

Abstract
Developing catalysts that produce each stereoisomer of a desired product selectively is a longstanding synthetic challenge. Biochemists have addressed this challenge by screening nature’s diversity to discover enzymes that catalyze the formation of complementary stereoisomers. We show here that the same approach can be applied to a new-to-nature enzymatic reaction, alkene cyclopropanation via carbene transfer. By screening diverse native and engineered heme proteins, we identified globins and serine-ligated “P411” variants of cytochromes P450 with promiscuous activity for cyclopropanation of unactivated alkene substrates. We then enhanced their activities and stereoselectivities by directed evolution: just 1–3 rounds of site-saturation mutagenesis and screening generated enzymes that transform unactivated alkenes and electron-deficient alkenes into each of the four stereoisomeric cyclopropanes with up to 5,400 total turnovers and 98% enantiomeric excess. These fully genetically encoded biocatalysts function in whole Escherichia coli cells in mild, aqueous conditions and provide the first example of enantioselective, intermolecular iron-catalyzed cyclopropanation of unactivated alkenes.
Short abstract
Using their native heme cofactor, engineered globins and serine-ligated cytochromes P450 catalyze carbene transfer to unactivated and electron-deficient alkenes to give four stereodivergent products.
Introduction
The biological world is a marvelous ensemble of chiral molecules. From the amino acid and nucleoside building blocks that form proteins and DNA to intricate natural products produced by living organisms, chirality dictates how molecules interact with living systems.1 Modern medicines often draw inspiration from chiral natural products.2 Because alternate stereoisomers can have very different biological effects,3 characterization of novel bioactive compounds during drug candidate screening should include testing each stereoisomer.4 Developing stereodivergent syntheses, where a set of complementary catalysts can generate every possible stereoisomer of the product, is therefore useful and is actively sought after in catalysis.5 Enzymes are green, sustainable options for stereoselective catalysis, and stereocomplementary enzymes can often be found in nature’s diversity: lipases,6 ketoreductases,7 and transaminases8 chosen using genome mining,9 for example, have all afforded products with different stereoselectivities. We show here that natural protein diversity can be leveraged in a similar fashion to achieve stereodivergence for a new, non-natural enzyme-catalyzed reaction, cyclopropanation of unactivated alkenes via carbene transfer. Cyclopropanes are found in a variety of natural products. They are commonly biosynthesized through intramolecular cyclization of cationic or radical intermediates catalyzed by terpene cyclases, desaturases, or SAM-dependent enzymes. The formation of cyclopropane moieties via carbene transfer to alkenes, widely used in synthetic chemistry, is not known in nature.10
Previous work from this group and others has shown that iron–porphyrin (heme) proteins can be engineered to catalyze the cyclopropanation of styrenyl alkenes with ethyl diazoacetate (EDA, 1).11−14 This new-to-nature carbene transfer reaction has been applied in the synthesis of key pharmaceutical intermediates such as levomilnacipran,15 ticagrelor,16,17 and tasimelteon.17 Work from several groups has also shown that cytochromes P450 and myoglobins can be engineered to switch their diastereo- and/or enantioselectivity preference toward styrenyl olefin cyclopropanation,11,13,17 but none of these precedents yielded all possible stereoisomers in high turnover and selectivity (Supplemental Table 1a). In addition, alkene cyclopropanation by heme proteins with the native iron cofactor has been limited to styrenyl and other activated alkenes. Unactivated, aliphatic alkenes are attractive feedstocks for chemical synthesis, but their transformation to higher value chiral products is challenging due to their inert nature, high degree of conformational flexibility, and limited steric and electronic bias to guide stereocontrol.18
State-of-the-art methods for enantioselective unactivated alkene cyclopropanation often rely on noble metals,19−21 with only a few examples of Co-22 and Cu-based23 catalysts (Supplemental Table 1b); no iron-based catalyst for the enantioselective intermolecular cyclopropanation of unactivated alkenes has been reported. Recent work by Hartwig and others has shown that heme-binding proteins metalated with an artificial iridium-porphyrin cofactor can be leveraged for these20,21,24 and other challenging carbene transfer reactions,25 where the protein active site enhances the stereoselectivity and rate of the iridium-catalyzed reaction. Our group has previously used directed evolution of heme proteins to access carbene transfer reactions performed with noble-metal catalysts, such as carbon–silicon bond formation26 and intermolecular C–H amination.27 We therefore set out to create a collection of genetically encoded biocatalysts equipped with the native heme cofactor for stereodivergent unactivated alkene cyclopropanation, taking advantage of the natural diversity of heme proteins to identify suitable starting activity and stereoselectivity.
Results and Discussion
In order to find initial enzymatic activity for unactivated alkene cyclopropanation, we collected a panel of 11 heme proteins from thermophilic and hyperthermophilic bacteria and archaea (Supplemental Table 2), as thermostable proteins can better withstand the potentially destabilizing effects of mutations and are therefore more “evolvable”.28 They are also often easier to work with and better tolerate polar organic solvents used to solubilize substrates. These heme proteins were initially tested with mutations to the putative distal ligand and active-site entrance loop (similar mutations have been found by Fasan and co-workers to enhance myoglobin-catalyzed cyclopropanation of vinylarenes12) for the cyclopropanation of 1,7-octadiene (Supplemental Table 2). Protein variants that showed catalytic activity were subsequently tested with their mutations removed for 1-octene cyclopropanation. Through these experiments, wild-type Aeropyrum pernix protoglobin (ApePgb WT, UniProt ID: Q9YFF4) and wild-type Rhodothermus marinus nitric oxide dioxygenase (RmaNOD WT, UniProt ID: D0MGT2) were found to have low but measurable cyclopropanation activity on 1-octene (2a), catalyzing the reaction with 18 and 27 total turnovers per enzyme active site (TTN). Notably, ApePgb WT and RmaNOD WT displayed complementary diastereoselectivity, preferentially producing cis (1R,2S)-3a and trans (1S,2S)-3a, respectively.
In addition to searching globin protein diversity for this novel reactivity, we also investigated heme proteins obtained in previous directed evolution studies. A panel of 36 variants of a Bacillus megaterium cytochrome (P450BM3) engineered for other non-natural carbene and nitrene transfer reactions14 was tested for the ability to cyclopropanate 2a and 4-phenyl-1-butene (2b). 2b was chosen as a substrate for library screening, because the UV–visible phenyl group enables screening by HPLC-UV. BM3 variant P411-CIS L437F T438Q L75Y L181I (P411-UA, DNA sequence in Supplemental Table 12) showed significant activity and selectivity for production of cis (1S,2R)-3a, the third of the four possible isomers. This variant of a serine-ligated “P411” (P411-CIS29) had been engineered for cyclopropanation reactivity on electron-rich, non-styrenyl alkenes such as N-vinyl amides.30
Site-saturation mutagenesis libraries were generated and screened to increase the activities and selectivities of the different enzymes. Because crystal structures of ApePgb and RmaNOD have not been reported, homology models were built to help us identify residues within the putative distal heme pocket, where carbenoid formation and substrate binding are predicted to take place (Supplemental Figure 2). P411-UA residues were selected based on the crystal structure of its P411-CIS predecessor (PDB ID: 4H23). Individual site-saturation libraries were screened for increased activity and diastereoselectivity using 2b and 1 as substrates. Variants with enhanced diastereoselectivity in the production of 3b were regrown in larger scale, and their activities were tested in the cyclopropanation of 2b and 2a with 1. Enzyme variants with the greatest overall selectivity enhancements against 3a and 3b were used as parents in the next rounds of site-saturation mutagenesis and screening. A single mutation (Q52V) gave RmaNOD near-perfect stereoselectivity for producing trans (1S,2S)-3a. Three mutations (W59A Y60G F145W, or “AGW”) gave ApePgb the ability to make cis (1R,2S)-3a with 89:11 diastereomeric ratio (dr) and 99% enantiomeric excess (ee). During screening to increase P411-UA’s cis diastereoselectivity, a single mutation, V87F, was found to completely invert the diastereoselectivity from 89:11 cis (1S,2R)-3a to 4:96 trans (1R,2R)-3a, affording the fourth and final stereoisomer we needed. Residue 87 is known to modulate the stereoselectivity of P450BM3 for oxygenation of various substrates.31
With initial screening of 11 new and 36 previously engineered heme proteins, followed by just one to three rounds of site-saturation mutagenesis, we discovered four protein variants capable of cyclopropanating unactivated alkenes (RmaNOD Q52V, ApePgb W59A Y60G F145W (= ApePgb AGW), P411-UA-V87C, and P411-UA-V87F), each of which produced a distinct stereoisomer of the desired product 3a with 89:11 to <1:99 dr and 96% to >99% ee (Figure 1). The enzyme activities against unactivated alkenes are comparable to those of the state-of-the-art catalysts, with 100–490 TTN for 3a and as high as 2,400 TTN for 3b, the substrate against which the enzymes were screened. The system is straightforward and easy to use: the protein-expressing bacterial cells need only be resuspended to the desired concentration and the alkene and diazo ester added directly under an anaerobic atmosphere. When the reaction is complete, the product is extracted into organic solvents for analysis or purification. While these enzymes were optimized for use in whole cells, they also function to some degree in lysates and as purified proteins (Supporting Information).
Figure 1.
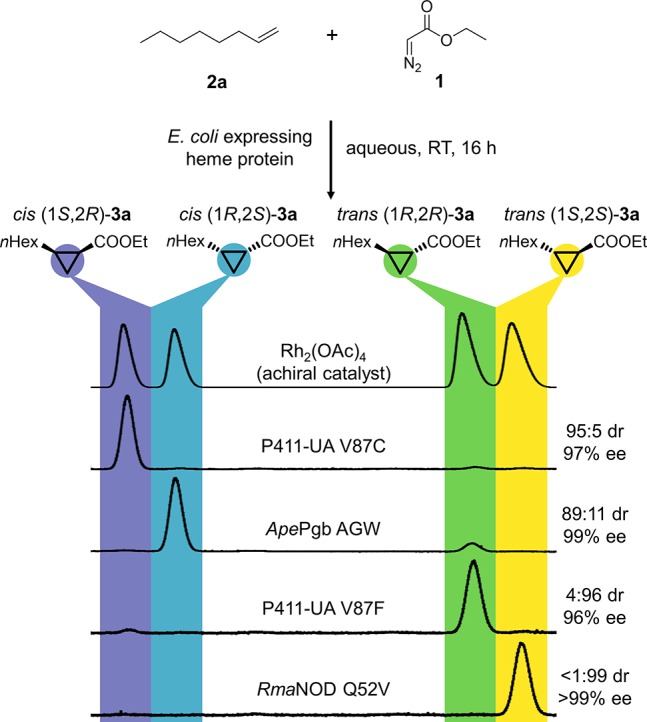
Stereoselective enzymatic cyclopropanation of the aliphatic alkene 2a and 1 to obtain each of four stereoisomers of cyclopropane product 3a with diastereoselectivies from 89:11 to <99:1 dr and enantioselectivies from 96% to >99% ee. Reaction conditions: whole E. coli cells in M9-N buffer, 25 mM glucose, 10 mM 2a, direct addition of 20 mM 1 under anaerobic conditions, 5% ethanol cosolvent. The diastereoselectivity ratio (dr) is given as cis:trans, and the enantiomeric excess (ee) is given for the major diastereomer. Catalysts used: rhodium acetate dimer (Rh2(OAc)4) to form the racemic authentic standard, two variants of the engineered, serine-ligated cytochrome P450BM3 (P411-UA-V87C and P411-UA-V87F), Aeropyrum pernix protoglobin W59A Y60G F145W (ApePgb AGW), and Rhodothermus marinus nitric oxide dioxygenase Q52V (RmaNOD Q52V). Protein sequences are available in the Supporting Information. Abbreviations used: RT, room temperature; nHex, n-hexyl.
The four engineered biocatalysts were tested on a range of alkenes. Their activities and selectivities were high on unbranched aliphatic alkenes similar to those for which they were engineered, but their substrate scope extends to sterically hindered and electron-deficient alkenes as well (Figure 2). Though the activity and stereoselectivity differed on different substrates, each catalyst accepted most of the substrates tested. It is likely that activity on specific substrates can be optimized further, if desired, as has been shown in many other directed evolution studies.32,33
Figure 2.

Cyclopropanation substrate scope. (a) Activity and selectivity of each protein variant against 3a, 3b, and 3c. (b) Activity and selectivity against a variety of alkenes. The diastereoselectivity ratio (dr) is given as cis:trans, and the enantiomeric excess (ee) is given for the major diastereomer. Enzyme variant used is denoted by superscripts: 1, ApePgb AGW; 2, RmaNOD Q52V. General reaction conditions: whole E. coli cells (OD600 = 5 (ApePgb AGW, RmaNOD Q52V), OD600 = 20 (P411-UA-V87C, P411-UA-V87F)) in M9-N buffer, 25 mM glucose, 10 mM alkene, direct addition of 20 mM 1 under anaerobic conditions, 5% ethanol cosolvent. Modified reaction conditions: 3g OD600 = 20; 3e OD600 = 10. RmaNOD Q52V 3a, OD600 = 10. Analytical yields for these reactions are given in Supplemental Table 8. The absolute configurations of products 3b and 3c are assigned by analogy to the 3a products (see the Supporting Information section “Determination of absolute configurations of the cyclopropane products” for details). Chiral separation conditions reported in the Supporting Information. *The benzyl ester of 3c has IUPAC naming priority, and therefore the chiral carbon numbering is reversed for these compounds.
We have shown that these protein variants function with the commonly used diazo carbene precursor EDA, which is especially useful because the ester moiety can be further derivatized, e.g., to form amides and other esters. It can also be reduced to alcohols and aldehydes, which are versatile synthetic handles for accessing a broad range of functional groups. Reports of cyclopropanation via trifluoromethylcarbene transfer to alkenes,34 as well as carbene transfer to silanes and boranes have shown that enzymes can also be engineered to use different carbene precursors.26,35
The small-molecule-catalyzed enantioselective preparation of cyclopropyl esters from electron-deficient alkenes has previously been limited to making the trans-cyclopropanes,36 whereas strategies to directly access 1-keto-2-ester or 1,2-diester cis-cyclopropanes (or their corresponding carboxylates) via enantioselective cyclopropanation are unknown. The biocatalysts, in contrast, enable access to the cis-1-keto-2-ester and cis-1,2-diester products in a single, intermolecular step using an Escherichia coli based platform (cis-3c, cis-3g, Figure 2). Some of these products are precursors to valuable compounds: cyclopropyl esters of unbranched, aliphatic alkenes are used in fragrances, for example, including the essential odorants in frankincense.37 Notably, the enzymes catalyze the reaction on 2-vinylpyridine (2h), which is a difficult substrate for many catalysts due to pyridine’s propensity to coordinate to and inhibit metal centers. This cyclopropanation product is a precursor for an orphan GPR88 agonist.38 A similar vinylpyridine substrate was used in myoglobin-catalyzed cyclopropanation.17
Enzymes are chemoselective and can generate desired products without additional steps to protect and deprotect other reactive functional groups on the same molecule. As shown in Figure 3, the enzymes described here, for example, can selectively cyclopropanate terminal alkenes in the presence of alcohol and carboxylic acid functional groups which often undergo competitive O–H insertion reactions with small-molecule carbene transfer catalysts like rhodium acetate dimer.39ApePgb AGW performed particularly well with unprotected 7-octen-1-ol (2i) and 7-octen-1-oic acid (2j), yielding products cis-3i and cis-3j at 77% and 64% isolated yield, respectively, in preparative-scale reactions. When functional groups cannot be protected easily, chemo- and regioselectivity is even more important. In the cases of (E)-penta-1,3-diene (2k) and (Z)-penta-1,3-diene (2l), all four engineered proteins cyclopropanate the terminal alkene with perfect regioselectivity, likely due to higher accessibility of this double bond in each enzyme’s active site. The diastereoselectivity varied for 3k and 3l, though the enantioselectivity for the major isomer remained high. As the electronic properties of 2k and 2l are similar, the difference in stereoselectivity likely reflects steric constraints of the enzyme active sites.
Figure 3.

Selective, preparative-scale cyclopropane syntheses from various aliphatic alkenes and dienes. Preparative-scale reactions against substrates with free alcohol (7-octen-1-ol, 2i), free carboxylic acid (7- octen-1-oic acid, 2j), the two geometric isomers of 1,3-pentadiene (2k, 2l), and ketone (5-hexen-2-one, 2m). The diastereoselectivity ratio (dr) is given as cis:trans, and the enantiomeric excess (ee) is given for the major diastereomer. The yields are reported for isolated products. Enzyme variant used is denoted by superscripts: 1, ApePgb AGW; 2, RmaNOD Q52V. Reaction and chiral separation conditions are available in the Supporting Information.
Citing the need for a greater reactivity of the metal center to cyclopropanate unactivated alkenes, Hartwig, Clark, and co-workers showed that heme proteins could bind an artificial iridium cofactor in place of iron heme for carbene transfer chemistry.20 They showed that a protein’s active site can confer selectivity to noble-metal, small-molecule catalysts that can already catalyze the reaction.20,21,25 Use of an artificial iridium cofactor (Ir(Me)PIX) required the lysis, purification, and in vitro metalation of the apoprotein with the Ir(Me)PIX, all of which add time and cost to catalyst preparation. Though it may be possible to incorporate the new metal in vivo,40 the synthetic, noble-metal cofactor is more expensive than the native heme, which is manufactured by the cell and loaded into the catalyst during protein expression in vivo. The use of iridium is also not ideal due to the negative impact mining and refining precious metals has on the environment.41 The Ir(Me)PIX system, however, has the advantage that it can cyclopropanate internal alkenes21 and catalyze carbene insertion into C–H bonds,25 neither of which have been reported yet with heme proteins.
There is precedent that iron–porphyrin catalysts are capable of unactivated alkene cyclopropanation. Two decades ago, Woo and co-workers showed that iron meso-tetrakis(pentafluorophenyl)porphyrin chloride (Fe(PFP)Cl) can catalyze the reaction of 2-ethyl-1-butene and EDA with 390 TTN; they reported the formation of cyclopropane products using 1-decene as well.42 In fact, we observed that heme in aqueous buffer, with no protein, can catalyze the formation of 3a, albeit with only 0.4 TTN. This basal activity is greatly enhanced and stereoselectivity is enforced by the protein environment, allowing the heme proteins described here to cyclopropanate a range of alkenes from electron-rich conjugated dienes to electron-deficient vinyl ketones and acrylates with high diastereo- and enantioselectivity. The primary factor in determining activity appears to be the binding of the alkene in a productive configuration: the heme’s local protein environment can be molded to enhance activity and selectivity by optimizing the substrate binding modes. Different local heme environments can be accessed by screening natural and engineered protein diversity. Directed evolution then fine-tunes these features.
Metalloporphyrin catalysts have been used in synthetic chemistry for decades, but nature has used them for millions of years. Present in all forms of life on Earth, heme-binding proteins have diverse functions as well as promiscuous activities for which they were never selected, such as the ability to form reactive carbene intermediates. We have taken advantage of this natural diversity to find catalysts for reactions not known to be catalyzed in biology, but that are synthetically useful and are driven by a synthetic carbene precursor (EDA).
While biocatalysts often possess very high selectivity, this selectivity can be synthetically limiting. A single enzyme may make only a single isomer, but access to other isomers may be equally important. Natural diversity can be leveraged effectively for this challenge. A combination of natural diversity and directed evolution let us realize the stereodivergent cyclopropanation of unactivated and electron-deficient alkenes in mild, aqueous conditions with a fully genetically encoded heme protein expressed in bacteria. This set of biocatalysts can serve as starting points for green, sustainable synthesis of valuable cyclopropanated products.
Acknowledgments
This work was supported by the National Science Foundation Division of Molecular and Cellular Biosciences (Grant MCB-1513007) and the Office of Chemical, Bioengineering, Environmental and Transport Systems SusChEM Initiative (Grant CBET-1403077). The authors thank Dr. Nathan Dalleska, Aurapat Ngamnithiporn, and Dr. Scott C. Virgil for analytical chiral GC support, and Dr. Stephan C. Hammer and Dr. Xiongyi Huang for helpful discussions and critical reading of the manuscript. A.M.K. gratefully acknowledges support from Caltech’s Center for Environmental Microbial Interactions and the NSF Graduate Research Fellowship (Grant No. DGE-1745301). R.D.L. is supported by an NIH–National Research Service Award training grant (5 T32 GM07616). O.F.B. acknowledges support from the Deutsche Forschungsgemeinschaft (Grant No. BR 5238/1-1) and the Swiss National Science Foundation (Grant No. P300PA-171225). A provisional patent application has been filed through the California Institute of Technology based on the results presented here.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00548.
Materials and experimental methods, detailed protein engineering strategies for each variant, and compound characterization (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Finefield J. M.; Sherman D. H.; Kreitman M.; Williams R. M. Enantiomeric natural products: occurrence and biogenesis. Angew. Chem., Int. Ed. 2012, 51, 4802–4836. 10.1002/anie.201107204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra B. B.; Tiwari V. K. Natural products: an evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. 10.1016/j.ejmech.2011.07.057. [DOI] [PubMed] [Google Scholar]
- Shi S.-L.; Wong Z. L.; Buchwald S. L. Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 2016, 532, 353–356. 10.1038/nature17191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks W. H.; Guida W. C.; Daniel K. G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. 10.2174/156802611795165098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautwald S.; Carreira E. M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 2017, 139, 5627–5639. 10.1021/jacs.6b13340. [DOI] [PubMed] [Google Scholar]
- Miller C. A. Advances in enzyme discovery technology: capturing diversity. Inform 2000, 11, 489–496. [Google Scholar]
- Kaluzna I. A.; Matsuda T.; Sewell A. K.; Stewart J. D. Systematic investigation of Saccharomyces cerevisiae enzymes catalyzing carbonyl reductions. J. Am. Chem. Soc. 2004, 126, 12827–12832. 10.1021/ja0469479. [DOI] [PubMed] [Google Scholar]
- Höhne M.; Schätzle S.; Jochens H.; Robins K.; Bornscheuer U. T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. 10.1038/nchembio.447. [DOI] [PubMed] [Google Scholar]
- Ferrer M.; Martínez-Abarca F.; Golyshin P. N. Mining genomes and ’metagenomes’ for novel catalysts. Curr. Opin. Biotechnol. 2005, 16, 588–593. 10.1016/j.copbio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Wessjohann L. A.; Brandt W.; Thiemann T. Biosynthesis and metabolism of cyclopropane rings in natural compounds. Chem. Rev. 2003, 103, 1625–1648. 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 2013, 339, 307–310. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Bordeaux M.; Tyagi V.; Fasan R. Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew. Chem., Int. Ed. 2015, 54, 1744–1748. 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gober J. G.; Rydeen A. E.; Gibson-O’Grady E. J.; Leuthaeuser J. B.; Fetrow J. S.; Brustad E. M. Mutating a highly conserved residue in diverse cytochrome P450s facilitates diastereoselective olefin cyclopropanation. ChemBioChem 2016, 17, 394–397. 10.1002/cbic.201500624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenberg O. F.; Fasan R.; Arnold F. H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 2017, 47, 102–111. 10.1016/j.copbio.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. J.; Renata H.; Peck N. E.; Farwell C. C.; Coelho P. S.; Arnold F. H. Improved cyclopropanation activity of histidine-ligated cytochrome P450 enables the enantioselective formal synthesis of levomilnacipran. Angew. Chem., Int. Ed. 2014, 53, 6810–6813. 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez K. E.; Renata H.; Lewis R. D.; Kan S. B. J.; Zhang C.; Forte J.; Rozzell D.; Mcintosh J. A.; Arnold F. H. Highly stereoselective biocatalytic synthesis of key cyclopropane intermediate to ticagrelor. ACS Catal. 2016, 6, 7810–7813. 10.1021/acscatal.6b02550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj P.; Sreenilayam G.; Tyagi V.; Fasan R. Gram-scale synthesis of chiral cyclopropane-containing drugs and drug precursors with engineered myoglobin catalysts featuring complementary stereoselectivity. Angew. Chem., Int. Ed. 2016, 55, 16110–16114. 10.1002/anie.201608680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs J. R.; Morken J. P. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem., Int. Ed. 2016, 55, 2636–2649. 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suematsu H.; Kanchiku S.; Uchida T.; Katsuki T. Construction of aryliridium–salen complexes: enantio- and cis-selective cyclopropanation of conjugated and nonconjugated olefins. J. Am. Chem. Soc. 2008, 130, 10327–10337. 10.1021/ja802561t. [DOI] [PubMed] [Google Scholar]
- Key H. M.; Dydio P.; Clark D. S.; Hartwig J. F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key H. M.; Dydio P.; Liu Z.; Rha J. Y.-E.; Nazarenko A.; Seyedkazemi V.; Clark D. S.; Hartwig J. F. Beyond iron: iridium-containing P450 enzymes for selective cyclopropanations of structurally diverse alkenes. ACS Cent. Sci. 2017, 3, 302–308. 10.1021/acscentsci.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Wang Y.; Cui X.; Wojtas L.; Zhang X. P. Metalloradical activation of α-formyldiazoacetates for the catalytic asymmetric radical cyclopropanation of alkenes. Chem. Sci. 2017, 8, 4347–4351. 10.1039/C7SC00658F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minuth T.; Boysen M. M. K. Carbohydrate-derived bis(oxazoline) ligand in the total synthesis of grenadamide. Synthesis 2010, 2010 (16), 2799–2803. 10.1055/s-0030-1258143. [DOI] [Google Scholar]
- Reynolds E. W.; Schwochert T. D.; McHenry M. W.; Watters J. W.; Brustad E. M. Orthogonal expression of an artificial metalloenzyme for abiotic catalysis. ChemBioChem 2017, 18, 2380–2384. 10.1002/cbic.201700397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Nazarenko A.; Rha J. Y.-E.; Seyedkazemi V.; Clark D. S.; Hartwig J. F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. 10.1126/science.aah4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan S. B. J.; Lewis R. D.; Chen K.; Arnold F. H. Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048–1051. 10.1126/science.aah6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Zhang R. K.; Buller A. R.; Brinkmann-Chen S.; Arnold F. H. Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme. Nat. Chem. 2017, 9, 629–634. 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J. D.; Labthavikul S. T.; Otey C. R.; Arnold F. H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 5869–5874. 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins. Nat. Chem. Biol. 2013, 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenberg O. F.; Prier C. K.; Chen K.; Knight A. M.; Wu Z.; Arnold F. H.. Stereoselective enzymatic synthesis of heteroatom-substituted cyclopropanes. ACS Catal. 2018,in press. DOI: 10.1021/acscatal.7b04423. [DOI] [Google Scholar]
- Vottero E.; Rea V.; Lastdrager J.; Honing M.; Vermeulen N. P. E.; Commandeur J. N. M. Role of residue 87 in substrate selectivity and regioselectivity of drug-metabolizing cytochrome P450 CYP102A1M11. JBIC, J. Biol. Inorg. Chem. 2011, 16, 899–912. 10.1007/s00775-011-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N. J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573. 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- Cobb R. E.; Chao R.; Zhao H. Directed evolution: past, present, and future. AIChE J. 2013, 59, 1432–1440. 10.1002/aic.13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco A.; Steck V.; Tyagi V.; Fasan R. Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via myoglobin-catalyzed transfer of trifluoromethylcarbene. J. Am. Chem. Soc. 2017, 139, 5293–5296. 10.1021/jacs.7b00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan S. B. J.; Huang X.; Gumulya Y.; Chen K.; Arnold F. H. Genetically programmed chiral organoborane synthesis. Nature 2017, 552, 132–136. 10.1038/nature24996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Ruppel J. V.; Zhang X. P. Cobalt-catalyzed asymmetric cyclopropanation of electron-deficient olefins. J. Am. Chem. Soc. 2007, 129, 12074–12075. 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]
- Cerutti-Delasalle C.; Mehiri M.; Cagliero C.; Rubiolo P.; Bicchi C.; Meierhenrich U. J.; Baldovini N. The (+)-cis- and (+)-trans-olibanic acids: key odorants of frankincense. Angew. Chem., Int. Ed. 2016, 55, 13719–13723. 10.1002/anie.201605242. [DOI] [PubMed] [Google Scholar]
- Jin C.; Decker A. M.; Huang X.-P.; Gilmour B. P.; Blough B. E.; Roth B. L.; Hu Y.; Gill J. B.; Zhang X. P. Synthesis, pharmacological characterization, and structure–activity relationship studies of small molecular agonists for the orphan GPR88 receptor. ACS Chem. Neurosci. 2014, 5, 576–587. 10.1021/cn500082p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noels A. F.; Demonceau A.; Petiniot N.; Hubert A. J.; Teyssié P. Transition-metal-catalyzed reaction of diazocompounds, efficient synthesis of functionalized ethers by carbene insertion into the hydroxylic bond of alcohols. Tetrahedron 1982, 38, 2733–2739. 10.1016/0040-4020(82)80031-8. [DOI] [Google Scholar]
- Sreenilayam G.; Moore E. J.; Steck V.; Fasan R. Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts. Adv. Synth. Catal. 2017, 359, 2076–2089. 10.1002/adsc.201700202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss P.; Eckelman M. J. Life cycle assessment of metals: a scientific synthesis. PLoS One 2014, 9, e101298. 10.1371/journal.pone.0101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J. R.; Hamaker C. G.; Djukic J.; Kodadek T.; Woo L. K. Shape and stereoselective cyclopropanation of alkenes catalyzed by iron porphyrins. J. Am. Chem. Soc. 1995, 117, 9194–9199. 10.1021/ja00141a011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.