-time profiles (resulting from the naive-pooled analysis) described the observed (total drug) plasma concentration-time profiles as shown in Figs. 6 and 7. The volume of distribution of ponatinib in the central compartment was estimated to be 1478.3 ml/kg, intercompartmental rate constants (k21 and k12) were 2.34 and 2.15 hours−1, respectively, elimination rate constant from the central compartment (k10) was 0.47 hours−1, and the absorption rate constant (ka) was 0.28 hours−1. All parameter estimates had a CV of less than 20% (Table 5). Using these estimated parameters, the volume of distribution of unbound (free) drug in the central compartment (Vu,central = 645.8 l/kg using eq. 7), terminal elimination rate constant from the body (kelim = 0.21 hours−1 using eq. 4), clearance of ponatinib from systemic circulation (CLsystemic = 310.4 ml/h per kilogram), and half-life (3.3 hours) for ponatinib elimination from systemic circulation were calculated (Table 5).
TABLE 5.
Pharmacokinetic parameters obtained from an open two-compartment model that described the total plasma concentration-time profiles from naive-pooled analysis of all genotypes (N = 4 at each time point) after a single intravenous bolus (3 mg/kg) or oral dose (30 mg/kg)
The parameters are presented as the mean estimate.
| Mean | CV% | 95% Confidence Interval | |
|---|---|---|---|
| Estimated parameters | |||
| Vcentral (ml/kg) | 1478.3 | 8.1 | (964.8, 1991.9) |
| K10 (h−1) | 0.47 | 6.4 | (0.34, 0.60) |
| K21 (h−1) | 2.34 | 19.2 | (0.41, 4.3) |
| K12 (h−1) | 2.15 | 10.4 | (1.2, 3.1) |
| Kelim (h−1) | 0.21 | 1.7 | (0.20, 0.23) |
| Ka (h−1) | 0.28 | 11.5 | (0.20, 0.36) |
| Calculated parameters | |||
| Vu,central (l/kg) | 645.8 | ||
| CLsystemic (ml/min per kilogram) | 5.2 | 1.7 | (4.8, 5.6) |
| Half-life (h)a | 3.3 | 1.7 | (3.0, 3.5) |
Half-life of ponatinib (total drug) from the systemic circulation (body).
Fig. 6.

Observed (red square) and model-predicted (red solid line) total plasma concentration-time profiles, and observed (black circle) and model-predicted (black dashed line) total brain concentration-time profiles in FVB wild-type (A) and Mdr1a/b(−/−)Bcrp1(−/−) (B) mice (N = 3 to 4 at each time point) after a single intravenous bolus (3 mg/kg). The observed data are presented as the mean ± S.D.
Fig. 7.

Observed (red square) and model-predicted (red solid line) total plasma concentration-time profiles, and observed (black circle) and model-predicted (black dashed line) total brain concentration-time profiles in FVB wild-type (A), Bcrp1(−/−) (B), Mdr1a/b(−/−) (C), and Mdr1a/b(−/−)Bcrp1(−/−) (D) mice (N = 4 at each time point) after a single oral dose (30 mg/kg). The observed data are presented as the mean ± S.D. The observed data for the wild-type were previously reported (Laramy et al., 2017) and were included in this present study to compare with the three other genotypes that lack efflux transporters.
The BBB model (Fig. 1) described the observed plasma and brain concentration-time data well, as displayed in Figs. 6 and 7. The individual total plasma concentration-time profile for each genotype visually matches the model-predicted plasma concentration-time profile resulting from fitting the model to the naive-pooled plasma data from all genotypes (Figs. 6 and 7). Upon initial fitting of the BBB model, the kin value for ponatinib in each genotype was similar to the kin of the Mdr1a/b(−/−)Bcrp1(−/−) genotype. Therefore, the kin value, which describes the influx processes at the BBB for ponatinib, was assumed to be the same across the four genotypes, and the kin estimate from the Mdr1a/b(−/−)Bcrp1(−/−) genotype was subsequently used and fixed for the three other genotypes for the remaining model-fitting procedures to make the fitting routines more tractable. The resultant pharmacokinetic parameter estimates from the BBB model are displayed in Table 6.
TABLE 6.
Pharmacokinetic parameters obtained from the compartmental BBB model describing the brain and plasma concentration-time after administration of a single intravenous bolus (3 mg/kg, i.v.; N = 3 to 4 at each time point) or oral dose (30 mg/kg, PO; N = 4 at each time point) in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and/or Mdr1a/b(−/−)Bcrp1(−/−) mice
Fixed values are left as blank (—). The parameters are presented as the mean estimate.
| Route of Administration | Genotype | Parameters |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kin (h−1) |
kout (h−1) |
MTTbrain (h)a |
CLin (ml/h per kilogram) |
CLout (ml/h per kilogram) |
CLu,in (ml/h per kilogram) |
CLu,out (ml/h per kilogram) |
||||||||
| Mean | CV (%) | Mean | CV (%) | Mean | Mean | CV (%) | Mean | CV (%) | Mean | CV (%) | Mean | CV (%) | ||
| Intravenous | WT | 4.0 × 10−5 | — | 7.7 | 10.2 | 0.13 | 0.059 | — | 0.066 | 36.6 | 25.8 | — | 225.8 | 22.5 |
| TKO | 4.0 × 10−5 | 11.2 | 0.64 | 14.0 | 1.6 | 0.059 | 13.8 | 0.0055 | 37.9 | 25.8 | 11.2 | 18.7 | 24.4 | |
| By mouth | WT | 5.5 × 10−5 | — | 12.4 | 2.7 | 0.081 | 0.081 | — | 0.11 | 35.3 | 35.2 | — | 361.6 | 20.2 |
| Bcrp-KO | 5.5 × 10−5 | — | 8.2 | 18.2 | 0.12 | 0.081 | — | 0.070 | 39.6 | 35.2 | — | 239.4 | 27.0 | |
| Pgp-KO | 5.5 × 10−5 | — | 2.9 | 26.0 | 0.34 | 0.081 | — | 0.025 | 43.8 | 35.2 | — | 85.1 | 32.8 | |
| TKO | 5.5 × 10−5 | 16.2 | 0.65 | 15.7 | 1.5 | 0.081 | 18.1 | 0.0056 | 38.5 | 35.2 | 16.2 | 19.0 | 25.5 | |
Bcrp-KO, Bcrp1(−/−); KO, knockout; Pgp-KO, Mdr1a/b(−/−); TKO, Mdr1a/b(−/−)Bcrp1(−/−); WT, wild-type.
Calculated by 1/kout.
Based on the implemented BBB model, each genotype had a considerably different tissue transfer rate constant out of the brain (kout) and considerably different clearances out of the brain (CLout and CLu,out) for ponatinib, regardless of the route of administration (intravenous bolus or oral). Clearance values of ponatinib out of the brain (CLout and CLu,out) were reduced by 1.5-, 4.2-, and approximately 19-fold in Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−), respectively (Table 7). Consistent with the magnitude differences in the clearances out of the brain (CLout and CLu,out), the estimated kout values were the greatest in the wild-type genotype and lowest in the Mdr1a/b(−/−)Bcrp1(−/−) genotype. Likewise, the MTTbrain of ponatinib (calculated by eq. 11) was the shortest in the wild-type genotype (approximately 5 minutes), followed by the Bcrp1(−/−) and Mdr1a/b(−/−) genoypes, and was longest in the Mdr1a/b(−/−)Bcrp1(−/−) genotype (approximately 1.5 hours). These values indicate that transporter-mediated drug efflux at the BBB can significantly reduce the therapeutic exposure time of ponatinib in the brain (Table 6).
TABLE 7.
The Kp,pred and Kp,uu,pred values and the DA,pred values, using the pharmacokinetic parameters obtained from the compartmental BBB model describing the brain and plasma concentration-time profiles after administration of a single intravenous bolus (3 mg/kg, i.v.; N = 3 to 4 at each time point) or oral dose (30 mg/kg, PO; N = 4 at each time point) in FVB wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and/or Mdr1a/b(−/−)Bcrp1(−/−) mice
The parameters are presented as the mean estimate.
| Route of Administration | Genotype | Parameters |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC(0→∞),predicted (Mean Estimate) h*μg/ml |
Transient Steady State (Mean Estimate) μg/ml |
Clearance Based (Mean Estimate) |
||||||||||
| AUC(0→∞),plasma | AUC(0→∞),brain | Kp,preda | DA,predb | Cp,tss(µg/ml) | Cmax, brain | Kp,preda | DA,predb | Kp,preda | Kp,uu,predc | DA,predb | ||
| Intravenous | WT | 4.0 | 3.6 | 0.89 | 1.2 | 1.1 | 0.89 | 0.89 | 0.11 | |||
| TKO | 4.0 | 43.1 | 10.7 | 12 | 0.5 | 5.3 | 10.8 | 12.0 | 10.7 | 1.4 | 12.1 | |
| By mouth | WT | 10.9 | 8.2 | 0.76 | 1.2 | 0.95 | 0.76 | 0.76 | 0.097 | |||
| Bcrp-KO | 10.6 | 12.2 | 1.1 | 1.5 | 1.1 | 1.2 | 1.1 | 1.5 | 1.1 | 0.15 | 1.5 | |
| Pgp-KO | 11.2 | 35.9 | 3.2 | 4.2 | 1.1 | 3.4 | 3.2 | 4.2 | 3.2 | 0.41 | 4.2 | |
| TKO | 10.7 | 152.3 | 14.3 | 18.8 | 1.0 | 12.9 | 13.4 | 17.7 | 14.4 | 1.9 | 19.1 | |
Bcrp-KO, Bcrp1(−/−); KO, knockout; Pgp-KO, Mdr1a/b(−/−); TKO, Mdr1a/b(−/−)Bcrp1(−/−); WT, wild-type.
Kp,pred, calculated by 
DA,pred, calculated by Kp,pred,knockout/Kp,pred,wild-type, or Kp,uu,pred,knockout/Kp,uu,pred,wild-type.
Kp,uu,pred, calculated by  .
.
Using the pharmacokinetic parameters estimated from the BBB model, and the resultant metrics (predicted AUCs), the total and free brain-to-plasma ratios (i.e., Kp,pred and Kp,uu,pred), and distribution advantages (i.e., DA,pred) were calculated. These values are presented in Table 7. The Kp,pred, or CLin/CLout, values resulting from the compartmental analysis (BBB model) (Table 7) closely matched the observed Kp from the NCA (Tables 1–4). Likewise, the resultant free derivative of Kp (i.e., Kp,uu) from the NCA also closely aligned with the ratio of CLu,in to CLu,out, as anticipated. Therefore, the Kp,pred, Kp,uu,pred, and DA,pred values were consistent with the NCAs, regardless of the calculation method (AUC, transient steady state, and clearance based), as shown in Table 7. These predicted values were similar to the observed (experimental) data (Tables 1–4), supporting the idea that the model-related assumptions were reasonable, and the data were well characterized by the BBB model.
Discussion
Functionally cooperative drug transport by the BBB efflux transporters has been previously reported for several tyrosine kinase inhibitors with important implications of such restricted brain delivery having a negative impact on efficacy (Parrish et al., 2015b). Consistent with our recent publication that reported restricted orthotopic efficacy of ponatinib in a PDX GBM model (Laramy et al., 2017), the present study showed the transporter-mediated drug efflux at the BBB, which supports its contribution to impaired free drug delivery of ponatinib to the brain. The DA due to the absence of both P-gp and Bcrp transporter activities was greater when compared with the absence of a single transporter. This suggests that ponatinib is a dual substrate of P-gp and Bcrp, where these two transporters functionally cooperate to restrict the brain distribution of ponatinib (Agarwal et al., 2011a,b). Eliminating the gatekeeper functions of these efflux transporters decreased the tissue transfer rate constant (i.e., kout) and clearance of ponatinib out of the brain (i.e., CLout and CLu,out), resulting in a greater tissue exposure (AUCbrain) as well as a greater MTT or therapeutic exposure time in the brain in the efflux-deficient mice.
The magnitude of differences in the transport of ponatinib out of the brain among the four genotypes (i.e., brain exit rate constants and clearances out of the brain) can be described by the Kp and Kp,uu estimates. However, the CL and Kp estimates based on total drug can be misleading because this does not consider free (active) drug concentration (Laramy et al., 2017). A Kp value near unity, as seen with ponatinib, does not always indicate that drug in plasma moves effectively across the BBB, because the Kp values can be confounded by the relative magnitude of drug-binding affinities between plasma and brain. Instead, only free drug is available to move across the BBB, so that the assessment of BBB penetration needs to use the parameters that reflect the movement of free drug, including Kp,uu and Vu,brain. These results are consistent with those from a previous publication (Laramy et al., 2017) that reported a compromised efficacy of ponatinib in the PDX of GBM due to the heterogeneous tissue binding and drug distribution into the intracranial tumor. This discrepancy between Kp and Kp,uu highlights the importance of considering the relative drug binding in plasma and brain, because a high Kp value (i.e., near unity) can misrepresent the extent of brain delivery of active compound, and therefore may not accurately predict the delivery of an efficacious drug concentration. The ratio of free fraction in brain homogenate to that in plasma (fu,brain/fu,plasma) matched the DA (Kp,knockout/Kp,wild-type), which is consistent with the findings of a previous report (Kalvass et al., 2007) that assessed such a correlation for the purpose of predicting the extent of CNS penetration of investigational compounds. The extent of brain penetration (Kp,uu) of a compound in the Mdr1a/b(−/−)Bcrp1(−/−) genotype will presumably equal unity (1) if all transporter-mediated efflux was absent and the clearances into and out of the brain were equivalent. However, the observed Kp,uu values were slightly greater than 1, ranging from 1.1 to 1.7, in the Mdr1a/b(−/−)Bcrp1(−/−) mice. Although experimental errors could also lead to a Kp,uu of greater than 1, the existence of a weak influx system that transports ponatinib into the brain could also lead to this result. In light of this Kp,uu in the triple knockout mice, the substrate status of ponatinib regarding possible BBB uptake transporters is of interest. These transporters may include various organic anion and especially cation transport systems, and nutrient influx transports, such as the amino acid influx systems (Ohtsuki and Terasaki, 2007). It is known that influx systems can influence the extent of partitioning of free drug across the BBB. Further investigation of such an influx mechanism is necessary to determine whether this possible small influx component of ponatinib can affect the resultant efficacy in a tumor-bearing brain.
The use of transporter knockout mice has been useful in elucidating functionally cooperative transporter-mediated drug efflux at the BBB for investigational compounds. The Kp value in the triple knockout mice [Mdr1a/b(−/−)Bcrp1(−/−)] had a greater than additive increase in the Kp in a single knockout mice missing either Bcrp or P-gp, regardless of the route of administration and analysis approaches (i.e., NCAs and compartmental analyses). This suggests that the absence of one efflux transporter leads to functional compensation by another efflux transporter for ponatinib, as previously described for many compounds in the literature (Kodaira et al., 2010). Such functional cooperation of BBB efflux transporters is not accompanied by compensatory changes in the expression of P-gp and Bcrp in the isolated brain capillaries of the four genotypes—wild-type, Bcrp1(−/−), Mdr1a/b(−/−), and Mdr1a/b(−/−)Bcrp1(−/−)—according to a previous quantitative proteomic study (Agarwal et al., 2012). This proteomic study also reported that the quantitative expression level of P-gp (Mdr1a/b) is approximately 4.6-fold higher than that of Bcrp in the brain capillary endothelial cells isolated from wild-type, Bcrp1(−/−), and Mdr1a/b(−/−)Bcrp1(−/−) mice. Therefore, genetic deletion of P-gp would lead to a higher brain-to-plasma ratio (Kp or Kp,uu) than that of Bcrp, especially when the drug transport capacity (relative expression levels) of these efflux transporters, not differences in relative affinity (binding affinity to P-gp vs. Bcrp), dictates the transporter-mediated efflux. Ponatinib had a higher brain-to-plasma ratio in the absence of P-gp [Mdr1a/b(−/−)] compared with Bcrp [Bcrp1(−/−)] in the present study. This suggests that functional capacity, in relation to the cooperative efflux of P-gp and Bcrp, drives transporter-mediated efflux of ponatinib at the BBB.
Efficient net efflux of a drug at the BBB will lead to a Kp,uu value below 1, where the efflux capability exceeds influx of a free drug at the BBB (CLu,in < CLu,out). Most tyrosine kinase inhibitors that have been examined for the treatment of brain tumor have total and free brain-to-plasma ratios below 1 (Ballard et al., 2016; Heffron, 2016). Other processes in the brain, including drug metabolism within the brain and bulk flow (extracellular fluid drainage), may contribute to the clearance of a drug out of the brain, but transporter-mediated efflux is often a key main contributor of such elimination mechanisms relative to other processes. The unbound drug concentration in plasma drives the movement of drug across the BBB into the brain, but the relative difference between kout and kelim determines the effective half-life of drug in the brain. The present study showed that the kout value was greater than the kelim value for ponatinib, which will influence the time dependence of drug partitioning into the brain (Tables 5 and 6).
In conclusion, this study showed that the two major efflux transporters, P-gp and Bcrp, cooperate to modulate the brain exposure of ponatinib without affecting systemic exposure to ponatinib. Genetically modified mice lacking both P-gp and Bcrp displayed a brain-to-plasma ratio that was higher than what would be anticipated from brain-to-plasma ratios in the single knockout genotype [Mdr1a/b(−/−) or Bcrp1(−/−)], indicating functionally cooperative transport of ponatinib out of the brain. The compartmental analyses and NCAs resulted in similar parameter estimates describing the extent of brain penetration, and the compartmental BBB model described the observed data well (plasma and brain concentration-time profiles) providing insight into the transport of ponatinib across the BBB. Transporter-mediated efflux transport at the BBB reduced the extent of brain penetration (both Kp and Kp,uu) of ponatinib, and such a transport mechanism further compromised the therapeutic exposure time of the MTTbrain value. Ponatinib brain distribution is an exemplary case to appreciate how drug binding may influence efficacy and how the total Kp may be misleading. Combined with the previous data regarding heterogeneous binding and drug distribution in the intracranial tumor (Laramy et al., 2017), transporter-mediated efflux of ponatinib at the BBB further compromises its therapeutic potential for the treatment of GBM.
Acknowledgments
The authors thank Jim Fisher, Clinical Pharmacology Analytical Laboratory, University of Minnesota, for his support in the development of the LC-MS/MS assay for ponatinib; and David Hottman, a graduate student in the department of Experimental and Clinical Pharmacology, University of Minnesota, for hands-on demonstration on how to use a Vibratome.
Abbreviations
- aECF
artificial extracellular fluid
- AUC
area under the curve
- AUC(0→∞)
trapezoidal rule integration to the time infinity including the extrapolated area
- AUC(0→∞),brain
trapezoidal rule integration to the time infinity including the extrapolated area of brain concentration-time profile
- AUC(0→∞),plasma
trapezoidal rule integration to the time infinity including the extrapolated area of plasma concentration-time profile
- AUC(0→t)
trapezoidal rule integration to the last time point
- BBB
blood-brain barrier
- Bcrp
breast cancer resistance protein
- Cbrain
total drug concentration in brain
- CL
clearance
- CLapparent
apparent clearance
- CLin
clearance of total drug into the brain
- CLout
clearance of total drug out of the brain
- CLsystemic
clearance of total drug from the systemic circulation (body)
- CLu,in
clearance of unbound (free) drug into the brain
- CLu,out
clearance of unbound (free) drug out of the brain
- Cmax,brain
Cmax of brain concentration
- CNS
central nervous system
- Cplasma
total drug concentration in plasma
- Cp,tss
plasma concentration corresponding to Cmax for brain
- Cu,brain
unbound (free) drug concentration in brain
- Cu,plasma
unbound (free) drug concentration in plasma
- DA
distribution advantage
- DA,pred
predicted distribution advantage
- F
bioavailability
- fu,brain
unbound (free) drug fraction in brain homogenate
- fu,plasma
unbound (free) drug fraction in plasma
- FVB
Friend leukemia virus strain B
- GBM
glioblastoma
- ka
absorption rate constant
- Ka
absorption rate constant after oral dosing
- Kelim
terminal elimination rate constant
- kin
tissue transfer rate constant into the brain
- kout
tissue transfer rate constant out of the brain, Kp, total brain-to-plasma ratio
- Kp,pred
model-predicted total brain-to-plasma ratio
- Kp,uu
unbound (free) brain-to-plasma ratio
- Kp,uu,pred
model-predicted unbound (free) brain-to-plasma ratio
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- Mdr1
murine p-glycoprotein
- MTT
mean transit time
- MTTbrain
mean transit time in the brain
- NCA
noncompartmental analysis
- PDX
patient-derived xenograft
- P-gp
p-glycoprotein
- Tmax
transient steady state at the time of Cmax
- Vbrain
volume of distribution of total drug in the brain
- Vcentral
volume of distribution of total drug in the central compartment
- Vi
residual volume of buffer that can remain on the slice surface
- Vu,brain
volume of distribution of unbound (free) drug
- Vu,central
volume of distribution of unbound (free) drug in the central compartment
Authorship Contributions
Participated in research design: Laramy, Kim, Parrish, Sarkaria, and Elmquist.
Conducted experiments: Laramy, Kim, and Parrish.
Contributed new reagents or analytic tools: Laramy.
Performed data analysis: Laramy, Kim, and Elmquist.
Wrote or contributed to the writing of the manuscript: Laramy, Kim, Parrish, Sarkaria, and Elmquist.
Footnotes
This work was supported by the National Institutes of Health [Grants R01-CA-138437, R01-NS-077921, U54-CA-210180, and P50-CA-108961]. J.K.L. was supported by the Edward G. Rippie, Rory P. Remmel, and Cheryl L. Zimmerman in Drug Metabolism and Pharmacokinetics, and American Foundation for Pharmaceutical Education Pre-Doctoral Fellowships.
This work was presented, in part, in J.K.L.’s doctoral dissertation (University of Minnesota, Minneapolis, MN) “Permeability, Binding and Distributional Kinetics of Ponatinib, a Multi-Kinase Inhibitor: Implications for the Treatment of Brain Tumors.”
References
- Abbott NJ, Ronnback L, Hansson E. (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Hartz AM, Elmquist WF, Bauer B. (2011a) Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des 17:2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. (2011b) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. (2012) Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos 40:1164–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, Pickup K, Jordan A, Hickey M, Grist M, et al. (2016) Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res 22:5130–5140. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- De Falco V, Buonocore P, Muthu M, Torregrossa L, Basolo F, Billaud M, Gozgit JM, Carlomagno F, Santoro M. (2013) Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab 98:E811–E819. [DOI] [PubMed] [Google Scholar]
- De Witt Hamer PC. (2010) Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncol 12:304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, McAllister CB, Inoue M, Wilkinson GR. (1989) Plasma binding and transport of diazepam across the blood-brain barrier. No evidence for in vivo enhanced dissociation. J Clin Invest 84:1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friden M, Bergstrom F, Wan H, Rehngren M, Ahlin G, Hammarlund-Udenaes M, Bredberg U. (2011) Measurement of unbound drug exposure in brain: modeling of pH partitioning explains diverging results between the brain slice and brain homogenate methods. Drug Metab Dispos 39:353–362. [DOI] [PubMed] [Google Scholar]
- Friden M, Ducrozet F, Middleton B, Antonsson M, Bredberg U, Hammarlund-Udenaes M. (2009) Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab Dispos 37:1226–1233. [DOI] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D. (1998) Pharmacokinetics, Marcel Dekker, Inc., New York. [Google Scholar]
- Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM. (2012) Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 11:690–699. [DOI] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Friden M, Syvanen S, Gupta A. (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25:1737–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Paalzow LK, de Lange EC. (1997) Drug equilibration across the blood-brain barrier–pharmacokinetic considerations based on the microdialysis method. Pharm Res 14:128–134. [DOI] [PubMed] [Google Scholar]
- Heffron TP. (2016) Small molecule kinase inhibitors for the treatment of brain cancer. J Med Chem 59:10030–10066. [DOI] [PubMed] [Google Scholar]
- Kakee A, Terasaki T, Sugiyama Y. (1996) Brain efflux index as a novel method of analyzing efflux transport at the blood-brain barrier. J Pharmacol Exp Ther 277:1550–1559. [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, Pollack GM. (2007) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666. [DOI] [PubMed] [Google Scholar]
- Keir S, Saling J, Roskoski M, Friedman H, Bigner D. (2012) Efficacy of combination therapy with ponatinib (AP24534) +/− bevacizumab against pediatric glioblastoma. Neuro-oncol 14:i56–i68. [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796. [DOI] [PubMed] [Google Scholar]
- Kong AN, Jusko WJ. (1988) Definitions and applications of mean transit and residence times in reference to the two-compartment mammillary plasma clearance model. J Pharm Sci 77:157–165. [DOI] [PubMed] [Google Scholar]
- Laramy JK, Kim M, Gupta SK, Parrish KE, Zhang S, Bakken KK, Carlson BL, Mladek AC, Ma DJ, Sarkaria JN, et al. (2017) Heterogeneous binding and central nervous system distribution of the multitargeted kinase inhibitor ponatinib restrict orthotopic efficacy in a patient-derived xenograft model of glioblastoma. J Pharmacol Exp Ther 363:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loryan I, Friden M, Hammarlund-Udenaes M. (2013) The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. (2012) Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther 342:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi RK, Mittapalli RK, Elmquist WF. (2013) Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J Pharmacol Exp Ther 347:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki S, Terasaki T. (2007) Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 24:1745–1758. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. (2016) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro-oncol 18:v1–v75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish KE, Cen L, Murray J, Calligaris D, Kizilbash S, Mittapalli RK, Carlson BL, Schroeder MA, Sludden J, Boddy AV, et al. (2015a) Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther 14:2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish KE, Pokorny J, Mittapalli RK, Bakken K, Sarkaria JN, Elmquist WF. (2015b) Efflux transporters at the blood-brain barrier limit delivery and efficacy of cyclin-dependent kinase 4/6 inhibitor palbociclib (PD-0332991) in an orthotopic brain tumor model. J Pharmacol Exp Ther 355:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinoso RF, Telfer BA, Rowland M. (1997) Tissue water content in rats measured by desiccation. J Pharmacol Toxicol Methods 38:87–92. [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN. (2011) Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, Wolters Kluwer Health/Lippincott William & Wilkins, Philadelphia. [Google Scholar]
- Suzuki Y, Tanaka K, Negishi D, Shimizu M, Yoshida Y, Hashimoto T, Yamazaki H. (2009) Pharmacokinetic investigation of increased efficacy against malignant gliomas of carboplatin combined with hyperbaric oxygenation. Neurol Med Chir (Tokyo) 49:193–197, discussion 197. [DOI] [PubMed] [Google Scholar]
- Vaidhyanathan S, Wilken-Resman B, Ma DJ, Parrish KE, Mittapalli RK, Carlson BL, Sarkaria JN, Elmquist WF. (2016) Factors influencing the central nervous system distribution of a novel phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor GSK2126458: implications for overcoming resistance with combination therapy for melanoma brain metastases. J Pharmacol Exp Ther 356:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Welty DF. (1996) The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res 13:398–403. [DOI] [PubMed] [Google Scholar]