

Abstract
Anthracnose causes severe losses of tea production in China. Although genes and biological processes involved in anthracnose resistance have been reported in other plants, the molecular response to anthracnose in tea plant is unknown. We used the susceptible tea cultivar Longjing 43 and the resistant cultivar Zhongcha 108 as materials and compared transcriptome changes in the leaves of both cultivars following Colletotrichum fructicola inoculation. In all, 9015 and 8624 genes were differentially expressed between the resistant and susceptible cultivars and their controls (0 h), respectively. In both cultivars, the differentially expressed genes (DEGs) were enriched in 215 pathways, including responses to sugar metabolism, phytohormones, reactive oxygen species (ROS), biotic stimuli and signalling, transmembrane transporter activity, protease activity and signalling receptor activity, but DEG expression levels were higher in Zhongcha 108 than in Longjing 43. Moreover, functional enrichment analysis of the DEGs showed that hydrogen peroxide (H2O2) metabolism, cell death, secondary metabolism, and carbohydrate metabolism are involved in the defence of Zhongcha 108, and 88 key genes were identified. Protein–protein interaction (PPI) network demonstrated that putative mitogen-activated protein kinase (MAPK) cascades are activated by resistance (R) genes and mediate downstream defence responses. Histochemical analysis subsequently validated the strong hypersensitive response (HR) and H2O2 accumulation that occurred around the hyphal infection sites in Zhongcha 108. Overall, our results indicate that the HR and H2O2 are critical mechanisms in tea plant defence against anthracnose and may be activated by R genes via MAPK cascades.
Plant defence: pathways for tea protection
A comparison of gene activity in resistant and susceptible tea cultivars provides insights into mechanisms of defence against one of the most devastating tea diseases: anthracnose. Tea (Camellia sinensis) is an important crop in many tropical countries; however, yields can be devastated by the fungus Colletotrichum, the causal agent of anthracnose. Xinchao Wang and Yajun Yang, of the Chinese Academy of Sciences’ Tea Research Institute in Hangzhou, explored the defence response in tea cultivars infected with Colletotrichum. Discrepancies in the genes activated in resistant and susceptible cultivars suggest resistance is mediated by specific molecular signalling pathways, known resistance (R) genes, and involves the production of hydrogen peroxide and initiation of programmed cell death around sites of infection. These mechanisms could be harnessed to develop tea cultivars with greater resistance to anthracnose.
Introduction
Tea plant (Camellia sinensis (L.) O. Kuntze) is a perennial evergreen woody plant that is widespread throughout tropical and subtropical areas, such as China, India, Kenya and Sri Lanka. As an important commercial product, the fresh shoots of tea plant provide an wide variety of nutrition for the human body, including flavonoids, alkaloids and theanine. Long-term tea drinking can protect against different diseases; therefore, tea has become the most popular healthy, non-alcoholic beverage in the world1,2. However, tea plant is frequently affected by many kinds of disease. Of these diseases, anthracnose, which is caused by Colletotrichum, is one of the most devastating diseases to tea plant3. Colletotrichum damages mature tea plant leaves, affecting the growth and yield of the plant4.
Host plants have evolved various defence mechanisms during interactions with plant pathogens. The first type involves pathogen- or microbial-associated molecular pattern (PAMP or MAPM, respectively)-triggered immunity (PTI)5. Pattern recognition receptors (PRRs) located on host plasma membranes recognise PAMPs or MAMPs, and these complexes induce mitogen-activated protein kinases (MAPKs) and/or calcium signalling; these MAPKs and/or calcium signalling trigger a series of defence responses, which results in the suppression of pathogen colonisation6. However, for successful invasion, adapted pathogens have evolved numerous virulence proteins called effectors to suppress or escape PTI in order to achieve infection. In turn, hosts also have evolved genes that encode intracellular nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins that can specifically recognise effectors; this recognition activates a second host immune response named effector-triggered immunity (ETI) to restrict pathogen growth7,8. Interestingly, ETI is a faster and stronger version of PTI, and usually regulates the generation of host programmed cell death (PCD) in addition to the production of reactive oxygen species (ROS) at the site of pathogen infection9.
Colletotrichum is one of the largest genera of pathogens. Colletotrichum species are pathogenic to more than 3200 plants and cause large economic losses10. The hypersensitive response (HR) is a phenomenon of PCD. The HR is one of the most well-known resistance reactions and is associated with host resistance to Colletotrichum. For example, O’Connell et al.11 compared differences in resistance reactions between incompatible and compatible Arabidopsis plants in response to Colletotrichum destructivum at different infection phases, the results demonstrated that the incompatible Arabidopsis plants produced a rapid HR and deposited both callose and papillae in the infected epidermal cells following C. destructivum inoculation. In general, generation of the HR is associated with the activation of NBS-LRRs by pathogen effectors, the NBS-LRRs act together with multiple defence-related genes in plants to defend against Colletotrichum9,12,13. The resistance mechanism of tea plants has been loosely explained based on the results of several studies. For example, by studying tea plant defence to grey blight disease caused by Pestalotiopsis species, Senthilkumar et al.14, who used suppressive subtractive hybridisation techniques, suggested that the HR and ROS play crucial roles in tea plant resistance to P. theae. Moreover, Palanisamy and Mandal15 reported that the antioxidative enzymes associated with ROS in resistant tea cultivars have higher activity than those in susceptible cultivars following Pestalotiopsis sp. infection. Another important leaf disease of tea plant is blister blight, which is caused by Exobasidium. The results of Jayaswall et al.16 based on transcriptome analysis indicated that numerous defence-related genes are upregulated upon induction by Exobasidium vexans in tea plant, and a great number of well-known NBS-LRR genes are involved in the defence response. Despite many studies on tea plant resistance to pathogens, information on the molecular mechanism of resistance against Colletotrichum in tea plant is poorly understood.
To elucidate the resistance mechanism of tea plant to anthracnose, we previously used conidial suspensions of Colletotrichum to inoculate resistant (cultivar Zhongcha 108, ZC108) and susceptible (cultivar Longjing 43, LJ43) Ca. sinensis cultivars. ZC108 was produced by irradiating the offspring of LJ43. We speculated that the resistance mechanism of tea plant may be associated with NBS-LRR genes as well as the phenylpropanoid and flavonoid pathways based on our previous results using microarray data17. At the same time, we proved that flavonoid and caffeine biosynthesis are involved in tea plant defence to Colletotrichum fructicola infection4. Therefore, in the present study, to further reveal the resistance mechanism of tea plants to anthracnose, we comparatively analysed the changes in transcription levels in the leaves of both cultivars following C. fructicola inoculation using RNA-sequencing (RNA-Seq) and revealed a possible resistance mechanism in the tea plant response against anthracnose.
Materials and Methods
Plant material, fungal isolates and treatment
Tea plants (Camellia sinensis (L.) O. Kuntze) of the resistant cultivar ZC108 and the susceptible cultivar LJ43 as well as isolates of C. fructicola L33 were maintained as described previously4,17. Three-year-old plants were used as experimental materials. The plants were maintained in the glasshouse (28 °C, 14 h light, 80% humidity) and inoculated with conidial suspensions of C. fructicola (106 spores/mL). For pathogenicity tests, wound inoculation of plants was performed in vitro based on the method described by Wang et al.17. The third leaves of ZC108 and LJ43 were sampled at 3, 7 and 11 days post inoculation (dpi). The non-wound inoculation of plants in vivo was used for RNA-Seq analysis as described by Jayaswall et al.16. Leaf tissues (first-fourth leaves) of ZC108 and LJ43 were sampled at 0 h (before inoculation), 24 and 72 h post inoculation (hpi).
RNA isolation, library construction and sequencing
A total of 18 RNA samples were isolated using the cetyl-trimethylammonium bromide method described by Hao et al.18. The RNA quality was verified by a 1% denaturing agarose gel and a NanoDrop 2000 system (Thermo Scientific, Delaware, USA). Total RNA was used to construct cDNA libraries using a TruSeq RNA Sample Prep Kit (Illumina, San Diego, USA) according to the manufacturer’s protocol. The cDNA library was sequenced on an Illumina HiSeqTM 2000 platform, and paired-end reads in 150 bp length were yielded.
Data analysis
The detailed processes of de novo assembly, functional annotation and differentially expressed gene (DEG) identification were performed in accordance with the methods of Hao et al.18. Briefly, de novo assembly was performed using the Trinity (v2.2.0) programme19. By BlastX analysis (Basic Local Alignment Tool (BLAST) 2.2.30+) with the non-redundant (NR) database and TAIR database (Athaliana_167_TAIR10.protein.fa and Athaliana_167_TAIR10. annotation_info.txt) (http://www.arabidopsis.org/), the best hits (with a significant E-value of <1e−5) were assigned to the assembled transcripts of tea plant. RSEM v1.2.11 programme was used to analyse the RNA-Seq data for alignment and expression calculation20. The expression patterns and posterior probability of differential expression ‘posterior probability of differential expression’ (PPDE) values of each gene/contig were estimated by EBSeq v1.1.5 21, and the DEGs were identified with PPDE = 1. The RNA-Seq raw data have been deposited in the NCBI Sequencing Read Archive database and can be accessed with the following accession numbers: SRR5986350; SRR5986349; SRR5986352; SRR5986351; SRR5986337; SRR5986353; SRR5986348; SRR5986347; SRR5986346; SRR5986345; SRR5986343; SRR5986339; SRR5986340; SRR5986338; SRR5986342; SRR5986344; SRR5986354; and SRR5986341. A Venn diagram was constructed using software that is available online (http://bioinformatics.psb.ugent.be/webtools/Venn/). Gene Ontology (GO) term enrichment was analysed by Gene Ontology Enrichment Analysis Software Toolkit (GOEAST), and statistical enrichment was considered when P < 0.0522. The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed by the Database for Annotation, Visualisation and Integrated Discovery23. The short time-series expression miner software (STEM) software was used for analysing the different patterns of the shared DEG expression in both the resistant and susceptible tea plant cultivars24. Besides, the protein–protein interaction (PPI) network was constructed based on the data produced by the Search Tool for the Retrieval of Interacting Genes database (http://string-db.org/) and visualised using Cytoscape software (version 3.5.1)25.
Microscopic observations
The leaves were collected at 12, 24, 48, 72 and 96 hpi and were cut into 0.5 cm segments, which were then used for observation of hypersensitive cell death and hydrogen peroxide (H2O2) accumulation. Trypan blue (Sangon Biotech Company, Shanghai, China) was used for the detection of hypersensitive cell death, as described by Faoro et al.26. The segments were boiled for 5 min in 1 mg/mL trypan blue solution (phenol:lactic acid:glycerol:distilled water = 1:1:1:1), after which the tissues were decolourized by chloral hydrate (Sangon Biotech Company, Shanghai, China). Diaminobenzidine (DAB) (Sangon Biotech Company, Shanghai, China) was used to visualise H2O2 accumulation in accordance with the method of Hao et al.27. The segments were infiltrated with 1 mg/mL DAB solution (pH = 3.8) and were vacuum-infiltrated for 10 min. After 12 h, the segments were cleared in saturated chloral hydrate and then discoloured in boiling 95% ethanol. Above treated samples were mounted on glass slides in 50% glycerol, and were observed under a Nikon 80i microscope (Japan).
Gene expression validation by quantitative real-time PCR (qRT-PCR)
For qRT-PCR, 1 µg of total RNA used in the previous RNA-Seq library construction was used for cDNA synthesis. A PrimeScript RT enzyme with a gDNA eraser (Takara, Japan) was used for cDNA synthesis. qRT-PCR was performed on an Applied Biosystems 7500 Sequence Detection System using SYBR Premix Ex Taq™ II (Takara, Japan). The primers in this step are listed in Supplementary Table S1. The polypyrimidine tract-binding protein (CsPTB1) gene was used as an internal control28. The relative expression levels were calculated using the 2−ΔΔCt method29.
Results
Pathogenicity tests
The resistant cultivar ZC108 is significantly more resistant to anthracnose in the field than is the susceptible cultivar LJ43 (Fig. 1a). To confirm these results, we inoculated the wounded mature leaves of ZC108 and LJ43 in vitro with conidial suspensions of C. fructicola (106 spores/mL). Pathogenicity tests showed that LJ43 leaves displayed the typical brown lesions of anthracnose disease around wounded areas during pathogen infection (3, 7 and 11 dpi). However, the ZC108 leaves did not exhibit clear disease symptoms during inoculation (Fig. 1b). The in vitro results were consistent with the field results.
Fig. 1. Altered disease resistance of resistant (ZC108) and susceptible (LJ43) tea plant leaves to anthracnose.

a The images were taken from the same areas under natural conditions; b differential disease resistance of LJ43 and ZC108 leaves to C. fructicola infection in vitro
Sequencing, assembly and DEG identification
In total, 878 273 717 raw data reads were generated from 18 samples. After the raw read sequences were filtered and passed through quality control, 851 249 406 clean data were obtained (Supplementary Table S2). Based on the high-quality clean data, a total of 864 790 transcripts were assembled across all 18 samples. After their de novo assembly by the Trinity (version 2.2.0) pipeline, the transcripts had an average length of 611 bp and an N50 of 788 (Supplementary Table S3). A total of 497 332 unigenes were generated, of which 234 496 and 134 208 were annotated by BLAST analysis using the NR database and The Arabidopsis Information Resource (TAIR10) according to significant hits (E-value < 1e−5), respectively (Supplementary Table S3). All unigenes were grouped into 203 expression patterns (Supplementary Table S4). Total of 49 784 and 49 661 DEGs were detected in the resistant and susceptible cultivars, respectively, following the elimination of genes that were not differentially expressed (Supplementary Table S4). In our study, the significant DEGs annotated by TAIR10 (E-value < 1e−5) were used to further analyse their function in both cultivars in response to C. fructicola. Total of 9015 and 8624 DEGs were identified in the inoculated leaves of ZC108 and LJ43, respectively (PPDE = 1; Supplementary Fig. S1). Compared with the unigene transcription at 0 h (control), at 24 hpi, 4581 DEGs were upregulated and 4254 were downregulated in ZC108; however, 4825 DEGs were upregulated and 3590 were downregulated in LJ43; at 72 hpi, the number of upregulated DEGs reached 5945 in ZC108, and 3058 DEGs were downregulated; in LJ43, 4554 upregulated and 4051 downregulated DEGs were identified (Supplementary Fig. S1). To identify changes in the resistance mechanism of Ca. sinensis during the C. fructicola infection progress, the DEGs of ZC108 and LJ43 annotated by the Arabidopsis database were used for the further analysis. Relevant information on the DEGs is listed in Supplementary Table S5.
Investigation of DEGs and pathways involved in both resistance and susceptible cultivars
To identify the resistance mechanisms in response to anthracnose in both the resistant and susceptible tea cultivars, all the DEGs were assessed in the two cultivars at 24 and 72 hpi. The Venn diagram illustrated that the 3250 up- and 2024 downregulated DEGs in both ZC108 and LJ43 leaves are involved in the response to C. fructicola during the infection progress (Fig. 2a). Based on the GO analysis, these DEGs were enriched in 216 terms, including 176 biological process (the upregulated DEGs involved in 111 pathways and downregulated DEGs in 102 pathways), 8 cellular component (the upregulated DEGs involved in 4 pathways and downregulated DEGs in 8 pathways) and 32 molecular function (the upregulated DEGs involved in 25 pathways and downregulated DEGs in 16 pathways); among them, the upregulated DEGs were mainly enriched in the functional pathway that were mostly associated with disease resistance, such as the response to sugar metabolism (sucrose, disaccharides, hexose, monosaccharides, fructose and mannitol), phytohormones (ethylene, auxin and cytokinin), ROS, biotic stimulus and signalling, transmembrane transporter activity, protease activity and signalling receptor activity (Fig. 2b, the complete results of the GO enrichment are listed in Supplementary Fig. S2). Using the STEM, the 3250 up- and 2024 downregulated shared DEGs were clustered into 3 profiles (Fig. 2c). Each profile represents a group of genes that exhibit similar expression trends. For the upregulated DEGs, profile 1 had the most genes (1877), followed by profiles 2 (1238) and 3 (135) in the resistant cultivar ZC108; in contrast, the most genes in the susceptible cultivar LJ43 were classified into profile 3 (2409), followed by profiles 1 (807) and 2 (34). More genes whose expression continuously increased (profiles 1 and 2) were identified in ZC108 (3115) than in LJ43 (841). For the downregulated DEGs, profile 3 had the most genes (1243), followed by profiles 2 (712) and 1 (69) in the ZC108; correspondingly, the most genes were enriched in profile 2 (1441) in the LJ43, and the profile 3 was least enriched in the number of genes (206). In addition, 35 up- and 25 downregulated DEGs associated with disease resistance were observed during the infection process; the fold changes of these genes were markedly greater in ZC108 than in LJ43 (Supplementary Table S6). Together, various types of defence in both the resistant and susceptible tea plant cultivars were involved in the response to anthracnose, but the effectiveness of the expression of the shared genes in the resistant cultivar was better than that in the susceptible cultivar.
Fig. 2. Shared genes in both resistant and susceptible Ca. sinensis leaves.

a Venn diagram of the up- and downregulated DEGs in ZC108 and LJ43 leaves at 24 and 72 hpi, respectively; b GO functional classification of the 3250 up- and 2024 downregulated shared genes in both ZC108 and LJ43 leaves; c DEG patterns of the shared genes in both the ZC108 and LJ43 leaves. The cluster analysis was performed using the STEM Clustering method; the top number indicates the profile ID number, and the bottom number indicates the number of genes. The number of up- and downregulated DEGs is shown in red and blue, respectively
Identification of specific pathways in resistant cultivars
The Venn diagram of the up- and downregulated DEG data of both cultivars shows that 584 and 828 upregulated DEGs as well as 1235 and 571 downregulated DEGs were specifically expressed at 24 hpi in ZC108 and LJ43, respectively (Fig. 3a). More specific upregulated DEGs were identified in ZC108 (2235 DEGs) than in LJ43 (844 DEGs) at 72 hpi, while the number of specific downregulated DEGs decreased in the ZC108 (800) dramatically, reversely increased in the LJ43 (1793; Fig. 3b). The GO analysis indicated that these DEGs were enriched in multiple pathways associated with disease resistance. The number of DEGs clearly changed during the infection process (P < 0.05; Supplementary Fig. S3). In particular, DEGs were enriched in ROS (H2O2 metabolic process, organic hydroxyl compound catabolic process and ROS metabolic process), cell death (host PCD and cell death), cytoskeleton (cytoskeleton organisation, actin filament-based process and microtubule-based process), secondary metabolic process, regulation of biological process (regulation of catabolic process, regulation of cellular process, regulation of cellular component organization, negative regulation of biological process, cellular response to stimulus and cellular component organization) and protease activity (protein kinase activity, protein serine/threonine kinase activity, cation transmembrane transporter activity and peroxidase activity) in ZC108, and the number of DEGs significantly increased during C. fructicola infection. In contrast, these DEGs were not enriched in the susceptible cultivar, and the number of DEGs decreased (Fig. 3c). Taken together, the results revealed that cell death and ROS play important roles in tea plant defence to anthracnose.
Fig. 3. Change in DEGs in resistant (ZC108) and susceptible (LJ43) Ca. sinensis cultivars inoculated with C. fructicola during the infection process in comparison with their respective controls (0 h).

a, b Venn diagram of the DEGs in ZC108 and LJ43 leaves at 24 and 72 hpi, respectively; c heatmap of DEGs specific to ZC108 and their respective homologous genes in LJ43 and of enriched GO terms related to disease resistance identified from DEGs. Terms are considered enriched when P < 0.05
Specific DEGs identified at different times after inoculation in resistant cultivars
The data in Fig. 2a demonstrated that the 3007 DEGs (1751 upregulated and 1256 downregulated) and the 2146 DEGs (960 upregulated and 1186 downregulated) specific to the resistant and the susceptible Ca. sinensis cultivar were induced by the C. fructicola infection process, respectively. To identify the function of these DEGs, the KEGG analysis was used. In the susceptible Ca. sinensis cultivar, 2146 DEGs were only enriched in 8 pathways; comparatively, 3007 DEGs in the resistant cultivar were enriched in 23 pathways, of which flavonoid biosynthesis and phenylalanine metabolism were markedly enriched (P < 0.01 and fold enrichment < 0.4; Fig. 4a), suggesting that these two pathways play important roles in tea plant defence against anthracnose. Among the 3007 DEGs, 88 key genes associated with disease resistance were identified, including genes involved with pathogen receptors (23); peroxidase (4); signalling transduction, including Ca2+ signalling (7) and MAPK signalling systems (2); sugar metabolism (6); secondary metabolites (2); fatty acids (8); glutathione (3); transcription factors (TFs) (19); cell death (3); wall-associated kinases (2); protein kinases (1); and cytochrome P450 (3), as well as other defence function genes (5). The expression of all these genes were significantly changed in ZC108, but no change or opposite expression patterns were observed in LJ43 during C. fructicola infection (Fig. 4b). Information of the 88 key genes associated with disease resistance were listed in Supplementary Table S7.
Fig. 4. DEGs specific to resistant Ca. sinensis inoculated with C. fructicola.

a KEGG classifications of the 3007 and 2146 DEGs specific to resistant and susceptible Ca. sinensis inoculated with C. fructicola; b heatmap of 88 key DEGs expressed specifically in the resistant Ca. sinensis cultivar
Regulatory network analysis of selected genes involved in the resistance response
To explore the possible signal pathway in tea plant defence to C. fructicola, the putative 88 key DEGs specific to the resistant cultivar (ZC108) and 60 shared DEGs were used to build a PPI network with Arabidopsis. A total of 126 unique TAIR IDs from the putative 148 genes were identified, and 76 of these TAIR IDs directly interacted with each other. The network analysis was used for treating the network as directed; the average number of direct neighbours in the network for each gene was 4.974. In this network, MAPKs; Ca2+ signalling; TFs; and pathogen receptors (resistance genes, R genes), likely RLKs, RLPs, cysteine-rich RLKs and NBS-LRRs, were significantly correlated. Seven hub nodes (MAPK) were involved in tea plant defence against C. fructicola, which suggests that the MAPK signalling pathway is the crucial signal transduction pathway; R genes were the major pathogen receptors to C. fructicola stimulation, as 18 nodes interacted with major signal pathway and defence genes (Fig. 5).
Fig. 5. Interaction analysis of putative DEGs that are associated with disease resistance and are involved in Ca. sinensis defence to C. fructicola.

The analysis was generated with a PPI network of Arabidopsis thaliana
HR and H2O2 accumulation observation in inoculated leaves
To confirm whether the HR and H2O2 are involved in tea plant defence to C. fructicola, the leaves of ZC108 and LJ43 were inoculated with conidial suspensions and then stained with trypan blue and DAB solution, respectively. As shown in Fig. 6a, ZC108 leaves exhibited clear HR generation during C. fructicola infection, whereas LJ43 leaves did not. Also, the ZC108 leaves exhibited thickening of the cell walls and epidermal cell necrosis at 24 hpi; these symptoms gradually increased as infection time increased (72 and 96 hpi), and mesophyll cells became necrotic. In contrast, mesophyll cell necrosis was observed only in LJ43 leaves at 72 and 96 hpi, but the epidermal cells were not necrotic. These results suggest that hyphae successfully infected LJ43 leaves in vivo but did not elicit a HR. Therefore, the HR is associated with Ca. sinensis resistance to C. fructicola.
Fig. 6. Microscopic detections of the HR and H2O2 accumulation in the leaves of ZC108 and LJ43 following C. fructicola infection at different times.
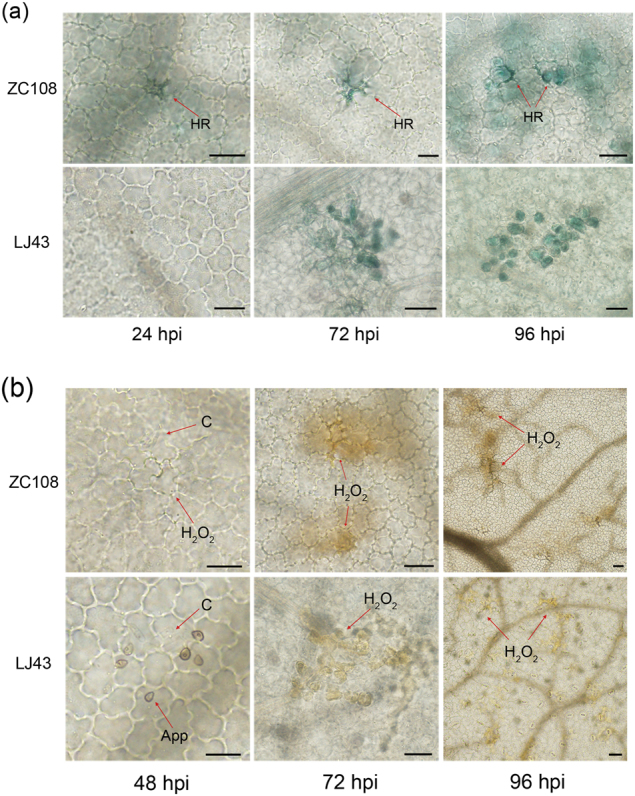
a Trypan blue staining showing HR lesions in the leaves of ZC108 following C. fructicola infection; b H2O2 accumulation in the leaves of ZC108 and LJ43 visualised by DAB staining. C conidia, App appressorium, HR hypersensitive response. Scale bars: 10 μm
Microscopic analysis indicated that brown precipitate occurred more on the ZC108 leaves than on the LJ43 leaves. The epidermal cells of ZC108 leaves exhibited H2O2 accumulation and cell wall thickening, but H2O2 accumulation was observed only in the mesophyll cells of LJ43 (Fig. 6b). Together, these results illustrated that the HR and H2O2 accumulation are involved in the resistance of tea plant against C. fructicola infection, and the structure of the cell wall in tea plant cells could play an important role in the defence against the pathogen.
Validation of differential expression data
To validate the RNA-Seq results, eight DEGs that were randomly selected from RNA-Seq data were analysed using qRT-PCR. The expression results showed that the expression patterns were similar between the qRT-PCR and RNA-Seq data at different times post inoculation, suggesting reliable expression data by RNA-Seq (Supplementary Fig. S4).
Discussion
Anthracnose causes severe damage to tea production. However, little is known about the molecular mechanisms of tea plant against Colletotrichum. In this study, we used RNA-Seq to analyse the upregulated DEGs in a resistant tea plant cultivar during the different stages of the C. fructicola infection process. The experimental results revealed that the HR and H2O2 play crucial roles in disease resistance; these defence responses might be mediated by multiple R genes, Ca2+ signalling and MAPK signalling; at the same time, sugar metabolism and secondary metabolites are also involved in tea plant defence to anthracnose.
R genes specifically recognise pathogenic secretions
At the initial stage of infection, Colletotrichum species secrete various virulence factors into host cells to facilitate successful invasion. With respect to the plant response, R genes are responsible for specifically recognising pathogenic secretions, and these complexes trigger the plant immune system12,30,31,32. In our study, 23 R genes (7 NBS-LRRs and 16 PRRs) specific to the resistant tea plant as well as 15 genes shared between the resistant and susceptible cultivars were identified, and the expression of these genes was significantly induced by C. fructicola (Supplementary Table S6, S7 and Fig. 4b). Jayaswall et al.16 reported that 25 NBS-LRRs are also involved in tea plant defence against E. vexans using RNA-Seq. These findings suggest that multiple R genes are involved in tea plant defence. In general, pathogen effectors can disturb or inhibit PAMP-PRR complex-activated defence signalling networks for successful infection33–36, and NBS-LRRs can sequester effectors and reactivate the plant immune system37,38. Interestingly, we observed that these 23 R genes were significantly expressed in the resistant tea plant but were not expressed in the susceptible cultivar. Therefore, we hypothesised that Colletotrichum effectors may inhibit the PAMP-PRR complexes in the susceptible cultivar, resulting in the successful invasion. On the other hand, seven NBS-LRR genes may specifically recognise the related effectors in the resistant cultivar and activate downstream signalling to reactivate the innate immunity to restrict Colletotrichum infection.
MAPK, Ca2+ signalling pathways are meditated by R gene and triggers PCD
MAPK cascades, which consist of MAPKKK, MAPKK and MAPK, are among the most important signalling pathways and are activated by R genes39. In our study, we obtained seven MAPK genes that were significantly induced by C. fructicola, and the PPI network showed that these candidate MAPK and R genes can directly interact with each other; meanwhile, seven Ca2+ signalling-related genes that were upreregulated and associated with plant defence were identified40, but only four of these genes interacted with R genes and other defence-related genes in the PPI network (Supplementary Table S7, and Figs. 4b and 5). These results demonstrated that the MAPK and Ca2+ signalling may be mediated by R genes following Colletotrichum infection, and MAPK cascades constitute the main defence signalling pathway. In addition, the expression of MAPK5 was significantly upregulated in the resistant tea plant cultivar following C. fructicola infection but was downregulated in the susceptible cultivar (Fig. 4b). Interestingly, the proteome data of Li et al.41 indicated that the expression of MAPK5 is downregulated in susceptible wheat leaves after Blumeria graminis f. sp. tritic infection. These results suggest that the specific upregulated expression of MAPK5 may play an important role in tea plant defence against C. fructicola. In addition, MAPK phosphatase 2 (MKP2) can regulate MAPK signalling and cell death to enhance Arabidopsis defence against biotrophic and necrotrophic pathogens by regulating MAPK3 and MAPK642,43. We observed that MKP2 and MAPK3 expression also clearly increased in resistant tea plant cultivars after C. fructicola infection. On the other hand, only one unigene of MAPK6 was identified, and its expression was lower. Moreover, the results of the GO analysis demonstrated that PCD was also a defensive pathway that contained enriched DEGs in the resistant cultivar (Fig. 3c), and microscopic observations also showed that the HR significantly accumulated around the C. fructicola hyphal infection site in the resistant tea plant cultivar (Fig. 6). These results were similar to those of Vilela et al.43. Furthermore, we observed the upregulated expression of three genes associated with PCD in the resistant tea plant: two Development and Cell Deaths and one Long Chain Base Biosynthesis Protein 1 (Fig. 4b)44,45. We therefore speculated that MKP2 may regulate MAPK3 and MAPK5, and positively activate the three genes associated with PCD in tea plant in defence against C. fructicola.
ROS bursts regulate multiple defence responses
In general, ROS bursts constitute one of the earliest plant responses to pathogen invasion. As a signalling molecule, ROS can regulate PCD in plants during pathogen infection and can mutually regulate MAPK signalling39,46,47. Senthilkumar et al.14 reported that the HR and ROS bursts are involved in tea plant defence against P. theae. In our study, we observed that DEGs were enriched in the H2O2 catabolic process (Fig. 3c), and the histochemistry results indicated that H2O2 levels are significantly higher in the resistant cultivar than in the susceptible following C. fructicola infection (Fig. 6b). These results showed that H2O2 plays an important role in tea plant defence against multiple diseases, including anthracnose, and H2O2 production may be regulated by MAPK signalling. Li et al.48 recently reported that a C2H2-type TF can affect H2O2 levels by suppressing peroxidase to enhance the broad-spectrum blast resistance of rice. We discovered that peroxidase 2 (PA2) (TR123656|c4_g4), which is clearly induced by C. coccodes in Capsicum annuum49, is enriched in H2O2 catabolic processes and that the expression of this gene is distinctly upregulated in the susceptible tea plant cultivar but not in the resistant cultivar (Fig. 4b)50. Hence, we suggested that PA2 may be a negative regulator of H2O2 production and that PA2 is suppressed by an unknown TF to increase H2O2 accumulation in the resistant tea plant; in turn, this increased H2O2 accumulation limits Colletotrichum infection. ROS metabolism, which is localised in the peroxisome, is usually controlled by the protein peroxin 11a (PEX11a) under stress conditions51. Notably, we observed that PEX11a was distinctly upregulated in the resistant tea plant during C. fructicola infection and may also be associated with H2O2 production. In addition, thickening of cell walls, which constitute an important of physical barrier to invading pathogens, is induced by H2O2 generation and is associated with wall-associated kinase 352,53. In the present study, the cell walls were significantly reinforced at the penetration sites of C. fructicola hyphae, and this reinforcement was accompanied by H2O2 accumulation in both epidermal and mesophyll cells. At the same time, one wall-associated kinase 3, a cell signalling receptor, was upregulated in the resistant cultivar; on the other hand, H2O2 was generated only in mesophyll cells in the susceptible cultivar. These results suggested that H2O2 may regulate cell wall strengthening and activate signalling to resist C. fructicola attack. Overall, we suggested that H2O2 plays a significant role in tea plant defence to C. fructicola, H2O2 generation may be directed by PEX11a under pathogen stress and mediated by MAPK cascades, and PA2 may be inhibited by other genes to maintain higher levels of H2O2 in tea plant to defend against C. fructicola. Therefore, the function of ROS during the interaction between tea plant and Colletotrichum needs further clarification.
Multiple metabolic pathways are involved in disease resistance
During plant interactions with pathogens, carbohydrate metabolism increases in the host not only to supply massive energy to defence responses but also to regulate the expression of resistance-related genes54. More DEGs were enriched in carbohydrate metabolism in the resistant tea plant cultivar than in the susceptible tea plant cultivar following C. fructicola infection (Figs. 2b and 3c), and the expression of six specific genes associated with carbohydrate metabolism was significantly upregulated (Fig. 4b). Among these genes, 6-phosphogluconate dehydrogenase, which encodes a protein positively induced by powdery mildew in wheat leaves, has been reported to activate host defences41, and three UDP-glycosyltransferase genes that are closely related to the mechanism of galloylated catechins and flavonol 3-O-guycosides in tea were induced55. We previously demonstrated that the content of (-)-epigallocatechin-3-gallate and caffeine rapidly accumulated after C. fructicola infection; at the same time, the expression levels of key genes associated with flavonoids and the caffeine metabolism pathway were clearly upregulated, including the phenylalanine ammonia-lyase (PAL) and S-adenosylmethionine synthetase (SAMS)4. In the present study, DEGs involved with phenylpropanoid and its downstream metabolism of flavonoids were enriched in the resistant tea plant, and the expression levels of PAL (TR106249|c2_g2) and SAMS (TR316985|c0_g1) also increased by C. fructicola (Fig. 4b). These results were similar to those of Figueiredo et al.56, in which SAMS and PAL were involved in grapevine species defence against multiple pathogen attack. In general, sugars, which are sources of carbon, are mainly acquired by pathogens from their hosts57. Based on their biosynthesis and transference, fatty acids have recently been reported to be important organic nutrients between microbes and hosts; RAM2 and ATP-binding cassette transporters play critical roles in these processes58. Our results showed that eight genes associated with the fatty acid metabolism were detected, and the expression of these genes was higher in the resistant tea plant than in the susceptible tea plant cultivar. In contrast, these genes were either downregulated or not regulated in the susceptible cultivar (Fig. 4b). Despite these results being similar to those of Jiang et al.58, the role of fatty acids in the interaction between tea plant and Colletotrichum needs to be further explained16,59.
Plant hormones associated with tea plant defence to Colletotrichum
In addition, plant hormones play important roles in plant defence against pathogens. In our study, the shared genes in the two cultivars were enriched in various phytohormone-related pathways, including responses to ethylene, cytokinin and auxin (Fig. 2b). Meanwhile, six related genes (ethylene response factor 1, like AUXIN RESISTANT 2, one auxin-responsive protein family gene, two gibberellin-regulated family genes, and one SAUR-like auxin-responsive protein family gene) were induced by C. fructicola (Supplementary Table S6). At the same time, the expression levels of six key genes that are specific to the resistant tea plant increased, including two gibberellin-regulated family gene, one auxin efflux carrier family gene, two SAUR-like auxin-responsive protein family gene and one ethylene-responsive element binding protein-coding gene (Supplementary Table S7), suggesting that these genes are involved in the regulation of plant hormones to activate plant defence responses60,61.
Conclusion
Tea plant has a complex defence network to defend against various pathogens. We constructed a possible model of Ca. sinensis defence against anthracnose based on the results of our study (Fig. 7). During the early stage of infection, Colletotrichum appressoria successfully penetrate the cells of tea plant. The formed primary hyphae then secrete and transport diverse virulence factors into the cell, and the pathogen receptors (RLKs, RLPs and NBS-LRRs) specifically recognise pathogenic secretions in the host. The activated pathogen receptors subsequently trigger the defence signalling, which includes MAPK and Ca2+ signalling pathways. At the same time, the ROS (H2O2) production is regulated by these two signalling pathways. Activated R genes also regulate the thickening of cell wall tissue to defend against hyphal growth. The activated signalling pathways trigger the expression of genes associated with the interdependency metabolism pathway, including those involved with sugar, phenylpropanoid, flavonoids and lipid metabolisms; these metabolites, most likely EGCG, suppress the invading pathogen. The activated R genes also trigger a sustained induction of both MAPK and Ca2+ signalling as well as the continuous production of H2O2 in the peroxisome. This signalling and production regulate HR-associated cell death and H2O2 accumulation around the hyphal infection sites. Furthermore, the functional elimination of peroxidase was suppressed by an unknown factor, extending the H2O2 inhibition (Fig. 7).
Fig. 7. Hypothetical model for Ca. sinensis defence against anthracnose based on the results of our study.

The defence function is shown in red
Electronic supplementary material
Acknowledgements
This work was supported by the Earmarked Fund for China Agriculture Research System (CARS-19), the Chinese Academy of Agricultural Sciences through an Innovation Project for Agricultural Sciences and Technology (CAAS-ASTIP-2017-TRICAAS), China Postdoctoral Science Foundation Funded Project (Project No.: 2017M620970); Zhejiang Province Postdoctoral Science Foundation Funded Project.
Authors’ contributions
Conceived and designed the experiments: Y.C.W.; X.Y.H.; X.C.W.; and Y.J.Y. Performed the experiments: Y.C.W.; X.Y.H.; and Q.H.L. Analysed the data: Y.C.W.; X.Y.H.; L.W.; and X.C.W. Contributed reagents/materials/analysis tools: Y.C.W.; X.Y.H.; Q.H.L.; W.J.Q.; X.C.W.; and Y.J.Y. Wrote the paper: Y.C.W.; X.Y.H.; and X.C.W.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Yuchun Wang, Xinyuan Hao.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41438-018-0025-2.
Contributor Information
Xinchao Wang, Phone: +86 571-8665 316, Email: wangxinchao@caas.cn.
Yajun Yang, Phone: +86 571-8665 0226, Email: yjyang@tricaas.com.
References
- 1.Cabrera C, Artacho R, Giménez R. Beneficial effects of green tea-a review. J. Am. Coll. Nutr. 2006;25:79–99. doi: 10.1080/07315724.2006.10719518. [DOI] [PubMed] [Google Scholar]
- 2.Yang CS, Zhang J, Zhang L, Huang J, Wang Y. Mechanisms of body weight reduction and metabolic syndrome alleviation by tea. Mol. Nutr. Food Res. 2016;60:160–174. doi: 10.1002/mnfr.201500428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang YC, et al. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 2016;6:35287. doi: 10.1038/srep35287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang YC, et al. Metabolic changes of caffeine in tea plant (Camellia sinensis (L.) O. Kuntze) as defense response to Colletotrichum fructicola. J. Agric. Food Chem. 2016;64:6685–6693. doi: 10.1021/acs.jafc.6b02044. [DOI] [PubMed] [Google Scholar]
- 5.Kunkel BN, Brooks DM. Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 2002;5:325–331. doi: 10.1016/S1369-5266(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 6.Schwessinger B, Zipfel C. News from the frontline: recent insights into PAMP-triggered immunity in plants. Curr. Opin. Plant Biol. 2008;11:389–395. doi: 10.1016/j.pbi.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Thomma BP, Nurnberger T, Joosten MH. Of PAMPs and effectors: the blurred PTI-ETI dichotomy. Plant Cell. 2011;23:4–15. doi: 10.1105/tpc.110.082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui HTK, Parker JE. Effector-triggered immunity: from pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015;66:487–511. doi: 10.1146/annurev-arplant-050213-040012. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg JT, Yao N. The role and regulation of programmed cell death in plant-pathogen interactions. Cell. Microbiol. 2004;6:201–211. doi: 10.1111/j.1462-5822.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- 10.Cannon PF, Damm U, Johnston PR, Weir BS. Colletotrichum-current status and future directions. Stud. Mycol. 2012;73:181–213. doi: 10.3114/sim0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connell R, et al. A novel Arabidopsis-Colletotrichum pathosystem for the molecular dissection of plant-fungal interactions. Mol. Plant Microbe Interact. 2004;17:272–282. doi: 10.1094/MPMI.2004.17.3.272. [DOI] [PubMed] [Google Scholar]
- 12.Bhadauria V, et al. Identification of Lens culinaris defense genes responsive to the anthracnose pathogen Colletotrichum truncatum. BMC Genet. 2013;14:31. doi: 10.1186/1471-2156-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padder BA, Kamfwa K, Awale HE, Kelly JD. Transcriptome profiling of the Phaseolus vulgaris-Colletotrichum lindemuthianum pathosystem. PLoS ONE. 2016;11:e0165823. doi: 10.1371/journal.pone.0165823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senthilkumar P, Thirugnanasambantham K, Mandal AK. Suppressive subtractive hybridization approach revealed differential expression of hypersensitive response and reactive oxygen species production genes in tea (Camellia sinensis (L.) O. Kuntze) leaves during Pestalotiopsis thea infection. Appl. Biochem. Biotechnol. 2012;168:1917–1927. doi: 10.1007/s12010-012-9907-1. [DOI] [PubMed] [Google Scholar]
- 15.Palanisamy S, Mandal AK. Susceptibility against grey blight disease-causing fungus Pestalotiopsis sp. in tea (Camellia sinensis (L.) O. Kuntze) cultivars is influenced by anti-oxidative enzymes. Appl. Biochem. Biotechnol. 2014;172:216–223. doi: 10.1007/s12010-013-0529-z. [DOI] [PubMed] [Google Scholar]
- 16.Jayaswall K, et al. Transcriptome analysis reveals candidate genes involved in blister blight defense in tea (Camellia sinensis (L) Kuntze) Sci. Rep. 2016;6:30412. doi: 10.1038/srep30412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, et al. Transcriptome analysis of an anthracnose-resistant tea plant cultivar reveals genes associated with resistance to Colletotrichum camelliae. PLoS ONE. 2016;11:e0148535. doi: 10.1371/journal.pone.0148535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao X, et al. Comprehensive transcriptome analyses reveal differential gene expression profiles of Camellia sinensis axillary buds at para-, endo-, ecodormancy, and bud flush stages. Front. Plant Sci. 2017;8:553. doi: 10.3389/fpls.2017.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leng N, et al. EBSeq-HMM: a Bayesian approach for identifying gene-expression changes in ordered RNA-seq experiments. Bioinformatics. 2015;31:2614–2622. doi: 10.1093/bioinformatics/btv193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Q, Wang XJ. GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008;36:358–363. doi: 10.1093/nar/gkn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ernst J, Barjoseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faoro F, Maffi D, Cantu D, Iriti M. Chemical-induced resistance against powdery mildew in barley: the effects of chitosan and benzothiadiazole. BioControl. 2008;53:387–401. doi: 10.1007/s10526-007-9091-3. [DOI] [Google Scholar]
- 27.Hao X, et al. Histochemical studies on the accumulation of H2O2 and hypersensitive cell death in the nonhost resistance of pepper against Blumeria graminis f. sp. tritici. Physiol. Mol. Plant Pathol. 2011;76:104–111. doi: 10.1016/j.pmpp.2011.07.003. [DOI] [Google Scholar]
- 28.Hao X, et al. Identification and evaluation of reliable reference genes for quantitative real-time PCR analysis in tea plant (Camellia sinensis (L.) O. Kuntze) Int. J. Mol. Sci. 2014;15:22155–22172. doi: 10.3390/ijms151222155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 30.Ferrier-Cana E, et al. Characterization of expressed NBS-LRR resistance gene candidates from common bean. Theor. Appl. Genet. 2003;106:251–261. doi: 10.1007/s00122-002-1032-z. [DOI] [PubMed] [Google Scholar]
- 31.Li J, et al. Genome-wide identification and comparative expression analysis of NBS-LRR-encoding genes upon Colletotrichum gloeosporioides infection in two ecotypes of Fragaria vesca. Gene. 2013;527:215–227. doi: 10.1016/j.gene.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Takahara H, et al. Colletotrichum higginsianum extracellular LysM proteins play dual roles in appressorial function and suppression of chitin-triggered plant immunity. New Phytol. 2016;211:1323–1337. doi: 10.1111/nph.13994. [DOI] [PubMed] [Google Scholar]
- 33.Jones DA, Takemoto D. Plant innate immunity-direct and indirect recognition of general and specific pathogen-associated molecules. Curr. Opin. Immunol. 2004;16:48–62. doi: 10.1016/j.coi.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 34.Bonardi V, Dangl JL. How complex are intracellular immune receptor signaling complexes? Front. Plant Sci. 2012;3:237. doi: 10.3389/fpls.2012.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monaghan J, Zipfel C. Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 2012;15:349–357. doi: 10.1016/j.pbi.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Zipfel C. Plant pattern-recognition receptors. Trends Immunol. 2014;35:345–351. doi: 10.1016/j.it.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 38.Tsuda K, Katagiri F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol. 2010;13:459–465. doi: 10.1016/j.pbi.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Meng X, Zhang S. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013;51:245–266. doi: 10.1146/annurev-phyto-082712-102314. [DOI] [PubMed] [Google Scholar]
- 40.Steinhorst L, Kudla J. Calcium and reactive oxygen species rule the waves of signaling. Plant Physiol. 2013;163:471–485. doi: 10.1104/pp.113.222950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, et al. Proteomic analysis of the compatible interaction of wheat and powdery mildew (Blumeria graminis f. sp. tritici) Plant Physiol. Biochem. 2017;111:234–243. doi: 10.1016/j.plaphy.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Lumbreras V, et al. MAPK phosphatase MKP2 mediates disease responses in Arabidopsis and functionally interacts with MPK3 and MPK6. Plant J. 2010;63:1017–1030. doi: 10.1111/j.1365-313X.2010.04297.x. [DOI] [PubMed] [Google Scholar]
- 43.Vilela B, Pagès M, Lumbreras V. Regulation of MAPK signaling and cell death by MAPK phosphatase MKP2. Plant Signal. Behav. 2010;5:1497–1500. doi: 10.4161/psb.5.11.13645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tenhaken R, Doerks T, Bork P. DCD-a novel plant specific domain in proteins involved in development and programmed cell death. BMC Bioinformatics. 2005;6:169. doi: 10.1186/1471-2105-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi L, et al. Involvement of sphingoid bases in mediating reactive oxygen intermediate production and programmed cell death in Arabidopsis. Cell Res. 2007;17:1030–1040. doi: 10.1038/cr.2007.100. [DOI] [PubMed] [Google Scholar]
- 46.Gechev TS, Van Breusegem F, Stone JM, Denev I, Laloi C. Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. Bioessays. 2006;28:1091–1101. doi: 10.1002/bies.20493. [DOI] [PubMed] [Google Scholar]
- 47.Daudi A, et al. The apoplastic oxidative burst peroxidase in Arabidopsis is a major component of pattern-triggered immunity. Plant Cell. 2012;24:275–287. doi: 10.1105/tpc.111.093039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell. 2017;170:114–126. doi: 10.1016/j.cell.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Choi HW, Hwang BK. The pepper extracellular peroxidase CaPO2 is required for salt, drought and oxidative stress tolerance as well as resistance to fungal pathogens. Planta. 2012;235:1369–1382. doi: 10.1007/s00425-011-1580-z. [DOI] [PubMed] [Google Scholar]
- 50.Ascencio-Ibáñez JT, et al. Global analysis of Arabidopsis gene expression nncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 2008;148:436–454. doi: 10.1104/pp.108.121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodríguez-Serrano M, Romero-Puertas MC, Sanz-Fernández M, Hu J, Sandalio LM. Peroxisomes extend peroxules in a fast response to stress via a reactive oxygen species-mediated induction of the peroxin PEX11a. Plant Physiol. 2016;171:1665–1674. doi: 10.1104/pp.16.00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vellosillo T, Vicente J, Kulasekaran S, Hamberg M, Castresana C. Emerging complexity in reactive oxygen species production and signaling during the response of plants to pathogens. Plant Physiol. 2010;154:444–448. doi: 10.1104/pp.110.161273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kubicek CP, Starr TL, Glass NL. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014;52:427–451. doi: 10.1146/annurev-phyto-102313-045831. [DOI] [PubMed] [Google Scholar]
- 54.Trouvelot S, et al. Carbohydrates in plant immunity and plant protection: roles and potential application as foliar sprays. Front. Plant Sci. 2014;5:592. doi: 10.3389/fpls.2014.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui L, et al. Identification of UDP-glycosyltransferases involved in the biosynthesis of astringent taste compounds in tea (Camellia sinensis) J. Exp. Bot. 2016;67:2285–2297. doi: 10.1093/jxb/erw053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Figueiredo A, et al. Transcriptional and metabolic profiling of grape (Vitis vinifera L.) leaves unravel possible innate resistance against pathogenic fungi. J. Exp. Bot. 2008;59:3371–3381. doi: 10.1093/jxb/ern187. [DOI] [PubMed] [Google Scholar]
- 57.Trépanier M, et al. Dependence of arbuscular-mycorrhizal fungi on their plant host for palmitic acid synthesis. Appl. Environ. Microbiol. 2005;71:5341–5347. doi: 10.1128/AEM.71.9.5341-5347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Y, et al. Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science. 2017;356:1172–1175. doi: 10.1126/science.aam9970. [DOI] [PubMed] [Google Scholar]
- 59.Rubio M, et al. Analysis of gene expression changes in peach leaves in response to Plum pox virus infection using RNA-Seq. Mol. Plant Pathol. 2015;16:164–176. doi: 10.1111/mpp.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo W, et al. An ethylene response-related factor, GbERF1-like, from Gossypium barbadense improves resistance to Verticillium dahliae via activating lignin synthesis. Plant Mol. Biol. 2016;91:305–318. doi: 10.1007/s11103-016-0467-6. [DOI] [PubMed] [Google Scholar]
- 61.Becker MG, et al. Transcriptome analysis of the Brassica napus-Leptosphaeria maculans pathosystem identifies receptor, signaling and structural genes underlying plant resistance. Plant J. 2017;90:573–586. doi: 10.1111/tpj.13514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.