

Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an arrhythmogenic disorder caused by mutations in the cardiac ryanodine receptor (RyR2) that increase diastolic Ca2+ leak from the sarcoplasmic reticulum (SR). Calmodulin (CaM) dissociation from RyR2 has been associated with diastolic Ca2+ leak in heart failure.
Objective
Determine if tetracaine-derivative, EL20, inhibits abnormal Ca2+ release from RyR2 in a CPVT model and the underlying mechanism of inhibition.
Methods
Spontaneous Ca2+ sparks in cardiomyocytes and inducible VT were assessed in a CPVT mouse model, RyR2-R176Q/+ (R176Q) in the presence of EL20 or vehicle. Single-channel studies using sheep cardiac SR or purified RyR2 reconstituted into proteoliposomes with and without exogenous CaM were used to assess mechanisms of inhibition.
Results
EL20 potently inhibits abnormal Ca2+ release in R176Q myocytes (IC50=35.4nM) and diminishes arrhythmia in R176Q mice. EL20 inhibition of single-channel activity of purified RyR2 occurs in a similar range as seen in R176Q myocytes (IC50=8.2nM). Inhibition of single-channel activity for cardiac SR or purified RyR2 supplemented with 100nM or 1µM CaM shows a 200 to 1000-fold reduction in potency.
Conclusion
This work provides a potential therapeutic mechanism for the development of antiarrhythmic compounds that inhibit leaky RyR2 resulting from CaM dissociation often associated with failing hearts. Our data also suggest that CaM dissociation may contribute to the pathogenesis of arrhythmias in the CPVT-linked R176Q mutation.
Keywords: Calmodulin, Cardiac Ryanodine Receptor, CPVT, Antiarrhythmic, Reconstituted Single-Channel, Calcium Sparks
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited disorder which results from mutations in proteins associated with the cardiac ryanodine receptor (RyR2).1–3 The predominant form (CPVT1) arises from point mutations in RyR2 itself.4–6 These mutations result in a gain-of-function, where adrenergic stimulation leads to a heightened activity of RyR2 and diastolic Ca2+ leak. This leak can trigger an inward Na+ current resulting in delayed afterdepolarizations and triggered arrhythmias.7 Left untreated ventricular tachycardia (VT) can degenerate into ventricular fibrillation and sudden cardiac death. The mechanism that underlies this leak of Ca2+ remains controversial. The Ca2+-binding protein, calmodulin (CaM), is a known RyR2-stabilizer, and reduced binding has been associated with an RyR2 diastolic leak in heart failure8–11 and CPVT.12 This suggests that altered binding of CaM to RyR2 may potentiate diastolic release of Ca2+.
CPVT is a difficult disease to manage with limitations in the efficacy of current treatments. β-blockers are commonly used to prevent the onset of symptoms but have limited long-term effectiveness. Studies have indicated that 19% of patients prescribed β-blockers experienced arrhythmogenic events after 4 years and that number doubled at 8 years.13 The antiarrhythmic drug, flecainide, has been shown to reduce onset of CPVT symptoms,14 but also has proarrhythmic properties in patients with structural heart disease.15 Flecainide is also a direct inhibitor of RyR2,16 but whether this mechanism accounts for its antiarrhythmic properties is controversial.17 ICDs are often used in conjunction with medication or in situations where medication is not effective. ICDs are invasive, often not an option for children, and subject to failure.
As the source of Ca2+ leak, direct targeting of RyR2 offers a potential therapeutic approach to CPVT. Here we investigate the therapeutic effects of a novel compound, 2-(diethylamino)ethyl 4-(butylamino)-2-methoxybenzoate (EL20), which diminishes abnormal Ca2+ handling and arrhythmogenesis in an established CPVT mouse model (RyR2-R176Q/+) by inhibiting RyR2 at low nanomolar concentrations. We demonstrate that normal excitation-contraction (EC) coupling remains intact in cellular and whole-animal studies. Single-channel measurements carried out in the presence and absence of added CaM indicate that CaM binding to RyR2 blocks inhibition by EL20. From these results, we conclude that channels depleted of CaM become leaky, allowing EL20 to block the leak. Normal RyR2 channels and cardiac function is not altered in the presence of EL20 due to CaM association.
A preliminary report of the work presented here has appeared in abstract form.18
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich. Lipids for the bilayer studies were purchased from Avanti Polar Lipids. Calmodulin was obtained from Calbiochem.
Compound synthesis
The novel tetracaine derivative, 2-(diethylamino)ethyl 4-(butylamino)-2-methoxybenzoate (EL20) was synthesized as described in the supplemental information.19 Two hydrolysis products of EL20 were also used in this study: 4-(butylamino)-2-methoxybenzoic acid20 (2) was synthesized as described in the supplemental information and 2-(diethylamino)-ethanol (3) was purchased from Sigma-Aldrich.
Animals
This study conformed to the Guide for the Care and Use of Laboratory Animals. Mice harboring RyR2-R176Q/+ mutation were used in cellular and whole-animal studies as described previously.21 Sheep hearts were provided by Oregon Health & Science University. Animal protocols were approved by the Institutional Animal Care and Use Committees of Baylor College of Medicine and Portland State University.
Ca2+ sparks measurement
Ca2+ sparks were imaged in ventricular myocytes from R176Q mice pre-loaded with Fluo-4-AM (2µM) using a LSM510 confocal microscope (Zeiss) as previously described.22 Cells were treated with 100nM isoproterenol to induce an increase in spontaneous Ca2+ spark activity. Pre-loaded myocytes were exposed to vehicle or test compound for 2hrs. Ca2+ spark frequency (CaSF) was calculated using SparkMaster.
Electrophysiology study
Intracardiac electrophysiology studies were performed in R176Q and WT littermates as previously described.21,23,24 Simultaneous recordings of 6-lead ECG as well as intracardiac electrograms were simultaneously recorded in adult (2–3-month-old) mice. After induction of anesthesia (1.5% isoflurane in 95% O2), baseline and intracardiac electrogram recordings were made. Inducibility of VT was determined using extrastimuli pacing after injections of epinephrine (2mg/kg) and caffeine (120mg/kg). These measurements were carried out both with and without intraperitoneal (i.p) injection of test compound.
SR preparation and RyR2 purification
SR vesicles were prepared from sheep hearts following a method previously described.25 RyR2 was purified from SR vesicles as previously described.26 Briefly, SR vesicles were solubilized in CHAPS/PC mixture for 1hr. followed by centrifugation on a discontinuous sucrose gradient. Fractions containing RyR2 were dialyzed and reconstituted into proteoliposomes.
Single-channel recordings
Bilayers were formed using a 5:3:2 DOPE:DOPS:DOPC mix of lipids (Avanti-Cat. #790304) on an aperture separating cis and trans chambers. Purified RyR2 reconstituted into proteoliposomes or cardiac SR vesicles (0.02mg/ml final) were added to the cis chamber (250mM KCl, 15mM HEPES, 100µM CaCl2, pH 7.4 w/KOH). Upon fusion, trans chamber (25mM KCl, 15mM HEPES, pH 7.4) was equilibrated with cis using concentrated KCl. Cis-Ca2+ was buffered to 25µM using EGTA, calculated using the chelation program bound and determined.27 To ensure proper orientation of the purified RyR2, changes in open channel probability were measured following well-defined changes in the cis-Ca2+ concentration. Channels that did not respond to changes in the cis-Ca2+ (3 out of 37) were not used in analysis.
Following addition of test compound to the cis chamber, cis and trans chambers were stirred for 30 seconds and channel activity was recorded for ≥1.5min. Data was filtered at 1-kHz and held at +36mV using a Warner Instruments BC-535 amplifier. Analysis of single-channel activity was performed using pClamp (Axon Instruments).
3H-Ryanodine binding
Equilibrium ryanodine binding to cardiac SR vesicles (0.5mg/ml) was carried out over 3hrs. in buffer: 50µM CaCl2, 250mM KCl, 2nM 3H-ryanodine, 13nM ryanodine, 20mM HEPES, 37°C, pH 7.4, ± test compound. Non-specific binding was assayed in the presence of 200nM ryanodine and 4mM EGTA and subtracted from measurements. Binding was quenched through rapid filtration (Brandel 48-well harvester). Ca2+-dependent binding was carried out over a range of free Ca2+ concentrations controlled by EGTA. Rates were measured in an identical manner with the following modifications: unlabeled ryanodine was not present, non-specific binding measurements were not made, and experiments were quenched at specified time points.
Statistics
Data are presented as mean ± SEM. Statistical significance was determined using paired or unpaired Student’s T-test where appropriate. Data was determined significant at P<0.05.
Results
Compound synthesis
Tetracaine analog EL20 (Fig. 1A) was synthesized via a two-step synthesis route (Supplemental). The first step involves the reductive amination of the commercially available 4-amino-2-methoxybenzoic acid with benzaldehyde using sodium acetoxy borohydride as the reducing agent, to produce 4-(butylamino)-2-methoxybenzoic acid, 2 (Fig. 1B) in 57% yield. Subsequent esterification of compound 2 with 2-(diethylamino)ethan-1-ol, 3 (Fig. 1C) using EDCI/DMAP as the coupling agents affords compound EL20 in 58% yield.
Figure 1.
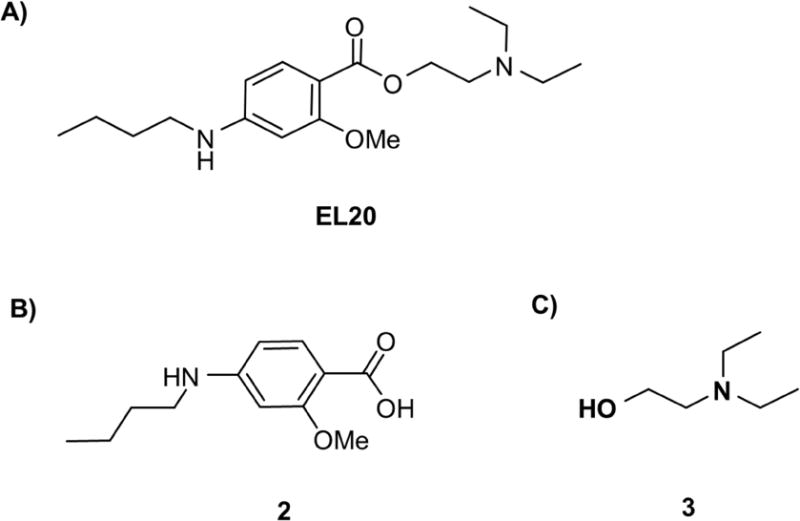
Chemical structures of compounds of study. (A) 2-(diethylamino)ethyl 4-(butylamino)-2-methoxybenzoate (EL20), a novel tetracaine-derivative. (B) 4-(butylamino)-2-methoxybenzoic acid (2) (C) 2-(diethylamino)-ethanol (3).
EL20 reduced Ca2+ spark frequency in R176Q myocytes
To determine whether EL20 selectively alters abnormal Ca2+ release at the cellular level of R176Q channels more so than WT channels, Ca2+ sparks were assessed (Fig. 2A). Isoproterenol (100nM) was used to mimic adrenergic stimulation leading to abnormal Ca2+ release as previously described.21 The results show that EL20 (500nM) did not significantly alter the CaSF in WT myocytes. In contrast, EL20 reduced CaSF by ~70% compared to vehicle (P<0.01). R176Q spark parameters given in Supplemental Table 1. The results show that EL20 normalizes excessive SR Ca2+ release through defective RyR2 channels in myocytes from R176Q mice to normal levels seen in WT mice.
Figure 2.
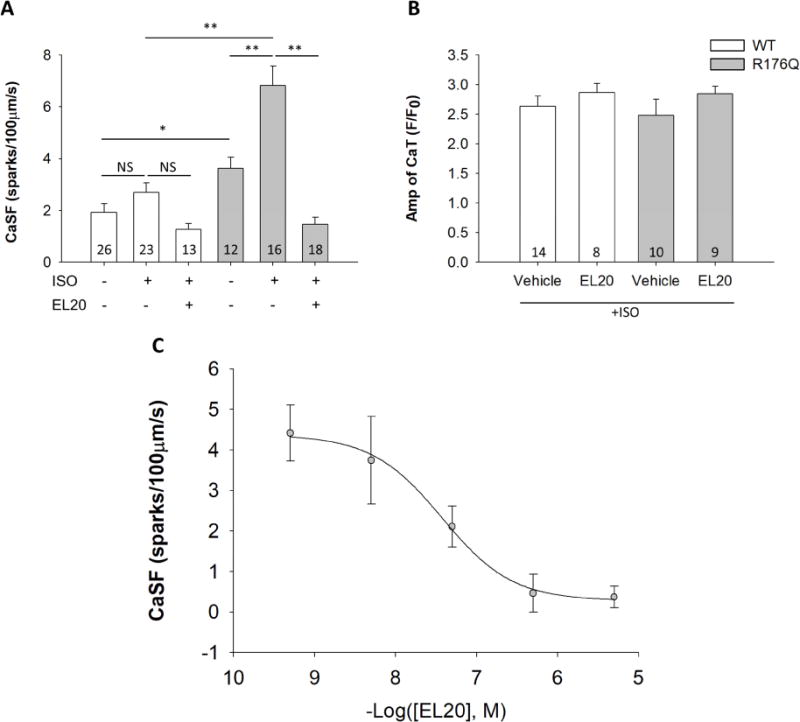
EL20 inhibition of abnormal Ca2+ release events at the cellular level. (A) Summary data showing EL20 (500nM) reduced CaSF in R176Q (grey bars) ventricular myocytes compared to vehicle (and little effect on WT (white bars) myocytes). (B) Summary data showing that EL20 (500nM) did not alter the Ca2+ transient amplitude in both WT and R176Q ventricular myocytes. (C) Dose-response relationship and IC50 determination of EL20 using CaSF inhibition in R176Q myocytes (n=6–10 cells per group, 3 mice). Results summarized in Table 1. Data presented as mean ± SEM (*P<0.05, **P<0.01).
Absence of detrimental side effects of EL20 on systolic SR Ca2+ handling
It is important to consider if the experimental compound, EL20, might alter systolic SR Ca2+ transients. A reduction of Ca2+ transients could potentially lead to unwanted side effects, consequently leading to abnormal cardiac contractility. To address this, we measured the Ca2+ transient amplitude induced by 1-Hz field stimulation (Fig. 2B). EL20 at the high dose of 500nM did not alter the Ca2+ transient amplitude in ventricular myocytes of WT mice (2.7 ± 0.2 F/Fo) compared to vehicle controls (2.6 ± 0.2). In addition, EL20 did not alter the Ca2+ transient amplitude in R176Q myocytes (2.8 ± 0.3) compared to the vehicle-treated cells (2.4 ± 0.1) at the cellular level, indicating no detrimental side effects at the whole-organism level.
Dose-response of Ca2+ sparks inhibition by EL20
Dose-dependent inhibition of CaSF by EL20 (0.5nM-5µM) was assessed to determine the concentrations of EL20 necessary to inhibit abnormal Ca2+ release (Fig. 2C). Sigmoidal fitting of the dose-response was used to determine the half-maximal inhibition concentration (IC50), taken here as the measure of potency. EL20 yielded an IC50 = 35.4nM, the hydrolysis product, compound 2 inhibited spark activity with an IC50 = 200nM (not shown).
EL20 reduces incidence of VT in R176Q mice
Whether inhibition of spontaneous Ca2+ release in the R176Q myocytes translates to diminish ventricular arrhythmias at the whole-animal level, EL20 was further characterized in R176Q mice. β-adrenergic stimulation was mimicked by administering epinephrine (2mg/kg) and caffeine (120mg/kg) by i.p. injection, and induction of VT by extra-stimuli protocol was assessed as previously described.21
Following pacing protocols, none of the vehicle-treated WT mice developed VT (0 of 9) (Fig. 3A and E), whereas, 73% of the vehicle-treated R176Q mice developed bidirectional and/or polymorphic VT (8 of 11, P <0.01 compared to WT) (Fig. 3B, C and E). In contrast, R176Q mice treated with EL20 (2.5µg/kg, i.p.) 15 minutes prior to pacing showed a significant reduction of inducible VT (1 of 7, P<0.05 compared to vehicle) (Fig. 3D and E). Importantly, EL20 did not alter any ECG parameters (RR, HR, PR, QRS, QTc) after injection in the R176Q mice (Supplemental Table 2), suggesting that EL20 does not affect cardiac conduction properties.
Figure 3.
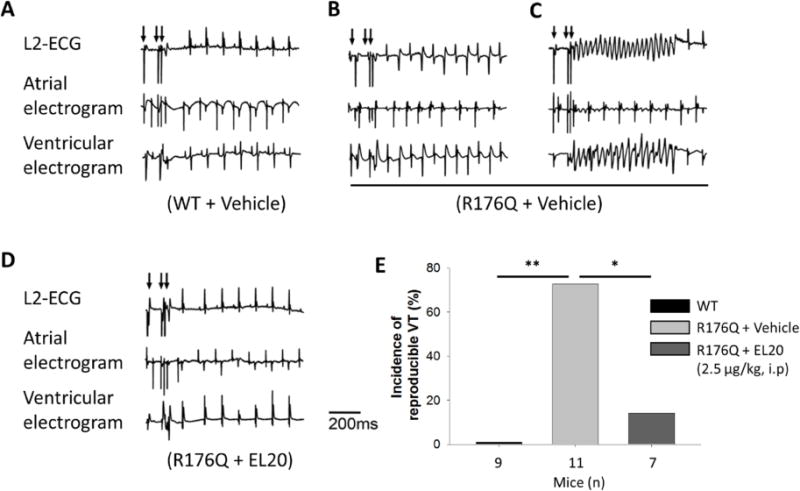
EL20 suppresses the induction of VT in R176Q mice. Representative simultaneous recordings of surface ECG and intracardiac electrograms after S1–S2 extra stimuli (indicated by arrows) revealed: (A) sinus rhythm in WT mice treated with vehicle, (B) bidirectional and (C) sustained VT in R176Q mice treated with vehicle, (D) sinus rhythm in R176Q mice treated with EL20 (2.5µg/kg). (E) Bar graph summarizing incidence of VT in each set. (*P<0.05, **P<0.01).
EL20 inhibits purified RyR2 in planar lipid bilayers
To understand the mechanism that leads to inhibition by EL20, single-channel activity was examined in a planar lipid bilayer. RyR2 from sheep was purified and then reconstituted into proteoliposomes to remove endogenous proteins to identify direct interactions between the channel and EL20. We also considered the effects of the two metabolites of EL20 which may be present following hydrolysis in vivo. To best assess the inhibitory effects, recordings were carried out under conditions of high single-channel activity (25µM Ca2+).
In the absence of test compound the average open probability (Po) = 0.84 ± 0.02 (n=34). Representative recordings of single-channel activity show that EL20 and compound 2 decreased activity with increasing concentration (Fig. 4A and B). Figure 4C shows the normalized Po for the accumulated data plotted as a function of compound concentration on a logarithmic scale. The IC50 (determined from four-parameter logistic fits) for EL20 and compound 2 were between 8 and 10nM (Table 1). At concentrations above 100nM, channel activity was completely inhibited. Compound 3 did not inhibit channel activity at all concentrations tested (10nM-100µM). These results indicate that EL20 directly inhibited RyR2 at nanomolar concentrations. Furthermore, compound 2 showed near-identical inhibition to EL20 indicating that inhibition is retained following the hydrolysis of EL20.
Figure 4.
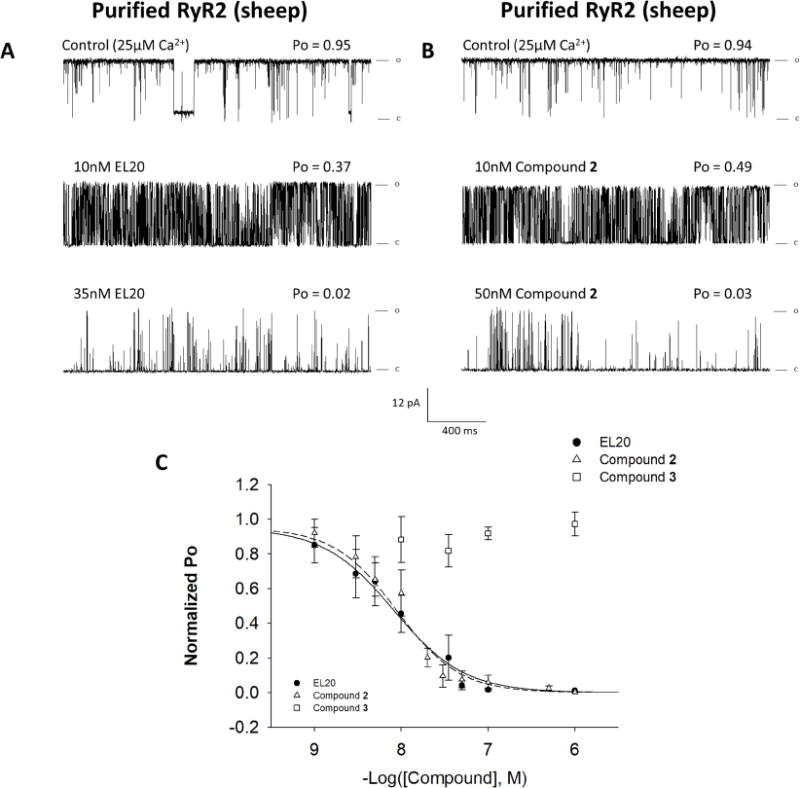
Single-channel activity of purified RyR2 in the presence of EL20 and metabolites. Recordings were made at +36mV in symmetric 250mM KCl, 20mM HEPES, pH 7.4. (A and B) 2sec traces of single-channel activity at increasing concentrations of EL20 and compound 2, respectively. Channel openings are indicated by deflections upward. (C) Dose-response of EL20 (black circles), compound 2 (open triangles), and compound 3 (open squares). Results summarized in Table 1. Data is normalized to each channel’s control Po and then grouped and plotted as the mean ± SEM.
Table 1.
Dose-Response Summaries
| Assay | Condition | −Log([IC50], M) | IC50 (nM) | n |
|---|---|---|---|---|
| Single-Channel | ||||
| Cardiac SR | ||||
| EL20 | 5.09±0.44 | 8,110 | 4–8 | |
| Purified RyR2 | ||||
| EL20 + 1μM CaM | 5.45±0.38 | 3,560 | 4–9 | |
| EL20 + 100nM CaM | 5.73±0.29 | 1,860 | 4–8 | |
| EL20 | 8.09±0.14 | 8.21 | 4–13 | |
| Compound 2 | 8.02±0.10 | 9.45 | 3–12 | |
| Compound 3 | NA | NA | 3–5 | |
|
| ||||
| CaSF | ||||
| R176Q | ||||
| EL20 | 7.43±0.86 | 35.4 | 6–10 | |
EL20 shows weakened inhibition of RyR2 from cardiac SR
We next assessed EL20 inhibition of the more native RyR2 macromolecular complex to better understand why EL20 did not alter systolic function in the R176Q cells while purified RyR2 was completely inhibited at the single-channel level. Sheep cardiac SR vesicles that retain associated proteins28 were incorporated into planar lipid bilayers and measurements of single-channel activity were carried out under identical conditions as experiments involving the purified receptor.
The mean Po in the absence of test compound (0.69 ± 0.06, n=8) was 18% lower than the mean control activity of purified RyR2 (Supplemental Fig. 1A). Representative single-channel traces (Fig. 5A) and the dose-response (Fig. 5C) indicated mild inhibition of RyR2 from SR by EL20. The potency of EL20 inhibition of non-purified RyR2 decreased by a factor of 1000 compared to identical experiments carried out with purified RyR2.
Figure 5.
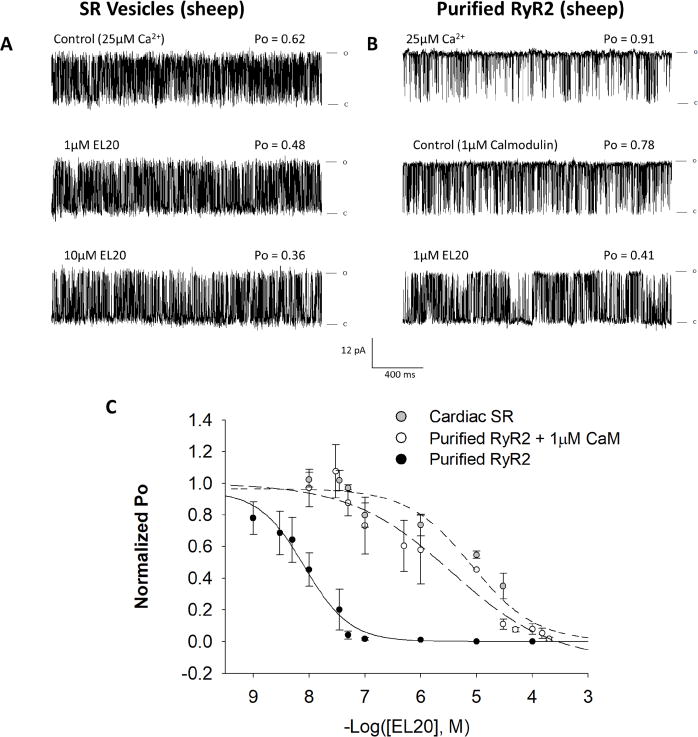
Inhibition of single-channel activity by EL20. Recordings were made at +36mV in symmetric 250mM KCl, 20mM HEPES, pH 7.4. (A) 2sec traces of single-channel activity of RyR2 from SR vesicles at increasing concentrations of EL20. (B) Purified RyR2 single-channel recording with the addition of 1µM CaM, followed by the addition of EL20. (C) Corresponding dose-response curves for EL20 inhibition of: RyR2 from SR vesicles (grey circles), purified RyR2 + 1µM CaM (open circles), and purified RyR2 without CaM (black circles) (replotted from Fig. 4C). Results are summarized in Table 1. Data is normalized to each channel’s control Po and then grouped and plotted as the mean ± SEM.
3H-Ryanodine binding to sheep cardiac SR confirmed that EL20 does not inhibit the RyR2 macromolecular complex at therapeutic concentrations. Inhibition in the rate of 3H-ryanodine binding in the presence of EL20 could only be seen at concentrations >1µM, while equilibrium binding over 3 hours required higher concentrations (>10µM) (Supplemental Fig. 2). Equilibrium binding indicated similar inhibition between compound 2 and EL20 while compound 3 had no effect (Supplemental Fig. 2B). EL20 also did not shift the Ca2+-dependence of 3H-ryanodine binding to SR at therapeutic concentrations (Supplemental Fig. 2C).
Effects of calmodulin on inhibition of purified RyR2 by EL20
To determine if the presence of CaM, whose absence from RyR2 has been linked to CPVT,12 may alter EL20’s inhibition of RyR2, we added exogenous CaM to purified RyR2 incorporated into planar lipid bilayers. The addition 100nM or 1µM CaM decreased channel Po by 19% (n=9) and 24% (n=9), respectively (Supplemental Fig. 1B), confirming the absence of endogenous CaM. Exogenous CaM did not significantly alter activity of cardiac SR under identical conditions (Supplemental Fig. 1B).
Representative single-channel traces of the purified RyR2 with exogenous 1µM CaM (Fig. 5B) and the dose-response (Fig. 5C) demonstrate inhibition by EL20 is decreased in the presence of CaM. Decreasing the concentration of CaM by a factor of 10 did not significantly alter EL20’s inhibition (Table 1). The inhibition of either cardiac SR fused to the bilayer membrane or purified RyR2 supplemented with CaM resulted in IC50 values that are 200 to 1000 times higher than what was observed in CaM-free RyR2 (Fig. 5C).
Discussion
As the source of improper Ca2+ handling, RyR2 has been suggested as a therapeutic target for treatment of CPVT and other cardiac disorders.29 Compounds designed for this purpose need to inhibit the diastolic Ca2+ leak while not affecting normal channel function.11 Recently, the Wehrens’ lab reported on a novel tetracaine-derivative, EL9, that inhibited arrhythmogenic states while not altering healthy cardiac function in an R176Q mouse model.30 Here we demonstrated the therapeutic effects of another novel tetracaine-derivative, EL20, that diminished abnormal Ca2+ handling at low nanomolar concentrations and prevented arrhythmogenesis in a CPVT model by targeted inhibition of RyR2. We further explored the mechanism of selective inhibition of RyR2-Ca2+ leak showing that EL20 only inhibited RyR2 single-channels in the absence of the stabilizing protein calmodulin.
EL20 prevents arrhythmogenic states
EL20 inhibited arrhythmogenic events in mice harboring a human CPVT mutation at both the cellular and whole-animal level. Using an R176Q mouse model, EL20 potently inhibited the diastolic Ca2+ leak in myocytes (IC50 = 35.4nM). At the whole-animal level, EL20 (2.5µg/kg) significantly reduced ventricular tachycardia in the R176Q mice compared to vehicle-treated littermates (Fig. 3).
In addition to its antiarrhythmic properties, EL20, showed no adverse effects on EC coupling. The Ca2+ transient amplitude was not altered by a high concentration of EL20 (500nM), indicating that systolic SR Ca2+ release from RyR2 was not inhibited. More importantly, ECG parameters (RR, QRS, QTc) from the R176Q mice were not affected post-treatment with EL20 (Supplemental Table 2). These results indicate that conduction property and contractility were not affected by EL20, suggesting that EL20 possesses cardiac safety for use in treating cardiac diseases.
Selective inhibition of CaM deficient channels
Experiments at the single-channel level demonstrated that EL20’s potency increased by 1000-fold in purified RyR2 compared to RyR2 from SR (IC50 = 8 nM and 8 µM, respectively). Interestingly, EL20’s inhibition of the purified channel closely resembled the concentrations necessary to diminish abnormal diastolic CaSF in the R176Q myocytes (IC50 = 35nM). Given that single-channel experiments were carried out at 25µM Ca2+, which is significantly higher than the intracellular Ca2+ concentration, it is unlikely that EL20’s mechanism of inhibition is directly Ca2+-dependent. This is further evidenced by the fact that EL20 did not alter the Ca2+-dependence of 3H-ryanodine binding to cardiac SR (Supplemental Fig. 2C). Therefore, EL20’s mechanism of inhibition appears to be dependent on some conformational change that is common to both the leaky R176Q and purified RyR2.
The open probabilities of the purified RyR2 were significantly higher than those from cardiac SR (Supplemental Fig. 1A). This was likely due to the absence of stabilizing proteins that are present in the SR,28 and may indicate that EL20 only inhibits channels absent of a stabilizing protein. Calmodulin is removed following purification of RyR2, but its endogenous effects remain in cardiac SR.33 Exogenous CaM added to the purified RyR2 reduced single-channel activity (Supplemental Fig. 1B) and reduced inhibition by EL20 (Fig. 5C), more closely resembling the characteristics observed in cardiac SR.
Whether the reduced inhibition observed with single-channels from cardiac SR is due to the presence of CaM is unclear. A recent study showing that exogenous CaM was required for dantrolene inhibition of RyR2 suggests that CaM dissociates readily from RyR2 in lipid bilayers.35 In the present study, exogenous CaM added to cardiac SR did not affect single-channel activity, suggesting the presence endogenously bound CaM. This discrepancy may be due to experimental factors such as our use of high concentrations of cis-Ca2+ that decreases CaM-RyR2 dissociation.32,33 However, we cannot rule out the possibility that other proteins in the RyR2 macromolecular complex may also alter EL20’s inhibition.
It has been proposed that CaM may play a role maintaining a closed state during diastole and terminating SR Ca2+ release.31 FRET32 and radiolabeled33 studies have indicated that CaM-RyR2 affinity in SR vesicles is weaker at diastolic Ca2+, while single-channel studies have shown the inhibitory effects to be greater at diastolic Ca2+.33,34 Therefore, loss of CaM during diastole may contribute to Ca2+ leak. Mutations in RyR2 may potentiate the dissociation of CaM. In knock-in mice with CPVT-linked mutation, RyR2-R2474S, the addition of cAMP both increased spontaneous Ca2+ sparks and reduced CaM-RyR2 affinity compared to WT myocytyes.12
To the best of our knowledge, no study has directly linked CaM dissociation to the pathogenesis of the R176Q Ca2+ leak. Our data showing that CaM alone blocks EL20’s inhibition of RyR2 suggests that leaky R176Q channels are likely CaM deficient. We propose that adrenergic stimulation of R176Q mice leads to the dissociation of CaM from RyR2 and under these circumstances, EL20 decreases the Ca2+ leak and restores normal function.
Conclusion
This study characterized the antiarrhythmic properties of a novel tetracaine-derivative, EL20, and provided a working model for the mechanism of selective inhibition of leaky RyR2 channels. Ca2+ leak via RyR2 that is potentiated by the loss of CaM can be corrected by EL20, thereby reducing the induction of ventricular tachycardia. Our data suggest that CaM dissociation may be a contributing factor in the pathogenesis of arrhythmias in the CPVT-linked, RyR2-R176Q/+ mutation. This work provides a potential therapeutic mechanism for the development of antiarrhythmic compounds that inhibit leaky RyR2 channels resulting from CaM dissociation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R42-HL114206 to J.J.A., R01-HL136389 to N.L., R01-HL089598, R01-HL091947, R01-HL117641, and R41-HL129570 to X.H.T.W.) and the American Heart Association (14SDG20080008 to N.L. and 13EIA14560061 to X.H.T.W.). Sheep hearts were graciously provided by Dr. Kent Thornburg at Oregon Health and Science University. R176Q mice were kindly provided by Dr. Susan Hamilton at Baylor College of Medicine.
Abbreviations
- CaM
calmodulin
- CaSF
Ca2+ spark frequency
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- EC
excitation-contraction
- RyR2
cardiac ryanodine receptor
- SR
sarcoplasmic reticulum
- VT
ventricular tachycardia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
Drs. Abramson, Strongin, and Wehrens are co-founders and co-owners of ELEX Biotech, a biotech company dedicated to the development of drug molecules for the treatment of heart disease.
References
- 1.Hwang HS, Nitu FR, Yang Y, et al. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124. doi: 10.1161/CIRCRESAHA.114.303391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff J-M, Da Costa A, Sebillon P, Mannens MMAM, Wilde AAM, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 3.Nyegaard M, Overgaard MT, Søndergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the Cardiac Ryanodine Receptor (RyR2) Gene in Familial Polymorphic Ventricular Tachycardia. Circulation. 2001;103:485– 490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 6.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 7.Scoote M, Williams AJ. The cardiac ryanodine receptor (calcium release channel)Emerging role in heart failure and arrhythmia pathogenesis. Cardiovasc Res. 2002;56:359–372. doi: 10.1016/s0008-6363(02)00574-6. [DOI] [PubMed] [Google Scholar]
- 8.Ono M, Yano M, Hino A, et al. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM. Cardiac Myocyte Z-Line Calmodulin Is Mainly RyR2-Bound, and Reduction Is Arrhythmogenic and Occurs in Heart FailureNovelty and Significance. Circ Res. 2014;114:295–306. doi: 10.1161/CIRCRESAHA.114.302857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J Clin Invest. 2007;117:1344–1353. doi: 10.1172/JCI29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, Chen-Izu Y, Cornea RL, Wehrens XHT, Bers DM. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72. doi: 10.1016/j.yjmcc.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X, Yano M, Uchinoumi H, et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem Biophys Res Commun. 2010;394:660–666. doi: 10.1016/j.bbrc.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Werf C, Zwinderman AH, Wilde AAM. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2012;14:175–183. doi: 10.1093/europace/eur277. [DOI] [PubMed] [Google Scholar]
- 14.van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide Therapy Reduces Exercise-Induced Ventricular Arrhythmias in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. J Am Coll Cardiol. 2011;57:2244–2254. doi: 10.1016/j.jacc.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7:76–85. doi: 10.4330/wjc.v7.i2.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehra D, Imtiaz MS, van Helden DF, Knollmann BC, Laver DR. Multiple modes of ryanodine receptor 2 inhibition by flecainide. Mol Pharmacol. 2014;86:696–706. doi: 10.1124/mol.114.094623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bannister ML, Thomas NL, Sikkel MB, Mukherjee S, Maxwell C, MacLeod KT, George CH, Williams AJ. The Mechanism of Flecainide Action in CPVT Does Not Involve a Direct Effect on RyR2. Circ Res. 2015;116:1324–1335. doi: 10.1161/CIRCRESAHA.116.305347. [DOI] [PubMed] [Google Scholar]
- 18.Klipp RC, Li N, Wang Q, Sibrian-Vazquez M, Strongin RM, Wehrens XHT, Abramson JJ. Novel Compounds Inhibit Calmodulin Deficient RyR2 Activity and Arrhythmias in a CPVT Mouse Model. Biophys J. 2016;(110):97a. [Google Scholar]
- 19.Strongin RM, Abramson JJ, Sibrian-Vazquez M, Wehrens X. Compounds for treatment of cardiac arrhythmias. 2016 Available from: https://www.google.com/patents/US20160311760.
- 20.Clinton RO, Salvador UJ, Laskowski SC, Wilson M. Derivatives of 4-Amino-2-hydroxybenzoic Acid. II. J Am Chem Soc. 1952;74:592–598. [Google Scholar]
- 21.Kannankeril PJ, Mitchell BM, Goonasekera SA, et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XHT. Inhibition of CaMKII Phosphorylation of RyR2 Prevents Induction of Atrial Fibrillation in FKBP12.6 Knockout Mice. Circ Res. 2012;110:465–470. doi: 10.1161/CIRCRESAHA.111.253229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional Ventricular Tachycardia and Fibrillation Elicited in a Knock-In Mouse Model Carrier of a Mutation in the Cardiac Ryanodine Receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Wehrens XHT. Programmed electrical stimulation in mice. J Vis Exp JoVE. 2010 doi: 10.3791/1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem. 1987;262:3065–3073. [PubMed] [Google Scholar]
- 26.Lee HB, Xu L, Meissner G. Reconstitution of the skeletal muscle ryanodine receptor-Ca2+ release channel protein complex into proteoliposomes. J Biol Chem. 1994;269:13305–13312. [PubMed] [Google Scholar]
- 27.Brooks SPJ, Storey KB. Bound and determined: A computer program for making buffers of defined ion concentrations. Anal Biochem. 1992;201:119–126. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 28.Marks AR, Marx SO, Reiken S. Regulation of Ryanodine Receptors via Macromolecular Complexes. Trends Cardiovasc Med. 2002;12:166–170. doi: 10.1016/s1050-1738(02)00156-1. [DOI] [PubMed] [Google Scholar]
- 29.Santonastasi M, Wehrens XHT. Ryanodine receptors as pharmacological targets for heart disease. Acta Pharmacol Sin. 2007;28:937–944. doi: 10.1111/j.1745-7254.2007.00582.x. [DOI] [PubMed] [Google Scholar]
- 30.Li N, Wang Q, Sibrian-Vazquez M, Klipp RC, Reynolds JO, Word TA, Scott L, Jr, Salama G, Strongin RM, Abramson JJ, Wehrens XHT. Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int J Cardiol. 2017;227:668–673. doi: 10.1016/j.ijcard.2016.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu L, Meissner G. Mechanism of Calmodulin Inhibition of Cardiac Sarcoplasmic Reticulum Ca2+ Release Channel (Ryanodine Receptor) Biophys J. 2004;86:797–804. doi: 10.1016/S0006-3495(04)74155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo T, Fruen BR, Nitu FR, Nguyen TD, Yang Y, Cornea RL, Bers DM. FRET detection of calmodulin binding to the cardiac RyR2 calcium release channel. Biophys J. 2011;101:2170–2177. doi: 10.1016/j.bpj.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin Binding and Inhibition of Cardiac Muscle Calcium Release Channel (Ryanodine Receptor) J Biol Chem. 2001;276:20144–20153. doi: 10.1074/jbc.M010771200. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi N, Xu L, Pasek DA, Evans KE, Meissner G. Molecular Basis of Calmodulin Binding to Cardiac Muscle Ca2+ Release Channel (Ryanodine Receptor) J Biol Chem. 2003;278:23480–23486. doi: 10.1074/jbc.M301125200. [DOI] [PubMed] [Google Scholar]
- 35.Oo YW, Gomez-Hurtado N, Walweel K, van Helden DF, Imtiaz MS, Knollmann BC, Laver DR. Essential Role of Calmodulin in RyR Inhibition by Dantrolene. Mol Pharmacol. 2015;88:57–63. doi: 10.1124/mol.115.097691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.