

Abstract
GNAS mutations leading to constitutively active stimulatory G protein alpha-subunit (Gsα) cause different tumors, fibrous dysplasia of bone, and McCune-Albright syndrome, which are typically not associated with short stature. Enhanced signaling of the parathyroid hormone/parathyroid hormone-related peptide receptor, which couples to multiple G proteins including Gsα, leads to short bones with delayed endochondral ossification. It has remained unknown whether constitutive Gsα activity also impairs bone growth. Here we generated mice expressing a constitutively active Gsα mutant (Gsα-R201H) conditionally upon Cre recombinase (cGsαR201H mice). Gsα-R201H was expressed in cultured bone marrow stromal cells from cGsαR201H mice upon adenoviral-Cre transduction. When crossed with mice in which Cre is expressed in a tamoxifen-regulatable fashion (CAGGCre-ER™), tamoxifen injection resulted in mosaic expression of the transgene in double mutant offspring. We then crossed the cGsαR201H mice with Prx1-Cre mice, in which Cre is expressed in early limb-bud mesenchyme. The double mutant offspring displayed short limbs at birth, with narrow hypertrophic chondrocyte zones in growth plates and delayed formation of secondary ossification center. Consistent with enhanced Gsα signaling, bone marrow stromal cells from these mice demonstrated increased levels of c-fos mRNA. Our findings indicate that constitutive Gsα activity during limb development disrupts endochondral ossification and bone growth. Given that Gsα haploinsufficiency also leads to short bones, as in patients with Albright’s hereditary osteodystrophy, these results suggest that a tight control of Gsα activity is essential for normal growth plate physiology.
Keywords: Skeletal development, cAMP, G protein, GNAS, stimulatory G protein
1. Introduction
The alpha-subunit of the stimulatory G protein (Gsα), which is essential for the actions of many hormones, stimulates adenylyl cyclase to generate the ubiquitous intracellular second messenger cAMP [1–5]. Heterozygous inactivating mutations in the gene encoding Gsα (GNAS) cause multiple human disorders characterized by hormone resistance and skeletal and developmental abnormalities (e.g. Albright’s hereditary osteodystrophy and pseudohypoparathyroidism) [6–8]. GNAS mutations that cause constitutive activation of Gsα, on the other hand, are found in many different benign and malignant tumors and cause McCune-Albright syndrome (MAS), a disorder characterized by fibrous dysplasia of bone (FD), hyperpigmented skin lesions, and hyperactivity of multiple endocrine organs [9–12].
The stimulatory G protein signaling downstream of PTH-related peptide (PTHrP, encoded by PTHLH) is an important regulator of endochondral bone formation, acting via the cAMP/protein kinase A (PKA) pathway [13]. Accordingly, inactivating mutations of GNAS, PTHLH, PDE4D (encoding cAMP phosphodiesterase type 4D), and PRKAR1A (encoding type 1A regulatory subunit of PKA), cause disorders characterized by short stature and brachdactyly type E [7, 8, 14–18]. Interestingly, patients who carry activating mutations of the parathyroid/parathyroid-related peptide receptor (PTHR) (i.e. Jansen’s metaphyseal chondrodysplasia) are also short with disproportionately short limbs [19]. Thus, it appears that both inactivation and hyperactivation of the PTHrP/Gsα/cAMP/PKA signaling pathway disrupts long bone growth. However, the PTHR couples to additional heterotrimeric G proteins and G protein-independent pathways [20], some of which are known to play roles in growth plate physiology based on mouse studies [21, 22]. It is thus conceivable that these additional signaling pathways contribute, fully or partially, to the bone growth phenotypes associated with increased PTHR signaling. Moreover, patients with MAS, who carry constitutively active Gsα mutants, do not typically present with short stature or brachdactyly. Only few cases with short stature have been reported in the literature [23, 24], and these may reflect some of the associated hormonal abnormalities, such as precocious puberty. In addition, patients with Carney-Complex (CNC), who have mutations in PRKAR1A that cause increased PKA activity, also lack short stature as a typical clinical feature [25]. Although both MAS and CNC patients can sometimes have excess growth hormone, which may mask direct effects of increased Gsα/cAMP/PKA signaling on the growth plate, the paucity of patients with short stature may suggest that the endochondral bone formation is preserved despite enhanced activation of this signaling pathway. It has therefore remained uncertain whether constitutive Gsα activity directly impairs bone growth.
In this study, we generated mice in which a constitutively active Gsα mutant (Gsα-R201H) can be expressed upon the action of Cre recombinase (cGsαR201H mice). Activation of the transgene expression in early limb-bud mesenchyme resulted in short limbs with growth plate abnormalities and delayed endochondral ossification.
2. Materials and methods
2.1. Generation of the cGsαR201H mice and in vivo activation of transgene expression
The HA-tagged rat Gsα-R201H cDNA, described previously [26], was cloned into pBSApBpACAGftILn [27], using SalI and SbfI restriction sites. This plasmid was then digested with AscI and StuI to obtain a 8637-bp fragment containing the transgene. After gel purification, this DNA fragment was microinjected into C57BL/6×129/SvJ F1-hybrid embryos (the Gene Modification Facility, Harvard University). The founders were identified by PCR using a forward primer annealing to the HA-containing portion of Gsα cDNA (5′-GACGTGCCGGATTACGCGTC-3′) and a reverse primer annealing to the native Gsα sequence (5′-TCTCAGGGTTGGCCAGCTCC-3′). The founders were crossed with wild-type C57BL/6 mice to generate F1 and F2 heterozygous offspring, which were used for analysis of transgene expression. The cGsaR201H mice were crossed with either Prx1-Cre or CAGGCre-ER™ mice to activate transgene expression in developing limb mesenchyme and in widespread tissues, respectively. Single mutant (Cre-only or transgenic-only) and wild-type littermates among the offspring of these crosses did not show gross differences from one another and were used as control mice. Both males and females were used for the analyses and comparisons were made among littermates. Prx1-Cre and CAGGCre-ER™ mice were obtained from Jackson Laboratories (Stock No: 005584 and 004682, respectively). Control and double mutant offspring from cGsαR201H and CAGGCre-ER™ matings were injected intraperitoneally with 75 mg/kg/daily tamoxifen (Sigma-Aldridge) from P14 through P18 for 5 consecutive days and euthanized for tissue analyses seven days later. All the mouse studies received approval from the Massachusetts General Hospital Institutional Animal Care and Use Committee.
2.2. BMSC isolation, qRT-PCR analysis, and Western blots
Bone marrow stromal cells (BMSCs) were isolated from femurs of 4 to 6 week-old mice and cultured in alpha-MEM medium containing 10% fetal bovine serum, as previously described [28]. After two-weeks of culture, cells were trypsinized and subcultured in 12- or 24-well plates. The following day, cells were transduced with adenovirus encoding Cre recombinase, yellow fluorescent protein (YFP), lacZ, or HA-tagged native Gsα (Gsα-HA). Three days after transduction, total RNA was isolated from transduced BMSCs using the RNeasy Plus Mini Kit (QIAGEN), and cDNA was synthesized with the ProtoScript II First stand cDNA synthesis kit (New England Biolabs). qRT-PCR analysis was performed with specific primers and FastStart Universal SYBR Green Master (Roche) with β-actin as a reference gene. Primer sequences for amplification of the HA-tagged Gsα-R201H were the same as those used for genotyping (see above). Primer sequences for amplifying c-fos and β-actin transcripts have been previously described [29, 30]. In separate experiments, whole cell lysates were prepared three days after transduction, followed by quantification of protein concentration by using the BCA reagent (Pierce). Equal amounts of total proteins for each cell lysate were separated by 9% SDS-PAGE, blotted on nitrocellulose filters by using a semi-dry blotter, and immunoreacted to a mouse monoclonal anti-HA antibody (1:1000; Abcam). After washing and subsequent incubation with anti-mouse IgG-HRP (Santa Cruz Biologicals), immunoreactive proteins were detected through the use of enhanced chemiluminescence. β-actin immunoreactivity, detected by a monoclonal antibody (Santa Cruz Biologicals), was used subsequently to verify comparable amount of protein loading among the wells.
2.3. Skeleton staining
Skeletal preparations of cartilage and bone were stained with alcian blue/alizarin red as described [31], but with minor modifications. Briefly, following euthanasia, the neonates were skinned, eviscerated, and fixed in 95% ethanol overnight, and followed by incubation in acetone for 24 hours to remove the fat. Intact skeletons were then stained overnight in alcian blue (0.3 mg/ml alcian blue 8GX, 80% ethanol, 20% acetic acid). The next day, skeletons were cleaned in 70% ethanol for 6-8 h and then transferred to 1% KOH for 24 hours until mostly clear. Subsequently, the skeletons were counterstained overnight in alizarin red (alizarin red 50μg/ml, 1% KOH), followed by cleaning in 1% KOH and 20% glycerol for 2 days or more.
2.4. X-Gal Staining and Histological analyses
Adult mice were initially perfused with intracardiac administration of 2% paraformaldehyde in phosphate-buffered saline, and their organs were dissected and further fixed overnight at 4°C. After washing with phosphate-buffered saline several times, organs were cryoprotected with 30% sucrose and embedded in the OCT compound (Tissue-Tek). Frozen sections were prepared at 12 micrometers on Shandon cryostat machine. Cultured BMSCs were fixed at room temperature for 20 min with 0.2% gluteraldehyde in phosphate-buffered saline containing 0.02% Nonidet-P40 and subsequently washed with phosphate-buffered saline containing 0.02% Nonidet-P40, 0.01% sodium-deoxycholate, and 2 mM MgCl2. Fixed and washed tissue sections and cultured BMSCs were incubated at 37C overnight in the X-gal staining solution, as described [32]. For light microscopy, femurs were removed from double mutant and control littermates and fixed in 4% paraformaldehyde overnight at 4 °C. Femurs of newborn and three-week-old mice were subsequently decalcified in 20% EDTA (pH 7.4) for a week or 3 weeks, respectively. Decalcified femurs were embedded in paraffin blocks by standard histological procedures, sectioned (5–6 μm thickness) at several different levels, and stained with hematoxylin and eosin (Center for Skeletal Research Histology Core, Endocrine Unit, Massachusetts General Hospital). X-gal stained BMSCs in culture dishes were analyzed by Leica MZ16 dissecting microscope and captured by using Leica DFC420 camera and Leica Application Suite software (Houston, TX). Stained slides were analyzed by Nikon eclipse Ni microscope (Tokyo, Japan), and images captured by using Diagnostic Instruments SPOT RT-SE™ Digital Camera (Houston, TX) and SPOT software. The ImageJ software [33] was employed to measure the relative lengths of hypertrophic and columnar proliferating chondrocyte zones in distal femur growth plates.
2.5. Statistical analyses
Mean and standard errors were calculated from multiple independent qRT-PCR experiments and growth plate measurements. Statistical significance of differences between two group means was determined by using the two-tailed Student’s t-test.
3. Results
3.1. Generation of transgenic mice conditionally expressing a constitutively active Gsα mutant
To investigate the role of enhanced stimulatory G protein signaling in multiple tissues, we generated transgenic mice in which a constitutively active Gsα mutant (Gsα-R201H) is expressed conditionally upon the action of Cre recombinase (cGsαR201H mice). The mutant Gsα cDNA included sequences for a HA epitope tag, inclusion of which was previously shown to be inert with respect to Gsα function [34]. The transgene also included the lacZ gene, which included a nuclear localization signal and was positioned downstream of the mutant Gsα cDNA following an internal ribosomal entry sequence (Fig. 1A). This allowed utilization of β-galactosidase activity as a reporter for the transgene expression.
Figure 1. Generation and validation of transgenic mice conditionally expressing a constitutively active Gsα mutant.
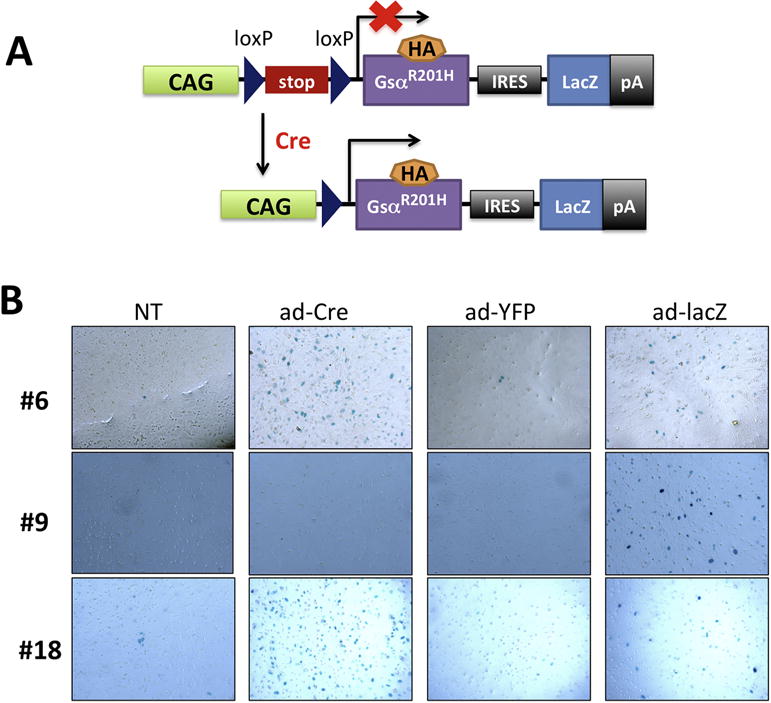
(A) Schematic diagram of the construct used for generation of cGsαR201H transgenic mice. The Gsa-R201H cDNA, as well as the lacZ gene, is expressed upon Cre recombinase activity. (B) Detection of β-galactosidase activity in BMSCs upon adenovirus transduction. Four to six-week-old F1 or F2 generation mice from each of the five founder lines were used for validation. Results are shown for lines #6, #9, and #18 only. Ad-LacZ was used as positive control for the staining. NT, non-transduced. Ad-YFP was used as negative control. Note that lines #6 and #18, but not line #9, were able to express the transgene upon Cre recombinase activity.
We identified five transgenic founders and investigated these in F1 and F2 generation to determine whether the transgene expression can be induced. To that end, we isolated BMSCs from the five founder lines and transduced these cells in culture with adenovirus expressing Cre (ad-Cre). X-gal staining revealed positive beta-galactosidase activity in BMSCs from lines #6 and #18, but not in the remaining three lines (Fig. 1B; only one of the negative lines, #9, is shown). Ad-lacZ transduction was used as a positive control for staining, and ad-YFP and non-transduced (NT) cells served as negative controls (Fig. 1B). These results indicated that the transgene could be expressed in a Cre-dependent manner in lines #6 and 18. Accordingly, qRT-PCR experiments using primers specific for the HA tag in the mutant Gsα cDNA showed that, following ad-Cre transduction, the transgene-derived Gsα mRNA was present in BMSCs isolated from lines #6 and #18, but not from line #9 (Fig. 2A). Cells transduced with ad-Gsα-HA were used as positive control. Moreover, Western blots using an antibody against the HA-tag detected the transgene-derived Gsα mutant in lysates of ad-Cre-transduced but not of ad-YFP-transduced or non-transduced BMSCs (Fig. 2B). These results indicated that the transgene is expressed in a conditional manner upon Cre expression.
Figure 2. qRT-PCR and Western blot analysis of Gsα-R201H expression in BMSCs.
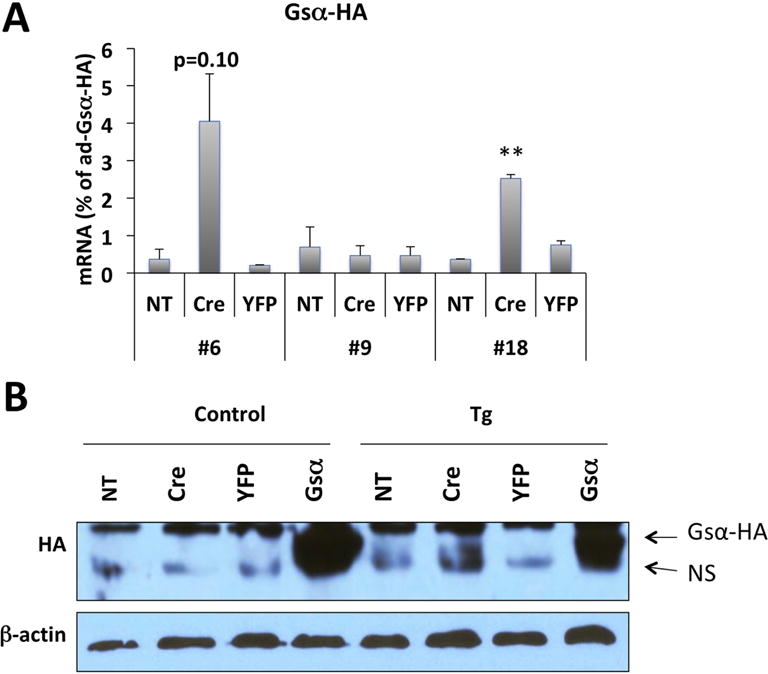
(A) qRT-PCR analysis of Gsα-R201H mRNA level in BMSCs upon adenovirus transduction. BMSCs were isolated from 4-6-week-old F1 or F2 generation of 3 founder lines. Primers were designed to detect the transgene-derived mutant Gsα transcript by taking advantage of the sequences of the HA tag. mRNA levels were normalized against the samples transduced with adenovirus-Gsα-HA. Data represent mean ± SEM of two-to-three independent experiments; **, p<0.01. (B) Western blot analysis of protein lysates from BMSCs upon adenovirus transduction. BMSCs were isolated from 4-6-week-old F1 or F2 generation cGsαR201H line #6 or control littermates. The Gsα-R201H protein was detected using an antibody against the HA tag; note the immunoreactivity above the non-specific (NS) band in the sixth lane from left. Cells transduced with ad-Gsα-HA were used as positive control. Ad-YFP was used as negative control. NT, non-transduced. β-actin immunoreactivity was used to ensure comparable gel loading.
3.2. Global, mosaic expression of the transgene in vivo
To examine whether the transgene can be expressed in vivo, we crossed our transgenic mouse strains to CAGGCre-ER™ mice, in which tamoxifen injection can induce the Cre expression ubiquitously [35]. Double mutant or control offspring were injected postnatally with tamoxifen and subsequently sacrificed for analysis. While X-gal staining failed to detect any transgene-expressing cells in double mutant offspring from line #6 (Fig. 3A,B), it showed positive β-galactosidase activity in some cells of the liver from line #18 double mutants but not controls (Fig. 3C–F). Specific X-gal staining was also observed in renal sections, both some tubular cells and glomeruli, from double mutants of line #18 (Fig. 3G,H). These results suggested that line #18, but not line #6, could be employed in vivo to study the role of constitutive Gsα activity in a Cre-inducible manner.
Figure 3. Tamoxifen induced-lacZ expression in offspring of crosses between cGsαR201H and CAGGCre-ER™ mice.
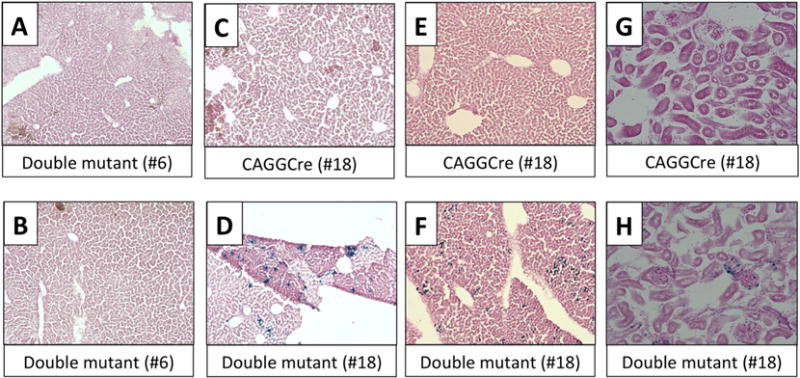
Cre-only and double mutant littermates from #6 and #18 lines were injected with Tamoxifen from postnatal day 14 to 18 for five consecutive days and liver and kidney were harvested at postnatal day 25. Following X-gal staining, the sections were counter-stained with Nuclear Fast Red. Liver sections were captured at 10X and kidney sections at 20X magnification. (A,B) Double mutants from line #6; (C,E,G) single mutants (CAGGCre) from line #18; (D,F,H) double mutants from line #18. Note the lacZ positive cells in liver (D,F) and kidney (H) of line #18 double mutants.
3.3. Impaired bone growth resulting from Gsα-R201H expression in limb-bud mesenchyme
As Gsα signaling pathway regulates the actions of PTHrP during endochondral bone formation, we crossed the cGsαR201H mice with Prx1-Cre mice, in which Cre is expressed in early limb-bud mesenchyme [36]. Double mutant neonates from line #18 (Prx1-Cre;Tg) displayed markedly short limbs, as judged by the gross appearance of these mice and the alizarin red/alcian blue staining of the skeleton showing mineralized tissue and cartilage, respectively (Fig. 4A,B). While no marked differences in the length of the axial skeleton was observed between double mutants and control littermates, the double mutant pups displayed short and thin long bones in all the extremities (Fig. 4C–F). Moreover, compared to controls, the double mutants appeared to have small heads and short snouts (Fig. 4A,B,G), consistent with the expression pattern of Prx1-Cre [36]. Histological analyses revealed alterations in the long bone growth plates of Prx1-Cre;Tg newborns, with a shortened hypertrophic chondrocyte zone relative to the columnar proliferating zone (Fig. 5A–E).
Figure 4. Abnormal limb development in neonatal Prx1-Cre;Tg mice.
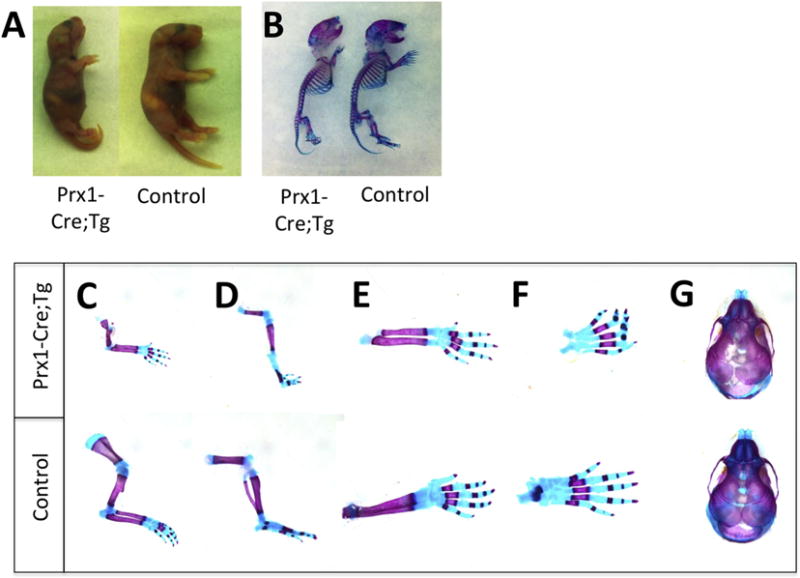
(A) Gross appearance of Prx1-Cre;Tg and littermate control at birth. (B) Whole-mount alizarin red/alcian blue staining of Prx1-Cre;Tg neonate and littermate control. Note the shortened long bones and the relatively normal length of the axial skeleton in Prx1-Cre;Tg. (C) Alizarin red/alcian blue staining of forelimbs and (D) hind limbs of Prx1-Cre;Tg neonate and littermate control. (E,F,G) Alizarin red/alcian blue staining of radius, ulna and forefeet (E), hind feet (F), and skull (G) of Prx1-Cre;Tg neonate and littermate control. Note the markedly shorter and thinner long bones and the shorter snout in Prx1-Cre;Tg mice. Images are representative of at least three different pups of the Prx1-Cre;Tg and control genotype from two independent litters.
Figure 5. Hypertrophic to columnar proliferating chondrocyte zone length is reduced in Prx1-Cre;Tg compared to control littermates.
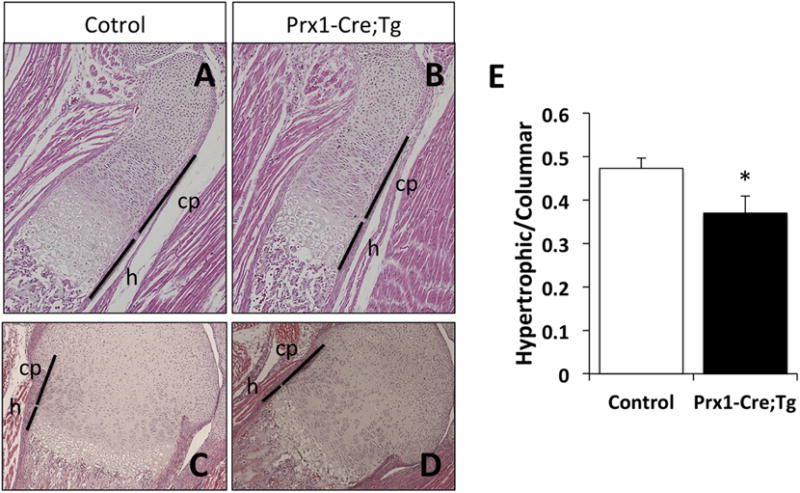
(A–D) Hematoxylin and eosin staining of control and Prx1-Cre;Tg growth plates at birth. Tibia (A,B) and distal femur (C,D) captured at 10× magnification. Lines indicate the columnar proliferative (cp) and hypertrophic (h) chondrocyte zones. (E) Ratio of the length of hypertrophic to columnar proliferating zones in control versus Prx1-Cre;Tg neonates. Data represent mean ± SEM (n=5-6); *, p<0.05 vs control (two-tailed Student’s t-test).
The Prx1-Cre;Tg mice continued to be grossly smaller than control littermates at age three weeks (Fig. 6A). Histological analyses also revealed differences in the epiphyseal regions of double mutant and control littermates, with delayed formation of the secondary ossification center in Prx1-Cre;Tg mice (Fig. 6B). A marker of enhanced Gsα/cAMP/PKA signaling in endocrine and bone cells is elevated expression of c-fos proto-oncogene [37, 38]. In fact, the FD lesions in patients carrying constitutively activating Gsα mutations show dramatically increased c-fos expression [39]. We thus cultured BMSCs isolated from double mutant and control mice and analyzed these cells with respect to the level of c-fos mRNA. qRT-PCR analyses showed significantly higher c-fos mRNA levels in Prx1-Cre;Tg mice than in control mice, consistent with the overactive Gsα signaling in these cells (Fig. 6F).
Figure 6. Prx1-Cre;Tg mice continues to be smaller than control littermates at postnatal day 21, with delayed endochondral ossification and increased c-fos mRNA levels in BMSCs.
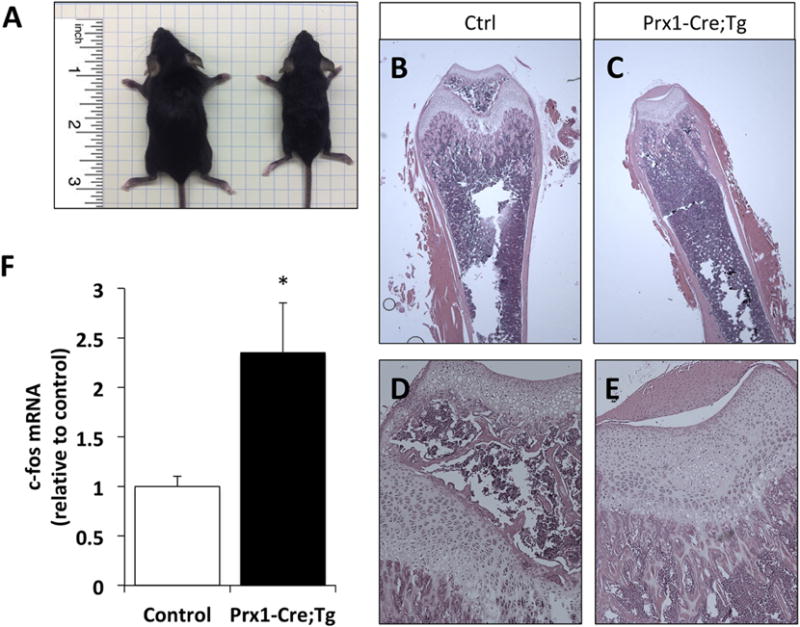
(A) Gross phenotypes of 3-week old control (on the left) and Prx1-Cre; Tg (on the right) littermates. (B–E) Hematoxylin and eosin stained femur sections of control (B,D) and Prx1-Cre;Tg mice (C,E). Images were captured at 2× (B,C) or 10× (D,E) magnification. Note the absence of secondary ossification center in Prx1-Cre;Tg samples (Panels C and E). (F) Elevated levels of c-fos mRNA in BMSCs from Prx1-Cre; Tg compared to control littermates, measured by using qRT-PCR and β-actin mRNA as a reference control. Data represent mean ± SEM (n=7); *, p<0.05 vs control (two-tailed Student’s t-test).
4. Discussion
We developed mice in which a constitutively active Gsα mutant is expressed in a Cre-dependent manner. Through the use of a lacZ reporter located downstream of the Gsα cDNA we could easily observe transgene expression in cultured cells or in tissues following Cre expression. To examine the effect of Gsα hyperfunction in limb development we then activated the expression of the transgene in early limb-bud mesenchyme, using the Prx1 promoter-driven Cre. The Prx1-Cre;Tg mice displayed short limbs with delayed endochondral ossification, as well as modest craniofacial defects, consistent with the expression profile of Prx1.
Gsα haploinsufficiency in humans causes premature closure of epiphyses, short stature, and brachydactyly, as seen in patients with Albright’s hereditary osteodystrophy [1–5]. Studies using different mouse models have also indicated that ablation of Gsα in chondrocytes causes defective epiphyseal development and growth plates, and accelerates hypertrophic chondrocyte differentiation [40, 41]. PTHrP plays an important role in hypertrophic chondrocyte differentiation in the growth plate by binding its G protein-coupled receptor located primarily in the prehypertrophic chondrocytes [13]. The PTHR couples to both Gsα and Gq/11α in this tissue, and both signaling pathways are essential for the physiological regulation of chondrocyte differentiation [21, 22]. Both mice overexpressing PTHrP in growth plate chondrocytes [42] and mice expressing a constitutively active PTHR mutant found in patients with Jansen’s metaphyseal chondrodysplasia in the same tissue [43] present with abnormal and delayed endochondral bone formation, leading to short limbs. Our results from the Prx1-Cre;Tg mice suggest that enhanced Gsα signaling plays a major role in the growth plate phenotypes observed in those models.
MAS and CNC are both diseases of enhanced Gsα/PKA signaling, and accordingly, clinical features of these disorders are broadly similar [44]. CNC is characterized by abnormalities of skin pigmentation, myxomas, endocrine tumors/overactivity, and schwannomas. Both MAS and CNC patients, however, typically lack short stature or short limbs. In MAS, the patients express a constitutively active Gsα mutant (the gsp oncogene) but are mosaic for wild-type and mutation-bearing cells [10, 45, 46]. The mosaicism is likely to contribute to the expressivity of short bone phenotype in those patients. It is possible that, in most cases, the mutation-bearing cells are too few to cause an appreciable defect in endochondral ossification. It is also likely that the increased basal cAMP generation is counteracted by adaptive mechanisms. For example, compensatory elevations in the levels and activity of certain cAMP phosphodiesterases can occur, as demonstrated previously in cultured cells expressing a constitutively active Gsα mutant [47]. CNC patients have germline heterozygous inactivating mutations in PRKAR1A, which encodes the type-Iα regulatory subunit of PKA [25]. PRKAR1A acts as a classic tumor suppressor gene, so that the function of this PKA regulatory subunit is abolished when a somatic “second hit” occurs on the normal allele. Therefore, CNC patients are predicted to have increased PKA activity only in cells in which the normal PRKAR1A allele is also mutated or entirely lost (loss of heterozygosity). Thus, as in MAS, the number of aberrant cells may be too few to cause a significant growth plate phenotype. In addition, at least three other PKA regulatory subunits have been described, showing different tissue specificity [48]. It is possible that those, rather than the type-Iα subunit, are expressed in the growth plate and, therefore, prevent PKA overactivity in this tissue. A compensatory increase in the level of one or more of those subunits may also occur, as described previously [49].
One of the unusual manifestations of CNC (~1% of the cases) is osteochondromyxomas that can be present in craniofacial or long bones [50]. Heterozygous loss of Prkar1a in mice also leads to neoplastic fibro-osseous lesions that display characteristic features of osteochondromyxoma [51]. These rare tumors, however, are histologically distinct from FD, which is the most frequent abnormality in patients with MAS and can also occur as an isolated clinical feature. The difference in the prevalence of bone involvement between CNC and MAS may reflect the respective genetic mechanisms for these disorders (i.e. tumor suppressor inactivation vs. proto-oncogene activation) and the existing compensatory and redundant pathways, as suggested above for the growth plate phenotype.
Neither the double mutant offspring from cGsαR201H and CAGGCre-ER™ crosses nor the Prx1-Cre;Tg mice exhibited any evidence of FD based on our histological analyses up to age two years. A model of FD through the transgenic expression of Gsα-R201C, another constitutively active Gsα mutant, has been previously described, in which the mutant cDNA was expressed under the control of two different ubiquitously active promoters [52]. In those mice, the complete FD phenotype was observed at age one year or older, although early signs of FD could be detected after several months of age. On the other hand, the expression of the same Gsα mutant under the control of 2.3-kb Col1a1 promoter, a marker of osteoblasts, led to a high bone mass phenotype rather than FD [53]. While we were completing our manuscript, another mouse model has been reported, in which R201H was conditionally knocked into the endogenous mouse Gnas gene [54]. On the use of Prx1-Cre, these mice displayed deformed long bones and dramatically increased trabecular bone. The mice also displayed evidence of fibrosis in the marrow space, reminiscent of FD features. In addition, a marked delay in endochondral bone formation was observed. This delay, however, was more drastic than what we observed in our Prx1-Cre;Tg mice, which also lacked deformed long bones and increased trabecular bone. Our mouse model uses the CAG promoter to express the Gsα mutant. The mild phenotype in our mouse model could reflect the differences between the CAG promoter and the endogenous Gsα promoter with respect to strength and temporal and spatial regulation. While our manuscript was under peer-review, another mouse model using Gsα-R201C was reported [55]. Upon the use of Prx1-Cre, these mice developed dysmorphic limbs and bone lesions consistent with FD. This study used a tetracycline-induced minimal promoter to express the constitutively active Gsα mutant, further highlighting the importance of the promoter activation profile for the development of FD-like lesions.
GNAS mutations leading to constitutive Gsα activity are also found in many tumors, ranging from various adenomas and intrahepatic mucinous neoplasms to certain cancers [3]. Thus, our cGsαR201H mice could also serve as a valuable tool for investigating, in vivo, the pathogenesis of these tumors and the role of Gsα signaling in oncogenesis.
Highlights.
- Transgenic mice was generated, expressing, conditionally, a constitutively active Gsα mutant (Gsα-R201H) found in patients with different tumors, McCune-Albright Syndrome, and fibrous dysplasia of bone.
- The transgene was expressed in a mosaic manner in vivo upon crossing these mice to another mouse model in which Cre is expressed in a tamoxifen-induced fashion.
- Expression of Gsα-R201H in the developing limb bud mesenchyme led to short limbs with disrupted endochondral ossification.
- The results highlight the importance of Gsα signaling endochondral bone formation and the requirement for tight regulation of Gsα activity for normal bone growth.
- This mouse model provides a valuable tool for studying Gsα signaling in physiology and disease.
Acknowledgments
We thank Lauren Toyomi Shumate for technical assistance in maintenance of the mouse colonies and some of the histological analyses. We also thank the researchers at the Endocrine Unit, particularly Drs. Tatsuya Kobayashi and Henry Kronenberg, for helpful discussions during the study.
Funding: This study was funded in part by grants from The Fibrous Dysplasia Foundation, The Milton Fund, and the National Institutes of Health (NIDDK RO1 073911 to MB and NIAMS RO3 AR059942 to JYW). This study was also supported by a Sanofi-Genzyme Innovation Award (to MB). Tissue embedding and sectioning were performed at the Massachusetts General Hospital, Endocrine Unit, Center for Skeletal Research Histology Core, which is funded by NIH/NIAMS P30 AR066261.
Abbreviations
- Gsα
stimulatory G protein alpha-subunit
- MAS
McCune-Albright Syndrome
- FD
fibrous dysplasia of bone
- CNC
Carney-Complex
- PTHrP
PTH-related peptide
- PTHR
PTH/PTHrP receptor
- BMSCs
bone marrow stromal cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: All the authors state that they have no conflict of interest.
Author contributions: MB, QH, and JYW designed the research; AK, VRM, YZ, OT, EP, QH performed the experiments and analyzed the data; AK, VRM, YZ, and MB wrote the manuscript.
References
- 1.Weinstein LS, Yu S, Warner DR, Liu J. Endocrine Manifestations of Stimulatory G Protein alpha-Subunit Mutations and the Role of Genomic Imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- 2.Plagge A, Kelsey G, Germain-Lee EL. Physiological functions of the imprinted Gnas locus and its protein variants Galpha(s) and XLalpha(s) in human and mouse. J Endocrinol. 2008;196:193–214. doi: 10.1677/JOE-07-0544. [DOI] [PubMed] [Google Scholar]
- 3.Turan S, Bastepe M. GNAS Spectrum of Disorders. Curr Osteoporos Rep. 2015;13:146–58. doi: 10.1007/s11914-015-0268-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemos MC, Thakker RV. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders. Hum Mutat. 2015;36:11–9. doi: 10.1002/humu.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsalpha-cAMP-linked disorders: current view and open issues. Nat Rev Endocrinol. 2016;12:347–56. doi: 10.1038/nrendo.2016.52. [DOI] [PubMed] [Google Scholar]
- 6.Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidism - an example of “Seabright-Bantam syndrome”. Endocrinology. 1942;30:922–932. [Google Scholar]
- 7.Patten JL, Johns DR, Valle D, Eil C, Gruppuso PA, Steele G, Smallwood PM, Levine MA. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. New Engl J Med. 1990;322:1412–1419. doi: 10.1056/NEJM199005173222002. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein LS, Gejman PV, Friedman E, Kadowaki T, Collins RM, Gershon ES, Spiegel AM. Mutations of the Gs alpha-subunit gene in Albright hereditary osteodystrophy detected by denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:8287–90. doi: 10.1073/pnas.87.21.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. New Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 10.Schwindinger WF, Francomano CA, Levine MA. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci U S A. 1992;89:5152–6. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune-Albright fibrous dysplasia of bone. J Bone Miner Res. 2000;15:120–8. doi: 10.1359/jbmr.2000.15.1.120. [DOI] [PubMed] [Google Scholar]
- 12.Weinstein LS. G(s)alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21(Suppl 2):120–4. doi: 10.1359/jbmr.06s223. [DOI] [PubMed] [Google Scholar]
- 13.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 14.Lee H, Graham JM, Jr, Rimoin DL, Lachman RS, Krejci P, Tompson SW, Nelson SF, Krakow D, Cohn DH. Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet. 2012;90:746–51. doi: 10.1016/j.ajhg.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, Motte E, Pinto G, Chanson P, Bougneres P, Clauser E, Silve C. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med. 2011;364:2218–26. doi: 10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 16.Michot C, Le Goff C, Goldenberg A, Abhyankar A, Klein C, Kinning E, Guerrot AM, Flahaut P, Duncombe A, Baujat G, Lyonnet S, Thalassinos C, Nitschke P, Casanova JL, Le Merrer M, Munnich A, Cormier-Daire V. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012;90:740–5. doi: 10.1016/j.ajhg.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, Blom E, Gillerot Y, Weigel JF, Kruger G, Hiort O, Seemann P, Mundlos S. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet. 2010;86:434–9. doi: 10.1016/j.ajhg.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maass PG, Wirth J, Aydin A, Rump A, Stricker S, Tinschert S, Otero M, Tsuchimochi K, Goldring MB, Luft FC, Bahring S. A cis-regulatory site downregulates PTHLH in translocation t(8;12)(q13;p11.2) and leads to Brachydactyly Type E. Hum Mol Genet. 2010;19:848–60. doi: 10.1093/hmg/ddp553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schipani E, Kruse K, Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 20.Gensure RC, Gardella TJ, Jüppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328:666–78. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 21.Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3:183–94. doi: 10.1016/s1534-5807(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 22.Chagin AS, Vuppalapati KK, Kobayashi T, Guo J, Hirai T, Chen M, Offermanns S, Weinstein LS, Kronenberg HM. G-protein stimulatory subunit alpha and Gq/11alpha G-proteins are both required to maintain quiescent stem-like chondrocytes. Nat Commun. 2014;5:3673. doi: 10.1038/ncomms4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itonaga T, Goto H, Toujigamori M, Ohno Y, Korematsu S, Izumi T, Narumi S, Hasegawa T, Ihara K. Three-Quarters Adrenalectomy for Infantile-Onset Cushing Syndrome due to Bilateral Adrenal Hyperplasia in McCune-Albright Syndrome. Horm Res Paediatr. 2017;88:285–290. doi: 10.1159/000473878. [DOI] [PubMed] [Google Scholar]
- 24.Mamkin I, Philibert P, Anhalt H, Ten S, Sultan C. Unusual phenotypical variations in a boy with McCune-Albright syndrome. Horm Res Paediatr. 2010;73:215–22. doi: 10.1159/000284365. [DOI] [PubMed] [Google Scholar]
- 25.Stratakis CA. Carney complex: A familial lentiginosis predisposing to a variety of tumors. Rev Endocr Metab Disord. 2016;17:367–371. doi: 10.1007/s11154-016-9400-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Turan S, Wehbi VL, Vilardaga JP, Bastepe M. Extra-long Galphas variant XLalphas protein escapes activation-induced subcellular redistribution and is able to provide sustained signaling. J Biol Chem. 2011;286:38558–69. doi: 10.1074/jbc.M111.240150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–50. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 28.Wu JY, Aarnisalo P, Bastepe M, Sinha P, Fulzele K, Selig MK, Chen M, Poulton IJ, Purton LE, Sims NA, Weinstein LS, Kronenberg HM. Gsalpha enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J Clin Invest. 2011;121:3492–504. doi: 10.1172/JCI46406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faubert BL, Kaminski NE. AP-1 activity is negatively regulated by cannabinol through inhibition of its protein components, c-fos and c-jun. J Leukoc Biol. 2000;67:259–66. doi: 10.1002/jlb.67.2.259. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Y, He Q, Aydin C, Rubera I, Tauc M, Chen M, Weinstein LS, Marshansky V, Juppner H, Bastepe M. Ablation of the Stimulatory G Protein alpha-Subunit in Renal Proximal Tubules Leads to Parathyroid Hormone-Resistance With Increased Renal Cyp24a1 mRNA Abundance and Reduced Serum 1,25-Dihydroxyvitamin D. Endocrinology. 2016;157:497–507. doi: 10.1210/en.2015-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 32.Ma W, Rogers K, Zbar B, Schmidt L. Effects of different fixatives on beta-galactosidase activity. J Histochem Cytochem. 2002;50:1421–4. doi: 10.1177/002215540205001015. [DOI] [PubMed] [Google Scholar]
- 33.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levis MJ, Bourne HR. Activation of the alpha subunit of Gs in intact cells alters its abundance, rate of degradation, and membrane avidity. J Cell Biol. 1992;119:1297–307. doi: 10.1083/jcb.119.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 36.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 37.Clohisy JC, Scott DK, Brakenhoff KD, Quinn CO, Partridge NC. Parathyroid hormone induces c-fos and c-jun messenger RNA in rat osteoblastic cells. Mol Endocrinol. 1992;6:1834–42. doi: 10.1210/mend.6.11.1480173. [DOI] [PubMed] [Google Scholar]
- 38.Gaiddon C, Boutillier AL, Monnier D, Mercken L, Loeffler JP. Genomic effects of the putative oncogene G alpha s. Chronic transcriptional activation of the c-fos proto-oncogene in endocrine cells. J Biol Chem. 1994;269:22663–71. [PubMed] [Google Scholar]
- 39.Candeliere GA, Glorieux FH, Prud’homme J, St-Arnaud R. Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. N Engl J Med. 1995;332:1546–51. doi: 10.1056/NEJM199506083322304. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto A, Chen M, Kobayashi T, Kronenberg HM, Weinstein LS. Chondrocyte-specific knockout of the G protein G(s)alpha leads to epiphyseal and growth plate abnormalities and ectopic chondrocyte formation. J Bone Miner Res. 2005;20:663–71. doi: 10.1359/JBMR.041210. [DOI] [PubMed] [Google Scholar]
- 41.Bastepe M, Weinstein LS, Ogata N, Kawaguchi H, Jüppner H, Kronenberg HM, Chung UI. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci U S A. 2004;101:14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes skeletal dysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schipani E, Lanske B, Hunzelman J, Kovacs CS, Lee K, Pirro A, Kronenberg HM, Jüppner H. Targeted expression of constitutively active PTH/PTHrP receptors delays endochondral bone formation and rescues PTHrP-less mice. Proc Natl Acad Sci USA. 1997;94:13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: An overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clin Genet. 1986;29:321–4. doi: 10.1111/j.1399-0004.1986.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 46.Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, Van Wyk JJ, Merino MJ, Feuillan PP, Spiegel AM. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J Pediatr. 1993;123:509–18. doi: 10.1016/s0022-3476(05)80943-6. [DOI] [PubMed] [Google Scholar]
- 47.Piersanti S, Remoli C, Saggio I, Funari A, Michienzi S, Sacchetti B, Robey PG, Riminucci M, Bianco P. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. 2010;25:1103–16. doi: 10.1359/jbmr.091036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front Biosci. 2000;5:D678–93. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- 49.Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS. Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem. 1997;272:3993–8. doi: 10.1074/jbc.272.7.3993. [DOI] [PubMed] [Google Scholar]
- 50.Golden T, Siordia JA. Osteochondromyxoma: Review of a rare carney complex criterion. J Bone Oncol. 2016;5:194–197. doi: 10.1016/j.jbo.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirschner LS, Kusewitt DF, Matyakhina L, Towns WH, 2nd, Carney JA, Westphal H, Stratakis CA. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005;65:4506–14. doi: 10.1158/0008-5472.CAN-05-0580. [DOI] [PubMed] [Google Scholar]
- 52.Saggio I, Remoli C, Spica E, Cersosimo S, Sacchetti B, Robey PG, Holmbeck K, Cumano A, Boyde A, Bianco P, Riminucci M. Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J Bone Miner Res. 2014;29:2357–68. doi: 10.1002/jbmr.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Remoli C, Michienzi S, Sacchetti B, Consiglio AD, Cersosimo S, Spica E, Robey PG, Holmbeck K, Cumano A, Boyde A, Davis G, Saggio I, Riminucci M, Bianco P. Osteoblast-specific expression of the fibrous dysplasia (FD)-causing mutation Gsalpha(R201C) produces a high bone mass phenotype but does not reproduce FD in the mouse. J Bone Miner Res. 2015;30:1030–43. doi: 10.1002/jbmr.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan SK, Yadav PS, Elliott G, Hu DZ, Xu R, Yang Y. Induced Gnas(R201H) expression from the endogenous Gnas locus causes fibrous dysplasia by up-regulating Wnt/beta-catenin signaling. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1714313114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao X, Deng P, Iglesias-Bartolome R, Amornphimoltham P, Steffen DJ, Jin Y, Molinolo AA, de Castro LF, Ovejero D, Yuan Q, Chen Q, Han X, Bai D, Taylor SS, Yang Y, Collins MT, Gutkind JS. Expression of an active Galphas mutant in skeletal stem cells is sufficient and necessary for fibrous dysplasia initiation and maintenance. Proc Natl Acad Sci U S A. 2018;115:E428–E437. doi: 10.1073/pnas.1713710115. [DOI] [PMC free article] [PubMed] [Google Scholar]