

Abstract
Rift Valley fever (RVF) is an acute, febrile zoonotic disease that is caused by the RVF virus (RVFV). RVF is mainly prevalent on the Arabian Peninsula, the African continent, and several islands in the Indian Ocean near southeast Africa. RVFV has been classified by the World Organisation for Animal Health (OIE) as a category A pathogen. To avoid biological safety concerns associated with use of the pathogen in RVFV neutralization assays, the present study investigated and established an RVFV pseudovirus-based neutralization assay. This study used the human immunodeficiency virus (HIV) lentiviral packaging system and RVFV structural proteins to successfully construct RVFV pseudoviruses. Electron microscopy observation and western blotting indicated that the size, structure, and shape of the packaged pseudoviruses were notably similar to those of HIV lentiviral vectors. Infection inhibition assay results showed that an antibody against RVFV inhibited the infective ability of the RVFV pseudoviruses, and an antibody neutralization assay for RVFV detection was then established. This study has successfully established a neutralization assay based on RVFV pseudoviruses and demonstrated that this method can be used to effectively evaluate antibody neutralization.
Keywords: Rift Valley fever virus, antibody neutralization assay, pseudovirus
Introduction
Certain viruses, such as the severe acute respiratory syndrome-associated coronavirus (SARS-CoV), Ebola virus (EBOV), and H5N1 subtype avian influenza virus, are highly infectious and highly pathogenic, presenting tremendous difficulties and dangers during research [8,13,19]. In contrast, pseudovirus technology is a very safe and effective research tool [18]. Briefly, a pseudovirus is a type of retrovirus that can integrate with the envelope glycoprotein of another type of virus to form a new virus with an exogenous viral envelope and a genome that maintains the characteristics of the retroviral genome. Compared with natural viruses, pseudoviruses integrate with the envelope protein of another virus by using an envelope protein-encoding gene within a modified nucleic acid molecule; pseudoviruses can only accomplish one infection cycle because they lose their self-replicating ability. Thus, pseudoviruses are more biologically safe than infectious viruses. Pseudoviruses also have extensive host ranges and higher transfection efficiencies, can be more easily concentrated than the original viruses, can defend against the deactivation function of serum complement, have a non-cell-cycle-dependent integration feature, and can efficiently transfect quiescent cells. Pseudovirus preparation is thus an effective, safe, and reliable method for application in the study of viruses with high pathogenicity that are difficult to culture in vitro. Because pseudoviruses are relatively safe and have the advantages of extensive host range, non-cell-cycle-dependence, and highly efficient transfection of quiescent cells, they can have particularly important roles in studies of virus-host cell interactions, neutralizing antibody titer evaluation, and gene therapy. Thus, pseudoviruses can be used as a novel, safe experimental tool.
Rift Valley fever virus (RVFV) is an RNA virus in the Phlebovirus genus of the Bunyaviridae family [10]. In particular, RVFV is a high-risk pathogen that can induce fatal encephalitis and hemorrhagic fever in humans and ruminants [5,11]. Due to its strong pathogenicity and fast dissemination, RVFV has attracted considerable worldwide attention. Currently, viral isolation, hemagglutination inhibition assays, enzyme-linked immunosorbent assays (ELISAs), agar gel immunodiffusion assays, immunofluorescence assays, radioimmunoassays, and complement fixation assays are the primary methods used to detect RVFV [16,17]. Additionally, clinical detection of anti-RVFV antibodies is primarily accomplished by using the ELISA method [16]. Commonly used coating antigens include the envelope protein (Gn) and inactivated RVFV. However, this method can only preliminarily detect antibody levels in humans, and those results do not truly reflect whether the antibodies have pathogen-neutralizing, anti-RVFV, and anti-infection functions. Therefore, the virus neutralization assay has become an important clinical method for measuring antibody activity and evaluating immune status in humans. The neutralization assay typically uses natural viruses, which can cause infections in the assay operators and pose a risk of viral spread; thus, this assay is considered very dangerous. To avoid the potential risks associated with the neutralization assay, the present study used a lentiviral packaging system and targeted RVFV structural proteins to construct RVFV pseudoviruses to replace live viruses. The pseudoviruses were then used to establish an RVFV neutralization assay method to allow effective evaluation of human antibody titers.
Materials and Methods
Bacteria, plasmids, and cells
Escherichia coli DH5α competent cells, the PUC57-Simple-M recombinant plasmid (containing the GN and GC sequences of the RVFV M gene), HEK293T cells, and the pcDNA3.1 eukaryotic expression plasmid were obtained from the Animal Virology and Special Disease Research Laboratory, Military Veterinary Research Institute, Academy of Military Medical Sciences, China.
Construction of recombinant plasmids expressing RVFV structural proteins
To enhance expression, codon optimization was performed on the G-protein gene sequence of the RVFV ZH501 strain. The cloning primers were designed based on optimized sequences, and Not I and Kpn I restriction sites were introduced at the 5′ ends of the upstream (5′-CGGGGTACCATGGCTGGTATCGCTATGAC-3′) and downstream (5′-ATTTGCGGCCGCTTAAGAGGCTTTCTTTGTGGC-3′) primers. The primers were synthesized by Sangon Biotech (China).
The pUC57-Simple-M recombinant plasmid was used as the template, and polymerase chain reaction (PCR) amplification was performed with the synthesized primers. The obtained DNA fragment was subjected to double digestion and was then ligated into a double-digested eukaryotic expression plasmid (pcDNA3.1). The recombinant plasmid was confirmed to have the correct ligation by gene sequencing performed by Beijing Genomics Institute (China). Finally, the recombinant expression plasmid was extracted for future use and named pcDNA3.1-M-rvfv.
Pseudovirus packaging
The plasmid used for transfection was extracted by using an endotoxin-free plasmid extraction reagent kit (Endo Free Plasmid DNA Mini Kit II; OMEGA, China). Twenty-four hours before transfection, 293T cells were inoculated onto 10-cm plates (Corning, USA) at a density of 4 − 5 × 106 cells/mL in 10 mL of Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) containing 10% fetal bovine serum (FBS; Thermo, USA). The cells were then cultured at 37℃ with 5% CO2. When cell growth reached 80% to 90% confluence, 5 µg of the lentiviral plasmid pLV-EGFP-C (Invitrogen, USA), 3.75 µg of the PH1 plasmid (Invitrogen), and 1.25 µg of the recombinant plasmid pcDNA3.1-M-rvfv (the PH2 plasmid was used as a positive control) were transfected into the 293T cells by using Lipofectamine 2000 transfection reagent (Invitrogen). After the cells were cultured at 37℃ with 5% CO2 for 4 to 6 h, the culture medium was replaced with 10 mL of fresh 293T culture medium. After 24 h of transfection, the culture medium was replaced with 10 mL of fresh virus culture medium.
After 48 to 72 h of transfection, the cell culture supernatant was collected, placed into a 50 mL centrifuge tube, and centrifuged at 4℃ and 5,000 × g for 10 min. The supernatant was then collected, filtered through a 0.45 µm filter, and transferred into a new 50 mL centrifuge tube. Finally, the filtrate was transferred into centrifugal filter devices and centrifuged at 4℃ and 5,000 × g for 10 min. The liquid in the bottom layer was discarded, and the filtrate further centrifuged at 4℃ and 5,000 × g for 30 min. The solution in the top layer of the filter device was the virus concentrate. The viruses were aliquoted and stored at −80℃.
Detection and analysis of the pseudoviruses
The RVFV pseudovirus packaging conditions were identified by using electron microscopy and western blotting. During the electron microscopy detection process, the harvested pseudoviruses or pseudovirus concentrates were first adsorbed to a copper grid and then stained with 1% phosphotungstic acid (Sigma, USA) for 2 to 3 min. After the residual staining solution was absorbed by using filter paper, the pseudovirus particles were observed by using an electron microscope. Additionally, the cell supernatant collected after transfection was mixed with 5× protein loading buffer (used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis; Thermo) at a 4:1 dilution and added to a 1.5 mL centrifuge tube. After the samples were mixed thoroughly and boiled for 5 min, 15 µL of each sample was used for western blotting detection and analysis. During the experimental western blotting process, membranes were blocked with a 0.5% Tween-20 (Thermo) membrane-blocking solution containing 5% bovine serum albumin for 2 h at room temperature. The membranes were next incubated with a rabbit polyclonal anti-RVFV-MP12 primary antibody (1:4,000 dilution; IBT Bioservices, USA) for 2 h at room temperature and then with an HRP-labeled goat anti-mouse IgG secondary antibody (Invitrogen) for 1 h at room temperature. The blots were developed with a chemiluminescent developing solution (Thermo), and the experimental data were stored in the LAS-4000 system (Fujifilm, Japan).
Determination of pseudovirus infectious titers
To confirm the multiplicity of infection of the pseudoviruses and to obtain a stable infection effect, we measured the RVFV pseudovirus titers. Specifically, the harvested and purified virus solution was used to determine the viral titers (although frozen virus solution could have been used). Notably, very large differences were detected in the viral titers measured using different cell lines and methods; thus, very large differences might have existed between the viral titer values and the viral titers required to infect target cells. To avoid this issue, the same virus solution-based measurement method was used to determine the viral titers in order to provide more accurate measurement of the virus packaging effects. Next, 293T cells were cultured to the logarithmic growth phase and dissociated with 0.25% trypsin (Gibco). After counting, the cells were seeded into 96-well culture plates at a density of 5 × 104 cells/well and cultured at 37℃ with 5% CO2. For virus inoculation, pseudovirus solution stored in a −80℃ freezer was thawed in an ice bath and then subjected to a series of gradient dilutions using DMEM containing 10% FBS (10−1, 10−2, 10−3, 10−4, 10−5, 10−6, 10−7, 10−8, and 10−9). Each dilution was added to three replicate wells. After the cells were cultured at 37℃ with 5% CO2 overnight, the culture medium was replaced with 100 µL of DMEM containing 10% FBS, and the cells were continuously cultured for 48 to 72 h. The cells were then observed via fluorescence microscopy, and the number of cells with positive green fluorescence in each well was calculated. The virus titer was the number of cells with positive green fluorescence multiplied by the corresponding fold dilution.
Inhibition of pseudovirus infection
A polyclonal antibody against the RVFV G-protein (1:4,000 dilution; IBT Bioservices) was diluted 20-fold and 40-fold. Healthy mouse serum was used as a negative control. The antibody and RVFV/vesicular stomatitis virus (VSV)-G pseudovirus solution (500 IU) were mixed at equal volumes, added to 96-well cell culture plates, and incubated at 37℃ for 1 h. Next, the mixture was added to 293T cells in the logarithmic growth phase at 100 µL/well (containing 5 × 104 cells) and mixed thoroughly. After the cells were cultured at 37℃ with 5% CO2 for 72 h, the number of cells with positive fluorescence in each well was determined via fluorescence microscopy.
Establishment of the pseudovirus neutralization assay
Serum samples collected from RVFV G-protein-immunized mice were used to generate 2-fold serial dilutions. Next, 50 µL of diluted antibody and 50 µL of RVFV pseudovirus/VSV pseudovirus (used as the negative control) (500 IU) were mixed at equal volumes and incubated at 37℃ for 1 h. Subsequently, 293T cells (5 × 104 cells) were added to each well and cultured at 37℃ with 5% CO2 for 72 h. Finally, the experimental results were collected by observation under a fluorescence microscope. The antibody neutralization activity was calculated according to the formula:
Antibody neutralization activity (%) = (number of cells with positive green fluorescence in the control group − number of cells with positive green fluorescence in the experimental group)/number of cells with positive green fluorescence in the control group × 100.
Results
RVFV target gene amplification
The RVFV structural M gene was obtained by applying the PCR amplification method. The amplified fragment length was 3218 bp. As shown in Fig. 1, the pUC57-Simple-M recombinant plasmid was used as the amplification template, and the results were assessed by carrying out agarose gel electrophoresis. One band was present at approximately 3200 bp, which was consistent with the size (3218 bp) of the expected target fragment. These results indicated that the correct target gene band was amplified by PCR.
Fig. 1. The M gene polymerase chain reaction (PCR) product. M, marker; Lane 1, the PCR products of the Rift Valley fever virus structural M gene.
Identification of the RVFV recombinant plasmid
Gene sequencing analysis and restriction endonuclease digestion were used to validate the construction of the recombinant plasmid pcDNA3.1-M-rvfv. The pcDNA3.1-M-rvfv plasmid was digested with the Not I and Kpn I enzymes and evaluated by performing agarose gel electrophoresis (Fig. 2). The results showed two nucleic acid bands, at approximately 3200 bp and 5500 bp, which were consistent with the target gene fragment (3218 bp) and the pcDNA3.1 empty plasmid (5428 bp), respectively. This result verified the correct insertion of the target fragment. The gene fragment obtained in this experiment and the target gene sequence obtained by using gene sequencing were 100% homologous, indicating that the pcDNA3.1-M-rvfv recombinant plasmid was correctly constructed.
Fig. 2. Enzyme digestion of pcDNA3.1-M-rvfv. M, marker; Lane 1, polymerase chain reaction products of the restriction endonuclease digestion of recombinant plasmid pcDNA3.1-M-rvfv.
Pseudovirus acquisition and identification
The RVFV pseudovirus packaging conditions were observed via electron microscopy. In the virus solution harvested after 48 to 72 h of co-transfection of mixed lentiviral plasmids and 293T cells, spherical particles with an envelope of 80 to 120 nm diameter were observed by using an electron microscope. Both particle morphology and structure were consistent with previously reported descriptions of human immunodeficiency virus (HIV) lentiviral vectors [1,12]. The results indicated that the pcDNA3.1-M-rvfv recombinant plasmid obtained in this study could be packaged in 293T cells to produce pseudoviruses (Fig. 3).
Fig. 3. Detection of pseudoviruses by transmission electron microscopy. Arrows indicate RVFV pseudoviruses. RVFV, Rift Valley fever virus. Scale bar = 200 nm.
To confirm that the acquired viral particles were RVFV pseudoviruses, western blotting was performed to determine whether the pseudovirus envelope protein was composed of RVFV structural proteins. The supernatants of cells transfected with pcDNA3.1-M-rvfv were examined by using a rabbit polyclonal anti-RVFV-MP12 antibody (Fig. 4). The western blotting results showed two specific bands, with molecular weights of approximately 65 kDa and 56 kDa, consistent with the sizes of the RVFV G1 and G2 proteins, respectively. The negative control group did not show specific bands. These results indicated that the target protein was correctly expressed. Moreover, the western blotting detection results both indicated that the obtained RVFV pseudoviruses contained the RVFV G1 and G2 proteins and confirmed that pseudoviruses were successfully established.
Fig. 4. Identification of pseudoviruses by using western blotting.
Determination of pseudovirus infectious titers
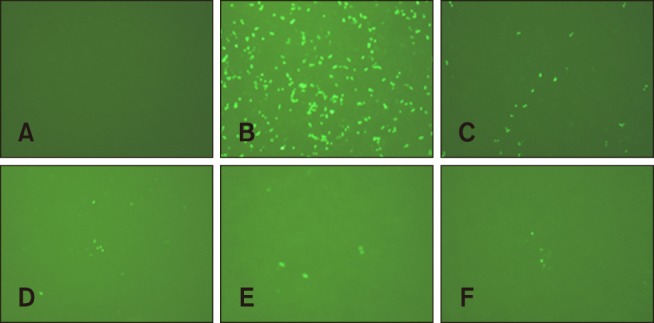
The obtained RVFV pseudoviruses were serially diluted to determine the infectious titers. The detection results showed that the RVFV pseudovirus titer was 1 × 106 IU/mL. Fig. 5 shows positive fluorescence expression in 293T cells incubated with different fold dilutions of the RVFV pseudoviruses.
Fig. 5. Determination of Rift Valley fever virus pseudovirus infectious titers. (A) Control. (B–F) Expression of fluorescence in 293T cells infected with different fold dilutions of pseudoviruses. (B) 10−1, (C) 10−2, (D) 10−3, (E) 10−4, (F) 10−5.
Pseudovirus infection inhibition assay
A virus infection inhibition assay was performed to evaluate whether the infective ability of the pseudoviruses could be inhibited by an anti-RVFV antibody. As shown in Fig. 6, after incubation with a polyclonal antibody against the RVFV protein, the RVFV pseudoviruses could not effectively infect 293T cells, whereas the VSV pseudoviruses and the polyclonal antibody against the RVFV protein could not effectively defend against virus infection in 293T cells. Therefore, the antibody inhibition assay indicated that the infective ability of the RVFV pseudoviruses could be inhibited by specific antibodies. These results further indicated that the obtained RVFV pseudoviruses could bind to specific antibodies and could be used as the antigen in a neutralization assay; thus, the pseudoviruses could be effectively applied in an antibody neutralization assay.
Fig. 6. Rift Valley fever virus (RVFV) pseudovirus infection inhibition assay. (A) Expression of fluorescence in 293T cells after incubation with RVFV pseudoviruses and negative serum reaction mixture. (B) Expression of fluorescence in 293T cells after incubation with the vesicular stomatitis virus (VSV) pseudovirus and anti-RVFV polyclonal antibody reaction mixture. (C) Expression of fluorescence in 293T cells after incubation with RVFV pseudoviruses and anti-RVFV polyclonal antibody reaction mixture.
Establishment of the pseudovirus neutralization assay
RVFV pseudoviruses were reacted with serial dilutions of positive mouse serum to measure the neutralizing antibody titer. As shown in Table 1, positive serum samples from mice immunized with RVFV proteins all had 100% neutralization activity in the 2- to 16-fold dilution range (indicating inhibition of RVFV pseudovirus infectious activity). Serum that was diluted 32-fold neutralized 70% of the virus infection, while 64-fold serum dilution neutralized approximately 45% of the virus infection, and 128-fold serum dilution neutralized approximately 15% of the virus infection. However, the RVFV-positive mouse serum did not inhibit VSV pseudovirus infection. These results indicated that the RVFV pseudovirus neutralization assay could be used to effectively measure antibody neutralization; thus, this assay could be used to evaluate the anti-infection immune status in humans.
Table 1. Detection of the neutralization activity of mouse serum against RVFV pseudoviruses.

RVFV, Rift Valley fever virus; VSV, vesicular stomatitis virus; −, negative results (no neutralization activity).
Discussion
RVFV is a high-risk pathogen that can be transmitted through insect bites or direct contact. Therefore, its physical manipulation poses enormous safety concerns for veterinary workers and laboratory personnel. This issue severely limits studies on the genomic structure and pathogenic mechanism of RVFV. Under normal circumstances, vaccines can induce high neutralizing antibody titers in humans, so neutralizing antibody titers are an important indicator of the immune effect of vaccines. However, traditional RVFV virus neutralization assays involve tedious work, including culture of the virus, which can be performed only in biological safety level 3 laboratories, thus limiting the application of neutralization assays in the monitoring, detection, and diagnosis of RVFV infection and in the evaluation of the effects of vaccines on RVFV infection. Therefore, safety issues associated with RVFV is an important problem that has drawn considerable attention, and such issues need to be addressed in studies targeting high-risk viruses such as RVFV [1,2].
Recently, pseudovirus technology has become a very effective tool in scientific research [6,7,14] and has demonstrated several unique application advantages. For example, in experimental studies, pseudoviruses can be used to replace high-risk viruses with greater pathogenicity and infectivity, with RVFV being a typical example. Compared with natural viruses, pseudovirus particles have similar patterns of cell infection, and pseudoviruses can also effectively simulate the early cell infection process of natural viruses. Additionally, free viral DNA or reporter genes (i.e., EGFP and LacZ) can be artificially introduced into pseudoviruses, and this approach is conducive to rapid and timely detection and analysis in a clinic. Furthermore, pseudoviruses exhibit defective replication (ensuring safe manipulation), avoid the risk of infection posed by high-risk viruses, and eliminate the risk of serious safety concerns such as release from the laboratory. Therefore, pseudoviruses can be used in studies of the mechanism underlying viral infection of host cells. More importantly, pseudoviruses can be used to replace the natural viruses used in traditional virus neutralization assays. Currently, neutralization assays incorporating pseudoviruses are becoming a new, effective route for the detection of neutralizing antibodies. The many types of currently established pseudoviruses available both domestically and internationally include Middle East respiratory syndrome coronavirus (MERS-CoV), EBOV, hepatitis C virus, and SARS-CoV [4,12,20]. Some pseudoviruses have been successfully applied in drug screening, neutralizing antibody activity detection, and the evaluation of vaccine effects [3,9].
Currently, pseudovirus packaging systems are primarily developed based on HIV lentiviral vectors and the murine leukemia virus [1,9,15], which have the advantages of broad suitable host ranges and high packaging efficiencies. The present study used a lentiviral packaging system and recombinant expression plasmids encoding RVFV structural proteins to construct RVFV pseudoviruses. The electron microscopy observations and western blotting results all indicated the successful construction of RVFV pseudoviruses. Additionally, the infective activity of the obtained RVFV pseudoviruses was inhibited by specific antibodies, indicating that the pseudoviruses could be used to detect RVFV-neutralizing antibody titers. Moreover, the RVFV pseudovirus neutralization assay established in this study effectively evaluated antibody titers in positive mouse serum. The pseudovirus packaging experiment showed that the passage time and status of the 293T cells and the transfection reagents, plasmid purity, gene features of the structural proteins, and efficiency of the eukaryotic expression plasmids all greatly influenced the pseudovirus packaging efficiency and affected the titer levels and stability of the packaged pseudoviruses. Therefore, the pseudovirus neutralization assay established in the present study can be used for clinical monitoring and diagnosis of RVFV infection and for assessment of the effects of RVFV vaccines. Furthermore, the pseudovirus can be used to replace the natural virus in infection mechanism studies. However, the packaging conditions should be optimized to increase the lentiviral packaging efficiency and infection titers.
Acknowledgments
This study was supported by the National Key Technology R&D Program – Monitoring and Control Technology Research on Cross-Border Spread of Foreign Animal Diseases by Wildlife Animals (2013BAD12B04), and the China Postdoctoral Science Foundation – Research on Virus-Like Particle-Based Vaccine Candidate for Rift Valley Fever (2013M541089).
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Anderson GW, Jr, Slone TW, Jr, Peters CJ. The gerbil, Meriones unguiculatus, a model for Rift Valley fever viral encephalitis. Arch Virol. 1988;102:187–196. doi: 10.1007/BF01310824. [DOI] [PubMed] [Google Scholar]
- 2.Ashrafi GH, Piuko K, Burden F, Yuan Z, Gault EA, Müller M, Trawford A, Reid SW, Nasir L, Campo MS. Vaccination of sarcoid-bearing donkeys with chimeric virus-like particles of bovine papillomavirus type 1. J Gen Virol. 2008;89:148–157. doi: 10.1099/vir.0.83267-0. [DOI] [PubMed] [Google Scholar]
- 3.Bartosch B, Bukh J, Meunier JC, Granier C, Engle RE, Blackwelder WC, Emerson SU, Cosset FL, Purcell RH. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci U S A. 2003;100:14199–14204. doi: 10.1073/pnas.2335981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boshra H, Lorenzo G, Busquets N, Brun A. Rift valley fever: recent insights into pathogenesis and prevention. J Virol. 2011;85:6098–6105. doi: 10.1128/JVI.02641-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown BD, Sitia G, Annoni A, Hauben E, Sergi LS, Zingale A, Roncarolo MG, Guidotti LG, Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- 7.Giroglou T, Cinatl J, Jr, Rabenau H, Drosten C, Schwalbe H, Doerr HW, von Laer D. Retroviral vectors pseudotyped with severe acute respiratory syndrome coronavirus S protein. J Virol. 2004;78:9007–9015. doi: 10.1128/JVI.78.17.9007-9015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hilgenfeld R, Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann H, Hattermann K, Marzi A, Gramberg T, Geier M, Krumbiegel M, Kuate S, Uberla K, Niedrig M, Pöhlmann S. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikegami T. Molecular biology and genetic diversity of Rift Valley fever virus. Antiviral Res. 2012;95:293–310. doi: 10.1016/j.antiviral.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikegami T, Makino S. The pathogenesis of Rift Valley fever. Viruses. 2011;3:493–519. doi: 10.3390/v3050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YB, Lee MK, Han DP, Cho MW. Development of a safe and rapid neutralization assay using murine leukemia virus pseudotyped with HIV type 1 envelope glycoprotein lacking the cytoplasmic domain. AIDS Res Hum Retroviruses. 2001;17:1715–1724. doi: 10.1089/08892220152741414. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Bu Z, Chen H. Avian influenza vaccines against H5N1 ‘bird flu’. Trends Biotechnol. 2014;32:147–156. doi: 10.1016/j.tibtech.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nie Y, Wang G, Shi X, Zhang H, Qiu Y, He Z, Wang W, Lian G, Yin X, Du L, Ren L, Wang J, He X, Li T, Deng H, Ding M. Neutralizing antibodies in patients with severe acute respiratory syndrome-associated coronavirus infection. J Infect Dis. 2004;190:1119–1126. doi: 10.1086/423286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niklasson B, Peters CJ, Grandien M, Wood O. Detection of human immunoglobulins G and M antibodies to Rift Valley fever virus by enzyme-linked immunosorbent assay. J Clin Microbiol. 1984;19:225–229. doi: 10.1128/jcm.19.2.225-229.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paweska JT, Burt FJ, Anthony F, Smith SJ, Grobbelaar AA, Croft JE, Ksiazek TG, Swanepoel R. IgG-sandwich and IgM-capture enzyme-linked immunosorbent assay for the detection of antibody to Rift Valley fever virus in domestic ruminants. J Virol Methods. 2003;113:103–112. doi: 10.1016/s0166-0934(03)00228-3. [DOI] [PubMed] [Google Scholar]
- 18.Qiu C, Huang Y, Zhang A, Tian D, Wan Y, Zhang X, Zhang W, Zhang Z, Yuan Z, Hu Y, Zhang X, Xu J. Safe pseudovirus-based assay for neutralization antibodies against influenza A(H7N9) virus. Emerg Infect Dis. 2013;19:1685–1687. doi: 10.3201/eid1910.130728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivera A, Messaoudi I. Molecular mechanisms of Ebola pathogenesis. J Leukoc Biol. 2016;100:889–904. doi: 10.1189/jlb.4RI0316-099RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]