

ABSTRACT
Complete replication of the genome is an essential prerequisite for normal cell division, but a variety of factors can block the replisome, triggering replication stress and potentially causing mutation or cell death. The cellular response to replication stress involves recruitment of proteins to stabilize the replication fork and transmit a stress signal to pause the cell cycle and allow fork restart. We find that the ubiquitously expressed DNA damage response factor 53BP1 is required for the normal response to replication stress. Using primary, ex vivo B cells, we showed that a population of 53BP1−/− cells in early S phase is hypersensitive to short-term exposure to three different agents that induce replication stress. 53BP1 localizes to a subset of replication forks following induced replication stress, and an absence of 53BP1 leads to defective ATR-Chk1-p53 signaling and caspase 3-mediated cell death. Nascent replicated DNA additionally undergoes degradation in 53BP1−/− cells. These results show that 53BP1 plays an important role in protecting replication forks during the cellular response to replication stress, in addition to the previously characterized role of 53BP1 in DNA double-strand break repair.
KEYWORDS: 53BP1, replication, cell cycle, cancer biology
INTRODUCTION
Maintenance of genomic integrity relies on effective cellular responses to DNA damage. Although DNA damage can be a consequence of exogenous mutagens, such as ionizing radiation, a significant amount of damage also arises as a result of endogenous cellular processes, such as normal oxidative metabolism. One key cellular process that can contribute to DNA damage is DNA replication (1). During a typical S phase, cells encounter challenges to replication, such as extended repetitive genomic regions or exogenous replication poisons, which can cause replication forks to stall. Such obstacles to normal replication represent replication stress (2). Replication stress triggers an S-phase cell cycle checkpoint, during which late replication origins do not fire, and specific cellular responses stabilize the fork and prevent collapse and formation of DNA double-strand breaks (DSBs).
During the cellular replication stress response, a number of changes take place at the replication fork. The minichromosome maintenance (MCM) helicase uncouples from the replicative DNA polymerase, and a region of single-stranded DNA is formed, coated with replication protein A (RPA) (3). The fork protection complex, a heterodimer of Timeless and Tipin, binds to RPA at single-stranded DNA, helping to stabilize the fork and prevent dissociation of the replisome components (4). PCNA, which normally ensures processivity of the replication fork, becomes ubiquitylated and is displaced (5, 6). A large number of other proteins, including damage signaling molecules and repair factors, become enriched at stalled forks (7). In some cases, replication forks reverse, by unwinding of newly synthesized DNA from its homologous template followed by annealing to form a fourth regressed “arm” (8, 9). Loading of RAD51 at reversed forks helps protect newly synthesized DNA tracts from exonucleolytic digestion (10, 11). Many of these responses to replication stress are orchestrated by the kinase ATR (ataxia telangiectasia related) (12). ATR kinase is recruited to RPA at single-stranded DNA regions at stalled replication forks, along with its binding partner, ATRIP. ATR activity becomes active upon association with either TopBP1 or ETAA1, whereupon it phosphorylates multiple targets, including replisome components and the variant histone H2AX (13). Phosphorylated H2AX-Ser139 (γ-H2AX) helps recruit RAD51, which protects the stalled fork and assists with fork restart (14). Another key target of ATR activity is the checkpoint kinase Chk1. Activation of Chk1 is responsible for mediating a substantial subset of the downstream effects of ATR, including fork stabilization and enforcement of the S-phase checkpoint (15). The importance of ATR and Chk1 for normal replication is demonstrated by the high levels of chromosome instability and reduced cell viability observed when either of these factors is mutated or inhibited (16–19). Consequently, anything that contributes to ATR-Chk1 signaling is likely to have relevance for protecting the cell against replication stress.
The DNA damage response factor 53BP1 is a nuclear protein that has been implicated in the response to replication stress. 53BP1 was originally identified through a yeast two-hybrid screen as a binding partner of p53 (20). Studies on knockout mice subsequently suggested that 53BP1 acts during DNA double-strand break repair (21, 22). We found that deletion of 53BP1 rescues embryonic lethality and genomic instability in BRCA1-deficient mice (23, 24). This finding demonstrated that 53BP1 is a critically important regulator of the DNA damage response when BRCA1 is absent or mutated but does not fully explain the normal cellular function of 53BP1. 53BP1 regulates at least two separate cellular processes, specifically, double-strand break resection and class switch recombination at the immunoglobulin heavy chain locus in B lymphocytes. These processes are mediated through the formation of complexes involving 53BP1 and its downstream effectors, RIF1 and PTIP (25, 26). Several lines of evidence indicate that 53BP1 may also act in controlling the response to replication stress. Large nuclear bodies staining positively for 53BP1 appear in G1-phase cells, and the number of these nuclear bodies increases after treatment to induce replication stress (27, 28). 53BP1 colocalizes with p53 at sites of hydroxyurea (HU)-induced replication fork stalling (29) and is essential for normal p53 signaling (30).
We therefore set out to test whether 53BP1 impacts cell survival after replication stress. We found that B cells isolated from the spleens of 53BP1−/− mice show consistently reduced survival following HU treatment. A population of 53BP1−/− cells in early S phase showed persistent stress signaling after replication stress, and hyperactivation of caspase 3 was evident. These markers of cellular stress correlated with deficient activation of Chk1 and p53 and increased degradation of nascent DNA tracts at replication forks. 53BP1 was therefore revealed as a key regulator of the response to replication stress in mammalian cells, through its ability to activate the ATR-Chk1 signaling pathway at stalled forks.
RESULTS
Increased cell death following replication stress in 53BP1−/− B cells.
To test if 53BP1 is required for the normal cellular response to replication stress, we cultured primary B lymphocytes isolated from the spleens of wild-type (WT) and 53BP1−/− mice in vitro for 24 h and then added hydroxyurea (HU) for 3 h to induce replication fork stalling. This duration of HU exposure was selected because it causes replication fork stalling, but fork collapse and the widespread appearance of double-strand breaks take place only after HU treatments of 12 h or more or with inactivation of ATR (9, 31). We measured cell viability 18 h and 24 h after removal of HU (Fig. 1A and B). WT cells showed a small decrease in viability following HU treatment, but 53BP1−/− cells showed a significantly greater decline in viability. We also measured the viability of WT and 53BP1−/− B cells following short-term exposure to the DNA polymerase inhibitor aphidicolin or the replication chain terminator gemcitabine (Fig. 1C to E). In each case, 53BP1−/− cells showed increased death relative to that of the WT cells, consistent with a role for 53BP1 in protecting cells from the effects of replication stress.
FIG 1.

53BP1 is required for survival of B lymphocytes following transient replication stress. (A) Flow cytometry analysis of splenic B cells cultured 24 h in vitro and either not treated (NT) or treated with 4 mM hydroxyurea (HU) for 3 h. Cell death was assayed 24 h after removal of HU by quantifying the percentage of cells staining for propidium iodide (PI). Figures in gated regions indicate percentage of the cell population that remained viable. FSC, forward scatter of analyzed cells. (B) Quantification of data from panel A. The graph shows percentages of WT and 53BP1−/− cells that became inviable 18 h or 24 h after HU treatment (n = 3). Error bars show SDs. P values were calculated with Student's t test. (C) Flow cytometry analysis of B cells cultured for panel A and then either not treated or treated with 40 μM aphidicolin (APH) for 2 h. PI staining shows cells that became inviable measured 18 h post-APH treatment. Figures in gated regions indicate percentages of the cell populations that remained viable. (D) Flow cytometry analysis of B cells cultured as for panel A and then either not treated or treated with 250 nM gemcitabine (GEM) for 2 h. PI staining shows cells that became inviable measured 18 h post-GEM treatment. Figures in gated regions indicate percentages of the cell populations that remained viable. (E) Quantification of data from panels C and D. The graph shows percentages of WT and 53BP1−/− cells that became inviable 18 h after APH or GEM treatment (n = 5). Error bars show SDs. P values were calculated with Student's t test. (F) Colony assay showing survival of mouse embryonic fibroblasts (MEFs) after HU treatment. Cells used were 53BP1−/− MEFs stably transduced with a 53BP1ΔBRCT construct or GFP vector only. Colony numbers were normalized to the untreated sample. The chart shows means from 3 experiments. Error bars show SDs. (G) Colony assay showing survival of MEFs stably transduced with either shGFP or sh53BP1 shRNA constructs. Colony numbers were normalized to the untreated sample. The chart shows means from 2 experiments. Error bars show SDs.
To test if 53BP1 deficiency also causes increased cell death following replication stress in immortalized cell lines, we performed clonogenic colony formation assays to measure cell growth following hydroxyurea treatment. First we introduced constructs containing either a 53BP1 cDNA (53BP1ΔBRCT) (32) or a green fluorescent protein (GFP)-only vector into 53BP1−/− mouse embryonic fibroblasts (MEFs) (Fig. 1F). MEFs complemented with the 53BP1 expression construct grew at a rate equivalent to that of the control vector, indicating that the presence of 53BP1 does not have a strong effect on growth after replication stress in immortalized cell lines. As a second test, we knocked down 53BP1 in WT MEFs using short hairpin RNA (shRNA) (Fig. 1G). Cells expressing sh53BP1 showed growth similar to that of cells expressing a control shGFP, again suggesting that the impact of 53BP1 on cell growth after replication stress is relevant only in primary B cell populations and not in MEFs.
53BP1−/− cells show a defect in S-phase progression following replication stress.
To investigate why 53BP1−/− B cells are hypersensitive to replication stress, we looked for an increase in apoptosis induced by hydroxyurea treatment. 53BP1−/− cells showed a slight increase in subdiploid DNA content, corresponding to treatment-induced cell death or apoptosis, immediately after HU treatment (Fig. 2A and B). This difference is not sustained at 6 h posttreatment, however, and is too small to account for the difference in cell viability we had previously observed (Fig. 1). Measurements of caspase activity in the hours following HU treatment showed no difference between WT and 53BP1−/− B cells (Fig. 2C and D), suggesting that differences in cell viability must arise by some process other than immediate apoptosis induced by HU treatment.
FIG 2.

Defect in S-phase progression and persistent γ-H2AX induction in 53BP1−/− cells after replication stress. (A) Cell cycle analysis of B cells following HU treatment. Cells were either not treated or treated with HU at 2 mM for 2 h and then fixed with methanol. Propidium iodide was used in this case to measure DNA content in WT and 53BP1−/− cells either immediately after treatment or 6 h post-HU treatment. Figures show proportions of cells with sub-G1 DNA content. (B) Quantification of data from panel A, showing mean proportions of sub-G1 cells after HU treatment (n = 3). Error bars show SDs. P values were calculated with Student's t test. (C) Flow cytometry analysis of caspase 3/7 activity in B cells treated with HU (4 mM for 3 h) and then allowed to recover for the indicated periods. (D) Quantification of data from panel C. The graph shows means ± SDs. (E) Flow cytometry analysis of cell cycle in WT and 53BP1−/− splenic B cells following treatment with HU. Cells were cultured in vitro for 48 h, pulsed with ethynyl deoxyuridine (EdU) for 1 h, and then exposed to 2 mM HU for 2 h. Following fixation, PI staining was performed to measure DNA content and S-phase cells were revealed by EdU detection by click chemistry. Gated regions show early- and late-S-phase populations 0, 2, and 6 h after removal of HU. (F) Quantification of data from panel E (n = 4). Error bars show SDs. P values were calculated with Student's t test. (G) Flow cytometry analysis of γ-H2AX after HU treatment of WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then either not treated or treated with 2 mM HU for 2 h. Following recovery from HU treatment for up to 6 h, cells were fixed and stained with PI to reveal DNA content and with anti-γ-H2AX antibody. Gated regions show early- and late-S-phase populations. (H) Quantification of data from panel G. The percentages of cells in early S phase showing γ-H2AX staining 0 and 6 h post-HU treatment or without treatment are shown (n = 3). Error bars show SDs. P values were calculated with Student's t test.
As replication stress impacts S-phase cells, we tested how hydroxyurea treatment affected the progression of S-phase cells from WT and 53BP1−/− mice. Activated B cells were pulsed with the nucleoside analog 5-ethynyl-2-deoxyuridine (EdU) to allow detection of S-phase cells by flow cytometry (Fig. 2E and F). Although WT and 53BP1−/− cells initially showed equivalent cell cycle profiles, 53BP1−/− cells showed an increased proportion of cells in early S phase 6 h after treatment with hydroxyurea. This result suggests that 53BP1−/− cells have a deficiency in S-phase progression after replication stress. To further characterize this deficiency, we measured levels of γ-H2AX in B cells following treatment with HU (Fig. 2G and H). Phosphorylation of H2AX to form γ-H2AX is a characteristic chromatin response during replication stress (13). Both WT and 53BP1−/− cells showed a strong initial induction of γ-H2AX after HU treatment. In WT cells, the γ-H2AX signal mostly disappeared from early-S-phase cells 6 h after HU treatment, but the stress signal persisted in 53BP1−/− cells, consistent with a failure to recover from the effects of replication stress.
Defective ATR kinase signaling leads to caspase 3-mediated apoptosis in 53BP1−/− cells after replication stress.
53BP1 contributes to DNA damage signaling at DNA double-strand breaks by supporting ATM-mediated phosphorylation of Chk2, which triggers the DNA damage response, cell cycle checkpoint activation, and potentially apoptosis (22, 33, 34). We hypothesized that 53BP1 may also contribute to damage signaling under conditions of replication stress, which proceeds through ATR-mediated phosphorylation of Chk1 (12). We therefore measured levels of phosphorylated Chk1 in WT and 53BP1−/− cells after HU treatment. As expected, HU treatment strongly induced Chk1 phosphorylation in WT cells, dependent on ATR activity (Fig. 3A). Chk1 phosphorylation was substantially reduced in 53BP1−/− cells after HU treatment, indicating that 53BP1 is essential for normal signaling of replication fork stalling. RPA2-Ser33 is also a target for ATR activity, so we also measured levels of phosphorylated RPA-Ser33 in WT and 53BP1−/− cells following HU treatment (Fig. 3B). Although RPA-Ser33 phosphorylation is marginally lower in 53BP1−/− cells than in WT cells following HU treatment, the difference was very slight compared to what we observed with Chk1-pSer345 phosphorylation. These results indicate that 53BP1 modulates the activity of ATR on a subset of its potential targets.
FIG 3.

Western blot analysis of replication stress signaling pathways in 53BP1−/− cells (A) Western blot showing levels of Chk1-pSer345 and total Chk1 (pan-Chk1) in WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then treated as indicated with HU (4 mM for 3 h) either alone or with 3 μM VE821 (ATR inhibitor) or 20 μM Nu7026 (DNA-PK inhibitor). Protein lysates were prepared immediately after HU treatment. (B) Western blot showing levels of RPA32-pS33 and total RPA32 (pan-RPA) in WT and 53BP1−/− splenic B cells following HU treatment. Cell lysates were prepared as for panel A. (C) Western blot showing levels of p53 in WT and 53BP1−/− splenic B cells following HU treatment. Cell lysates were prepared as for panel A. (D) Western blot showing levels of p53-pSer15 in WT and 53BP1−/− splenic B cells following HU treatment. Cell lysates were prepared as for panel A. (E) Western blot showing levels of cleaved component of activated caspase 3 in WT and 53BP1−/− splenic B cells following HU treatment. Cell lysates were prepared as for panel A.
The tumor suppressor p53 is a major regulator of cell survival and apoptosis following stress (35, 36). HU treatment caused stabilization and phosphorylation of p53 in WT B cells (Fig. 3C and D). As with Chk1 activation, p53 activation was deficient in 53BP1−/− cells relative to that in WT cells after HU treatment. Consistent with the increased death observed in 53BP1−/− cells after HU treatment, we additionally observed heightened levels of the activated (cleaved) form of caspase 3 in 53BP1−/− cells after replication stress (Fig. 3E). Similar activation of caspase 3 has previously been shown to correlate with death in cells treated with Chk1 inhibitors during replication stress (37).
DNA damage can be signaled through the kinases ATM and DNA-dependent protein kinase (DNA-PK) in addition to ATR (38), so we measured the level of phosphorylation of substrates of these kinases to see if they may contribute to differences in survival of WT and 53BP1−/− cells following HU treatment. We found that the presence of 53BP1 does not affect the rate of phosphorylation of the ATM substrate Kap1-Ser824 following HU treatment (Fig. 4A). 53BP1 has previously been shown to modulate ATM signaling in response to double-strand breaks (22). This phenotype does not appear to be relevant with the short-term hydroxyurea treatment that we used, which does not induce measurable double-strand breakage (Fig. 4B and C) (39). We found, however, that phosphorylation of RPA-Ser4/8, a target of DNA-PK, was reduced in 53BP1−/− cells relative to that in the WT after HU treatment, suggesting that 53BP1 may promote activation of two stress signaling pathways based on ATR and DNA-PK (Fig. 4D and E).
FIG 4.

53BP1 mediates DNA-PK signaling but not ATM signaling following replication stress. (A) Western blot showing levels of Kap1-pS824 and tubulin in WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then treated as indicated with HU (4 mM for 3 h) either alone or with 5 μM KU55933 (ATM inhibitor [ATMi]). Protein lysates were prepared immediately after HU treatment. (B) Neutral comet assay to measure DNA double-strand breakage in B cells treated with HU (4 mM for 3 h) or camptothecin (CPT; 1 μM for 1 h). The scale bars represent 100 μm. (C) Quantification of data from panel B. Dots represent the percentage of signal in the tail for each treatment. The red lines indicate the median. At least 100 comets were scored from two experiments. P values were calculated using the Mann-Whitney U test. (D) Western blot showing levels of RPA2-pS4/8 following HU treatment (4 mM for 3 h) either alone or with 3 μM VE821 (ATR inhibitor [ATRi]) or 20 μM Nu7026 (DNA-PK inhibitor [DNAPKi]). Cell lysates were prepared as for panel A. (E) Western blot showing levels of RPA2-S4/8 in WT and 53BP1−/− splenic B cells following HU treatment either alone or with 5 μM KU55933. Cell lysates were prepared as for panel A.
53BP1 interacts with p53, and this interaction mediates p53 activation (30). p53 has additionally been reported to mediate replication fork stability under certain circumstances (35, 36). We therefore tested whether the 53BP1-p53 interaction is important for mediating recovery from replication stress. We found much less HU hypersensitivity in 53BP1−/− p53−/− double-knockout cells than in 53BP1−/− p53+/+ cells (Fig. 5A). Codeletion of 53BP1 does not impact cell survival after HU treatment in p53−/− cells, even though Chk1-Ser345 phosphorylation is lower than in p53−/− cells (Fig. 5B). We conclude that the 53BP1-p53 interaction does not increase cell survival after replication stress under the conditions we used and that p53 contributes to cell death after HU treatment in 53BP1−/− cells.
FIG 5.

p53 contributes to cell death in in 53BP1−/− cells following replication stress. (A) Percentages of WT, 53BP1−/−, p53−/−, and 53BP1−/− p53−/− cells that became inviable 18 h after HU treatment (4 mM for 3 h) (n = 4). Error bars show SDs. P values were calculated with Student's t test. N.S., not significant. (B) Western blot showing levels of Chk1-pS345 in WT, 53BP1−/−, p53−/−, and 53BP1−/− p53−/− B cells following HU treatment as for panel A. Levels of total Chk1 and tubulin are also shown.
53BP1 is recruited to a subset of replication forks under conditions of replication stress.
To test if 53BP1 is present at replication forks, we used isolation of proteins on nascent DNA (iPOND) (7, 40) to enrich for proteins at replication forks under normal conditions and after replication stress (Fig. 6A). The enrichment of cellular factors at replication forks was quantified using mass spectrometry. After hydroxyurea treatment, there was substantial enrichment of TopBP1 and ATR at replication forks, both of which have known roles in signaling the presence of replication stress (Fig. 6B; see also Tables S1 and S2 in the supplemental material) (12). PCNA was lost from the replication fork after HU treatment, consistent with unloading of PCNA after fork stalling as has previously been reported (5, 6). Significantly, 53BP1 was substantially enriched at nascent DNA following HU treatment, indicating that it is recruited to replication forks during conditions of replication stress. We next used immunofluorescence microscopy to investigate the subnuclear localization of 53BP1 after induction of replication stress. Replication forks were identified in fixed B cells by staining for RPA, which binds single-stranded regions of DNA that are exposed by the replicative helicases during S phase. To identify S-phase cells, we applied a pulse of EdU prior to fixation to allow us to label cells that were actively undergoing DNA replication. In untreated cells, we were able to identify a population of S-phase cells with nuclear foci of RPA and EdU staining (Fig. 6C). There was relatively little colocalization between nuclear foci of 53BP1 and RPA in untreated cells (median colocalization rate = 2.2%). Following treatment of the cells with hydroxyurea, a large number of small 53BP1 foci formed in RPA+ cells, as has previously been reported (29) (Fig. 6D). The extent of colocalization of these 53BP1 foci with RPA was variable, with a median colocalization rate of 7.47%. This difference was nonetheless extremely statistically significant (P < 0.00001, two-tailed Mann-Whitney U test) (Fig. 4C). Notably, the degree of colocalization of 53BP1 with RPA was very high in a subset of EdU+ RPA+ cells. As a comparison, we performed the same colocalization analysis between RPA and TopBP1, which has a well-characterized role in activation of ATR signaling at stalled replication forks. TopBP1 showed substantially greater colocalization with RPA than was the case with 53BP1 (median colocalization rate = 31.57%). These results suggest that whereas TopBP1 is a key signaling component at stalled replication forks, 53BP1 is recruited to a subset of stalled forks.
FIG 6.

Localization of 53BP1 to replication forks after replication stress. (A) Scheme of iPOND experiment to measure enrichment of 53BP1 after replication stress. Cells pulsed with 10 μM EdU for 10 min were either harvested to obtain nontreated samples or further pulsed with 2 mM HU for 2 h prior to performing click chemistry to biotinylate DNA regions containing EdU. (B) Results of iPOND analysis of proteins enriched at sites of DNA replication following HU treatment (2 mM for 2 h). Log2 ratios of peptide tags found in HU-treated versus nontreated samples of 293T cells are shown (n = 2). Error bars show SDs. (C) Confocal microscopy images of RPA, 53BP1, and EdU staining in fixed B cell nuclei without treatment. The merged image shows nuclear counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) in blue, RPA in red, and 53BP1 in green. The scale bars represent 4 μm. (D) Confocal microscopy images of RPA, 53BP1, and EdU staining in fixed B cell nuclei after treatment with 4 mM HU for 3 h. The merged image shows nuclear counterstaining with DAPI in blue, RPA in red, and 53BP1 in green. The scale bars represent 4 μm. (E) Quantification of images as shown in panels A and B. Colocalization is shown between RPA and 53BP1 in untreated and HU-treated cells and between RPA and TopBP1 in cells treated with HU. Only EdU+ cells were scored. Each dot represents one cell (n = 295 for RPA-53BP1 colocalization measurements in untreated cells, n = 213 for RPA-53BP1 colocalization in HU-treated cells, and n = 156 for RPA-TopBP1 colocalization in HU-treated cells). Red bars indicate medians. The P value was calculated by the two-tailed Mann-Whitney U test. (F) Analysis of iPOND data from WT and 53BP1−/− B cells after HU treatment (4 mM HU for 3 h). Total MS reads for each protein were corrected using the no-click control and normalized to reads for histone H4 in each sample. The graph shows the ratio of reads in the 53BP1−/− B cells divided by reads in the WT sample (n = 4). Means ± SDs are shown. *, P value of <0.05, consistent with statistical significance, calculated using Student's t test.
Using primary B cells from WT and 53BP1−/− mice, we next repeated our iPOND experiments to test if 53BP1 affects recruitment of stress signaling or repair factors to replication forks after HU treatment. We hypothesized that components of the ATR-Chk1 signaling apparatus would be reduced at the replication fork in 53BP1−/− cells relative to the case with WT cells after HU treatment, accounting for the observed reduction in Chk1 activation (Fig. 3A). Surprisingly, the levels of ATR at stalled forks were not different between B cells from WT and 53BP1−/− mice (Fig. 6F). Proteins involved in activation of ATR, including TopBP1, ATRIP, and Timeless, were also present at equivalent levels in WT and 53BP1−/− cells. These results suggest that 53BP1 does not affect the assembly or activation of ATR at stalled forks but may potentiate its ability to phosphorylate downstream targets such as Chk1. The Fanconi anemia protein FANCI was among the small number of proteins that showed a significant increase at stalled forks in 53BP1−/− cells relative to the value in WT cells. Both FANCI and FANCD2 showed increased abundance at stalled forks in each of four iPOND experiments, but FANCI was the only one that rose to the level of statistical significance. Notably, FANCI has been implicated in the response to replication stress (41).
53BP1 deficiency compromises the stability of nascent DNA at replication forks under stress conditions.
To further investigate the mechanism by which 53BP1 might regulate cell viability after replication stress, we used the DNA fiber assay to measure the stability of newly replicated DNA in WT and 53BP1−/− cells (Fig. 7A to D). 53BP1−/− cells showed a significant decrease in stability of the nascent replication tract following HU treatment. Despite this defect in fork protection in 53BP1−/− cells, replication forks appeared to restart normally in cells that lacked 53BP1 (Fig. 7E and F). This result indicates that 53BP1 is necessary to maintain newly replicated DNA after fork stalling and suggests that cell death under conditions of replication stress in 53BP1−/− cells arises at least in part as a consequence of deprotection of replication forks. Defects in fork protection have additionally been reported for cells deficient in members of the Fanconi anemia pathway, including BRCA2, or in cells lacking BRCA1, BOD1L, WRNIP, or Abro1 (39, 42–46). The nucleases Mre11 and Exo1 have been proposed to mediate degradation of nascent replication tracts in several of these genotypes. We therefore tested if these nucleases were relevant for fork degradation in 53BP1−/− cells. Mirin, an inihibitor of Mre11 nuclease activity, produced a highly statistically significant rescue of fork degradation in 53BP1−/− cells, supporting the idea that Mre11 targets newly replicated DNA in the absence of 53BP1 (Fig. 8A). Conversely, we did not find a difference in fork protection in 53BP1−/− Exo1−/− double-knockout B cells relative to that in 53BP1−/− cells, indicating that Exo1 is of lesser importance for fork degradation under these conditions (Fig. 8B).
FIG 7.

Replication fork shortening in the absence of 53BP1 after HU treatment. (A) Analysis of nascent DNA tract length in WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then pulsed with IdU, CldU, and HU as shown. Chromatin fibers were prepared and stained to reveal tracts containing IdU as green and CldU as red. (B) Quantification of data from panel A. IdU and CldU tract lengths were measured; ratios of CldU and IdU lengths are shown. Each point represents one fork as measured from 3 separate experiments. The red bars indicate the mean. Error bars represent SDs. P values were calculated using Student's t test. (C) DNA fiber analysis of replication fork restart in WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then pulsed with CldU, HU, and IdU as shown. Chromatin fibers were prepared and stained to reveal tracts containing CldU as red and tracts containing IdU as green. (D) Measurement of initial (CldU+) tract length from protocol as shown in panel C. Each point represents one fork as measured from 3 separate experiments. The red bars indicate the mean. Error bars represent SDs. P values were calculated using Student's t test. (E) Analysis of forks from chromatin fiber protocol as shown in panel C. Forks staining with both CldU+ and IdU+ regions were scored as “elongation” events. Tracts staining for CldU with no IdU staining were scored as “stalled.” Tracts staining for IdU only were scored as “new origin.” The graph shows the mean percentage of forks in each category. More than 2,000 forks were analyzed from 3 separate experiments for each genotype. Error bars represent SDs. (F) Analysis of forks from chromatin fiber protocol as shown in panel C, but with the IdU pulse reduced to 20 min.
FIG 8.
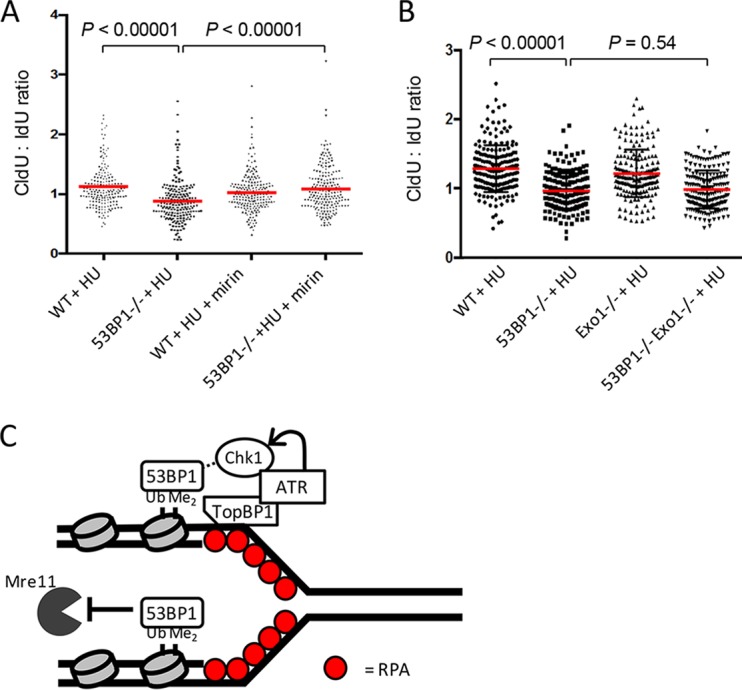
Replication fork shortening in 53BP1−/− cells after HU treatment is dependent on Mre11. (A) Analysis of nascent DNA tract length in WT and 53BP1−/− splenic B cells. Cells were cultured in vitro for 48 h and then pulsed with IdU (20 min), CldU (20 min), and HU (120 min) as for Fig. 7A. Where indicated, mirin was applied (50 μM) at the same time as the IdU and washed out at the end of the HU pulse. P values were calculated using the Mann-Whitney U test. (B) Analysis of nascent DNA tract length in WT, 53BP1−/−, Exo1−/−, and Exo1−/−53BP1−/− splenic B cells. Samples were prepared as for panel A. P values were calculated using the Mann-Whitney U test. (C) Model/summary for 53BP1 as a multifunctional mediator of stability at stalled replication forks. 53BP1 binds to nucleosomes containing H4-K20-Me2 and H2A-K15-Ub at stalled replication forks. 53BP1 binding facilitates ATR-mediated Chk1 phosphorylation, leading to stabilization of the fork. 53BP1 also inhibits exonuclease activity of Mre11 at the stalled fork.
DISCUSSION
Most studies on 53BP1 in the maintenance of genomic integrity have focused on its role in DNA double-strand break (DSB) repair (47). DSBs are signaled by the kinases ATM, ATR, and DNA-PK, which phosphorylate the variant histone H2AX to form γ-H2AX at a chromatin domain around the site of damage. γ-H2AX is a chromatin marker for DNA damage that triggers recruitment of additional DNA damage response factors, including the E3 ubiquitin ligases RNF8 and RNF168 (48). RNF168-mediated ubiquitylation of H2A creates a binding site for the 53BP1 ubiquitination-dependent recruitment (UDR) motif (49). This interaction, along with association of the 53BP1 Tudor domain with the ubiquitous H4-K20-Me2, allows 53BP1 to make a stable association with chromatin at damage sites. As is the case with DSB signaling, H2AX is phosphorylated in response to replication stress (13). RNF8 and RNF168 also become active under conditions of replication stress, as shown by their ubiquitylation of BLM after HU treatment (50). HU treatment also produces multiple nuclear foci of FK2 antibody staining, consistent with replication stress signaling by E3 ubiquitin ligases. 53BP1 may therefore be recruited to sites of replication stress by equivalent mechanisms to those operating in response to DSBs. Studies on 53BP1 in the early 2000s indicated that it had a role in transmitting an ATR-dependent signal for replication stress and in enforcing an intra-S checkpoint following ionizing radiation-induced DNA damage (29, 34). Immunoprecipitation results further showed that overexpressed 53BP1 interacts with RPA (51). 53BP1 also mediates ATM-dependent signaling, however, and studies on 53BP1−/− knockout mice supported the idea that 53BP1 acts downstream of ATM in the response to ionizing radiation and in class switch recombination in B cells (21, 22, 33, 52, 53). Our findings demonstrate that 53BP1 plays an important role in both ATM- and ATR-mediated DNA damage responses.
Our iPOND results are consistent with and extend those of Dungrawala et al., who performed a quantitative proteomic analysis which revealed that 53BP1 was one of a group of factors that become enriched at replication forks following hydroxyurea treatment (7). An earlier immunofluorescence analysis suggested that nuclear foci of 53BP1, which form after hydroxyurea treatment, correspond to sites of replication stress (29). In contrast, Yoo et al. reported that although 53BP1 relocalizes within the nucleus to form distinct foci following treatment with the topoisomerase I inhibitor camptothecin, these foci do not colocalize with RPA (51). We found that 53BP1 showed partial colocalization with areas of RPA staining in nuclei following HU treatment, supporting the idea that 53BP1 is recruited to a subset of replication forks following replication stress. As cells in early S phase show sustained γ-H2AX staining and a failure to progress after HU treatment, it is tempting to infer that 53BP1 protects forks emanating from early-firing replication origins, which are usually contained within gene-dense, open chromatin regions (54). Alternatively, 53BP1 may localize to replication origins in heterochromatic regions, which are late firing, stabilizing them or preventing them from firing during periods of replication stress. 53BP1 has been reported to act in repair of heterochromatic DSBs (55, 56), and a role in protecting underreplicated regions has been inferred from the appearance of G1 nuclear bodies staining positively for 53BP1 in cells after HU treatment (27, 28).
53BP1 has no known enzymatic activities and is considered to influence genomic integrity through chromatin effects or by interactions with other proteins in the nucleus. We and others have observed that 53BP1 recruitment limits the extent of resection of DSBs, potentially by blocking cellular activities that generate single-stranded DNA overhangs (23, 57–61). By establishing a chromatin environment that is refractory to resection, 53BP1 is proposed to inhibit repair by homologous recombination and to increase use of the competing nonhomologous end joining (NHEJ) pathway (58). A similar effect underpins the ability of 53BP1 to limit degradation of nascent replication forks. We found that binding of 53BP1 to newly replicated DNA regions protects stalled forks by limiting the ability of Mre11 to access the nascent DNA (Fig. 8A). Increased access of Mre11 to newly synthesized DNA appears to account for the fork degradation phenotype we observed in 53BP1−/− cells. 53BP1 has several binding partners, which may mediate its fork protection activity (26). Alternatively, 53BP1 may act by inducing global changes in the chromatin environment or nuclear position of stalled replication forks. Notably, binding of 53BP1 to the inner nuclear membrane proteins, SUN1 and SUN2, has been reported to contribute to the mobility of DSBs within the nucleus (62, 63). Timing of replication fork firing is furthermore dependent on nuclear positioning (54).
53BP1 becomes phosphorylated at an N-terminal site following DNA damage, allowing binding of RIF1 (64–67). RIF1 mediates a subset of 53BP1 functions at DSBs (26), but it has also been shown to participate in S-phase progression and cell survival following exposure to replication stress (68). As we observed with 53BP1 after replication stress, RIF1 foci associate with a subset of replication regions, potentially those that are close to pericentromeric heterochromatin. RIF1 is a key regulator of timing of replication fork firing; hence, its recruitment to stalled forks is very likely to contribute to the response to replication stress (69, 70). Surprisingly, our iPOND data did not show a big difference in RIF1 association with replication forks in 53BP1−/− cells (Fig. 6F). RIF1 may therefore have 53BP1-independent roles in replication. On the other hand, we did observe a consistent enrichment of FANCI at replication sites in 53BP1−/− cells after hydroxyurea treatment. FANCI becomes monoubiquitinated and recruited into distinct nuclear foci following hydroxyurea exposure (71). ATR-mediated phosphorylation of FANCI at chromatin sites subsequently regulates origin firing and repair of stalled replication forks (41). Based on our iPOND data, FANCI seems a likely candidate for the cellular response to replication stress in the absence of 53BP1. Further work will, however, be required to determine whether FANCI mediates increased cell death in the absence of 53BP1 or whether its recruitment to stalled forks in 53BP1−/− cells is a mechanism by which the cell compensates for deficient Chk1 activity.
Although the defect in fork protection in 53BP1−/− cells may be sufficient to explain the reduced viability of B cells after replication stress, the absence of 53BP1 is also linked to reduced ATR-Chk1 signaling after HU treatment. Loss of ATR-Chk1 signaling is known to be incompatible with cell survival, as has been shown by genetic studies with mice lacking either ATR or Chk1 (16–19). Targeting ATR or Chk1 has also emerged as a promising target for cancer therapy, based on the ability of inhibitors of these kinases to induce cytotoxicity by preventing appropriate responses to replication stress (72). Activated ATR and Chk1 stabilize replication forks, and inhibition of ATR signaling leads to fork collapse and CtIP/Mre11-mediated nucleolytic targeting of nascent DNA (39, 73). The defect in ATR signaling associated with loss of 53BP1 under conditions of replication stress may therefore account for the fork protection defect, even independently of any role of 53BP1 in repressing activity of Mre11 at the stalled fork. We favor a model in which 53BP1 is recruited to stalled replication forks, where it inhibits fork degradation, and promotes fork stabilization and repair by activation of Chk1 (Fig. 8C). This model is supported by previous work demonstrating coimmunoprecipitation of 53BP1 and TopBP1 and colocalization of these factors following ionizing radiation (74, 75). As 53BP1 shows variable expression in tumor cells (76, 77), a role as an adaptor mediating the response to replication stress would be likely to affect the rate of malignant cell growth and warrants further study as a potential therapeutic target.
MATERIALS AND METHODS
Animal experiments.
All mice (22) were housed in an exclusion barrier facility under protocol 12-024 from the Rutgers Institutional Animal Care and Use Committee (IACUC).
B cell culture.
Resting primary B cells were isolated from mouse spleen using ammonium-chloride-potassium (ACK) lysis of erythrocytes and negative selection using anti-CD43 magnetically activated cell sorting (MACS) microbeads (Miltenyi). Isolated B cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, minimum essential medium (MEM) nonessential amino acids, sodium pyruvate, 2-mercaptoethanol, and 10 mM HEPES. B cells were activated with 25 μg/ml of lipopolysaccharide (LPS; L2630; Sigma-Aldrich) and 50 U/ml of interleukin 4 (IL-4; I1020, Sigma-Aldrich) for 24 to 72 h.
Western blotting and immunofluorescence.
For Western blotting, primary antibodies to the following proteins were used: 53BP1 (Novus; NB100-304), γ-tubulin (Sigma-Aldrich), p53 (CST; 2524S), p53-pSer15 (CST; 9284S), CHK1-pSer345 (CST; 2341S), pan-CHK1 (Santa Cruz; sc-8408), RPA32-pSer4/8 (Bethyl; A300-245A), RPA2-pSer33 (Novus; NB100-544), pan-RPA32 (CST; 2208S), KAP1-pSer824 (Bethyl; A300-767A), pan-KAP1 (Abcam; ab10484), and cleaved caspase 3 (CST; 9661T).
For immunofluorescence, B cells were treated with 10 μM EdU for 10 min. After EdU was washed out, the cells for the nontreated sample were directly harvested, and the cells for the HU-treated sample were incubated with 2 mM hydroxyurea for 2 h before harvest. Collected B cells were attached to slides using CellTak (Corning) and preextracted with ice-cold Triton X-100 buffer (20 mM HEPES-KOH [pH7.5], 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100, 300 mM sucrose). Cells were fixed with 3% formaldehyde in phosphate-buffered saline (PBS) with 2% sucrose for 10 min, permeabilized in 0.5% Triton X-100 in PBS for 10 min, and incubated with antibodies. Primary antibodies used were antibodies to RPA32 (CST; 2208S), 53BP1 (Novus; NB100-304), and TopBP1 (Abcam; ab2402). After the binding of secondary antibody, EdU-positive cells were labeled with the Click-iT EdU Alexa Fluor 647 imaging kit. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and imaged using a Leica TCS SP5 II confocal microscope. More than 100 cells were analyzed using Leica Application Suite Advanced Fluorescence software for each experiment, using settings of 50% for threshold and 30% for background in the RPA/53BP1 and 50% for threshold and 40% for background in the RPA/TopBP1 channels.
Flow cytometry.
For the cell survival assay and caspase assay, B cells were cultured for 24 h and treated with a replication stress agent (4 mM hydroxyurea, 40 μM aphidicolin, or 250 nM gemcitabine; all from Sigma-Aldrich) for the desired times, subsequently washed, and allowed to recover for 0 to 24 h at 37°C. Cell survival was assayed by propidium iodide (PI) exclusion, and caspase was measured using a CellEvent caspase 3/7 green kit (Invitrogen; R37111). Cytometry was performed using a BD FACSCalibur running CellQuest with analysis in FlowJo. For analysis of γ-H2AX recovery, B cells cultured for 48 h were incubated in the presence of 2 mM hydroxyurea for 2 h, washed twice with PBS, and recovered for the desired time. Fixation was carried out with ice-cold methanol for 20 min, and γ-H2AX (Millipore; 05-636) antibody was used. DNA content was analyzed using PI. For checking the cell cycle profile following replication stress, B cells were labeled with 50 μM EdU for 1 h and treated with 2 mM hydroxyurea for 2 h. After removal of hydroxyurea, cells were released for 2 or 6 h and fixed with 70% ethanol. EdU was detected using the Click-iT EdU Alexa Fluor 647 imaging kit. Cells were stained with PI to measure DNA content.
DNA combing.
B cells were cultured for 48 h under activating conditions with LPS and IL-4. To measure the relative stability of nascent DNA after replication stress, cells were incubated with 25 μM 5-iododeoxyuridine (IdU; I7125; Sigma-Aldrich) for 20 min, followed by 20 min of incubation with 250 μM 5-chlorodeoxyuridine (CldU; C6891; Sigma-Aldrich). Cells were subsequently treated with 2 mM hydroxyurea for 2 h. In the case of mirin treatment, cells were treated with 50 μM mirin from the start of IdU treatment to the end of HU treatment. To examine fork length and elongation after replication stress, cells were labeled with 25 μM CldU for 20 min, then treated with 2 mM hydroxyurea for 2 h, followed by incubation with 250 μM IdU. DNA fiber spreads were prepared and analyzed by immunofluorescence as previously described (78). Briefly, the cell suspension was lysed with lysis buffer consisting of 50 mM EDTA and 0.5% SDS in 200 mM Tris-HCl (pH 7.5) for 2 min on a microscope slide. DNA fibers were prepared by tilting the slide and fixing it with methanol-acetic acid solution (3:1) for 10 min. After denaturation in 2.5 M HCl for 80 min, DNA fibers were stained with the following antibodies: anti-BrdU (rat; Abcam; ab6326) for IdU, anti-BrdU (mouse; BD; 347580) for CldU, and goat anti-rat antibody–Alexa Fluor 488 and sheep anti-mouse antibody–Alexa Fluor 555. Approximately 300 fiber lengths were analyzed per genotype in three independent experiments by measuring the lengths of IdU- and CldU-labeled tracts using ImageJ software (http://rsbweb.nih.gov/ij/).
Neutral comet assay.
Forty-eight hours after isolation, B cells were treated with 4 mM HU for 3 h or 1 μM camptothecin (CPT) for 1 h. Harvested cells were analyzed according to Trevigen's instructions for the neutral comet assay. Staining was done with SYBR gold, and image acquisition was done with a Nikon Eclipse E800 epifluorescence microscope using a 20× objective.
Colony formation assay.
One day after plating of each MEF lines, cells were treated with indicated concentrations of HU. After 10 days of incubation, cells were fixed with ice-cold methanol and stained with crystal violet. Dried colonies were counted for analysis.
iPOND.
iPOND was performed as previously described (79). In brief, 5 × 107 293T cells were plated for each condition 1 day before EdU incubation. In the case of B cells, 8 × 107 cells were isolated from mouse spleen for each condition 2 days before EdU incubation. Cells were pulsed with 10 μM EdU for 10 min. Nontreated (NT) cells were harvested immediately after EdU treatment. For HU treatment, 2 mM hydroxyurea was added to the medium for 2 h, followed by EdU removal and washing. Cells were fixed with 1% formaldehyde in PBS for 20 min and quenched by adding 1.25 M glycine. Then cells were permeabilized with 0.25% Triton X-100 in PBS for 30 min and subsequently subjected to a click reaction with biotin-azide (Invitrogen) for 2 h. After lysis with 1% SDS in 50 mM Tris (pH 8) and sonication, biotin-EdU-labeled DNA was incubated with streptavidin-agarose beads at 4°C for 20 h. The beads were washed with lysis buffer and 1 M NaCl and eluted with 2× NuPAGE LDS sample buffer containing 0.2 M dithiothreitol (DTT).
Mass spectrometry.
iPOND samples were subject to in-gel trypsin digestion followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Samples were first run ∼1 cm into a Novex gel (10%; Thermo Fisher). The entire gel plug was subjected to a standard in-gel digestion procedure, including reduction of disulfide bonds with 10 mM DTT for 30 min at 60°C, alkylation of free sulfhydryls with 20 mM iodoacetamide for 45 min at room temperature in the dark, and digestion with trypsin (sequencing grade; Thermo Scientific; catalog number 90058) overnight at 37°C. Peptides were extracted from the gel with 5% formic acid–60% acetonitrile and dried under vacuum. Peptide samples were analyzed by nano-LC-MS/MS (Dionex Ultimate 3000 RLSC nanosystem interfaced with Q Exactive HF [Thermo Fisher, San Jose, CA]). Samples were loaded onto a self-packed 100-μm by 2-cm trap (Magic C18AQ; 5 μm and 200 Å; Michrom Bioresources, Inc.) and washed with buffer A (0.1% trifluoroacetic acid) for 5 min at a flow rate of 10 μl/min. The trap was brought in-line with the analytical column (self-packed Magic C18AQ; 3 μm and 200 Å; 75 μm by 50 cm), and peptides were fractionated at 300 nl/min using a segmented linear gradient of 4 to 15% solution A in 30 min (solution A, 0.2% formic acid; solution B, 0.16% formic acid–80% acetonitrile), 15 to 25% solution B in 40 min, 25 to 50% solution B in 44 min, and 50 to 90% solution B in 11 min. Mass spectrometry data were acquired using a data-dependent acquisition procedure with a cyclic series of a full scan with a resolution of 120,000, followed by MS/MS (higher-energy C-trap dissociation; relative collision energy, 27%) of the 20 most intense ions and a dynamic exclusion duration of 20 s. LC-MS/MS peak lists (.mgf files) were generated using Thermo Proteome Discoverer v. 2.1 and searched against the human Ensembl database (v. 89) plus a database of common laboratory contaminants using an in-house version of the GPM software package containing the X!Tandem Alanine (2017.2.1.4) search engine. Search parameters were as follows: fragment mass error, 20 ppm; parent mass error, 7 ppm; fixed modification, carbamidomethylation on cysteine; potential modifications during initial search, methionine oxidation and acetylation on protein N termini; and up to one missed tryptic cleavage during the initial search and up to three missed cleavages during refinement. Potential modifications during refinement were as follows: pass 1, monoxidation at methionine and tryptophan and deamidation at asparagine and glutamine, and pass 2, dioxidation at methionine and tryptophan. Maximum valid expectation scores for proteins and peptides were 0.0001 and 0.01, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nanci Kane at the Waksman Institute Confocal Microscopy Core for assistance with microscopy and Peter Lobel at the Rutgers Biological Mass Spectrometry Facility for assistance with mass spectrometry.
This work was supported by NIH grant R01 CA190858 (S.F.B.) and a New Jersey Commission For Cancer Research postdoctoral award (J.H.). Resources at the Rutgers Biological Mass Spectrometry Facility are funded by NIGMS shared instrumentation grant S10OD016400 (Lobel).
There are no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00472-17.
REFERENCES
- 1.Branzei D, Foiani M. 2005. The DNA damage response during DNA replication. Curr Opin Cell Biol 17:568–575. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. 2005. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leman AR, Noguchi E. 2012. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 11:3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choe KN, Moldovan GL. 2017. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol Cell 65:380–392. doi: 10.1016/j.molcel.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, Zhang Z. 2014. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell 56:551–563. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. 2015. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- 9.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 10.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, Krejci L, Costanzo V. 27 July 2017. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saldivar JC, Cortez D, Cimprich KA. 2017. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18:622–636. doi: 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward IM, Chen J. 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 14.Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. 2009. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem 284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buisson R, Boisvert JL, Benes CH, Zou L. 2015. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol Cell 59:1011–1024. doi: 10.1016/j.molcel.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown EJ, Baltimore D. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 17.Brown EJ, Baltimore D. 2003. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14:1448–1459. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. 2009. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. 1994. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A 91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 2004. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 22.Ward IM, Minn K, van Deursen J, Chen J. 2003. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, Deng CX, Finkel T. 2009. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bunting SF, Nussenzweig A. 2013. End-joining, translocations and cancer. Nat Rev Cancer 13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, Wesemann DR, Lee JE, Tubbs A, Sleckman BP, Daniel JA, Ge K, Alt FW, Fernandez-Capetillo O, Nussenzweig MC, Nussenzweig A. 2013. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. 2011. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, Lukas J. 2011. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 29.Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, Halazonetis TD, Harris CC. 2004. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol 166:801–813. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuella-Martin R, Oliveira C, Lockstone HE, Snellenberg S, Grolmusova N, Chapman JR. 2016. 53BP1 integrates DNA repair and p53-dependent cell fate decisions via distinct mechanisms. Mol Cell 64:51–64. doi: 10.1016/j.molcel.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, Nussenzweig A, Nussenzweig MC. 2011. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 34.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 2002. 53BP1, a mediator of the DNA damage checkpoint. Science 298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 35.Hampp S, Kiessling T, Buechle K, Mansilla SF, Thomale J, Rall M, Ahn J, Pospiech H, Gottifredi V, Wiesmuller L. 2016. DNA damage tolerance pathway involving DNA polymerase iota and the tumor suppressor p53 regulates DNA replication fork progression. Proc Natl Acad Sci U S A 113:E4311–E4319. doi: 10.1073/pnas.1605828113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klusmann I, Rodewald S, Muller L, Friedrich M, Wienken M, Li Y, Schulz-Heddergott R, Dobbelstein M. 2016. p53 activity results in DNA replication fork processivity. Cell Rep 17:1845–1857. doi: 10.1016/j.celrep.2016.10.036. [DOI] [PubMed] [Google Scholar]
- 37.Myers K, Gagou ME, Zuazua-Villar P, Rodriguez R, Meuth M. 2009. ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS Genet 5:e1000324. doi: 10.1371/journal.pgen.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ III, Rothenberg E, Jallepalli PV, Huang TT. 2015. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58:323–338. doi: 10.1016/j.molcel.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, Stankovic T, Boulton SJ, Niedzwiedz W, Stewart GS. 2015. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell 59:462–477. doi: 10.1016/j.molcel.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Leuzzi G, Marabitti V, Pichierri P, Franchitto A. 2016. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J 35:1437–1451. doi: 10.15252/embj.201593265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu S, Wu X, Wu L, Castillo A, Liu J, Atkinson E, Paul A, Su D, Schlacher K, Komatsu Y, You MJ, Wang B. 2017. Abro1 maintains genome stability and limits replication stress by protecting replication fork stability. Genes Dev 31:1469–1482. doi: 10.1101/gad.299172.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daley JM, Sung P. 2014. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 34:1380–1388. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwertman P, Bekker-Jensen S, Mailand N. 2016. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat Rev Mol Cell Biol 17:379–394. doi: 10.1038/nrm.2016.58. [DOI] [PubMed] [Google Scholar]
- 49.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tikoo S, Madhavan V, Hussain M, Miller ES, Arora P, Zlatanou A, Modi P, Townsend K, Stewart GS, Sengupta S. 2013. Ubiquitin-dependent recruitment of the Bloom syndrome helicase upon replication stress is required to suppress homologous recombination. EMBO J 32:1778–1792. doi: 10.1038/emboj.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo E, Kim BU, Lee SY, Cho CH, Chung JH, Lee CH. 2005. 53BP1 is associated with replication protein A and is required for RPA2 hyperphosphorylation following DNA damage. Oncogene 24:5423–5430. doi: 10.1038/sj.onc.1208710. [DOI] [PubMed] [Google Scholar]
- 52.DiTullio RA Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 53.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 2004. 53BP1 is required for class switch recombination. J Cell Biol 165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hiratani I, Takebayashi S, Lu J, Gilbert DM. 2009. Replication timing and transcriptional control: beyond cause and effect—part II. Curr Opin Genet Dev 19:142–149. doi: 10.1016/j.gde.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kakarougkas A, Ismail A, Klement K, Goodarzi AA, Conrad S, Freire R, Shibata A, Lobrich M, Jeggo PA. 2013. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res 41:9719–9731. doi: 10.1093/nar/gkt729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, Goodarzi AA. 2010. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol 12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 57.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 2010. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman JR, Taylor MR, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 59.Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Mathe E, Liang G, Cobell J, Nakahashi H, Robbiani DF, Nussenzweig A, Hager GL, Nussenzweig MC, Casellas R. 2012. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. 2013. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc Natl Acad Sci U S A 110:2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sfeir A, de Lange T. 2012. Removal of shelterin reveals the telomere end-protection problem. Science 336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dimitrova N, Chen YC, Spector DL, de Lange T. 2008. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 2015. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 163:880–893. doi: 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, Nussenzweig A, Casellas R, Robbiani DF, Nussenzweig MC. 2013. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Feng L, Fong KW, Wang J, Wang W, Chen J. 2013. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buonomo SB, Wu Y, Ferguson D, de Lange T. 2009. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol 187:385–398. doi: 10.1083/jcb.200902039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31:3678–3690. doi: 10.1038/emboj.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. 2012. Rif1 regulates the replication timing domains on the human genome. EMBO J 31:3667–3677. doi: 10.1038/emboj.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sims AE, Spiteri E, Sims RJ III, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT. 2007. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 72.Dobbelstein M, Sorensen CS. 2015. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 14:405–423. doi: 10.1038/nrd4553. [DOI] [PubMed] [Google Scholar]
- 73.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. 2013. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cescutti R, Negrini S, Kohzaki M, Halazonetis TD. 2010. TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J 29:3723–3732. doi: 10.1038/emboj.2010.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamane K, Wu X, Chen J. 2002. A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol 22:555–566. doi: 10.1128/MCB.22.2.555-566.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bi J, Huang A, Liu T, Zhang T, Ma H. 2015. Expression of DNA damage checkpoint 53BP1 is correlated with prognosis, cell proliferation and apoptosis in colorectal cancer. Int J Clin Exp Pathol 8:6070–6082. [PMC free article] [PubMed] [Google Scholar]
- 77.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li M, Cole F, Patel DS, Misenko SM, Her J, Malhowski A, Alhamza A, Zheng H, Baer R, Ludwig T, Jasin M, Nussenzweig A, Serrano L, Bunting SF. 2016. 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO Rep 17:1532–1541. doi: 10.15252/embr.201642497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sirbu BM, Couch FB, Cortez D. 2012. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7:594–605. doi: 10.1038/nprot.2012.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.