

Abstract
For arrhythmia triggers that are secondary to dysfunctional intracellular Ca2+ cycling, there are few if any specific agents that target exactly the Ca2+ handling machinery. However, in the literature to date, several candidates have been proposed. We review here these agents with the idea that in the future these agents or those derived thereof will prove invaluable in clinical application.
INTRODUCTION
Under normal conditions for all cardiac cells, during systole, Ca2+ influx through the cardiac calcium channel provides a trigger for the calcium to be released from the sarcoplasmic reticulum (SR) through a large SR membrane, ligand operated, ion channel called the ryanodine receptor (RYR). The open probability of the RYR protein is increased by the elevation of cytoplasmic Ca2+ concentration [Ca2+]i. Thus, Ca2+ entry into the cell produces a small increase of Ca2+ which leads to an opening of the RYR and subsequent release of a larger amount of Ca2+ that is stored in the SR. This process is known as calcium induced calcium release (CICR) (Figure 1). Microscopic signals resulting from clusters of RYR openings generate Ca2+ signals called Ca2+ sparks. Spatial and temporal summation of action potential evoked Ca2+ sparks underlies the global Ca2+ transient which in contractile cells has a familiar rise and decay as Ca2+ is released is reuptaken into the SR ready for the next heartbeat. Any remaining cytosolic Ca2+ is pumped out of cell by sodium calcium exchanger protein (NCX). Under normal conditions, CICR that occurs does not propagate but rather remains controlled by L type Ca2+ channel influx.
Figure 1. Simple diagram of the excitation-contraction coupling system in the cardiac cell.
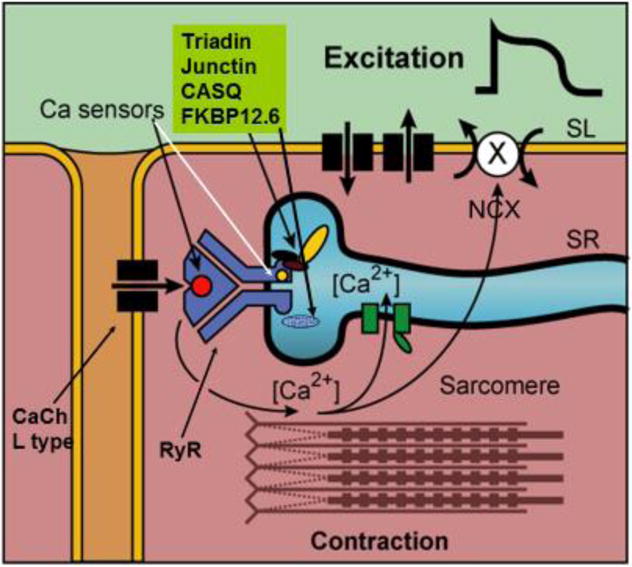
During the action potential Ca2+ enters the cell as a rapid influx followed by a maintained component of the slow inward Ca2+ current (Thick arrow). The rapid influx of Ca2+ via the T tubules is thought to induce release of Ca2+ from a release compartment in the SR, by triggering opening of Ca2+ channels via binding sites(sensors) on RYR protein. Relaxation follows when the cytosolic Ca2+ is sequestered again in an uptake compartment of the SR (SERCA pump, green boxes) and partly extruded through the cell membrane by the Na+/Ca++ exchanger (NCX). The process of NCX is electrogenic so that Ca2+ extrusion through NCX leads to a depolarizing current. From Ter Keurs and Boyden, Physiol Review, 200776.
So what is “Ca2+ leak” if the cell always has spontaneous Ca2+ sparks, albeit at low probability?
When a cell is “overloaded” with calcium the associated sequestration of Ca2+ by the SR can increase SR Ca2+ content to above normal levels, under these circumstances the Ca2+ leaks out of the SR in the form of Ca2+ waves. These are local Ca2+ release events that trigger a regenerative Ca2+ waves via the CICR process. The Ca2+ wave can propagate throughout the cell and in some cases can trigger a Ca2+ waves in an adjacent cell (Figure 2)(see also1). It appears that intracellular Ca2+ waves generally occur when the SR Ca2+ content is elevated above a threshold value2, 3, but other changes, such as altered Ca2+ sensitivity of the RyR can induced Ca2+ waves. Some of the Ca2+ in the wave is pumped out of the cell by the electrogenic NCX. The resulting current depolarizes the membrane (producing a delayed afterdepolarization (DAD) like membrane voltage change) and can be sufficient to initiate an action potential. Yet synchrony of Ca2+ releases between coupled cells is required for to provide sufficient depolarizing current within one region to initiate an arrhythmic action potential in an intact ventricle/atrium. The critical number of coupled cells experiencing a DAD is a topic of debate and research4–6.
Figure 2. Representative confocal line-scan images show spontaneous Ca2+ release events (SCaEs) in wild-type (WT) and R33Q (CPVT mutation in CASQ) cells in the presence of isoproterenol.
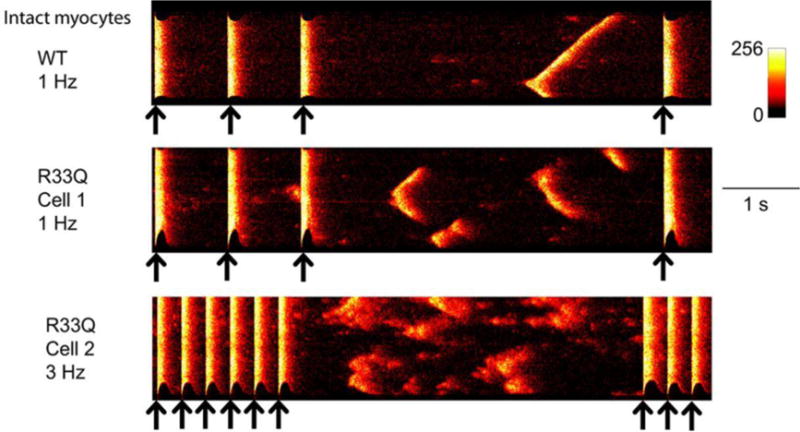
Black arrows indicate field stimulations. Spontaneous Ca events(SCaEs) in WT myocytes were usually due to a cell-wide wave that was initiated at 1 site (red arrow). SCaEs in diseased R33Q cells varied. Often, fragmented spontaneous Ca2+ waves occurred and slowly propagated (cells 1 and 2), and wavelets and Ca2+ sparks occurred before Ca2+ transients resume the diastolic level. From Liu N et al. Circulation Research 2013;113:142–15277.
SR Ca2+ leak is increased in numerous pathological conditions (eg. Heart failure (HF)7; post MI8, 9). SR Ca2+ leak, if persistent, decreases SR Ca2+ load and as explained above can lead to propagating Ca2+ waves and thus DADs (Figure 3).
Figure 3.
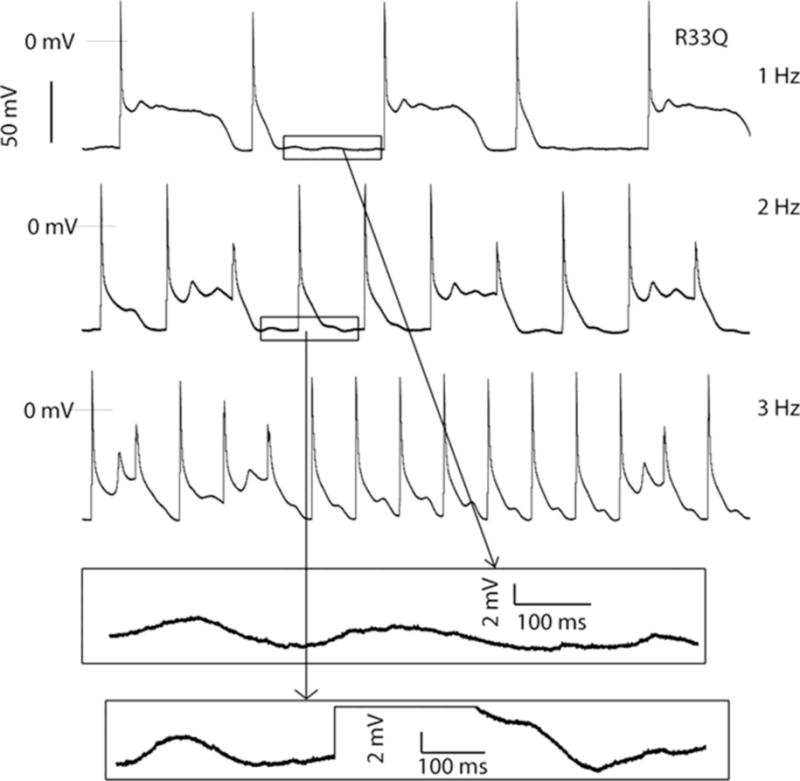
Action potentials recordings in a R33Q mouse cells in the presence of isoproterenol at 1- to 3-Hz pacing. Early afterdepolarizations occurred at lower pacing frequency; diverse patterns of action potential were shown in all pacing frequencies. Bottom, The enlarged membrane oscillations occurring between stimulated beats. From Liu N et al. Circulation Research 2013;113:142–15277.
While Ca2+ leak is an operational term, several mechanisms have been proposed to explain the altered RYR gating that leads to Ca2+ leak. An increased sensitivity of RYR to its ligand cytosolic Ca2+ may be due to enhanced protein kinase A (PKA) and/or CaMKII dependent RYR7 phosphorylation at specific sites10, 11–13. Recent data using human tissues favor one idea where Ca2+ handling abnormalities in HF are due to excessive CaMKII phosphorylation at a specific RYR residue14, 15. Other factors such as the oxidative state could change resulting in direct activation of the RYR protein16. Finally others have suggested that RYR gating may be altered when an abundance of endogenous proteins that modulate RYR are altered (eg. sorcin, S100A17, 18).
Finally mutations and dysregulation of RYR and other calcium binding proteins have been implicated in several gene-based arrhythmias; for example, RYR and CVPT, and Calsequestrin(CASQ) and CPVT19. Mechanisms of these arrhythmias are similar to those of acquired diseases above, that is the arrhythmic events are caused by abnormally propagating Ca2+ waves which cause NCX dependent membrane oscillations (DADs) and triggered beats (Figures 2,3).
Can Ca2+ leak be Managed?
As an antiarrhythmic, we would want an agent to modulate the mishandled Ca2+ so Ca2+ does not increase Ca2+ dependent currents to cause depolarization and elicit action potentials. If we target the spontaneous Ca2+ releases, then we would reduce the initiators of the Ca2+ waves, the delayed afterdepolarizations and thus triggering beats.
The arrhythmias mentioned above result when the cell’s SR Ca2+ content is increased above a threshold level at which waves are produced. Recent work suggests that a decrease of threshold (due to a sensitization to RYR Ca2+ release) also produces Ca2+ waves and DADs. For arrhythmias seen in heart failure the involvement of DADs in some ventricular arrhythmias has been shown20, 21. However, in heart failure the SR Ca2+ content is decreased suggesting that the threshold for Ca2+ release may be lower, such that Ca2+ waves would occur at a lower SR Ca2+ content. This may be a consequence of increased leakiness of the RYR during diastole, such that there is increased Ca2+ efflux at a given SR Ca2+ content. The exact molecular mechanisms responsible for this are controversial22, but as above, it may be associated with increased phosphorylation of the RYR due to PKA or CaM-Kinase14.
An example of the occurrence of DADs in the absence of increased SR Ca2+ content is provided by catecholaminergic polymorphic ventricular tachycardia (CPVT). This arrhythmia in patients is seen during exercise or other stress. The similarity of the abnormalities in the ECGs to those observed in digitalis toxicity led to the suggestion of similarities in an underlying mechanism. Genetic studies have shown that many CPVT patients have a mutation in RYR (eg R4496C) or the intrasarcoplasmic protein CASQ (eg. R33Q). The current hypothesis is that the mutated protein causes an increased leak of Ca2+ from the SR. Thus Ca2+ waves and DADs occur at a lower SR Ca2+ content than in controls23. (Figure 3)
Therapies for DAD-related arrhythmias
For these Ca2+ dependent (Ca2+ wave dependent) arrhythmias, the goal of therapy is to treat 1) to prevent the DAD from occurring and/or 2) to prevent the DAD from triggering an action potential.
The latter can potentially be achieved using sodium channel blockers. A better solution, however, would be to remove the underlying DAD directly. In the case of arrhythmias resulting from “calcium overload”, it may be possible to remove the underlying “overload”. For example, local anesthetics (eg. flecainide) reduce intracellular Na+ concentration as a consequence of decreasing Na+ entry (via inhibition of sodium current resulting in reduced excitability). Lowered intracellular Na+ concentration will act via NCX, to decrease intracellular Ca2+ load24. Antiarrhythmic effects of flecainide have been seen in murine models as well as patients with CPVT25, but the confirmation that the cellular basis is linked to intracellular Na+ levels has yet to be made.
Recently, the late Na+ current (INa-late) has gained interest since it is modulated in disease. A small fraction of cardiac sodium channels carry INa-late. For peak INa, sodium channels open quickly and close in a well-defined time and voltage dependent manner. INa-late current is formed when sodium channels remain open or reopen for 100s of ms after the peak current.
In HF and congenital long QT type3, INa-late is upregulated and provides enhanced Na+ influx during the AP26. This in turn alters Ca2+I which then could be arrhythmogenic27.
Ranolazine inhibits cardiac INa-late as well as other channels (eg IKr)28. Some have reported it also inhibits RyR directly29. It has been reported to have antiarrhythmic properties in various animal models (eg HF,30, 31). In recent clinical trials it was demonstrated to reduce arrhythmic events32, 33 although there appears to be a risk of Torsades de Pointes34. GS-967 is a newer more specific INa-late current inhibitor (lacks the IKr blockade seen with Ranolazine) that shows promise as an antiarrhythmic35, 36.
While Na+ channel blockade of the cardiac Na+ channel is considered now to be a viable therapy for Ca2+ mediated arrhythmias, new data suggest that selective blockade of Neuronal Na+ channels (eg. Nav1.1, Nav1.3 and Nav1.6s etc) in cardiac T-tubules using riluzole is anti-arrhythmic37. This suggests that there is a contribution of Na+ influx from overactive neuronal Na+ channels in RYR subcellular regions to more Ca2+ leak from SR (Figure 4).
Figure 4. Schematic diagram of a t-tubule and associated junctional SR.
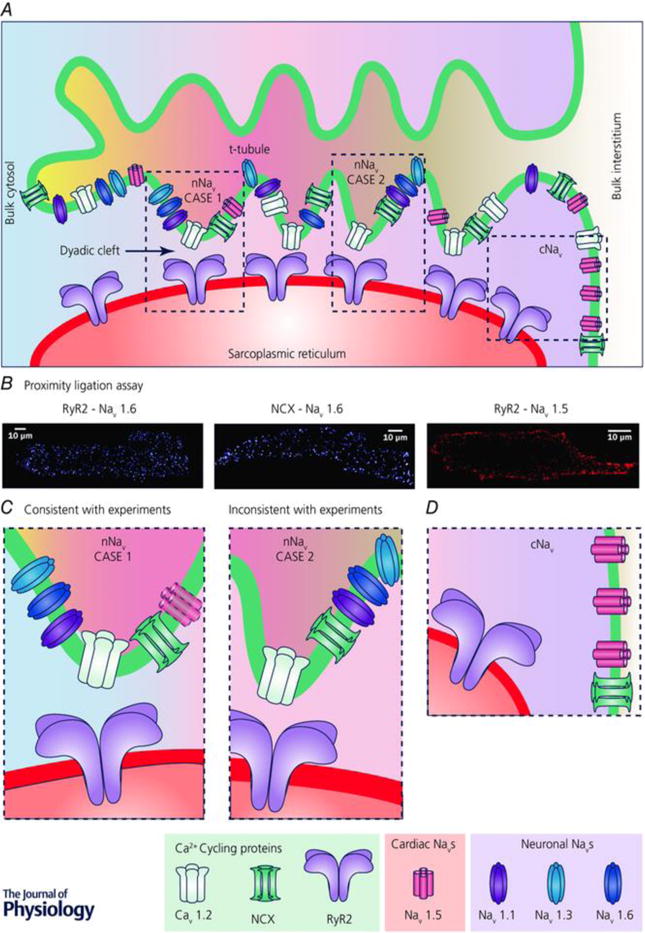
Microfolds in t-tubule are depicted based on recent findings. Different arrangements of Ca2+ cycling proteins and sodium channels are depicted along the t-tubule. Regions highlighted by the dashed boxes are presented at higher magnification in C and D. Note that differential shading of the interstitial space within the t-tubule and the cytoplasm within the dyadic cleft indicates local differences in ionic concentrations within these spaces due to their diffusional isolation from the bulk interstitial space and cytoplasm, respectively. B, results from Duolink assays (PLAs) show close association of nNaV isoform NaV1.6 with both RYR2 and NCX throughout myocytes, consistent with enrichment of nNaVs in t-tubules. In contrast, PLA signal corresponding to association between cNaV (NaV1.5) and RYR2 is only observed at the periphery of the cell, consistent with cNaV localization at the lateral membrane. Adapted from Radwański et al. 2016, doi:10.1016/j.jacbts.2016.04.004, Creative Commons Attribution-NonCommercial-No Derivatives License (CC BY NC ND). C, higher magnification views of regions from A showing two possible scenarios of nNaV localization within t-tubules. Left, case 1, very close association between nNaVs and RYRs, which is consistent with PLA results. A cNaV is depicted faded since experimental results including PLA results argue against cNaV enrichment in t-tubules. Right, case 2, nNaVs localized to t-tubules but not very closely associated with RYRs, which is not consistent with PLA results. D, higher magnification view of region from A showing cNaV (NaV1.5) localization at the lateral membrane. From Veeraraghavan et al, The Journal of Physiology, © 2017 The Physiological Society.37 Reproduced by permission of John Wiley and Sons, Inc.
Theoretically, it would be possible to modulate Ca2+ by affecting the membrane transports/channels involved in Ca2+ homeostasis. For example, L type Ca2+ channel pore blockers obviously decrease Ca2+ influx and in so doing would be expected to eventually reduce SR load, Ca2+i and diminish force. Thus Ca2+ channel pore blockers (eg. Verapamil) will affect intracellular Ca2+ and wave formation but at the expense of force generation. An alternative option to drugs acting directly on molecular targets of the SR is to modulate sarcolemma Ca2+ influxes that in turn reduce SR Ca2+ load and therefore associated Ca2+-leak related abnormalities. As with other targets, the risk associated with reduced SR Ca2+ load is that peak systolic Ca2+ will be reduced and associated inotropy. Currently accepted medications such as Ca2+ channel blockers and beta blockers reduce cardiovascular mortality partially via reduction of Ca+ influx to the heart through their effects on the L-type Ca2+ channel. But the relative contribution of SR unloading to the overall beneficial effect of these two classes of drugs is difficult to assess.
Alternatively, one might target the molecular mechanism involved in the inactivation of Ca2+ channel proteins or the Ca2+ dependent processes known to affect Ca2+ channel function (eg. CaMKII) or small proteins (eg.Gem) that are known to affect Ca2+ channel subunit assembly38.
Phosphorylation/dephosphorylation of the enzyme CaMKII is critical for cardiac excitability and function much like its well-known “neighborhood” protein, protein kinase A (PKA). Unlike PKA, CaMKII has the ability to become autophosphorylated and this is a Ca2+ independent process39. But like PKA, CaMKII activity is linked to the function of several intracellular cardiac proteins, for example, the L type Ca2+ channel40 and RYR41. Thus targeting inhibition of this enzyme to alter function of regulated proteins to ameliorate Ca2+ wave function and resulting DADs is a goal of both academia and industry42.
At this time, only three tools are available. KN-93 (and its inactive analog KN-62) are used frequently in experimental studies to illustrate the role of CaMKII in cardiac cell function. KN-93 does inhibit activation of CaMKII but not its autophosphorylation activity. But CaMKII inhibition prevents catecholamine induced VTs in CPVT mice43 and recently has proven useful on atrial arrhythmias secondary to Ca2+ leak44.
However, KN93 affects L type Ca 2+ channel function45 as well as some K+ channel function46. Experimentalists have also used autocamtide-3 inhibitor (AC3-1) peptides that inhibit CaMKII selectively over PKA, PKC47. AC3-1 is also a potent PKD inhibitor. These peptides remain as tools. There has also been a recent emergence of pharmacologically active agents designed after small endogenous proteins that inhibit CamKII, such as CaMKIIN48 and CaMKIINide49. Work continues to delineate how these inhibitors affect cardiac function.
Direct modulation of SR leak via actions on RYR channel
Designing drugs to bind to RYR directly to reduce the Ca2+ sensitivity of the channel is thought to be a valid anti-arrhythmic strategy, but no compounds to date have been approved for clinical use purely on this mechanism. While many drugs designed for other purposes have been found to alter RYR Ca2+ sensitivity, these have been used as tools to investigate the effects of drug-induced modulation of RYR. The anesthetics such as tetracaine, which reduces surface membrane excitability via Na+ channel inhibition is also known to reduce the sensitivity of Ca2+ induced SR Ca2+ release via a direct action on RYR50. Studies have shown tetracaine substantially reduces the frequency of both Ca2+ sparks and spontaneous Ca2+ waves51 but this effect is accompanied by an increased quantity of Ca2+ released from the SR at each spontaneous event (increased leak). Derivatives of tetracaine that block RyR appear to inhibit SR Ca2+ leak and prevent CPVT arrhythmias in mice52. Caffeine, a compound known to increase the Ca2+ sensitivity of RYR will increase the frequency of sparks and Ca2+ waves and each release event is smaller53. Interestingly, while these two compounds dramatically affect spontaneous Ca2+ release in different ways, the effect on systolic Ca2+ release (in the steady-state condition) is undetectable due to an autoregulatory mechanism involving Ca2+ influx via the L-type and Ca+ extrusion mainly via NCX27. Caffeine is known to increase ventricular premature beats in normal hearts via its ability to increase the incidence of spontaneous Ca2+ waves and thus generate a spontaneous diastolic depolarization which generates triggers the extra systole. Many aspects of this explanation still require clarification.
Drugs have been identified that have an almost exclusive effect on the RYR protein complex to reduce Ca2+ sensitivity.
The first drug candidate to emerge was the benzodiazepine derivative variously known as JTV519/K201. This molecule is similar in structure to L-type Ca2+ channel blockers, but was selected for its ability to reduce the effects of intracellular Ca2+ overload54. Subsequent work suggested that the drug’s mechanism was to bind to RYR and mimic the binding of the regulatory protein FK506 binding protein (FKBP12.6)55. FKBP12.6 is thought to bind to RYR and chronically reduce Ca2+ efflux through RYR. As part of the beta-adrenergic response, A-kinase mediated phosphorylation may alter the Ca2+ sensitivity of RYR via reduced binding of FKBP12.6. In HF, the associated altered status of the adrenergic signaling pathway in cardiac muscle is thought to result in hyper phosphorylation of RYR, reduced FKBP12.6 binding and thereby increase RYR-mediated Ca2+ leak from the SR, i.e. acting in an analogous way to caffeine56. These changes are thought to be responsible for the failing myocardium being more prone to DADs and subsequent pro-arrhythmic VPCs. In support of this, JTV519 improved outcome in an animal model of HF57. JTV519/K201 reduces Ca+ efflux via RYR at concentrations that allow reasonable specificity to RYR and appears to have an action in the absence of activation of the A-kinase pathway58. Therefore, regardless of the mechanism, the drug has the possibility to act via RYR as an antiarrhythmic agent on myocardium prone to arrhythmias due to dysfunctional RYR, e.g. Purkinje cells that survive in the infarcted heart8 (Figure 5). An alternative structure thought to be an even more potent inhibitor of RYR is variously known as S107 or RyCal10, 59, 60. Others are currently being sought.
Figure 5.
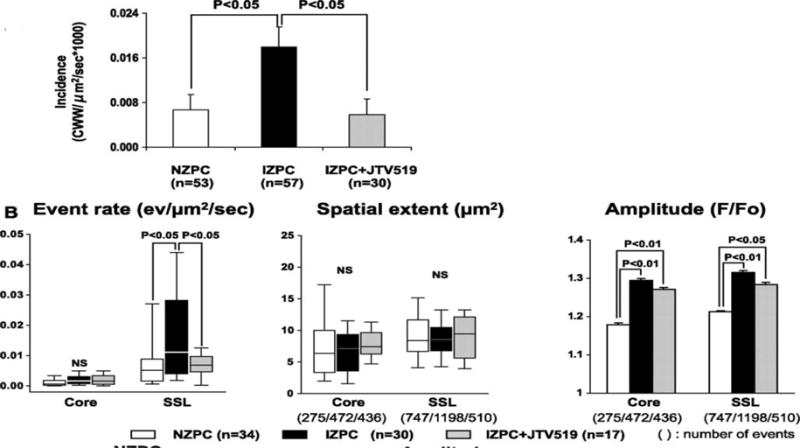
JTV519(K201) suppresses cell wide Ca waves in Purkinje cells from the infarcted heart A, Graph showing the incidence of cell wide Ca2+ waves in Normal Zone Purkinje Cells(NZPCs) and Infarct Zone Purkinjes (IZPCs) in the absence and presence of JTV519 (K201) 1 μmol/L (gray bar). B, Ca event rate, spatial extent, and amplitude in IZPCs in the absence and presence of JTV519 (K201) (gray bars). Total number of events used is shown in parentheses. From Hirose M et al. Circ Arrhythm Electrophysiol 2008;1:387-3958.
Dantrolene and dantrolene-like compounds while showing no effect on normal RyR function, inhibits Ca2+ leak in cells from failing hearts by promoting a stable RyR conformational state61–63.
Action on other targets that may aid anti-arrhythmic effects
One feature of small molecules is a lack of specificity that may benefit or counteract their ability to suppress spontaneous Ca2+ release. For example, JTV519/K201 was found to inhibit SERCA activity by a small amount (~10%) at drug levels that also significantly suppress RYR activity58. Suppression of SERCA is normally associated with smaller systolic Ca2+ releases and poor contractility in failing myocardium. Thus low levels of inhibition may not have the significant negative inotropic effects but could significantly suppress spontaneous Ca2+ waves and therefore arrhythmias. Data from several groups64–66 suggest that a burst of Ca2+-activated SERCA activity in regions of a cell adjacent to a region of spontaneous Ca2+ release could locally enhance SR load and increase the chance of spontaneous Ca2+ release propagating along the length of the cells. Thus mild SERCA inhibition may aid an anti-arrhythmic action through this route.
Studies on a CPVT mouse model and a limited number of human CPVT patients have shown that Flecainide (a Na+ channel blocker) can suppress arrhythmias associated with RYR dysfunction67, 68. The study suggests that the mode of anti-arrhythmic action is not via Na+ channel inhibition, but via an inhibitory effect of flecainide on RYR69. But this interpretation of flecainide’s action on RYR has been challenged by isolated RYR studies70 and in intact cell work71. Another example of a potential revision of the mode of action of a cardiovascular drug is the beta blocker Carvedilol. This drug is a potent inhibitor of RYR and may act to suppress arrhythmias through this route72. Further screens of beta-blockers have identified other examples of drugs that suppress RYR activity and therefore potentially possess anti-DAD and antiarrhythmic activity73. Such examples indicate the challenges in designing anti-arrhythmic therapy around a single target with a single molecule and in assigning mechanism to the observed anti-arrhythmic effect. However, in a recent publication, derivatives of tetracaine with high specificity to RyR were found to effectively suppress arrhythmias in a mouse model of CPVT52 and indicates that drug-based RyR inhibition can have powerful anti-arrhythmic effects.
Alternative anti-arrhythmic strategies
Alternative approaches being considered are for example, inhibition of the NCX. This may appear a good strategy since this exchanger provides the major Ca+ activated currents(Iti) in DADs73, 74. But tonic inhibition of this exchanger will reduce the Ca2+ efflux capacity of the myocardium, increase intracellular Ca2+ and SR load and potentially increase the probability of pro-arrhythmic Ca2+ release. A second approach that may indirectly be anti-arrhythmic is to use novel drugs designed to stimulate the sarcolemmal Na+/K+ pump/antiporter (NKA)75. These are designed to restore the intracellular Na+ concentration and in doing so, may reduce cellular Ca2+ load via NCX, SR load and associated pro-arrhythmic SR Ca2+-leak.
In summary, pharmacological strategies that specifically address abnormal SR Ca+ leak are at early stages of development but hold great promise as a means of providing novel anti-arrhythmic therapeutic options for a range of cardiac pathologies with associated high risk of sudden arrhythmic cardiac death.
Acknowledgments
PA Boyden supported by NIH HL114383.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
PAB: no conflicts
GS Smith has interests in Clyde Biosciences LTD UK, non-salaried
GLS: consult CLYDE BIOsciences
Contributor Information
Penelope A Boyden, Department of Pharmacology, Center for Molecular Therapeutics, Columbia University, New York NY.
Godfrey L. Smith, Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow UK
References
- 1.Bers DM. Cardiac Sarcoplasmic Reticulum Calcium Leak: Basis and Roles in Cardiac Dysfunction. Annu Rev of Physiol. 2014;76:107–27. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 2.Diaz ME, Trafford AW, O’Neil CL, Eisner DA. A measurable reduction of SR Ca content follows spontaneous Ca release in rat ventricular myocytes. Pfluegers Arch. 1997;434:852–4. doi: 10.1007/s004240050475. [DOI] [PubMed] [Google Scholar]
- 3.Chen-Izu Y, Ward CW, Stark W, Jr, Banyasz T, Sumandea MP, Balke CW, Izu LT, Wehrens XH. Phosphorylation of RyR2 and shortening of RyR2 cluster spacing in spontaneously hypertensive rat with heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H2409–H2417. doi: 10.1152/ajpheart.00562.2007. [DOI] [PubMed] [Google Scholar]
- 4.Houser SR. When does spontaneous sarcoplasmic reticulum CA(2+) release cause a triggered arrythmia? Cellular versus tissue requirements. Circ Res. 2000;87(9):725–7. doi: 10.1161/01.res.87.9.725. [DOI] [PubMed] [Google Scholar]
- 5.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99(5):1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local beta-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110(11):1454–64. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/Calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 8.Hirose M, Stuyvers BD, Dun W, Ter Keurs HED, Boyden PA. Function of Ca2+ release channels in Purkinje cells that survive in the infarcted canine heart; a mechanism for triggered Purkinje ectopy. Circ Arrhythmia Electrophys. 2008;1:387–95. doi: 10.1161/CIRCEP.107.758110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Györke S. Shortened Ca2+ Signaling Refractoriness Underlies Cellular Arrhythmogenesis in a Postinfarction Model of Sudden Cardiac Death. Circ Res. 2012;110:569–77. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta -adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–87. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belevych AE, Radwanäski PB, Carnes CA, Gyorke S. Ryanopathy: causes and manifestations of RyR2 dysfunction in heart failure. CARDIOVASC RES. 2013;98:240–7. doi: 10.1093/cvr/cvt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta -Adrenergic Enhancement of Sarcoplasmic Reticulum Calcium Leak in Cardiac Myocytes Is Mediated by Calcium/Calmodulin-Dependent Protein Kinase. Circ Res. 2007;100:391–8. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 14.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skårdal K, Wisløff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of RyR2 Phosphorylation at S2814 During Heart Failure Progression. Circ Res. 2012 May 24;110:1474. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Förster A, Schmitto JD, Gummert J, Schöndube FA, Hasenfuss G, Maier LS, Sossalla S. Ca2+/Calmodulin-Dependent Protein Kinase II and Protein Kinase A Differentially Regulate Sarcoplasmic Reticulum Ca2+ Leak in Human Cardiac Pathology. Circ. 2013;128:970–81. doi: 10.1161/CIRCULATIONAHA.113.001746. [DOI] [PubMed] [Google Scholar]
- 16.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca2+ Leak in Chronic Heart Failure. Circ Res. 2008;103:1466–72. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell EF, Antaramian A, Rueda A, Gomez AM, Valdivia HcH. Sorcin Inhibits Calcium Release and Modulates Excitation-Contraction Coupling in the Heart. J Biol Chem. 2003;278:34660–6. doi: 10.1074/jbc.M305931200. [DOI] [PubMed] [Google Scholar]
- 18.Most P, PLeger ST, Volkers M, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004;114:1550–63. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerrone M, Cummings S, Alansari T, Priori SG. A Clinical Approach to Inherited Arrhythmias. Circ: Cardiovasc Gene. 2012;5:581–90. doi: 10.1161/CIRCGENETICS.110.959429. [DOI] [PubMed] [Google Scholar]
- 20.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms Underlying Spontaneous and Induced Ventricular Arrhythmias in Patients With Idiopathic Dilated Cardiomyopathy. Circ. 1998;98:2404–14. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- 21.Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. CARDIOVASC RES. 2004;61:208–17. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic Reticulum Ca2+ and Heart Failure: Roles of Diastolic Leak and Ca2+ Transport. Circ Res. 2003;93:487–90. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- 23.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in Catecholaminergic Polymorphic Ventricular Tachycardia: Insights From a RyR2 R4496C Knock-In Mouse Model. Circ Res. 2006;99:292–8. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 24.Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. CARDIOVASC RES. 2013;98:286–96. doi: 10.1093/cvr/cvt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bjornstad H, Tande PM, Lathrop DA, Refsum H. Effects of temperature on cycle length dependent changes and restitution of action potential duration in guinea pig ventricular muscle. Cardiovasc Res. 1993;27(6):946–50. doi: 10.1093/cvr/27.6.946. [DOI] [PubMed] [Google Scholar]
- 27.Eisner DA, Trafford AW, Diaz ME, Overend CL, O’Neill SC. The control of Ca2+ release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc Res. 1998;38(3):589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 28.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological Effects of Ranolazine, a Novel Antianginal Agent With Antiarrhythmic Properties. Circ. 2004;110:904–10. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parikh A, Mantravadi R, Kozhevnikov D, Roche MA, Ye Y, Owen LJ, Puglisi JL, Abramson JJ, Salama G. Ranolazine stabilizes cardiac ryanodine receptors: A novel mechanism for the suppression of early afterdepolarization and torsades de pointes in long QT type 2. Heart Rhythm. 2012;9:953–60. doi: 10.1016/j.hrthm.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burashnikov A, Di Diego JM, Barajas-Martinez H, Hu D, Cordeiro JM, Moise NS, Kornreich BG, Belardinelli L, Antzelevitch C. Ranolazine Effectively Suppresses Atrial Fibrillation in the Setting of Heart Failure. Circ: Heart Failure. 2014;4:627–33. doi: 10.1161/CIRCHEARTFAILURE.114.001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burashnikov A, Di Diego JM, Sicouri S, Doss MX, Sachinidis A, Barajas-Martínez H, Hu D, Minoura Y, Sydney Moise N, Kornreich BG, Chi L, Belardinelli L, Antzelevitch C. A temporal window of vulnerability for development of atrial fibrillation with advancing heart failure. Eur J Heart Fail. 2014;16:271–80. doi: 10.1002/ejhf.28. [DOI] [PubMed] [Google Scholar]
- 32.Nieminen T, Scirica BM, Pegler JRM, Tavares C, Pagotto VP, Kanas AF, Sobrado MF3, Nearing BD, Umez-Eronini AA, Morrow DA, Belardinelli L, Verrier RL. Relation of T-Wave Alternans to Mortality and Nonsustained Ventricular Tachycardia in Patients With NonST-Segment Elevation Acute Coronary Syndrome from the MERLIN-TIMI 36 Trial of Ranolazine Versus Placebo. Am J Cardiol. 2014;114:17–23. doi: 10.1016/j.amjcard.2014.03.056. [DOI] [PubMed] [Google Scholar]
- 33.Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, Molhoek P, Verheugt FW, Gersh BJ, McCabe CH, Braunwald E. Effect of Ranolazine, an Antianginal Agent With Novel Electrophysiological Properties, on the Incidence of Arrhythmias in Patients With Non ST-Segment Elevation Acute Coronary Syndrome: Results From the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) Randomized Controlled Trial. Circ. 2007;116:1647–52. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 34.Gong M, Zhang Z, Fragakis N, Korantzopoulos P, Letsas KP, Li G, Yan GX, Liu T. Role of ranolazine in the prevention and treatment of atrial fibrillation: A meta-analysis of randomized clinical trials. Heart Rhythm. 2017;14(1):3–11. doi: 10.1016/j.hrthm.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Sicouri S, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Heart Rhythm. 2013;10:1036–43. doi: 10.1016/j.hrthm.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, Karpinski S, Li CH, Hu L, Li XJ, Crumb W, Wu L, Koltun D, Zablocki J, Yao L, Dhalla AK, Rajamani S, Shryock JC. A Novel, Potent, and Selective Inhibitor of Cardiac Late Sodium Current Suppresses Experimental Arrhythmias. JPET. 2013;344:23–32. doi: 10.1124/jpet.112.198887. [DOI] [PubMed] [Google Scholar]
- 37.Veeraraghavan R, Gyorke S, Radwanski PB. Neuronal sodium channels: emerging components of the nano-machinery of cardiac calcium cycling. J Physiol. 2017;595:3823–3934. doi: 10.1113/JP273058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puckerin AA, Chang DD, Subramanyam P, Colecraft HM. Similar molecular determinants on Rem mediate two distinct modes of inhibition of CaV1. 2 channels Channels. 2016;10:379–94. doi: 10.1080/19336950.2016.1180489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, et al. Oxidized Ca2+/Calmodulin-Dependent Protein Kinase II Triggers Atrial Fibrillation. Circ. 2013;128:1748–57. doi: 10.1161/CIRCULATIONAHA.113.003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grueter CE, Abiria SA, Wu Y, Anderson ME, Colbran RJ. Differential Regulated Interactions of Calcium/Calmodulin-Dependent Protein Kinase II with Isoforms of Voltage-Gated Calcium Channel Subunits. Biochemistry. 2008;47:1760–7. doi: 10.1021/bi701755q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Li N, Wang TW, et al. Inhibition of CaMKII Phosphorylation of RyR2 Prevents Induction of Atrial Fibrillation in FKBP12.6 Knockout Mice. Circ Res. 2012 Feb 3;110:465–70. doi: 10.1161/CIRCRESAHA.111.253229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellicena P, Schulman H. CaMKII Inhibitors: From Research Tools to Therapeutic Agents. Front Pharm. 2014;5 [Google Scholar]
- 43.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG. Calmodulin kinase II inhibition prevents arrhythmias in RyR2R4496C mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50:214–22. doi: 10.1016/j.yjmcc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, et al. CaMKII-mediated SR Ca leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–51. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson ME, Braun AP, Wu YLT, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca++ /Calmodulin-dependent protein kinase, Decreases early afterdepolarizations in rabbit heart. JPET. 1998;287:996–1006. [PubMed] [Google Scholar]
- 46.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-Hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a Calcium/Calmodulin-Dependent Protein Kinase II Inhibitor, Is a Direct Extracellular Blocker of Voltage-Gated Potassium Channels. JPET. 2006;317:292–9. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- 47.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. PNAS. 2009;106:2342–7. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang BH, Mukherji S, Soderling TR. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience. 2001;102:767–77. doi: 10.1016/s0306-4522(00)00520-0. [DOI] [PubMed] [Google Scholar]
- 49.Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual Mechanism of a Natural CaMKII Inhibitor. Mol Biol Cell. 2007;18:5024–33. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zahradnikova A, Palade P. Procaine effects on single sarcoplasmic reticulum Ca2+ release channels. Biophys J. 1993;64(4):991–1003. doi: 10.1016/S0006-3495(93)81465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Overend CL, Eisner DA, O’Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. J Physiol. 1997;502:471–9. doi: 10.1111/j.1469-7793.1997.471bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li N, Wang Q, Sibrian-Vazquez M, Klipp RC, Reynolds JO, Word TA1, Scott L, Jr, Salama G, Strongin RM, Abramson JJ, Wehrens XHT. Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int J Cardiol. 2017;227:668–73. doi: 10.1016/j.ijcard.2016.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007 Jan 5;100(1):105–11. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 54.Hachida M, Kihara S, Nonoyama M, Koyanagi H. Protective effect of JTV519, a new 1,4-benzothiazepine derivative, on prolonged myocardial preservation. J Card Surg. 1999;14(3):187–93. doi: 10.1111/j.1540-8191.1999.tb00977.x. [DOI] [PubMed] [Google Scholar]
- 55.Wehrens XH, Lehnart SE, Reiken S, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor stabilizing protein calstabin2. Science. 2004;304:292–6. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 56.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA Phosphorylation Dissociates FKBP12.6 from the Calium Release Channel (Ryanodine Receptor): Defective Regulation in Failing Hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 57.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA. 2005;102(27):9607–12. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N, Smith GL. K201 modulates excitation-contraction coupling and spontaneous Ca2+ release in normal adult rabbit ventricular cardiomyocytes. CARDIOVASc RES. 2007;76:236–46. doi: 10.1016/j.cardiores.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 59.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118(6):2230–45. [Google Scholar]
- 60.Sasaki K, Makiyama T, Yoshida Y, Wuriyanghai Y, Kamakura T, Nishiuchi S, Hayano M, Harita T, Yamamoto Y, Kohjitani H, Hirose S, Chen J, et al. Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a Potential Therapeutic Agent for Catecholaminergic Polymorphic Ventricular Tachycardia. PloS ONE. 2016;11:e0164795. doi: 10.1371/journal.pone.0164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maxwell JT, Domeier TL, Blatter LA. Dantrolene prevents arrhythmogenic Ca release in heart failure. AmPhysiology - Heart Circ Physiol. 2012;302:H953. doi: 10.1152/ajpheart.00936.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uchinoumi H, Yang Y, Oda T, Ca Li N, Alsina KM, Puglisi JL, Chen-Izu Y, Cornea RL, Wehrens XHT, Bers DM. MKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72. doi: 10.1016/j.yjmcc.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartmann N, Pabel S, Herting J, Schatter F, Renner A, Gummert J, Schotola H, Danner BC, Maier LS, Frey N, Hasenfuss G, Fischer TH, Sossalla S. Antiarrhythmic effects of dantrolene in human diseased cardiomyocytes. Heart Rhythm. 2017;14:412–9. doi: 10.1016/j.hrthm.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 64.Keller M, Kao JP, Egger M, Niggli E. Calcium waves driven by “sensitization” wave-fronts. Cardiovasc Res. 2007;74(1):39–45. doi: 10.1016/j.cardiores.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 65.Smith GL, O’Neill SC. A comparision of the effects of ATP and tetracaine on spontaneous Ca2+ release from rat permeabilised cardiac myocytes. J Physiol. 20011:534. 37–47. doi: 10.1111/j.1469-7793.2001.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luyanenko V, Gyorke I, Gyorke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pfluegers Arch. 1996;432:1047–54. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- 67.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15(4):380. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kannankeril PJ, Moore JP, Cerrone M. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: A randomized clinical trial. JAMA Cardiology. 2017;2:759–66. doi: 10.1001/jamacardio.2017.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BCl. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiology. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bannister ML, Thomas NL, Sikkel MB, Mukherjee S, Maxwell C, MacLeod KT, George CH, Williams AJ. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res. 2015;116(8):1324–35. doi: 10.1161/CIRCRESAHA.116.305347. [DOI] [PubMed] [Google Scholar]
- 71.Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Triple mode of action of flecainide in catecholaminergic polymorphic ventricular tachycardia: reply. Cardiovasc Res. 2013;98(2):327–8. doi: 10.1093/cvr/cvt068. [DOI] [PubMed] [Google Scholar]
- 72.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, Tian X, Jones PP, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17(8):1003–9. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan Z, Xiao Z, Wei J, Zhang J, Zhou Q, Smith CD, Nani A, Wu G, Song LS, Back TG, Fill M, Chen SR. Nebivolol suppresses cardiac ryanodine receptor-mediated spontaneous Ca2+ release and catecholaminergic polymorphic ventricular tachycardia. Biochem J. 2016;473(22):4159–72. doi: 10.1042/BCJ20160620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohajda Z, Farkas-Morvay N, Jost N, Nagy N, Geramipour A, Horváth A, Varga RS, Hornyik T, Corici C, Acsai K, Horváth B, Prorok J, et al. The Effect of a Novel Highly Selective Inhibitor of the Sodium/Calcium Exchanger (NCX) on Cardiac Arrhythmias in In Vitro and In Vivo Experiments. PloS ONE. 2016;11(11):e0166041. doi: 10.1371/journal.pone.0166041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuller W, Tulloch LB, Shattock MJ, Calaghan SC, Howie J, Wypijewski KJ. Regulation of the cardiac sodium pump. Cell Mol Life Sci. 2013;70(8):1357–80. doi: 10.1007/s00018-012-1134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ter Keurs HEDJ, Boyden PA. Calcium and Arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu N, Denegri M, Dun W, et al. Abnormal propagation of calcium waves and ultrastructural remodeling in recessive catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2013;113(2):142–52. doi: 10.1161/CIRCRESAHA.113.301783. [DOI] [PubMed] [Google Scholar]