

Abstract
IL-11 has been detected in inflamed joints; however its role in the pathogenesis of arthritis is not yet clear. Studies were conducted to characterize the expression and functional significance of IL-11 and IL-11Rα in rheumatoid arthritis (RA). IL-11 levels were elevated in RA synovial fluid (SF) compared to osteoarthritis (OA) SF and plasma from RA, OA and normal individuals (NLs). Morphologic studies established that IL-11 was detected in lining fibroblasts and macrophages in addition to sublining endothelial cells and macrophages at higher levels in RA compared to NL synovial tissues (STs). Since IL-11Rα was exclusively expressed in RA fibroblasts and endothelial cells, macrophages were not involved in IL-11 effector function. Ligation of IL-11 to IL-11Rα strongly provoked fibroblast infiltration into RA joint, while cell proliferation was unaffected by this process. Secretion of IL-8 and VEGF from IL-11 activated RA fibroblasts was responsible for the indirect effect of IL-11 on endothelial cell transmigration and tube formation. Moreover, IL-11 blockade impaired RA SF capacity to elicit endothelial cell transmigration and tube formation. We conclude that IL-11 binding to endothelial IL-11Rα can directly induce RA angiogenesis. In addition, secretion of proangiogenic factors from migrating fibroblasts potentiated by IL-11 can indirectly contribute to RA neovascularization.
Keywords: IL-11, RA synovial tissue, RA synovial fluid, endothelial migration and tube formation, RA synovial tissue fibroblasts
INTRODUCTION
IL-11 belongs to the IL-6 family of cytokines comprised of leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary inhibitory factor (CNTF), cardiotropin-1 (CT-1), cardiotrophin-like related cytokine factor 1/neurotrophin-1/B-cell stimulating factor 3 (NNT-1), neuropoietin (NPN), IL-27 and IL-31 [1, 2]. Initially, IL-11 was discovered in the culture media of a primate bone marrow derived stromal cell line, as a cytokine that induced formation and development of blood cells, and it was subsequently cloned in 1990 [3, 4]. A variety of cell types have been reported to secrete IL-11 including epithelial cells, fibroblasts, osteoblasts, lung smooth muscle cells, neurons, endothelial cells and endometrial cells [5]. IL-11 activates JAK/STAT and PI3K/AKT/mTORC1 pathways through heterodimerization of IL-11Rα and gp130 [5, 6].
IL-11 plays an important role in a number of physiological functions. IL-11 alone or in synergy with IL-3 and stem cell factor participates in production of red blood cells [7]. Moreover, IL-11 together with IL-13 and IL-4 can increase the frequency of myeloid progenitor cell differentiation into macrophages [8]. Consistent with these observations, IL-11 administration in a clinical trial resulted in increased bone marrow cellularity which was due to enhanced number of circulating immature red blood cells and myeloid progenitor cells [9]. Pathological overexpression of IL-11 occurs in cancers of epithelial cell origin as well as in a number of autoimmune diseases [10, 11]. Elevated levels of IL-11 and IL-11Rα were detected in colorectal and prostate carcinoma, and serum IL-11 has been proposed as a potential marker for prostate cancer [12, 13]. In multiple sclerosis (MS), IL-11 induces TH-17 cell differentiation and expansion of memory TH-17 cells [14].
Data concerning the role of IL-11 in preclinical models and RA are controversial. In mice, systemic treatment with IL-11 reduced collagen induced arthritis (CIA) disease severity; conversely local injection of IL-11 exacerbated joint swelling [15, 16]. However, RA patients in remission had lower serum IL-11 levels that correlated with disease activity score (DAS)28 improvement [17]. In phase I/II studies, treatment with IL-11 did not significantly modulate disease activity [18].
To address these controversies, we sought to elucidate the expression pattern, regulatory factors and the functional consequences of IL-11 and IL-11Rα expression in RA synovitis. We found that IL-11 and IL-11Rα are co-expressed in RA ST fibroblasts and endothelial cells and thus interconnect the function of these two cell types. Lining and sublining macrophages uniquely expressed IL-11; however because of IL-11Rα absence, macrophages are not IL-11 responder cells. The highest levels of IL-11 were secreted following IL-1 stimulation in RA ST fibroblasts compared to endothelial or RA myeloid cells activated by RA SF. Direct binding of IL-11 to IL-11Rα on RA fibroblasts provoked cell migration, which was not detected when supernatants of IL-11 treated RA fibroblasts or endothelial cells were used. In contrast, the effect of IL-11 on angiogenesis was mediated both through its direct interaction with endothelial IL-11Rα as well as indirectly via potent proangiogenic factors released from RA ST fibroblasts. Extending the in vitro results, IL-11 is strongly capable of forming new blood vessels in vivo in matrigel plugs. Our study suggests that IL-11 has a dual role in the pathogenesis of RA, both enhancing synovial fibroblast infiltration and further advancing disease severity by increasing the invasion of blood vessels into the RA pannus.
MATERIALS AND METHODS
Patient Recruitment and Ethics
Patients were recruited from the practices of orthopedic surgeons or rheumatologists in the group practice of the academic physicians of University of Illinois at Chicago. Synovial tissues and fibroblasts extracted from the synovium were obtained from individuals undergoing total joint replacement or synovectomy. All RA patients met the ACR 1987 Revised Classification of RA [19]. All studies were approved by the University of Illinois at Chicago Institutional Review Board and all donors provided written informed consent. All experiments were performed in accordance with these guidelines and regulation. STs from RA, OA and NLs were de-identified and were formalin fixed, paraffin-embedded and sectioned. Data recorded at the time of the tissue collection is date of collection and patient diagnosis. RA tissue samples or fluid submitted to our research required no special handling. RA peripheral blood was drawn into tubes containing citrate phosphate dextrose solution.
RA patient population
Blood was obtained from patients with RA, diagnosed according to the 1987 revised criteria of the American College of Rheumatology [19]. PB was obtained from 9 women and 1 man (mean age 57.1 ± 3.6 years). At the time of blood donation, patients were receiving no treatment (n=2), taking non-biological disease-modifying anti-rheumatic drug (DMARD)s (methotrexate, leflunomide, sulfasalazine azathioprine) alone (n=4), or taking a TNF-α inhibitor with a DMARD (n=4). These studies were approved by the University of Illinois at Chicago Institutional Ethics Review Board and all donors gave informed written consent.
Antibodies and Immunohistochemical analysis
STs were immunoperoxidase-stained using Vector Elite ABC kits (Vector Laboratories, Burlingame, CA), with diaminobenzidine (Vector Laboratories) as a chromogen. Briefly, slides were deparaffinized in xylene, followed by rehydration by transfer through graded alcohols. Antigens were unmasked by heat-induced retrieval in 10mM sodium citrate. STs were incubated with antibodies (Ab) to anti-human IL-11 (1:50; Santa Cruz biotechnology), anti-human IL-11Rα (1:250; Santa Cruz biotechnology) or an IgG control. For co-localization or immunofluorescence studies, RA ST sections were stained with Abs to CD68 (1:100 Dako Cytomation, Carpinteria, CA), VWF (1:1000, Dako Cytomation) or Vimentin (1:2000, Thermo Fisher) to determine which cell types express IL-11 and IL-11Rα. Proteinase K (20μg/mL) was used as an additional antigen retrieval step for CD68 sections. Species specific AffiniPure F(ab’)2 antibodies conjugated to Alexa Fluor® 488 or Alexa Fluor® 594 (Jackson ImmunoResearch) were utilized as secondary antibodies. Immunofluorescence was imaged on a Nikon A1A confocal microscope and Nikon Elements Acquisition software. STs were scored for lining, sublining and endothelial cell staining by blinded observers (SJK and MVV) [20–22]. Cell staining was scored on a 0–5 scale; where 0=no staining, 1=few cells stained, 2=some (less than half) cells stained, 3= around half of the cells were stained positively 4= majority or more than half of the cells were positively stained and 5= all cells were positively stained. Scored data were pooled, and the mean ± SE was calculated in each data group [20–22].
Western blot analysis
NL and RA peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll gradient (GE healthcare) thereafter macrophages were differentiated in vitro for 7 days as described previously [23, 24]. Fibroblasts from fresh RA ST were isolated by mincing and digestion in a solution of dispase, collagenase, and DNase and the cells were used between passages 3 and 9 [20, 25]. In vitro differentiated NL and RA macrophages, human umbilical vein endothelial cells (HUVEC)s and RA ST fibroblasts were lysed and cell lysates were examined by Western blot analysis. Blots were probed with IL-11Rα (1:50, Santa Cruz Biotechnology) and actin was used as the equal loading control (1:1000, BD transduction laboratories).
Cell treatment and ELISA
IL-11 concentrations were quantified in NL plasma (n=35), OA plasma (n=35), RA plasma (n=40), OA SF (n=40) and RA SF (n=40) by ELISA (R&D Systems). To determine which cell types produce IL-11; RA ST fibroblasts, HUVECs, RA PB monocytes and RA in vitro differentiated macrophages were either untreated (PBS) or stimulated with 100ng/ml of TNF, IL-1β, LPS, flagellin or IL-17 for 48h. In case of stimulation with 10% RA SF, IL-11 concentration in the medium containing 10% RA SF was considered as 1 fold and IL-11 levels secreted from each cell type in response to 10% RA SF was demonstrated as fold increase. Additionally, protein levels of IL-8, VEGF, Ang-1, CXCL1, CXCL5 or bFGF were quantified by ELISA (R&D Systems) in supernatants from RA ST fibroblasts or HUVECs that were untreated (PBS) or stimulated with IL-11 (100ng/ml) for 48h. Next, HUVECs and RA ST fibroblasts were stimulated with IL-11 (100ng/ml) for 48h and supernatants were analyzed for IL-6 and CCL2 secretion.
RA ST fibroblast scratch assay
A scratch was created in the middle of the wells that contained confluent RA ST fibroblasts [26]. Thereafter, RA ST fibroblasts (cultured in 5% FBS) were either untreated (PBS) or treated with IL-11 (1, 10, 100 and 200ng/ml; Gibco) or bFGF (+ control; 100ng/ml) for 24h. To determine the indirect effect of IL-11 on RA ST fibroblast migration; supernatants were obtained from 48h untreated (PBS sup) and IL-11 (100ng/ml) treated RA ST fibroblasts in the absence or presence of IL-11Rα-Fc chimera (added 1h prior to use) (R&D Systems, 10 μg/ml; an antagonist to IL-11[27]). To test the impact of IL-11 treated endothelial cells on fibroblast migration, supernatants obtained from untreated HUVECs (PBS sup) or those treated with IL-11 (100ng/ml) for 48h were assessed in fibroblast scratch assay in the absence or presence of soluble IL-11Rα-Fc chimera (10μg/ml). In all scratch assay experiments, cells were fixed with 10% formalin for 1h and were subsequently stained with 0.05% crystal violet for 1h prior to imaging. The number of cells in the scratch area was counted and compared to the untreated control.
RA ST Fibroblasts Proliferation assay
To determine the impact of IL-11 on RA ST fibroblast proliferation, MTT assay was performed. Cells were plated to 60–70% confluency in a 24 well plate and were either untreated (PBS) or treated with IL-11 (100ng/ml) or bFGF (+ control; 100ng/ml used only at 24h and 96h). Subsequent to 24h, 48h, 72h or 96h of treatment, MTT solution (5mg/ml) was added into each well for 3h. Thereafter, the MTT reaction was stopped [28] and the OD was determined at 570 nm. The data in each time point is shown as fold increase above the counterpart control.
Transwell endothelial cell migration assay
HUVEC transmigration was assessed using 8μm permeable inserts. To examine the effect of IL-11 on endothelial cell transmigration, PBS, IL-11 (1, 10, 100ng/ml) or VEGF (+ control; 100ng/ml) were added to the media (containing 0.5% BSA) in the wells of a 24 well plate and inserts containing endothelial cells (5×105 cells/insert) were immersed in the media for 24h. Next, to establish that IL-11 induced endothelial cell transmigration is dependent on IL-11Rα, HUVECs placed in inserts, were tested for migration in response to IL-11 (100ng/ml) alone or IL-11 plus IL-11Rα-Fc chimera protein (10μg/ml; added 1h prior to positioning the insert) for 24h. To investigate whether factors released from RA ST fibroblasts can impact endothelial cell migration, supernatants from RA fibroblasts that were untreated (PBS sup) or treated with IL-11 (100ng/ml) for 48h, were added to the wells in the absence or presence of IL-11Rα-Fc chimera protein (10μg/ml) and endothelial cell migration was tested for 24h. Soluble IL-11Rα-Fc chimera protein was added to the IL-11 supernatants to negate the direct effect of IL-11 on endothelial migration. In all experiments following 24h incubation, inserts were removed, the non-migrating endothelial cells on the upper surface were detached with a cotton swab and the cells that migrated to the lower surface were fixed with 10% formalin for 1h. Thereafter, the migrated cells were stained with 0.05% crystal violet and imaged. The number of migrated cells, were counted in 3 different 10× fields in each treatment condition.
Endothelial tube formation assay
To determine the role of IL-11 in endothelial tube formation, HUVEC capillary formation was examined in response to IL-11 (1, 10, 100ng/ml) or VEGF (+ control; 100ng/ml) for 18h. Endothelial tube formation was also examined in the presence of IL-11 (100ng/ml) or RA SF (10%) with or without IL-11Rα-Fc chimera (10μg/ml). To identify the proangiogenic factors released from IL-11 treated fibroblasts that are responsible for endothelial tube formation, HUVECs were pre-incubated with Abs against specific proangiogenic factor receptors, including anti-CXCR1 and anti-VEGFR2 (10μg/ml; R&D systems), alone or in combination, for 1h prior to addition of supernatants obtained from IL-11 stimulated RA fibroblasts. In the same experiment, the direct effect of IL-11 was neutralized by pre-incubation with IL-11Rα-Fc chimera (10μg/ml). The total number of tube branching points/4xfield was counted per well and the data represent average of 3 wells [29–31].
Matrigel plug assay in vivo
To examine the effect of IL-11 on angiogenesis in vivo, we used a matrigel plug assay. C57BL/6 mice were injected subcutaneously with 500μl matrigel containing PBS or bFGF (100ng) as negative or positive control and mouse IL-11 (4μg) served as the experimental condition. After 10 days, mice were sacrificed; matrigel plugs were removed and analyzed for vascular density. Histology slides from different treatment groups were examined by H&E staining. Vascular density was scored on a 0–5 scale, where 0 = no tubules, 1= few tubules, 2 = more dense tubules, 3= tubules with multilayer walls, 4= tubules with multilayer walls and red blood cells and 5= tubules with multilayer walls and connective tissues.
Data Sharing Plan
Materials generated under this project will be disseminated in accordance with the policies of the University of Illinois at Chicago and NIH. The results, techniques and the protocol generated in this study will be made available after being accepted for publication by depositing to Pubmed Central.
Statistical Analysis
One way analysis of variance was used for comparisons among multiple groups, followed by Student’s post hoc 2-tailed T-test. Students paired and unpaired 2-tailed test were used for comparisons between 2 groups. P values less than 0.05 were considered significant.
RESULTS
Elevated levels of IL-11 and IL-11Rα are detected in RA specimen
To understand the role of IL-11 in RA, we investigated the expression pattern of IL-11 and IL-11Rα in RA compared to OA and NL specimens. We found that expression of IL-11 was significantly elevated (2 fold) in the lining layer macrophages and fibroblasts as well as in the sublining macrophages and endothelial cells of RA relative to NL STs (Figs. 1A–B). In OA STs, IL-11 expression levels were lower (2.5 fold) in the lining cells and sublining macrophages compared to RA STs (Figs. 1A–B). Serial section and immunofluorescence staining of IL-11, CD68, VWF and vimentin in RA STs indicated that IL-11 is expressed by lining RA fibroblasts and macrophages in addition to sublining endothelial cells, macrophages and a number of fibroblasts (Figs. 1D–E). In agreement with these findings, IL-11 levels were significantly enhanced in RA SF compared to OA SF (47 fold) and plasma from RA (19 fold), OA (75 fold) and NL (18 fold) individuals (Fig. 1C). Similar to IL-11, IL-11Rα expression was markedly elevated in RA compared to NL ST lining fibroblasts (2.5 fold), sublining endothelial cells (2 fold) and fibroblasts (Figs. 2A–B). In contrast, IL-11Rα was absent in RA ST sublining and lining macrophages (Figs. 2C–E). Corroborating the histological studies, Western blot analysis authenticated that while IL-11Rα is expressed in endothelial cells (as shown in HUVECs) and RA ST fibroblasts, it is absent in NL and RA macrophages (Figs. 2A–E). The expression pattern of IL-11Rα indicates that fibroblasts and endothelial cells are the primary responders to IL-11 in RA ST.
Fig. 1. IL-11 is overexpressed in RA ST lining fibroblasts and macrophages as well as sublining endothelial cells and macrophages and its level in RA SF is significantly higher than OA SF and plasma from RA, OA and NL individuals.
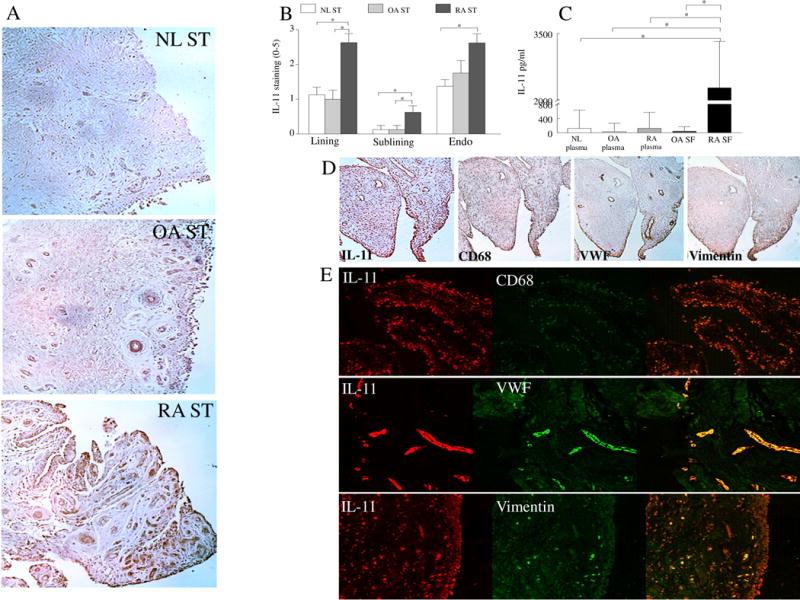
A. STs from NL, OA and RA were stained with anti-IL-11 Ab (original magnification x 200) and (B) staining was scored on a scale of 0–5 in the lining, sublining and endothelial cells, n=8−9. C. IL-11 protein concentration was determined in RA SF (n=40) and OA SF (n=40) as well as in plasma from RA (n=40), OA (n=35) and NL (n=35) individuals. D. RA ST serial sections were stained with Abs to anti-IL-11, CD68, VWF or Vimentin to establish which cell types produce IL-11, n=5. E. To confirm serial section studies, individual (red or green) as well as the overlapping (yellow) immunofluorescence staining was shown of RA STs that were stained with Abs to anti-IL-11 (green) or cell markers per slide including CD68 (red), VWF (red) or Vimentin (red) (original magnification x 400), n=5. Values are the mean ± SE. * represents p <0.05.
Fig. 2. IL-11Rα expression is markedly higher in ST lining fibroblasts and sublining fibroblasts and endothelial cells of RA compared to NL ST.
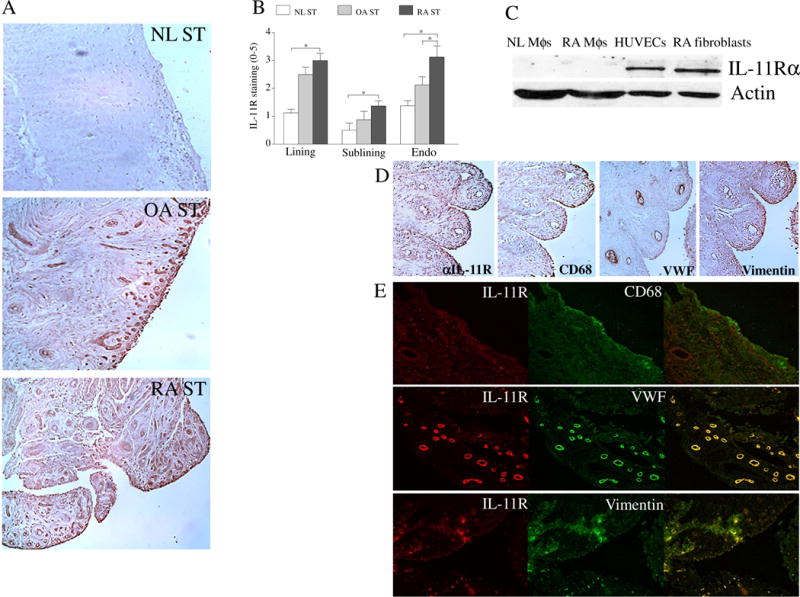
A. STs from NL, OA and RA were stained with anti-IL-11Rα Ab (original magnification x 200) and (B) staining was scored on a scale of 0–5 in the lining, sublining and endothelial cells, n=8−9. C. The IL-11Rα expression was quantified in NL and RA PB in vitro differentiated macrophages (MΦs), RA ST fibroblasts and HUVECs by Western blot analysis using anti-IL-11Rα Ab, n=5. D. RA ST serial sections were stained with Abs to anti-IL-11Rα, CD68, VWF or Vimentin to establish which cell types express IL-11Rα, n=5. E. To confirm serial section studies, individual (red or green) as well as the overlapping (yellow) immunofluorescence staining was shown of RA STs that were stained with Abs to anti-IL-11R (green) or cell markers per slide including CD68 (red), VWF (red) or Vimentin (red) (original magnification x 400), n=5. Values are the mean ± SE. * represents p <0.05.
IL-11 is produced from RA ST fibroblasts, endothelial cells, RA monocytes and macrophages
Next, different cell types were examined to determine the potential sources of IL-11 production. Consistent with the histology, RA ST fibroblasts secreted markedly higher levels of IL-11 upon stimulation with IL-1β (51 fold) and RA SF (6.5 fold; 42 vs 285 pg/ml) compared to the control treatment group (Figs. 3A, 3E). IL-11 was produced in endothelial cells (2.5 fold; 42 vs 105 pg/ml), RA PB monocytes (5.5 fold; 42 vs 236) and RA PB in vitro differentiated macrophages (3 fold; 42 vs 121) following RA SF stimulation (Fig. 3E). In contrast, endothelial cells, RA monocytes and RA in vitro differentiated macrophages were unable to secrete IL-11 in response to a number of proinflammatory factors including ligands to TLRs (Figs. 3B–D).
Fig. 3. Production of IL-11 was detected in RA ST fibroblasts, endothelial cells, RA PB monocytes and macrophages following stimulation.
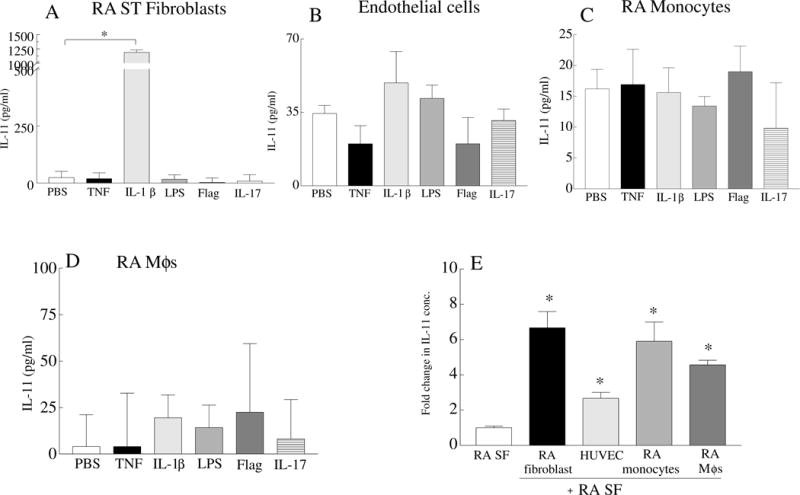
RA fibroblasts (A), HUVECs (B), RA PB monocytes (C) and RA in vitro differentiated macrophages (MΦs)(D) were untreated (PBS) or stimulated with 100 ng/ml of TNF, IL-1β, LPS and Flagellin (Flag), IL-17 for 48h. Supernatants were tested for IL-11 protein concentration by ELISA, n=6. E. RA fibroblasts, HUVECs, RA PB monocytes, and RA MΦs were stimulated with 10% RA SF for 48h. IL-11 concentration in the medium containing 10% RA SF was considered as 1 fold and IL-11 levels secreted from each cell type in response to 10% RA SF was demonstrated as fold increase above medium containing 10% RA SF. Values are the mean ± SD. * represents p <0.05.
Since hypoxia is an important mechanism that stimulates production of pro-angiogenic factors, RA myeloid cells and endothelial cells were exposed to hypoxia as well as IL-6 stimulation for 48h. Results from these studies also demonstrate that hypoxia was not capable of instigating secretion of IL-11 from RA macrophages or HUVECs. Likewise, IL-6 did not impact IL-11 release from RA macrophages or HUVECs under normoxia or hypoxia. In contrast the positive control, RA SF, markedly enhanced secretion of IL-11 from RA macrophages and HUVECs and the reduced oxygen level in hypoxic condition did not affect this process. Alternatively, there may be a combination of multiple factors present in RA SF that is responsible for IL-11 production from endothelial and RA myeloid cells. Given that the highest concentration of IL-11 secretion was induced in RA ST fibroblasts by IL-1β, the proliferating fibroblasts within the pannus may be an important source for IL-11 production.
Binding of IL-11 to IL-11Rα expressed on RA fibroblasts promotes cell migration but not cell proliferation
We found that expression of IL-11 and IL-11Rα is markedly enhanced in RA ST fibroblasts (Figs. 1D–E, 2D–E). Since fibroblast migration and hyperplasia contribute to pannus formation [32, 33], the impact of IL-11 and its receptor was examined in model systems relevant to these aspects of RA. Employing a scratch assay, we observed that IL-11 could dose dependently elevate RA fibroblast migration starting at 100ng/ml (Fig. 4A). The effect of IL-11 on RA fibroblast migration was abrogated when soluble IL-11Rα-Fc chimeric protein was utilized (Figs. 4B–C). In contrast, the supernatants obtained from IL-11 stimulated RA fibroblasts or HUVECs were unable to promote RA fibroblast migration (Figs. 4B–E). When the impact of IL-11 on RA ST fibroblast proliferation was evaluated, we found that while this manifestation was unaffected by IL-11 therapy, it was markedly accelerated by bFGF treatment (Fig. 4F). These findings suggest that IL-11 binding to IL-11Rα expressed on RA ST fibroblast directly provokes cell migration, and an indirect pathway is not involved in this process.
Fig. 4. Ligation of IL-11 to IL-11Rα induces RA ST fibroblasts migration.
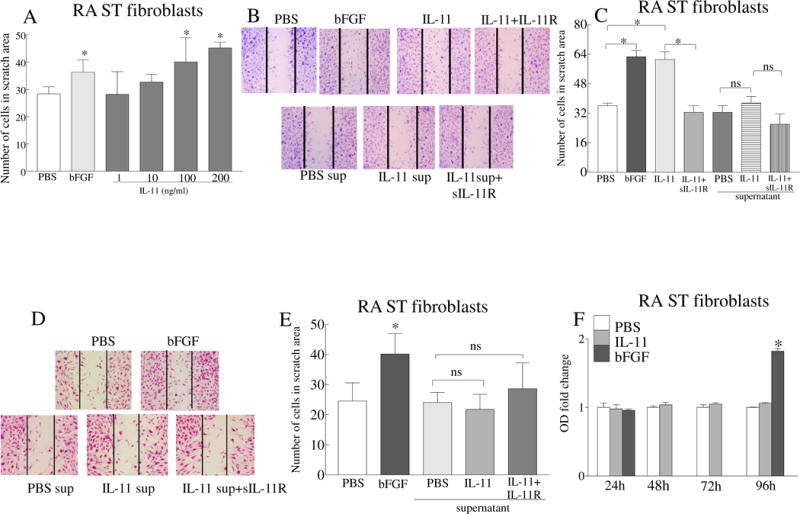
A. Effect of IL-11 (1–200ng/ml) on RA ST fibroblast migration was examined using an in vitro scratch assay for 24h. B. Representative images of RA ST fibroblast scratch assays performed on untreated cells (PBS) or cells treated with IL-11 (100ng/ml) with or without IL-11Rα-Fc chimera (10μg/ml, IL-11 antagonist). Additionally, supernatants obtained from RA ST fibroblasts that were untreated (PBS sup) or stimulated for 48h with IL-11 (100ng/ml) were tested in presence or absence of IL-11Rα-Fc chimera (10μg/ml). C. Number of fibroblasts counted in the scratch area shown in B. D. Representative images of the RA fibroblast scratch assay performed for 24h using the supernatants from HUVECs that were untreated (PBS sup) or treated with IL-11 (100ng/ml) in the absence or presence of IL-11Rα-Fc chimera (added 1h prior to use). E. Number of fibroblasts counted in the scratch area shown in D. F. MTT assay was performed in RA ST fibroblasts that were untreated (PBS) or treated with 100ng/ml of IL-11 or bFGF for 24–96h. OD was measured at 570 nm and the data in each time point is shown as fold increase above control. In all the experiments, bFGF (100ng/ml) was used as a positive control. All treatments were done in triplicate and each experiment was performed 3 independent times. Values are the mean ± SD. * represents p <0.05.
IL-11 contributes to endothelial cell migration and tube formation via both direct and indirect mechanisms
Angiogenesis is indispensable for initiation and progression of RA inflammation and bone destruction [34]. Since IL-11 and IL-11Rα were upregulated in RA ST vascular endothelium, we next investigated the impact of IL-11 on proangiogenic factors, endothelial transmigration and tube formation. HUVECs activated with IL-11 were screened for production of proangiogenic factors such as Ang-1, VEGF, FGF, CXCL1 and CXCL5. Results from these experiments demonstrated that HUVECs were only capable of producing modest levels of Ang-1 (22% increase) in response to IL-11 stimulation (Fig. 5A). IL-11 induced endothelial cell migration (3–15 fold) and tube formation (2–3 fold) in a dose dependent manner, starting at a low concentration of 1 ng/ml (Figs. 5B, 5E–F). This indicates that IL-11 (up to 3.7 ng/ml is expressed in RA SF) can contribute to angiogenesis at a physiologically relevant concentration. Endothelial transwell migration was significantly impaired in the presence of soluble IL-11Rα-Fc chimeric protein (50%), confirming that the migratory effect of IL-11 is modulated via endothelial IL-11Rα binding (Figs. 5C–D). Corroborating these findings, we found that ligation of SF IL-11 to endothelial IL-11Rα was involved in RA angiogenesis, as addition of soluble IL-11Rα-Fc chimeric protein significantly suppressed SF induced tube formation (Fig. 5G).
Fig. 5. IL-11 promotes endothelial cell migration and tube formation that is mediated through IL-11Rα ligation.
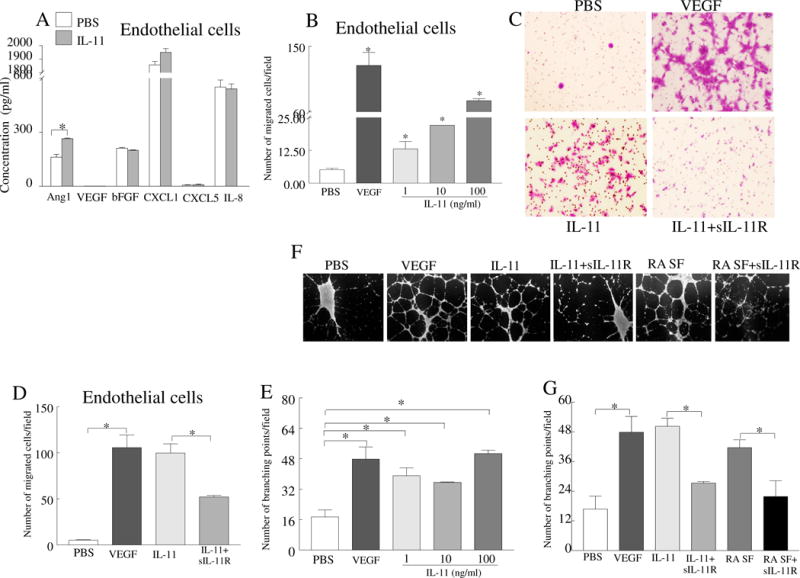
A. Protein levels of Ang-1, VEGF, bFGF, CXCL1, CXCL5 and IL-8 were quantified in supernatants obtained from HUVECs stimulated with PBS or IL-11 (100ng/ml) for 48h, n=5. B. Effect of IL-11 (1–100ng/ml) was determined on HUVEC migration using inserts of 8μm following 24h incubation. C. Representative images from HUVEC migration in response to IL-11 (100ng/ml) alone or IL-11 plus IL-11Rα-Fc chimera (10μg/ml). D. Number of endothelial cells that migrated in response to different treatment conditions shown in C. E. IL-11 (1–100ng/ml) effect was examined on HUVECs (25,000 cells/well) tube formation following 18h. The number of branching points were counted in each treatment and compared to negative control (PBS, 1% FBS in medium). F. Representative images of tube formation when HUVECs were treated with IL-11 (100ng/ml) or RA SF (10%) in absence or presence of IL-11Rα-Fc chimera (10μg/ml). G. The number of branching points formed in different treatment groups shown in F. In all experiments, VEGF (100ng/ml) was regarded as positive control. Experiments were performed in triplicates and repeated six times. Values are the mean ± SD. * represents p <0.05.
As RA fibroblasts secrete IL-8 and VEGF but not Ang1, CXCL1 or CXCL5 in response to IL-11 (Fig. 6A), we next explored whether these proangiogenic factors contribute to IL-11 induced angiogenesis. We found that supernatants from IL-11 treated RA fibroblasts facilitate endothelial cell transwell migration that was not affected by addition of soluble IL-11Rα-Fc chimeric protein (Figs. 6B–C), indicating that this process may be due to IL-8 and VEGF but not IL-11. We observed that endothelial tube formation was suppressed by neutralizing Abs to CXCR1 (IL-8 receptor alpha) and VEGFR2 (VEGF receptor)(Figs. 6D–E).
Fig. 6. VEGF and IL-8 produced from IL-11 treated RA ST fibroblasts, contribute to the indirect effect of IL-11 on angiogenesis.
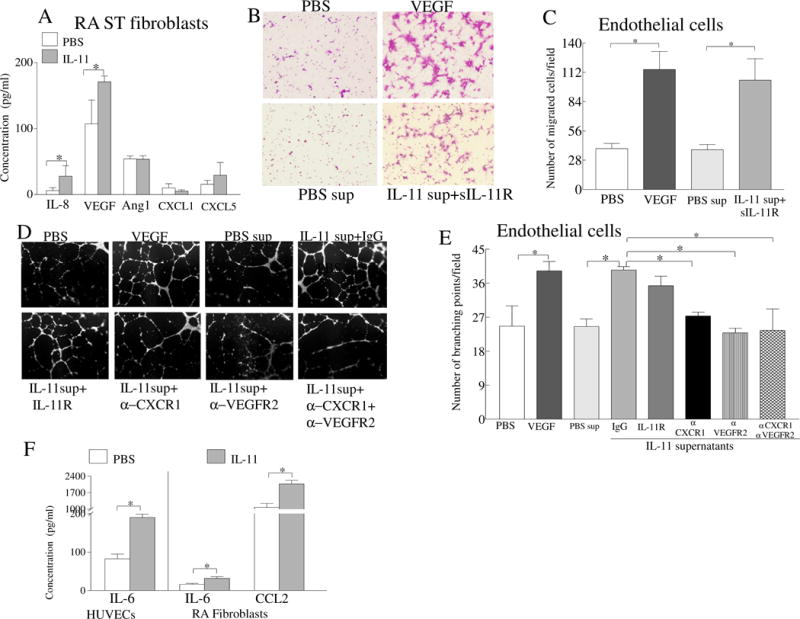
A. RA ST fibroblasts were either untreated (PBS) or stimulated with IL-11 (100ng/ml) for 48h and concentration of IL-8, VEGF, Ang1, CXCL1 and CXCL5 was determined by ELISA. B. Representative images of HUVECs that migrated in response to supernatants from RA ST fibroblasts that were untreated (PBS sup) or treated for 48h with IL-11 (100ng/ml). IL-11Rα-Fc chimera (10μg/ml) was incubated for 1h with the collected supernatants. C. Number of the HUVECs that migrated in different treatment groups shown in B. D. Endothelial tube formation assay was performed (18h) in response to supernatants from RA ST fibroblasts that were untreated (PBS sup) or treated with IL-11 (100ng/ml) for 48h, while HUVECs were preincubated with 10μg/ml of IgG, anti-CXCR1, anti-VEGFR2, anti-CXCR1 plus anti-VEGFR2 Abs. All the experiments were performed in triplicate and repeated three times, n=3. E. The branching points were counted in each treatment condition in D. F. RA PB in vitro differentiated macrophages were untreated (PBS) or stimulated with IL-11 (100ng/ml) for 48h and the supernatant levels of IL-6 and CCL2 were determined by ELISA, n=5. Values are the mean ± SD. * represents p <0.05.
Interestingly, HUVECs (IL-6) and RA ST fibroblasts (IL-6 and CCL2) secrete 2 fold higher levels of IL-6 and/or CCL2 in response to IL-11 activation (Fig. 6F). Although macrophages are nonresponsive to IL-11 stimulation due to absence of IL-11Rα on these cells, we anticipate that IL-11 can indirectly influence RA macrophage activation or migration via IL-6 and CCL2 production from endothelial cells and RA ST fibroblasts.
Taken together, we conclude that IL-11 directly promotes angiogenesis at concentrations present in RA joint; IL-11 can further amplify this effect by triggering the infiltrating fibroblasts to secrete potent proangiogenic factors.
IL-11 induces angiogenesis in vivo in matrigel plugs
The role of IL-11 on angiogenesis in vivo was assessed by determining its effect on blood vessel formation in matrigel plugs. Matrigel blood vessel formation was examined histologically by H&E staining following 10 days of inoculation. The histological analysis demonstrated that IL-11 markedly potentiates formation of blood vessel capillaries, increasing the vessel density by 2 fold compared to the control group (Figs. 7A–B). We observed red blood cell deposition in matrigel plugs containing bFGF, however these cells were absent in those containing IL-11. These results support the role of IL-11 in angiogenesis in vivo.
Fig. 7. IL-11 promotes formation of new blood vessels in matrigel plug in vivo.
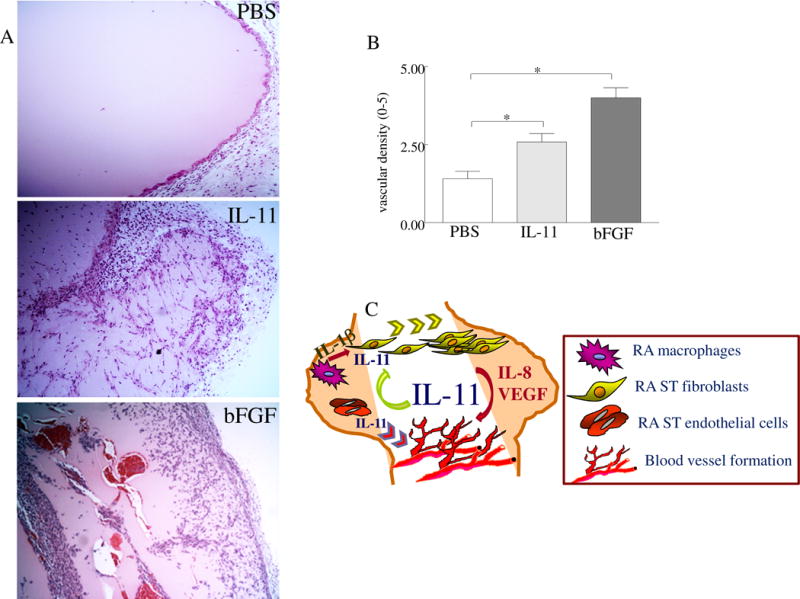
A. C57BL/6 mice were injected subcutaneously with 500μl matrigel containing PBS (-control), IL-11 (4μg) or bFGF (100ng; +control), n=5 mice per treatment group. After 10 days of inoculation, mice were sacrificed; matrigel plugs were removed and analyzed for vascular density. B. Histology slides from different treatment groups were examined by H&E staining and scored on a 0–5 scale, n=5. C. Schematic figure showing the mechanism of IL-11 function in RA. Values are the mean ± SE. * represents p <0.05.
DISCUSSION
To date, the unique roles of IL-11 in RA have not been defined. In this study we focused on uncovering the expression pattern of IL-11 and IL-11Rα in RA, and elucidating how their interaction regulates disease pathology. We determined that IL-11, released from RA ST fibroblasts and other cells, can bind to its receptor on fibroblasts to exert trafficking, without affecting cell proliferation. IL-11 also provokes the infiltrating RA ST fibroblasts to produce elevated levels of VEGF and IL-8 that promote joint angiogenesis. Moreover, at physiological concentrations, IL-11 induces formation of new blood vessels in vitro and in vivo in matrigel plugs, which can further potentiate neovascularization in the RA pannus (Fig. 7C).
Interestingly, a variety of mesenchymal cell types express IL-11, which include fibroblasts in connective tissues, lungs, uterus, osteosarcoma cell lines and osteoblasts [5]. In the majority of these cell types, IL-11 gene expression is regulated by IL-1α and phorbol myristate acetate (PMA) through induced mRNA stability [5]. Previous studies show that freshly isolated RA synoviocytes spontaneously produce IL-11 which is blunted by indomethacin, dexamethasone and IL-4 treatment [35, 36]. The same group of investigators found that IL-1α and TNF can synergistically promote IL-11 production from RA ST fibroblasts through a mechanism that is dependent on prostaglandin E2 (PGE2). IFN-γ inhibited IL-1 induced secretion of IL-11 from cultured RA fibroblasts, but not fresh RA synovial cells [35, 37].
Extending these observations, we document that IL-11 and IL-11Rα are co-localized in RA ST fibroblasts and that IL-11 production is regulated by IL-1β. We further observed that RA ST endothelial cells express IL-11 and are highly responsive to this cytokine due to expression of IL-11Rα. RA PB or ST macrophages do not express IL-11Rα and are therefore unresponsive to IL-11, despite myeloid cell production of IL-11 in response to RA SF. Others have shown that when macrophages and RA fibroblasts were isolated from fresh RA STs, spontaneous IL-11 was released from both cell types [38]. Our results indicate that while RA fibroblasts and endothelial cells are producers and responders to IL-11, RA macrophages are mainly producers of this factor.
We found that RA SF was the common stimulus that was capable of potentiating IL-11 secretion from RA ST fibroblasts, endothelial cells and RA myeloid cells; this may be in part due to the combined effect of multiple factors. In contrast to an earlier observation that demonstrates that SF from RA and OA have comparable levels of IL-11 [38], we document that IL-11 levels are significantly higher in RA SF compared to OA SF and plasma as well as RA and NL plasma. However, all previous studies are in agreement that IL-11 concentration is markedly increased in RA SF compared to RA serum, while IL-11 levels are even higher in RA ST relative to RA SF [38–40]. Interestingly, when compared to other potent proangiogenic factors expressed in RA patients, SF levels of IL-11 were higher than VEGF and comparable to bFGF, whereas IL-11’s concentration was lower than those reported for CXCL1 and CXCL5 [41–43].
The evidence regarding role of IL-11 in RA has been conflicting, since both pro- and anti-inflammatory effects have been reported in different cell types and preclinical models. When mouse bone marrow cells were co-cultured with cells extracted from calvaria, IL-11 together with parathyroid hormone significantly increased osteoclast maturation and pit formation [44]. Consistent with a role of IL-11 in bone erosion, IL-11 differentiated TH-17 cells independently of IL-6. However, IL-11 also synergizes with IL-6, IL-1β and IL-23 in amplifying TH-17 cell polarization [14]. Additionally, in in vitro and in vivo studies, IL-11 augmented antigen-specific Ab production from plasma cells, although this effect was felt to be indirect, through actions of IL-11 on CD4+ T cells and/or antigen-presenting cells [45]. Substantiating the pathogenic role of IL-11 in RA, we document that the density of the newly formed capillary tubules is 2 fold higher in the matrigel plugs containing IL-11 compared to the control plugs.
In contrast, several studies have documented the anti-inflammatory effect of IL-11. It is shown that in LPS treated mice as well as in thioglycollate elicited peritoneal macrophages; pretreatment with IL-11 impairs LPS induced TNF and IL-1 production by suppressing NF-κB signaling [46, 47]. Supporting these observations, anti-IL-11 Ab therapy alone or in combination with anti-IL-10 Ab increased TNF production from 2 to 22 fold in RA synovial membrane cells, suggesting that endogenous IL-11 and IL-10 can synergistically abrogate TNF production [40]. Interestingly, in the RA synovial membrane, IL-11 treatment had no effect on spontaneous TNF production; and these investigators document that the anti-inflammatory effect of IL-11 is only mediated in presence of its soluble receptor [40]. The fact that some cells including macrophages respond to IL-11 only in presence of soluble IL-11Rα is consistent with our finding that NL and RA macrophages do not express IL-11Rα. The lack of effect on macrophages limits the therapeutic use of IL-11 as an anti-inflammatory agent. The limited activity of IL-11, in the absence of its soluble receptor, may justify why IL-11 administration did not significantly ameliorate RA patients based on an American College of Rheumatology (ACR) 20% response [18].
Although numerous groups have reported that IL-11 is secreted from RA ST fibroblasts, the mechanism of IL-11 function is unclear in RA. We show for the first time that IL-11 plays an important role in migration of RA ST fibroblasts that is mediated through binding of IL-11 to its receptor. Factors released from IL-11 activated RA ST fibroblasts or endothelial cells do not promote fibroblast migration, however these cells secrete factors such as IL-6 and CCL2 that may play a role in myeloid cell activation or infiltration. It was previously shown that arthritis induced by local injection of IL-1 is alleviated in IL-11Rα-/- mice or with use of neutralizing IL-11 Ab, suggesting that IL-1 driven arthritis is in part due to IL-11 induction [16]. These observations are in agreement with our findings that demonstrate that IL-1 enhanced production of IL-11 from RA ST fibroblasts is responsible for fibroblast trafficking.
Similar to IL-11, other pro-inflammatory factors including IL-17 as well as TH-17 associated cytokines IL-21 and IL-22, TNF, LPS, CCL2, CXCL10, CXCL12 and CX3CL1 provoke RA ST fibroblast migration [48–54]. However, unlike IL-1 that modulates RA fibroblast migration via IL-11 induction and binding of IL-11 to the IL-11Rα, other monokines such as TNF, CCL2, CXCL10 and CXCL12 can facilitate this process by engagement of chemokine receptors distinct from IL-11Rα [54]. Also, the inflammatory monokines mechanism of action is partially different from IL-11, as they trigger both migration and proliferation of fibroblasts, whereas IL-11 exclusively facilitates RA fibroblast trafficking [48–54].
We uncover for the first time that IL-11 plays a novel role in RA angiogenesis, as blockade of its signaling impacts RA SF induced capillary tube formation. We show that IL-11-activated RA ST fibroblasts release physiological levels of VEGF and IL-8 that further augment the direct effect of IL-11 on angiogenesis. We conclude that IL-11 therefore represents a novel connection between the synovial fibroblasts and the new invading blood vessels in the RA joint, adding a new dimension to our current understanding [55] of how RA synovial fibroblasts can deliver proangiogenic signals to synovial endothelial cells.
Acknowledgments
This work was supported in part by awards from Department of Veteran’s Affairs MERIT Award 1I01BX002286, the National Institutes of Health AR056099 and AR065778, funding provided by Department of Defense PR093477. We would like to acknowledge Dr. Rhonda Kineman for critically reviewing the paper and granting us access to use her laboratory microscope.
Footnotes
COMPETING INTERESTS:
None declared.
AUTHORS CONTRIBUTION:
Designed the research: HAE, SS
Performed the research: HAE, MVV, ABE, ZC, SJK, KV, KP, SS
Analyzed the data: HAE, ZC, MVV, KV, MK, HMA, SJK, IBM, DAF, SS
Provided reagents: SA, GZ
Writing the paper: All the authors contributed to writing the paper.
LITERATURE CITED
- 1.Heinrich PC, Behrmann I, Haan S, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Putoczki TL, Thiem S, Loving A, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell. 2013;24:257–271. doi: 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Paul SR, Bennett F, Calvetti JA, et al. Molecular cloning of a cDNA encoding interleukin 11, a stromal cell-derived lymphopoietic and hematopoietic cytokine. Proc Natl Acad Sci U S A. 1990;87:7512–7516. doi: 10.1073/pnas.87.19.7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul SR, Yang YC, Donahue RE, et al. Stromal cell-associated hematopoiesis: immortalization and characterization of a primate bone marrow-derived stromal cell line. Blood. 1991;77:1723–1733. [PubMed] [Google Scholar]
- 5.Du X, Williams DA. Interleukin-11: review of molecular, cell biology, and clinical use. Blood. 1997;89:3897–3908. [PubMed] [Google Scholar]
- 6.Xu DH, Zhu Z, Wakefield MR, et al. The role of IL-11 in immunity and cancer. Cancer Lett. 2016;373:156–163. doi: 10.1016/j.canlet.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Quesniaux VF, Clark SC, Turner K, et al. Interleukin-11 stimulates multiple phases of erythropoiesis in vitro. Blood. 1992;80:1218–1223. [PubMed] [Google Scholar]
- 8.Jacobsen SE, Okkenhaug C, Veiby OP, et al. Interleukin 13: novel role in direct regulation of proliferation and differentiation of primitive hematopoietic progenitor cells. J Exp Med. 1994;180:75–82. doi: 10.1084/jem.180.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orazi A, Cooper RJ, Tong J, et al. Effects of recombinant human interleukin-11 (Neumega rhIL-11 growth factor) on megakaryocytopoiesis in human bone marrow. Exp Hematol. 1996;24:1289–1297. [PubMed] [Google Scholar]
- 10.Putoczki T, Ernst M. More than a sidekick: the IL-6 family cytokine IL-11 links inflammation to cancer. J Leukoc Biol. 2010;88:1109–1117. doi: 10.1189/jlb.0410226. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26:54–74. doi: 10.1016/j.smim.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Yoshizaki A, Nakayama T, Yamazumi K, et al. Expression of interleukin (IL)-11 and IL-11 receptor in human colorectal adenocarcinoma: IL-11 up-regulation of the invasive and proliferative activity of human colorectal carcinoma cells. Int J Oncol. 2006;29:869–876. [PubMed] [Google Scholar]
- 13.Yamazumi K, Nakayama T, Kusaba T, et al. Expression of interleukin-11 and interleukin-11 receptor alpha in human colorectal adenocarcinoma; immunohistochemical analyses and correlation with clinicopathological factors. World J Gastroenterol. 2006;12:317–321. doi: 10.3748/wjg.v12.i2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Tao Y, Chopra M, et al. IL-11 Induces Th17 Cell Responses in Patients with Early Relapsing-Remitting Multiple Sclerosis. J Immunol. 2015;194:5139–5149. doi: 10.4049/jimmunol.1401680. [DOI] [PubMed] [Google Scholar]
- 15.Walmsley M, Butler DM, Marinova-Mutafchieva L, et al. An anti-inflammatory role for interleukin-11 in established murine collagen-induced arthritis. Immunology. 1998;95:31–37. doi: 10.1046/j.1365-2567.1998.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong PK, Campbell IK, Robb L, et al. Endogenous IL-11 is pro-inflammatory in acute methylated bovine serum albumin/interleukin-1-induced (mBSA/IL-1)arthritis. Cytokine. 2005;29:72–76. doi: 10.1016/j.cyto.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Chung SJ, Kwon YJ, Park MC, et al. The correlation between increased serum concentrations of interleukin-6 family cytokines and disease activity in rheumatoid arthritis patients. Yonsei Med J. 2011;52:113–120. doi: 10.3349/ymj.2011.52.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreland L, Gugliotti R, King K, et al. Results of a phase-I/II randomized, masked, placebo-controlled trial of recombinant human interleukin-11 (rhIL-11) in the treatment of subjects with active rheumatoid arthritis. Arthritis Res. 2001;3:247–252. doi: 10.1186/ar309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 20.Pickens SR, Chamberlain ND, Volin MV, et al. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63:914–922. doi: 10.1002/art.30232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickens SR, Chamberlain ND, Volin MV, et al. Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2011;63:2884–2893. doi: 10.1002/art.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chamberlain ND, Vila OM, Volin MV, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-alpha levels. J Immunol. 2012;189:475–483. doi: 10.4049/jimmunol.1102977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SJ, Chen Z, Essani AB, et al. Identification of a novel TLR7 endogenous ligand in RA synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2015 doi: 10.1002/art.39544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Kim SJ, Chamberlain ND, et al. The novel role of IL-7 ligation to IL-7 receptor in myeloid cells of rheumatoid arthritis and collagen-induced arthritis. J Immunol. 2013;190:5256–5266. doi: 10.4049/jimmunol.1201675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahrara S, Pickens SR, Mandelin AM, 2nd, et al. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol. 2010;184:4479–4487. doi: 10.4049/jimmunol.0901942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan R, Endres J, Behbahani-Nejad N, et al. Expression and function of aminopeptidase N/CD13 produced by fibroblast-like synoviocytes in rheumatoid arthritis: role of CD13 in chemotaxis of cytokine-activated T cells independent of enzymatic activity. Arthritis Rheumatol. 2015;67:74–85. doi: 10.1002/art.38878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis DJ, Hilton DJ, Roberts B, et al. Recombinant soluble interleukin-11 (IL-11) receptor alpha-chain can act as an IL-11 antagonist. Blood. 1997;90:4403–4412. [PubMed] [Google Scholar]
- 28.Ota F, Maeshima A, Yamashita S, et al. Activin A induces cell proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2003;48:2442–2449. doi: 10.1002/art.11249. [DOI] [PubMed] [Google Scholar]
- 29.Pickens SR, Volin MV, Mandelin AM, 2nd, et al. IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol. 2010;184:3233–3241. doi: 10.4049/jimmunol.0903271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickens SR, Chamberlain ND, Volin MV, et al. Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis Rheum. 2012;64:2471–2481. doi: 10.1002/art.34452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z, Kim SJ, Essani AB, et al. Characterising the expression and function of CCL28 and its corresponding receptor, CCR10, in RA pathogenesis. Ann Rheum Dis. 2015;74:1898–1906. doi: 10.1136/annrheumdis-2013-204530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefevre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elshabrawy HA, Chen Z, Volin MV, et al. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis. 2015;18:433–448. doi: 10.1007/s10456-015-9477-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taki H, Sugiyama E, Mino T, et al. Differential inhibitory effects of indomethacin, dexamethasone, and interferon-gamma (IFN-gamma) on IL-11 production by rheumatoid synovial cells. Clin Exp Immunol. 1998;112:133–138. doi: 10.1046/j.1365-2249.1998.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taki H, Sugiyama E, Kuroda A, et al. Interleukin-4 inhibits interleukin-11 production by rheumatoid synovial cells. Rheumatology (Oxford) 2000;39:728–731. doi: 10.1093/rheumatology/39.7.728. [DOI] [PubMed] [Google Scholar]
- 37.Mino T, Sugiyama E, Taki H, et al. Interleukin-1alpha and tumor necrosis factor alpha synergistically stimulate prostaglandin E2-dependent production of interleukin-11 in rheumatoid synovial fibroblasts. Arthritis Rheum. 1998;41:2004–2013. doi: 10.1002/1529-0131(199811)41:11<2004::AID-ART16>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto H, Yamamura M, Morita Y, et al. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 1997;40:1096–1105. doi: 10.1002/art.1780400614. [DOI] [PubMed] [Google Scholar]
- 39.Trontzas P, Kamper EF, Potamianou A, et al. Comparative study of serum and synovial fluid interleukin-11 levels in patients with various arthritides. Clin Biochem. 1998;31:673–679. doi: 10.1016/s0009-9120(98)00062-9. [DOI] [PubMed] [Google Scholar]
- 40.Hermann JA, Hall MA, Maini RN, et al. Important immunoregulatory role of interleukin-11 in the inflammatory process in rheumatoid arthritis. Arthritis Rheum. 1998;41:1388–1397. doi: 10.1002/1529-0131(199808)41:8<1388::AID-ART7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 41.Yamashita A, Yonemitsu Y, Okano S, et al. Fibroblast growth factor-2 determines severity of joint disease in adjuvant-induced arthritis in rats. J Immunol. 2002;168:450–457. doi: 10.4049/jimmunol.168.1.450. [DOI] [PubMed] [Google Scholar]
- 42.Koch AE, Kunkel SL, Shah MR, et al. Growth-related gene product alpha. A chemotactic cytokine for neutrophils in rheumatoid arthritis. J Immunol. 1995;155:3660–3666. [PubMed] [Google Scholar]
- 43.Koch AE, Kunkel SL, Harlow LA, et al. Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J Clin Invest. 1994;94:1012–1018. doi: 10.1172/JCI117414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girasole G, Passeri G, Jilka RL, et al. Interleukin-11: a new cytokine critical for osteoclast development. J Clin Invest. 1994;93:1516–1524. doi: 10.1172/JCI117130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin TG, Schendel P, Yang YC. Enhancement of in vitro and in vivo antigen-specific antibody responses by interleukin 11. J Exp Med. 1992;175:211–216. doi: 10.1084/jem.175.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trepicchio WL, Bozza M, Pedneault G, et al. Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J Immunol. 1996;157:3627–3634. [PubMed] [Google Scholar]
- 47.Trepicchio WL, Wang L, Bozza M, et al. IL-11 regulates macrophage effector function through the inhibition of nuclear factor-kappaB. J Immunol. 1997;159:5661–5670. [PubMed] [Google Scholar]
- 48.Lee SY, Kwok SK, Son HJ, et al. IL-17-mediated Bcl-2 expression regulates survival of fibroblast-like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Res Ther. 2013;15:R31. doi: 10.1186/ar4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xing R, Yang L, Jin Y, et al. Interleukin-21 Induces Proliferation and Proinflammatory Cytokine Profile of Fibroblast-like Synoviocytes of Patients with Rheumatoid Arthritis. Scand. J Immunol. 2016;83:64–71. doi: 10.1111/sji.12396. [DOI] [PubMed] [Google Scholar]
- 50.Zhu J, Jia E, Zhou Y, et al. Interleukin-22 Secreted by NKp44+ Natural Killer Cells Promotes Proliferation of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Medicine (Baltimore) 2015;94:e2137. doi: 10.1097/MD.0000000000002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calmon-Hamaty F, Combe B, Hahne M, et al. Lymphotoxin alpha stimulates proliferation and pro-inflammatory cytokine secretion of rheumatoid arthritis synovial fibroblasts. Cytokine. 2011;53:207–214. doi: 10.1016/j.cyto.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 52.Elias JA, Zheng T, Whiting NL, et al. IL-1 and transforming growth factor-beta regulation of fibroblast-derived IL-11. J Immunol. 1994;152:2421–2429. [PubMed] [Google Scholar]
- 53.Guo X, Pan Y, Xiao C, et al. Fractalkine stimulates cell growth and increases its expression via NF-kappaB pathway in RA-FLS. Int J Rheum Dis. 2012;15:322–329. doi: 10.1111/j.1756-185X.2012.01721.x. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Vicuna R, Gomez-Gaviro MV, Dominguez-Luis MJ, et al. CC and CXC chemokine receptors mediate migration, proliferation, and matrix metalloproteinase production by fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Rheum. 2004;50:3866–3877. doi: 10.1002/art.20615. [DOI] [PubMed] [Google Scholar]
- 55.Edhayan G, Ohara RA, Stinson WA, et al. Inflammatory properties of inhibitor of DNA binding 1 secreted by synovial fibroblasts in rheumatoid arthritis. Arthritis Res Ther. 2016;18:87. doi: 10.1186/s13075-016-0984-3. [DOI] [PMC free article] [PubMed] [Google Scholar]