

ABSTRACT
Glucocorticoid receptor β (GRβ) is associated with glucocorticoid resistance via dominant negative regulation of GRα. To better understand how GRβ functions as a dominant negative inhibitor of GRα at a molecular level, we determined the crystal structure of the ligand binding domain of GRβ complexed with the antagonist RU-486. The structure reveals that GRβ binds RU-486 in the same ligand binding pocket as GRα, and the unique C-terminal amino acids of GRβ are mostly disordered. Binding energy analysis suggests that these C-terminal residues of GRβ do not contribute to RU-486 binding. Intriguingly, the GRβ/RU-486 complex binds corepressor peptide with affinity similar to that of a GRα/RU-486 complex, despite the lack of helix 12. Our biophysical and biochemical analyses reveal that in the presence of RU-486, GRβ is found in a conformation that favors corepressor binding, potentially antagonizing GRα function. This study thus presents an unexpected molecular mechanism by which GRβ could repress transcription.
KEYWORDS: glucocorticoid receptor beta (GRβ), RU-486, dominant negative, glucocorticoid resistance, transrepression
INTRODUCTION
Glucocorticoids (GCs) function as essential primary stress hormones that regulate a broad range of physiological processes, including cardiovascular function, immunity, metabolism, neurobiological effects, and reproduction (1). GCs exert powerful anti-inflammatory actions on many specific immune responses mediated by T cells and B cells, as well as potent immunosuppressive effects on the effector functions of phagocytes. As such, they are widely used to treat diseases caused by an overactive immune system, such as allergies, asthma, sepsis, ulcerative colitis, multiple sclerosis, and rheumatoid arthritis (2, 3). Today, synthetic GCs are the most widely prescribed therapy in dermatology (4). They are also used to prevent organ transplant rejection and to treat cancers such as leukemias, lymphomas, and myelomas (2). Unfortunately, a significant number of patients fail to respond to GCs, suggesting resistance. Understanding the factors involved in GC resistance is critical to facilitating development of better therapeutic strategies. One proposed mechanism underlying this resistance involves prolonged exposure to cytokines. Proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) disproportionately increase the expression of dominant negative glucocorticoid receptor β (GRβ), reducing the responsiveness to glucocorticoid administration (5).
The action of GCs is exerted by binding to and activating the glucocorticoid receptor (GR) transcription factor. In the absence of GCs, GRα resides predominantly in the cytoplasm, where it is associated with chaperone proteins and immunophilins in a transcriptionally inactive complex. Upon GC binding, GRα undergoes a conformational change, resulting in release of associated binding partners followed by translocation to the nucleus, where it controls target gene expression. GRα may regulate gene expression in three primary ways: binding directly to DNA, indirectly binding to DNA by tethering itself to other DNA-bound transcription factors, or binding directly to DNA and interacting with other DNA-bound transcription factors simultaneously (2). After binding to DNA sequences called glucocorticoid-responsive elements (GREs) or negative GREs (nGREs), GRα can upregulate the expression of anti-inflammatory proteins (transactivation) or downregulate the expression of proinflammatory proteins (transrepression) by interacting with transcription factors, such as activator protein 1 and nuclear factor κB (2, 6, 7). For efficient transcriptional regulation, GRα recruits coregulators such as nuclear receptor coactivator or nuclear receptor corepressor 1 (NCoR1), depending on the bound ligand and the specific GRE sequences (8–10).
The GRα (NR3C1, nuclear receptor subfamily 3, group C, member 1) is composed of three major distinct domains: an unstructured N-terminal transactivation domain (NTD) followed by a central DNA binding domain (DBD) and a C-terminal ligand binding domain (LBD) (11). The NTD contains a transcriptional activation function (AF-1) domain that interacts with coregulators and transcription machinery proteins (12). The DBD contains two zinc finger motifs that bind GREs to regulate target gene expression. The LBD contains a GC binding pocket and an AF-2 domain that interacts with coregulators in a ligand-dependent manner (13).
In humans, GR is expressed from a single gene containing 9 exons (14). Alternative splicing on exon 9 can give rise to two isoforms, human GRα (hGRα) and hGRβ, with distinct C termini (15). These two proteins are identical through amino acid 727, after which GRα contains 50 amino acids while GRβ contains a unique 15-amino-acid sequence (Fig. 1A). The GRα LBD adopts a globular fold containing 12 α-helices and four short β-strands, which form a hydrophobic glucocorticoid binding pocket (13). The C-terminal 50 amino acids of hGRα form helix 11 (α11) and helix 12 (α12), and residues on these helices contribute to the binding of agonists such as dexamethasone (DEX). In addition, α12 contributes to forming part of the AF-2 domain, responsible for coactivator binding (13). In the presence of the antagonist RU-486, the C-terminal α12 of hGRα exhibits structural heterogeneity (16), which is consistent with results from hydrogen/deuterium exchange (HDX) experiments revealing a dynamic C terminus (17). It is proposed that α12 functions as a ligand-dependent on/off switch (16). In contrast, the unique 15-amino-acid C terminus of hGRβ hypothetically renders it unable to bind glucocorticoid agonists such as DEX (15, 18, 19), though the structural mechanism for this loss of function is currently unknown.
FIG 1.

Functional similarity of the chimeric hGRβ′ (residues 1 to 520)/nmrGRβ (residues 521 to 742) to hGRβ. (A) Schematic representation showing domain structures of hGRα, hGRβ, and hGR1–520/nmrGRβ521–742 (GRβ′). Domains are identified by name and color. Divergent sequences at the C termini of hGRα, hGRβ, and nmrGRβ are shown in pink, red, and dark brown, respectively. The chimeric construct GRβ′ contains human NTD, DBD, and hinge followed by nmrGRβ LBD (dark orange) with the 15 unique amino acid sequence (brown). Secondary structural elements of the hGRα C terminus are shown above its sequence in cyan. (B) Western blot analysis shows the expression level of three stably transfected GRβ′ clones, as well as hGRα and hGRβ in U-2 OS cells. (C) Each protein band intensity was normalized against actin, and the expression level is compared to that of hGRα. The experiment was performed in triplicate, and the data are represented as means ± SD. (D) Immunocytochemistry was conducted using U-2 OS cells stably expressing hGRα, hGRβ, and GRβ′. Representative images are shown for nuclear translocation of each protein in the absence and presence of 100 μM RU-486. (E) Scoring scheme for GR localization. The relative fluorescence intensity ratio of nucleus (N) to cytoplasm (C) was estimated. The lowest number was assigned when receptor was observed to be found predominately in the cytoplasm while the highest number was assigned for those cells whose receptors were found predominately in the nucleus. (F to H) Localization scores are plotted after at least 100 cells were counted from five different images for each treatment. The translocation assay was performed in triplicate, and the data are represented as means ± SD. (I and J) Fold change of mRNA levels of genes GILZ and S100P, regulated by hGRα (mock) in the presence of 100 nM DEX was calculated after hGRβ and GRβ′ were transiently transfected into U-2 OS cells stably expressing hGRα. Experiments were performed in triplicate, and the data are represented as means ± standard errors of the mean (*, P ≤ 0.05; **, P ≤ 0.01).
While hGRα is found mainly in the cytoplasm in the absence of ligands, the majority of hGRβ resides in the nucleus and inhibits the activity of hGRα in a tissue-specific manner. When coexpressed with hGRα, hGRβ functions as a dominant negative inhibitor by putatively forming a transcriptionally inactive hGRα-hGRβ heterodimer (15, 20). Overexpression of hGRβ decreases the response to glucocorticoids. In addition, elevated levels of hGRβ have been reported in GC-insensitive patients, suggesting that hGRβ is involved in glucocorticoid resistance (21).
The ratio of GRα to GRβ expression is critical to glucocorticoid responsiveness in a variety of cells; thus, the proposed physiological role of GRβ is as a key modulator of the progression of immune-related diseases and glucocorticoid resistance (21). For example, an increase in the hGRβ expression level is associated with the development of glucocorticoid-resistant forms of immune-related diseases, including glucocorticoid-resistant asthma (22, 23), fatal asthma (24), leukemia (25–27), ulcerative colitis (28–30), nasal polyposis (31), and rheumatoid arthritis (32, 33).
To overcome the limitations of GRβ functional studies mostly conducted using in vitro systems, the physiological role of hGRβ was recently assessed by transfecting adeno-associated virus harboring hGRβ into both wild-type and GR knockout mouse livers. In these studies, GRβ attenuated hepatic gluconeogenesis via downregulation of phosphoenolpyruvate carboxykinase only in wild-type mouse livers. In addition, GRβ is transcriptionally active and regulates unique genes in a GRα-independent manner (34).
Currently, there are no known endogenous ligands for GRβ. Although mainly found in the nucleus, cytoplasmic GRβ can be translocated into the nucleus and regulate gene expression upon binding the GR antagonist RU-486 (19). Recently, it was reported that the binding of RU-486 to GRβ can decrease cell proliferation in prostate cancer (35). However, the molecular basis for the selectivity of RU-486 by GRβ is unknown. To better understand the ligand selectivity and the role of the unique C-terminal residues of GRβ in dominant negative function, we solved the crystal structure of the ligand binding domain of Heterocephalus glaber (naked mole rat) GRβ (nmrGRβ) in complex with RU-486. The use of nmrGRβ as a model for hGRβ was validated in U-2 OS cells by confirming that the localization, nuclear translocation, dominant negative activity, and gene regulation in response to RU-486 of these two proteins are similar. The structure reveals that GRβ binds RU-486 in the same position and orientation as GRα, despite having the majority of the C-terminal residues disordered. Combined with the crystallography, in silico binding studies suggest that these residues do not contribute substantially to RU-486 binding. Despite the lack of a structured α12, hGRβ can preferentially bind to a corepressor but not coactivator peptide in the presence of RU-486, suggesting that α12 might be dispensable in GR antagonism and gene repression.
RESULTS
Validation of nmrGRβ as a model system for hGRβ.
Structural determination of the full-length human GR is difficult due to solubility issues and the unstructured nature of the N-terminal domain. Individual domains of GR have been structurally characterized, including many ligand-bound GRα LBD (GRα hereafter) complexes using recombinant proteins, which are currently available in the Protein Data Bank (PDB). However, to date, no crystal structure of GRβ has been reported. To obtain crystal structures of the LBD of human GRβ, we explored a variety of solubility-enhancing tags, truncation constructs with a solubility-enhancing mutation (F602S) (13), and expression conditions without success. As an alternative approach, we searched highly homologous GRβs that maintained the same length and high sequence identity to the C-terminal 15 amino acids of hGRβ. Among GRβ homologs found in other animals, GRβ from Heterocephalus glaber (nmrGRβ) was an ideal candidate as it shares 93% sequence identity to the LBD of hGRβ, and also the length of the unique C terminus is identical, with 11 out of the 15 amino acids of nmrGRβ conserved with hGRβ (73% sequence identity in the tail) (Fig. 1A). Mouse and rat GRβ possess a nonhomologous C terminus and a consecutive N-terminal glutamine repeat not present in human or nmrGRβ. To compare the functional similarities between hGRβ LBD and nmrGRβ LBD, we generated U-2 OS cell lines stably expressing a chimera consisting of the N-terminal, DBD, and hinge regions of hGR (residues 1 to 520, or hGR1–520) and the LBD region of nmrGRβ (residues 521 to 742, or nmrGRβ521–742), termed GRβ′. Western blot analysis of proteins extracted from three different clonal cell lines confirmed the expression of GRβ′ (Fig. 1B and C). In a translocation assay, the expressed GRβ′ was found mainly in the nucleus in the absence of RU-486 and translocated completely into the nucleus in the presence of 100 μM RU-486, consistent with hGRβ but distinct from hGRα (Fig. 1D to H). To test the dominant negative activity of GRβ′ on hGRα, reverse transcription-PCR (RT-PCR) was carried out on two genes controlled by hGRα, GILZ and S100P, after cells stably expressing hGRα were transiently transfected with GRβ′ and treated with 100 nM DEX. hGRβ and GRβ′ exhibited similar dominant negative activities for these genes (Fig. 1I and J). Moreover, both hGRβ and GRβ′ in the presence of 1 μM RU-486 upregulated expression of BECN1 and COX17, genes that were previously shown to be regulated by GRβ (19) (Fig. 2). These results suggest that the hGRβ LBD and nmrGRβ LBD are functionally indistinguishable under our experimental conditions. In addition, the equivalent solubility-enhancing mutants hGRβ (F602S) and nmrGRβ (F598S) helped to improve the stability of these proteins for our biochemical and structural characterizations. However, the mutation did not appear to affect the function of the protein as hGRβ (F602S) displayed dominant negative activity similar to that of the wild-type protein (Fig. 3), suggesting that nmrGRβ (F598S) represents a good model system for studying structure-function relationships of hGRβ.
FIG 2.

Gene regulation by GRβ. Relative mRNA levels of the genes BECN1 and COX17 regulated by hGRβ or GRβ′ were assayed in the absence and presence of 1 μM RU-486. Experiments were performed in triplicate, and the data are represented as means ± standard errors of the mean (*, P ≤ 0.05; **, P ≤ 0.01).
FIG 3.

Dominant negative activity of GRβ mutants. Fold changes of mRNA level of the genes GILZ and S100P, regulated by hGRα (mock) in the presence of 100 nM DEX, were calculated after transient transfection of hGRβ, hGRβ (F602S), hGRβ (V571R), and hGRβ (V575R) into U-2 OS cells stably expressing hGRα. Experiments were performed in triplicate, and the data are represented as means ± standard errors of the mean (***, P ≤ 0.001).
Crystal structure of GRβ/RU-486 complex.
Soluble nmrGRβ518–738 was expressed as a cleavable N-terminal hexahistidine-tagged SUMO (6His-SUMO) fusion protein in Escherichia coli in the presence of RU-486, with and without a solubility-enhancing mutation (F598S in nmrGRβ) (13). The crystal structure of the nmrGRβ (F598S)/RU-486 complex was solved at 2.35 Å (Table 1). The asymmetric unit contains two GRβ monomers, each with a bound RU-486 molecule, that are related by noncrystallographic 2-fold symmetry (Fig. 4A). The interface between the two GRβ molecules in the asymmetric unit involves interactions between the α3-helixes of both molecules, as well as interactions between α7 and the C terminus of each molecule (Fig. 4A). In addition, four partially ordered CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} molecules present in the crystal structure are located at this interface (Fig. 4A). A nonconserved residue, Tyr634, is found at the interface forming potential interactions with Val725 and the solubility-enhancing residue Ser598, which is a phenylalanine in the wild-type protein (Fig. 4B and C). This orientation places the two binding pockets adjacent to one another, such that the 17β-hydroxyl atoms from the two RU-486 molecules in each pocket lie 3.7 Å apart (Fig. 4C). Like hGRα, the nmrGRβ structure reveals a three-layered antiparallel α-helical sandwich, consisting of nine α-helixes, two short 310-helices, and two β-strands (Fig. 5A and B). The structure of nmrGRβ superimposes well with that of hGRα (PDB ID 1M2Z) (root mean square deviation [RMSD] of 1.2 Å for 188 Cα atoms). Two N-terminal residues (518 and 519) and a short loop region (residues 612 and 613) between α5 and β1 are disordered in both molecules. In addition, no visible density is observed for the C-terminal region beyond Thr726, suggesting that the structural element α11 present in hGRα is disordered in GRβ. The protein used for crystallization was confirmed to be intact via mass spectrometry analysis. β-Strand 3 of GRα is not formed in GRβ. Instead, this region (residues 672 to 675) forms a short 310-helix (Fig. 5A), possibly due to the lack of β4 residues which are not found in GRβ. Of the 16 residues that differ between the LBDs of hGRβ and nmrGRβ, 4 are on the disordered C-terminal tail, and all others, except Phe668 (buried in a hydrophobic core), map to regions on the surface of the protein distal from the ligand binding site, suggesting that RU-486 binding to hGRβ would be very similar (Fig. 5C).
TABLE 1.
Selected crystallographic data
| Parameter | Value(s) for nmrGRβ (F598S)/RU-486a |
|---|---|
| Data collection | |
| Space group | P61 |
| Cell dimensions | |
| a, b, c (Å) | 48.4, 48.4, 385.8 |
| α, β, γ (°) | 90, 90, 120 |
| Wavelength (Å) | 1.0 |
| X-ray source | APS SER-CAT 22ID |
| Resolution (Å) | 50–2.35 (2.39–2.35) |
| No. of reflections | 120,449 |
| No. of unique reflections | 20,767 |
| Completeness (%) | 97.7 (84.3) |
| Rmerge (%)b | 6.9 (45.3) |
| I/σ | 28.6 (2.5) |
| Redundancy | 5.8 (3.3) |
| Refinement | |
| Resolution (Å) | 35.0–2.35 (2.47–2.35) |
| No. of molecules/AUc | 2 |
| No. of amino acids/AU | 414 |
| No. of RU-486 molecules/AU | 2 |
| No. of waters/AU | 18 |
| Rwork (%)d | 21.0 |
| Rfree (%)e | 24.1 |
| Average B-factors (Å2) | |
| Protein | 60.3 |
| RU-486 | 50.9 |
| Water | 53.8 |
| RMSD from ideal | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.501 |
| Ramachandran plot (%) | |
| Favored | 98.26 |
| Allowed | 1.74 |
| Outlier | 0 |
PDB ID 5UC1. Values in parentheses are for the highest-resolution shell.
Rmerge = ∑∑|I − <I>|/∑∑|I|, where I is the observed intensity and <I> is the average intensity.
AU, asymmetric unit.
Rwork = ∑||Fo| − |Fc||/∑|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree = ∑||Fo| − |Fc||/∑|Fo| for 5% of the data not used at any stage of the structural refinement.
FIG 4.

Asymmetric unit of nmrGRβ/RU-486 complex. (A) The asymmetric unit dimer of nmrGRβ with the noncrystallographic 2-fold symmetry-related molecules shown in green and gray. The two RU-486 molecules (cyan spheres) are found in the ligand binding pocket at the α3-α3 interface of the dimer (top), and CHAPS detergent molecules are drawn in stick (orange). A 180° rotation around the vertical axis (bottom) displays the C terminus and α7. N and C termini are indicated. (B) Residues such as Ser598, Tyr634, and Val725 contributing to the interface at the C terminus are shown. Hydrogen bonding between Ser598 and Tyr634 is indicated by a yellow dotted line. Hydrophobic interactions between Tyr598 and Val725 are indicated by black dotted lines. (C) The 17β-hydroxyl groups of the two RU-486 molecules are located 3.7 Å away from each other (red dotted line).
FIG 5.

Crystal Structure of GRβ. (A) GR LBD sequences for human GRα522–777, GRβ522–742, and nmrGRβ518–738 were aligned using ClustalW. Divergent sequences at the C termini of the two splice variants of hGR after amino acid 727 are highlighted in blue and magenta for hGRα and hGRβ, respectively. Differences in human and nmrGRβ sequences are highlighted in orange, and the position of the solubility-enhancing mutation (F598S) used in the crystal structure of nmrGRβ is highlighted in red. The disordered C-terminal residues of nmrGRβ are underscored. Secondary structural elements for hGRα (yellow) and nmrGRβ (green) are shown above and below their sequences, respectively. The α- and 310-helices are shown as cylinders. β-Strands are shown as arrows. (B) Structure of nmrGRβ with bound RU-486 (spheres, cyan). Helices (α and 310) and β-strands are numbered consistent with the description for panel A. N and C termini are indicated with arrows. (C) Sequence differences between hGRβ and nmrGRβ are mapped on the surface in orange. Due to the orientation, residues including Gly546, Thr547, and Val616 and the solubility-enhancing mutation F598S (red) are shown by a 45° rotation around the vertical axis (right).
Ligand interaction.
RU-486 binds in the ligand binding pocket (LBP) of nmrGRβ via multiple hydrophilic and hydrophobic interactions (Fig. 6A to C). Side chains from residues Arg607 and Gln566 both lie within hydrogen bonding distance to the three-keto group of RU-486 bound deep in the pocket (Fig. 6B and C). Hydrophobic residues Met556, Leu559, Leu562, Met600, Leu604, Phe619, Met635, and Met642 surround the bulk of the ligand while residues Gly563 and Trp596 form the cleft for the dimethyl nitrophenol group. In addition, the 17α-propynyl group is stabilized by residues Leu559, Phe619, Gln638, Cys639, and Met642. Residues Trp553 and Met556 from the other subunit in the asymmetric unit help to bury RU-486 at the noncrystallographic interface. The RU-486 fits well in the LBP, which buries 80% (570 Å2) of the total LBP surface area (709 Å2).
FIG 6.

Comparison of GRα and β structures. (A) nmrGRβ/RU-486 (left), hGRα/RU-486 (PDB ID 3H52, middle), and hGRα/DEX (PDB ID 1M2Z, right) complex structures. Two CHAPS molecules bound to nmrGRβ are shown in orange. Corepressor and coactivator peptides bound to hGRα are shown in pink (middle) and purple (right), respectively. RU-486 (cyan) and DEX (cobalt) are shown as spheres. (B) Superposition of the ligand binding pockets of the hGRα/RU-486 (yellow) and nmrGRβ/RU-486 (green) complex structures. Selected residues interacting with the ligands are shown. Residue numbers from hGRα/RU-486 and nmrGRβ/RU-486 complex are shown in black and green, respectively. (C) Superposition of the ligand binding pockets of hGRα/DEX (white) and nmrGRβ/RU-486 (green) complex structures. M556B and W553B are from molecule B of the nmrGRβ asymmetric unit.
Based on the crystal structures, the orientation and conformation of the RU-486 molecules bound to nmrGRβ are very similar to those of RU-486 bound to hGRα, with most LBD residues superimposing well (Fig. 6A and B). However, nmrGRβ lacks some residues interacting with RU-486, such as Leu732, Tyr735, and Ile756 found on α11 and α12 of hGRα. A few other minor differences exist. The sulfur atom of Cys639 in nmrGRβ is located 3.9 Å away from atom C-32 of RU-486 (4.6 Å in hGRα), and the ε carbon of Met600 faces toward the steroid ring, while the same atom of Met604 in hGRα faces away. In contrast to GRβ interactions with RU-486, a number of residues that interact with the agonist dexamethasone (DEX) in hGRα are quite different (Fig. 6C). These differences are due mainly to structural heterogeneity between the two ligand molecules and the presence/absence of α11 and α12. The interaction of hGRα with DEX involves three hydrogen bonds and multiple hydrophobic interactions. Residues specific to the hGRα/DEX complex are Leu732, Tyr735, Cys736, Ile747, Phe749, and Leu753 that reside on either α11 or α12 and do not exist in the GRβ sequence. CHAPS molecules present in GRβ are located at two different sites. One molecule is found at the same location of α11 in hGRα, nestled between side chains from α5, α7, the end of α10, RU-486, and α7 from the neighboring molecule. The other CHAPS molecule is located against helices α3 and α4 at the coactivator/corepressor site for hGRα, as well as α3 of the neighboring GRβ molecule (Fig. 4A and 6A).
Ligand binding energy.
Although RU-486 is the only known ligand for GRβ, it is not clear why. Utilizing in vitro biochemical studies has proven difficult since soluble GRβ can only be obtained when GRβ is expressed in the presence of RU-486. Attempts to exchange RU-486 with other GCs such as DEX in as high as 50-fold excess have failed. To better understand the specificity of GRβ for RU-486, we turned to in silico methods utilizing the program FRED (36). The FRED docking score of GRβ for RU-486 is almost two times higher than that of DEX (Table 2). In contrast, the docking scores of RU-486 and DEX binding to GRα are very similar (Table 2). We expanded the docking experiments to include 10 agonists and 4 antagonists, which have been tested in previous nuclear translocation assays (19). RU-486 docking still displayed the lowest binding energy to GRβ, supporting the relatively low affinity of other GCs compared to that of RU-486.
TABLE 2.
FRED docking score
| Ligand type and name | Docking score of the receptor (kcal/mol) |
|
|---|---|---|
| hGRαa | nmrGRβ | |
| Agonists | ||
| Corticosterone | −18.8 | −8.0 |
| Hydrocortisol | −19.4 | −7.8 |
| Cortisone | −18.3 | −7.8 |
| Deltafludrocortisone | −19.7 | −8.0 |
| Desoximetasone | −16.6 | −7.1 |
| Dexamethasone | −19.7 | −7.8 |
| Prednisolone | −19.4 | −7.8 |
| RU-28362 | −16.3 | −8.9 |
| Triamcinolone | −19.4 | −8.2 |
| Triamicinolone acetonide | −16.4 | −6.2 |
| Antagonists | ||
| Cortexolone | −12.6 | −8.3 |
| Dexamethasone-21-mesylate | −11.5 | −6.6 |
| RU-486 | −18.0 | −12.6 |
| ZK98299 | −12.7 | −9.7 |
Finally, to calculate the binding energy of ligands, we performed molecular dynamics simulations using GRβ/RU-486, a GRβ/RU-486 deletion variant lacking the C-terminal 15 amino acid residues (Δ15), GRα/DEX (PDB ID 1M2Z) (13), and GRα/RU-486 complex (PDB ID 3H52) (16). As shown in Table 3, the total calculated binding energy of the GRβ/RU-486 complex is similar to that of the GRα/RU-486 complex, despite the lack of α12. In addition, removal of the 15 C-terminal residues of GRβ had no impact on the calculated binding energy. These in silico calculations are consistent with the previous observation of RU-486 being a preferred ligand for GR (19).
TABLE 3.
Binding energy calculationa
| Complexb | Total binding energy (kcal/mol) | Stability gain from protein solution (kcal/mol)c |
|---|---|---|
| hGRβ/RU-486 | −87.2 ± 4.7 | −14.3 |
| hGRβ/RU-486 (Δ15) | −87.1 ± 4.8 | −14.2 |
| hGRα/RU-486 (3H52_A) | −90.4 ± 5.4 | −17.5 |
| hGRα/RU-486 (3H52_B) | −89.1 ± 4.6 | −16.2 |
| hGRα/RU-486 (3H52_C) | −84.6 ± 3.9 | −11.7 |
| hGRα/DEX (1M2Z_A) | −96.7 ± 4.8 | − 4.3 |
Data are represented as means ± SD.
The initial structures for RU-486- and DEX-bound GRα structures were based on the structures from PDB codes 3H52 and 1M2Z, respectively. A, B, and C denote the three conformations of hGRα in the asymmetric unit.
Hydration energies of RU-486 (−72.9 ± 5.6 kcal/mol) and DEX (−92.4 ± 7.7 kcal/mol) were subtracted from the total binding energy.
Coactivator/corepressor peptide binding.
The crystal structure of the nmrGRβ/RU-486 complex displays disorder in the C-terminal region, resulting in a lack or disruption of the secondary structural elements α11 and α12 found in hGRα. As these regions are important in forming the AF-2 domain of hGRα, we wanted to determine how the lack of C-terminal structure might affect coactivator/corepressor binding. To assess the binding capability of coregulators to GR LBDs, we measured fluorescence polarization using fluorescein-labeled coregulator peptides NCoR (corepressor) and TIF2 (coactivator). Solubility-enhancing mutants were used in order to improve the solubility of some GR proteins. To validate that the mutation would not disrupt peptide binding, binding of peptide to the nmrGRβ (F598S) was compared to that of the wild-type nmrGRβ and displayed a similar Kd (dissociation constant) value (Fig. 7A and B). All forms of GR displayed higher affinity for the corepressor NCoR than for the coactivator TIF2 in the presence of RU-486 (Fig. 7), with hGRβ (Fig. 7C) exhibiting a slightly higher Kd value while nmrGRβ demonstrated a 2- to 3-fold lower Kd value than hGRα and nmrGRα (Fig. 7D and E, respectively). Conversely, the GRα constructs displayed higher affinity for TIF2 in the presence of the agonist DEX (Fig. 7F and G). These results suggest that although hGRβ lacks α12, it is capable of preferentially binding to corepressor peptides rather than coactivator peptides in the presence of the antagonist RU-486. In addition, the fact that GRβ (F598S) and GRβ (F598S)Δ15 displayed similar Kd values (Fig. 7B and H) suggests that the unique 15 C-terminal residues of GRβ do not participate substantially in corepressor binding.
FIG 7.

NCoR and TIF2 peptide interaction with GR LBDs. (A to J) Fluorescence polarization was measured using serially diluted ligand binding domains of the indicated proteins and ligands, in the presence of fluorescein-labeled NCoR (5FAM-ASNLGLEDIIRKALMGSFD) or TIF2 (5FAM-EKHKILHRLLQDSY) peptides. The change in millipolarization (mP) is plotted as a function of the concentration of GR with bound ligand in log scale. Data points are averages of triplicate measurements, and values are shown as means ± SD. Kd values are averaged from triplicate measurements and were estimated after fitting of the binding curves. Kd values that were not measurable due to undersaturation are indicated as >100 μM. (K) Model containing hGRβ with V571R and V575R (equivalent to V567R and V571R in nmrGRβ, respectively) was generated using the crystal structure of nmrGRβ with the corepressor peptide from hGRα (PDB ID 3H52) superimposed.
To better understand how corepressor peptide binding contributes to GRβ function, we generated two mutants (V567R and V571R) that disrupted NCoR peptide binding to nmrGRβ (Fig. 7I and J). Based on the crystal structure, these mutants could create a steric clash with the peptide, prohibiting it from binding (Fig. 7K). Despite the inability of these nmrGRβ mutants to interact with the NCoR peptide, the equivalent mutations in hGRβ (V571R and V575R) still displayed dominant negative activity (Fig. 3). This suggests that the dominant negative activity may be independent of corepressor binding or that the AF-2 region is not the predominant corepressor binding site in vivo.
DISCUSSION
Understanding the molecular mechanism of the cellular response to glucocorticoids via hGRβ as well as hGRα will shed light on improved therapeutic solutions to treat GC resistance. RU-486 is the only known ligand of hGRβ controlled gene expression (19). Consistent with this observation, we were able to express soluble GRβ in the presence of 50 μM RU-486, whereas the presence of other ligand molecules such as glucocorticoids (DEX) and estrogens (E1 and E2) did not produce soluble protein. This suggests that among these ligands, RU-486 selectively binds and stabilizes GRβ. The molecular basis for the ligand selectivity was originally attributed to the distinct C-terminally abbreviated sequence (19). However, our findings suggest it is more due to the lack of helices α11 and α12 present in GRα.
In this study, we utilized nmrGRβ as a model system studying molecular mechanisms underlying glucocorticoid resistance, specifically focusing on its role as a dominant negative regulator of GRα. To validate the use of nmrGRβ as a functional model of hGRβ, we generated the chimeric construct GRβ′, with the amino terminal sequence of human GR and carboxy-terminal LBD sequence of nmrGRβ (Fig. 1). Utilizing various assays in U-2 OS cells, GRβ′ behaved indistinguishably from hGRβ in both the presence and absence of RU-486, with respect to cellular localization (Fig. 1D to H), dominant negative activity of GRα function (Fig. 1I and J), and gene regulation (Fig. 2), validating the use of nmrGRβ LBD as a functional surrogate for hGRβ LBD.
The crystal structure of the GRβ/RU-486 complex reveals that the unique C-terminal 15 amino acid residues of GRβ are mostly disordered and not likely to interact with the bound ligand (Fig. 4, 5, and 6). The docking experiment and binding energy calculations from molecular dynamics simulations consistently suggest that RU-486 has the highest ligand binding affinity to GRβ among the GC ligands tested (Tables 2 and 3). RU-486 binding is stabilized by not only the interacting residues but also the shape of the ligand. The hydrophobic propynyl and dimethylnitrophenyl groups of RU-486 fit into hydrophobic pockets, likely enhancing the binding affinity and compensating for the lack of α11 and α12 interactions that appear to be required for high-affinity DEX binding (Table 3 and Fig. 8). Consistent with these observations, the shape score from the FRED docking of RU-486 is the highest among tested agonists and antagonists (−14.8 kcal/mol). The major consequence of RU-486 binding to GRβ is repression of gene transcription (19). This transcriptional repression might be achieved by recruiting corepressors such as NCoR (nuclear receptor corepressor) and SMRT (silencing mediator for retinoid and thyroid receptors), associated with histone deacetylase (HDAC) (37). Despite the absence of α12, the GRβ/RU-486 complex binds corepressor peptides with a slightly higher affinity than the GRα/RU-486 complex. Moreover, the calculated binding energies of RU-486 to hGRα and hGRβ are similar (Table 3). It should be noted our overall Kd values for hGRα binding to TIF2 peptide in the presence of DEX and RU-486 are greater than published previously (13), possibly due to differences in experimental conditions. Differences in protein construct (glutathione S-transferase [GST] versus no tag) and measuring method (surface plasmon resonance versus fluorescence polarization) could also contribute to the difference in Kd values. Our biochemical and structural data suggest that α12 is not likely to contribute to corepressor recruitment to AF-2. This concept is further supported by the dynamic behavior of α12 as α12 adopts multiple conformations in the crystal structure of the hGRα/RU-486 complex (PDB ID 3H52) (16) and is positioned away from the AF-2 region when the nonsteroidal antagonist 29M is bound (PDB ID 4MDD) (M. Coghlan and J. Luz, unpublished data) (Fig. 9). In addition, hydrogen/deuterium exchange experiments on the hGRα/RU-486 complex displayed increased exchange in the AF-2 region (17). These observations suggest that α12 might be dispensable in GR antagonism. While the binding of agonist positions α12 to stabilize the ligand and to recruit coactivator, the binding of antagonist appears to exclude α12 from its coactivator binding position, which favors corepressor binding. We so far have been unable to obtain a crystal structure of the GRβ-NCoR peptide complex. It is conceivable that the corepressor peptide binding site on GRβ is likely to be similar to that seen in GRα crystal structures (PDB IDs 3H52 and 4MDD). Mutations in nmrGRβ residues (V567R and V571R) at the predicted NCoR peptide binding site showed dramatically reduced binding affinity to NCoR peptide in the presence of RU-486 (Fig. 7I and J). Interestingly, equivalent hGRβ mutants (V571R and V575R) displayed wild-type-like dominant negative activity (Fig. 3). This suggests either that the corepressor is dispensable for dominant negative activity of GRβ or that the AF-2 binding site may not represent the major binding interaction in vivo.
FIG 8.
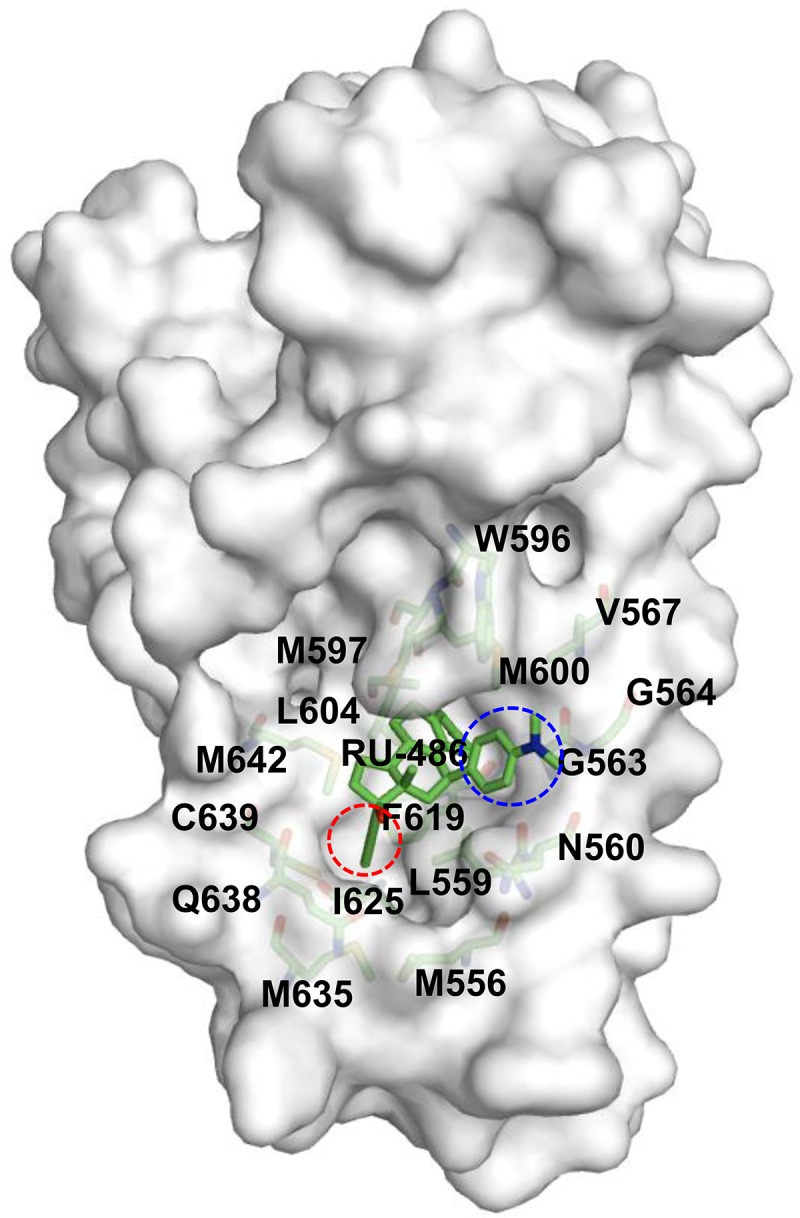
Hydrophobic ligand binding pocket of GRβ. Residues forming the hydrophobic environment for the propynyl group (red circle) are Met556, Leu559, Leu604, Phe619 Ile625 Met635, Gln638, Cys639, and Met642; for the dimethylnitrophenyl group (blue circle) of RU-486, the residues are Asn560, Gly563, Gly564, Val567, Trp596, and Met600.
FIG 9.

Conformational heterogeneity of GR C termini. Crystal structures of GR including nmrGRβ/RU-486 (A), hGRα/DEX (PDB ID 1M2Z) (B), hGRα/29M (PDB ID 4MDD) (C and D), and the chains from hGRα/RU-486 (PDB ID 3H52) (E to H) are shown in white cartoon with corresponding ligands and cofactors. Residues from 740 to the C-terminal end are highlighted in red, and the bound peptides are shown in magenta (TIF2) and pink (NCoR1), respectively. RU-486 (cyan), DEX (cobalt), and 29M (orange) are shown in sticks.
The precise mechanism by which GRβ functions as a dominant negative regulator of GRα and how RU-486 contributes to its function are unclear. In the absence of RU-486, GRβ is already found mainly in the nucleus, where it regulates the expression of many genes, perhaps through forming heterodimeric complexes with GRα (17). Our structure reveals that in the presence of RU-486, the C-terminal tail is disordered, leaving the corepressor binding site exposed and accessible. This observation is consistent with the secondary structure prediction of this region (38). One function of RU-486 might be to displace the C-terminal tail. In the apoprotein form of GRβ, the C-terminal tail may block corepressor binding but be displaced by the binding of RU-486. However, it is also conceivable that the apoprotein form may resemble the RU-486-bound structure and thus be capable of corepressor binding. Interestingly, a heme receptor, Rev-erbα (NR1D1), like hGRβ, also lacks α12 and represses gene activity by associating with NCoR (39, 40). Structural analysis of the Rev-erbα/NCoR complex suggests that Rev-erbα could mediate target gene repression via NCoR binding in the absence of heme (41). Similarly, GRβ might be a constitutive dominant negative repressor of GRα in the absence of ligand via corepressor binding. One effect of having RU-486 present could be to enhance the effective concentration of GRβ in the nucleus. It has been demonstrated that the addition of RU-486 causes slow translocation of transiently expressed cytoplasmic GRβ into the nucleus (19). In addition, we have been unable to obtain soluble recombinant GRβ without RU-486 present during expression or on mutants designed to disrupt RU-486 binding (R607A, Q566A, G563A, and G563S) in the presence or absence of RU-486. These observations suggest that RU-486 stabilizes GRβ, possibly extending its half-life and allowing for higher concentrations in the nucleus. This is also consistent with our modeling studies, which demonstrate enhanced stability of GRβ when RU-486 is bound (Table 3). Of course, one cannot rule out the possibility of a yet undiscovered endogenous ligand that is responsible for the dominant negative behavior of GRβ in the absence of RU-486.
Conclusion.
Our nmrGRβ/RU-486 structure demonstrates that the GR antagonist RU-486 binds in the same position and orientation in the ligand binding pocket as seen in hGRα. The lack of α11 and α12 in GRβ appears not to support high-affinity binding of agonist or proper formation of the AF-2 region for coactivator binding. The bound RU-486 supports an antagonistic conformation of GRβ, allowing for corepressor binding and transcriptional repression of GRα activity. Since the dominant negative activity of nmrGRβ LBD is functionally indistinguishable from that of hGRβ LBD, nmrGRβ may be a useful model system for future studies, including its use as a structural biology tool to delineate molecular mechanisms underlying glucocorticoid resistance.
MATERIALS AND METHODS
Reagents and cloning.
17β-Hydroxy-11β-(4-(dimethylamino)phenyl)-17α-(1-propynyl)estra-4,9-dien-3-one (mifepristone, or RU-486) was purchased from Carbosynth (Berkshire, United Kingdom). CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} was purchased from A. G. Scientific, Inc. (San Diego, CA). Arginine and glutamic acid were purchased from Sigma-Aldrich (St. Louis, MO). Oligonucleotide primers for PCR were synthesized by Eurofins MWG Operon LLC (Huntsville, AL). Heterocephalus glaber, or naked mole rat, GRβ (nmrGRβ) cDNA was synthesized by GenScript USA, Inc. (Piscataway, NJ). Ligand binding domains of hGR (residues 522 to 777 for hGRα and 522 to 742 for hGRβ) and nmrGR (residues 519 to 774 for nmrGRα and 518 to 738 for nmrGRβ) were subcloned into a pSUMO plasmid vector to encode an N-terminal ULP1 cleavable hexahistidine-tagged fusion protein. Site-directed mutagenesis was performed using a QuikChange II kit (Agilent Technologies).
Protein expression and purification.
For expression, Escherichia coli Rosetta2 (DE3) cells containing the pSUMO-GR constructs were grown on shakers overnight in LB medium containing kanamycin (Kan; 50 μg/ml) and chloramphenicol (Chlor; 100 μg/ml). Twenty milliliters of overnight culture was added to 12 flasks containing 1 liter of Terrific broth (Kan/Chlor) and incubated on shakers at 37°C until the optical density at 600 nm (OD600) reached 0.3. RU-486 was added to 50 μM, and the temperature was decreased to 10°C for 1 h. Cultures were induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated on shakers at 10°C overnight. Cells were harvested by centrifugation, and pellets were resuspended in buffer A (25 mM Tris, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.35% CHAPS, 100 mM Arg-Glu, and 1 mM dithiothreitol [DTT]) in the presence of 10 μM RU-486 and cOmplete protease inhibitor tablets (Roche, IN). To obtain dexamethasone-bound GRα, RU-486 was replaced with the same concentration of dexamethasone during the expression and purification steps. For purification, cells were ruptured by sonication, and the debris was removed by centrifugation. All proteins were purified from the soluble fraction by binding to Ni-nitrilotriacetic acid (NTA) affinity resin in batch. The proteins were then eluted with 400 mM imidazole in buffer A, followed by removal of the SUMO tag by ULP1 protease during overnight dialysis at 4°C against buffer A containing 10 μM RU-486 or dexamethasone. The cleaved SUMO and ULP1 protease were removed by passage through Ni-NTA resin. The proteins were further purified by gel filtration with a Superdex S200 HiLoad 26/600 column (GE Health Care) equilibrated with buffer A containing 10 μM RU-486 or dexamethasone.
Crystallization, data collection, and structure determination.
nmrGRβ (F598S) was concentrated to 5 mg/ml after buffer exchange with 25 mM Tris, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.2% CHAPS, 1 mM DTT, and 10 μM RU-486. Crystals for nmrGRβ (F598S) were obtained by sitting-drop vapor diffusion by mixing 250 nl of protein/RU-486 with 250 nl of mother liquor consisting of 0.1 M morpholineethanesulfonic acid (MES), pH 5.0, and 20% 2-methyl-2,4-pentanediol (MPD; vol/vol) at 4°C. For data collection, crystals were transferred to a cryo-solution consisting of an additional 15% ethylene glycol (vol/vol), prior to flash freezing in liquid nitrogen. Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory, at a wavelength of 1.0 Å. All data were processed with HKL2000 (42). To solve the structure, molecular replacement was carried out in Phenix (43) with Phaser (44), using a starting model of hGRα (PDB ID 1M2Z) truncated at residue 727 (13). The structure was refined in Phenix, and manual model building was carried out with Coot (45, 46) (Table 1). Ramachandran statistics were calculated using MolProbity (47).
Docking.
Using the FRED module of the OEDocking suite of the OpenEye software package, various ligands were screened at the ligand binding sites of GRα and GRβ to determine their relative binding affinities (Table 2). RU-486-bound GRα from PDB ID 3H52 and DEX-bound GRα from PDB ID 1M2Z, along with RU-486-bound GRβ, were used as the initial receptor complexes. The Chemgauss4 scoring function, based on shape complementarity and chemical feature alignments, was utilized on the optimized poses.
MD.
To determine in silico ligand binding energies to GRs, molecular dynamics (MD) simulations were carried out by generating solvated structures of GRα and GRβ complexed with RU-486 or DEX (Table 3). The initial structures for RU-486- and DEX-bound GRα structures were based on the X-ray crystal structures from PDB codes 3H52 and 1M2Z, respectively. Since the binding modes of RU-486 were slightly different in three molecules of GRα in the unit cell, we performed three separate simulations for RU-486-bound GRα. Initial structure of the RU-486-bound GRβ were obtained from the X-ray crystal structure with PDB ID 5UC1. The nmrGRβ sequence was converted into the human GRβ sequence. The disordered loops in the structures were generated using Modeller (48, 49). Each system described above was subjected to the following protocol to generate MD trajectories: (i) after addition of protons, solvation of each system in a box of water (between 20,000 to 30,000 water molecules, depending on various systems to accommodate at least 20-Å distance to the box boundary from the closest protein atom), (ii) 500-ps belly dynamics runs with fixed peptide and ligand, (iii) minimization, (iv) low-temperature constant pressure dynamics at fixed protein (and ligand) to ensure a reasonable starting density around 1 g/cc, (v) minimization, (vi) stepwise heating molecular dynamics at constant volume, and (vii) constant volume molecular dynamics for 5 ns for equilibration. All final unconstrained trajectories were calculated at 300 K under constant volume (for 60 to 100 ns, with a time step of 1 fs) using the PMEMD module of Amber, version 14, to accommodate long-range interactions. The amino acid parameters were taken from the Amber.ff14SB force field. Using the combination of Gaussian-09 (revision D01) (for charges at the B3LYP/6-31G* level) and the Antechamber module of Amber.14 (for all other parameters), the force field required for the ligands was created. In addition, following a similar protocol, two MD simulations of RU-486 and DEX solvated in water were performed to estimate the solvation energies of the ligands in water. Interaction energies of protons were estimated using the periodically selected structures from the final 40 ns of dynamics of corresponding trajectories.
Fluorescence polarization assay.
Fluorescence polarization experiments were conducted with a Polarstar Omega plate reader (BMG Labtech), using 480-nm excitation and 520-nm emission filters. The 5FAM-TIF2 (EKHKILHRLLQDSY; FAM is carboxyfluorescein) or 5FAM-NCoR (ASNLGLEDIIRKALMGSFD) peptide (5 nM) was added to serially diluted GR LBD in a total reaction volume of 50 μl. Due to the low solubility of hGR compared to that of nmrGR under the experimental conditions, the highest testable concentrations of hGR and nmrGR were ∼200 μM and ∼300 μM, respectively. Peptides and proteins were diluted in buffer A containing 10 μM dexamethasone or RU-486. The reaction mixture was incubated in a 96-well black, flat-bottom plate (Corning, Inc.) for 15 min on ice, and data were collected at room temperature. Each reaction was performed in triplicate. Data analysis was performed using Kaleidagraph (Synergy Software). The fluorescence polarization as a function of the increasing GR/ligand concentration (micromolar) was fit using the following equation: mP = {(mPbound − mPfree)(GR/ligand)/[Kd + (GR/ligand)]} + mPfree, where mP is the millipolarization (mP = P × 10−3) measured at a given concentration of GR/ligand, mPfree is the initial millipolarization of the free peptide, and mPbound is the maximum millipolarization of specifically bound peptide.
Generation of U-2 OS cell lines stably expressing the chimeric construct hGR1–520/nmrGRβ521–742.
All transfections were performed using a modified U-2 OS cell line that was stably transfected with the pTET-OFF plasmid as previously described, referred to here as U-2 OS cells (50). An nmrGRβ chimeric construct (GRβ′) was generated by ligating the hGR coding region (amino acids 1 to 520) to the nmrGRβ LBD coding region (amino acids 521 to 742) using the following primers: 5′-CGCGGATCCATGGACTCCAAAGAATCATTAACTCCTGGTAGAGAAGAAAACC, 5′-CGGGGTACCGGTACCGTCATATCCTGCATATAACACTTCAGGTTCAATAACC, 5′-CGGGGTACCGTGCCAGACACCACCTGGCGCATTATGACCACCCTGAACATGC, and 5′-ATAGTTTAGCGGCCGCTCACAGAGAGAAGTGGCTGGTGCTCTCAGGCTTCAGCC. After PCR amplification, the chimeric construct was ligated to pTRE2hyg vector digested with BamHI and NotI to make the pTRE2-GRβ′ plasmid. The pTRE2-GRβ′ plasmid was then transfected into U-2 OS cells, and clones that stably expressed GRβ′ were selected as previously described (19, 43, 50).
Immunocytochemistry and Western blot analysis.
Immunocytochemistry and Western blot analysis were performed as previously described (19) on U-2 OS cells stably expressing full-length hGRα, hGRβ, and GRβ′. For the translocation assay, U-2 OS cells were incubated at 37°C with and without 100 μM RU-486 for 3 h, and then cell images were captured using an epifluorescence microscope (Zeiss Axio Observer D1). Quantitative receptor localization analysis was manually evaluated using a scoring system described in a previous study (19). At least 100 cells per treatment condition per experiment were used to assess the fluorescence intensities for equivalently sized regions in the nucleus and cytoplasm. The experiment was performed in triplicate, and data are represented as means ± standard deviations (SD). For Western blot assays, anti-β-actin antibody (1:10,000 dilution; Chemicon, CA), glucocorticoid receptor (D8H2) XP rabbit monoclonal antibody (MAb) (1:2,000 dilution; Cell Signaling Technology), and Alexa Fluor 488-conjugated goat anti-rabbit IgG(H+L) secondary antibody (1:10,000 dilution; Invitrogen) were used as specified by the manufacturers. Protein band images were scanned and analyzed using an Odyssey system (Li-Cor Biosciences, NE) according to the manufacturer's instructions. Band intensities of hGRα, hGRβ, and GRβ′ were normalized against intensities of β-actin. The experiment was performed in triplicate, and data are represented as mean ± SD.
Transient transfections and quantitative RT-PCR analysis.
For the dominant negative assays, the U-2 OS cells stably expressing hGRα were maintained in Dulbecco's modified Eagle medium (DMEM)–F-12 (Invitrogen Life Technologies) supplemented with 5% heat-inactivated fetal calf serum, 50 units/ml penicillin, 0.05 mg/ml streptomycin, 2 mM l-glutamine, 0.2 mg/ml Geneticin (Invitrogen Life Technologies), and 0.2 mg/ml hygromycin B (Invitrogen Life Technologies). All cells were grown at 37°C and 5% CO2 in a humidified incubator and passaged every 3 to 7 days as they approached confluence. The U-2 OS hGRα stably expressing cell line was plated in six-well plates at approximately 70% confluence 1 day before transfection. Cells were transfected with FuGENE 6 transfection reagent as described by the manufacturer (Promega, Madison, WI) using 6 μl of FuGENE 6 and 1.0 μg of DNA (pTRE2-hGRβ or pTRE2-GRβ′) per well or mock transfected (no DNA). After 18 to 24 h, the transfection medium was removed and replaced with fresh DMEM–F-12 maintenance medium containing charcoal-stripped fetal bovine serum instead of fetal calf serum, and the cells were then treated with 100 nM dexamethasone or vehicle (H2O) for 6 h. Total RNA was isolated using a Qiagen RNeasy minikit. Real-time PCR was performed using a 7900HT sequence detection system with predesigned primer/probe sets available from Applied Biosystems (Foster City, CA), according to the manufacturer's instructions. The signal obtained from each gene primer/probe set was normalized to that of the unregulated housekeeping gene, cyclophilin B, primer/probe set (also available from Applied Biosystems). Each primer/probe set was analyzed with at least three different sets of RNA.
For the gene regulation assays, the U-2 OS cell lines stably expressing hGRβ and GRβ′ were plated in DMEM–F-12 charcoal-stripped fetal bovine serum and then treated for 6 h with 1 μM RU-486, and gene regulation was assayed as described above.
Accession number(s).
The atomic coordinates and structure factors of GRβ/RU-486 have been deposited in the RCSB Protein Data Bank under accession number 5UC1.
ACKNOWLEDGMENTS
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ZIA ES102645 to L.C.P.), Z01 ES043010 to L.P., and Z01 ES090057 to J.A.C., and the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences contract W-31-109-Eng-38. The NIEHS Mass Spectrometry Research and Support Group as well as the X-ray Crystallography Core and the Protein Expression Core Facility were utilized for this research.
We thank M. Negishi and R. Oakley for critical reading of the manuscript and J. Williams and R. Oakley for helpful discussions.
REFERENCES
- 1.Sapolsky RM, Romero LM, Munck AU. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21:55–89. [DOI] [PubMed] [Google Scholar]
- 2.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhen T, Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 4.Schacke H, Docke WD, Asadullah K. 2002. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43. doi: 10.1016/S0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 5.Webster JC, Oakley RH, Jewell CM, Cidlowski JA. 2001. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A 98:6865–6870. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beato M. 1989. Gene regulation by steroid hormones. Cell 56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 7.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. 2011. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 8.McKenna NJ, Lanz RB, O'Malley BW. 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 9.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS Jr, Louw A, Hapgood JP. 2009. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol 299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Kumar R, Thompson EB. 2005. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol 94:383–394. doi: 10.1016/j.jsbmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 12.Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R. 2002. RU486-induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem 277:26238–26243. doi: 10.1074/jbc.M203268200. [DOI] [PubMed] [Google Scholar]
- 13.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. 2002. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110:93–105. doi: 10.1016/S0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 14.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. 1985. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oakley RH, Sar M, Cidlowski JA. 1996. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem 271:9550–9559. [DOI] [PubMed] [Google Scholar]
- 16.Schoch GA, D'Arcy B, Stihle M, Burger D, Bar D, Benz J, Thoma R, Ruf A. 2010. Molecular switch in the glucocorticoid receptor: active and passive antagonist conformations. J Mol Biol 395:568–577. doi: 10.1016/j.jmb.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Frego L, Davidson W. 2006. Conformational changes of the glucocorticoid receptor ligand binding domain induced by ligand and cofactor binding, and the location of cofactor binding sites determined by hydrogen/deuterium exchange mass spectrometry. Protein Sci 15:722–730. doi: 10.1110/ps.051781406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. 1995. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. 2007. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol Cell Biol 27:2266–2282. doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oakley RH, Webster JC, Sar M, Parker CR Jr, Cidlowski JA. 1997. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology 138:5028–5038. doi: 10.1210/endo.138.11.5501. [DOI] [PubMed] [Google Scholar]
- 21.Lewis-Tuffin LJ, Cidlowski JA. 2006. The physiology of human glucocorticoid receptor beta (hGRβ) and glucocorticoid resistance. Ann N Y Acad Sci 1069:1–9. doi: 10.1196/annals.1351.001. [DOI] [PubMed] [Google Scholar]
- 22.Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, Chrousos GP, Szefler SJ, Leung DY. 1999. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med 159:1600–1604. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- 23.Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. 1997. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med 186:1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christodoulopoulos P, Leung DY, Elliott MW, Hogg JC, Muro S, Toda M, Laberge S, Hamid QA. 2000. Increased number of glucocorticoid receptor-beta-expressing cells in the airways in fatal asthma. J Allergy Clin Immunol 106:479–484. doi: 10.1067/mai.2000.109054. [DOI] [PubMed] [Google Scholar]
- 25.Longui CA, Vottero A, Adamson PC, Cole DE, Kino T, Monte O, Chrousos GP. 2000. Low glucocorticoid receptor alpha/beta ratio in T-cell lymphoblastic leukemia. Horm Metab Res 32:401–406. doi: 10.1055/s-2007-978661. [DOI] [PubMed] [Google Scholar]
- 26.Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, van Wering ER, Veerman AJ. 1998. Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia–implications for treatment of infants. Leukemia 12:1344–1348. doi: 10.1038/sj.leu.2401129. [DOI] [PubMed] [Google Scholar]
- 27.Shahidi H, Vottero A, Stratakis CA, Taymans SE, Karl M, Longui CA, Chrousos GP, Daughaday WH, Gregory SA, Plate JM. 1999. Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochem Biophys Res Commun 254:559–565. doi: 10.1006/bbrc.1998.9980. [DOI] [PubMed] [Google Scholar]
- 28.Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, Kohgo Y. 2000. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology 118:859–866. doi: 10.1016/S0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 29.Orii F, Ashida T, Nomura M, Maemoto A, Fujiki T, Ayabe T, Imai S, Saitoh Y, Kohgo Y. 2002. Quantitative analysis for human glucocorticoid receptor alpha/beta mRNA in IBD. Biochem Biophys Res Commun 296:1286–1294. doi: 10.1016/S0006-291X(02)02030-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Ouyang Q, Wen ZH, Fiocchi C, Liu WP, Chen DY, Li FY. 2005. Significance of glucocorticoid receptor expression in colonic mucosal cells of patients with ulcerative colitis. World J Gastroenterol 11:1775–1778. doi: 10.3748/wjg.v11.i12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilos DL, Leung DY, Muro S, Kahn AM, Hamilos SS, Thawley SE, Hamid QA. 2001. GRβ expression in nasal polyp inflammatory cells and its relationship to the anti-inflammatory effects of intranasal fluticasone. J Allergy Clin Immunol 108:59–68. doi: 10.1067/mai.2001.116428. [DOI] [PubMed] [Google Scholar]
- 32.Chikanza IC. 2002. Mechanisms of corticosteroid resistance in rheumatoid arthritis: a putative role for the corticosteroid receptor beta isoform. Ann N Y Acad Sci 966:39–48. doi: 10.1111/j.1749-6632.2002.tb04200.x. [DOI] [PubMed] [Google Scholar]
- 33.Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD. 2001. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol 28:2383–2388. [PubMed] [Google Scholar]
- 34.He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. 2015. Human glucocorticoid receptor beta regulates gluconeogenesis and inflammation in mouse liver. Mol Cell Biol 36:714–730. doi: 10.1128/MCB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ligr M, Li Y, Logan SK, Taneja S, Melamed J, Lepor H, Garabedian MJ, Lee P. 2012. Mifepristone inhibits GRβ coupled prostate cancer cell proliferation. J Urol 188:981–988. doi: 10.1016/j.juro.2012.04.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGann M. 2011. FRED pose prediction and virtual screening accuracy. J Chem Infect Model 51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 37.Glass CK, Rosenfeld MG. 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14:121–141. [PubMed] [Google Scholar]
- 38.Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. 2003. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol 23:4319–4330. doi: 10.1128/MCB.23.12.4319-4330.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazar MA, Jones KE, Chin WW. 1990. Isolation of a cDNA encoding human Rev-ErbAα: transcription from the noncoding DNA strand of a thyroid hormone receptor gene results in a related protein that does not bind thyroid hormone. DNA Cell Biol 9:77–83. doi: 10.1089/dna.1990.9.77. [DOI] [PubMed] [Google Scholar]
- 40.Yin L, Lazar MA. 2005. The orphan nuclear receptor Rev-erbα recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol 19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- 41.Phelan CA, Gampe RT Jr, Lambert MH, Parks DJ, Montana V, Bynum J, Broderick TM, Hu X, Williams SP, Nolte RT, Lazar MA. 2010. Structure of Rev-erbα bound to N-CoR reveals a unique mechanism of nuclear receptor-corepressor interaction. Nat Struct Mol Biol 17:808–814. doi: 10.1038/nsmb.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 47.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5:Unit 5.6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webb B, Sali A. 2014. Protein structure modeling with Modeller. Methods Mol Biol 1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- 50.Lu NZ, Cidlowski JA. 2005. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]