

Abstract
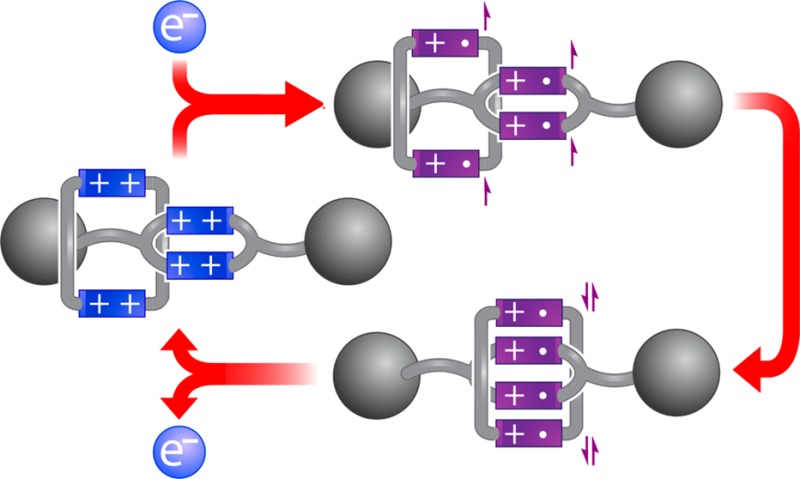
The trisradical recognition motif between a 4,4′-bipyridinium radical cation and a cyclo-bis-4,4′-bipyridinium diradical dication has been employed previously in rotaxanes to control their nanomechanical and electronic properties. Herein, we describe the synthesis and characterization of a redox-active ring-in-ring [2]rotaxane BBR·8PF6 that employs a tetraradical variant of this recognition motif. A square-shaped bis-4,4′-bipyridinium cyclophane is mechanically interlocked around the dumbbell component of this rotaxane, and the dumbbell itself incorporates a smaller bis-4,4′-bipyridinium cyclophane into its covalently bonded structure. This small cyclophane serves as a significant impediment to the shuttling of the larger ring across the dumbbell component of BBR8+, whereas reduction to the tetraradical tetracationic state BBR4(+•) results in strong association of the two cyclophanes driven by two radical-pairing interactions. In these respects, BBR·8PF6 exhibits qualitatively similar behavior to its predecessors that interconvert between hexacationic and trisradical tricationic states. The rigid preorganization of two bipyridinium groups within the dumbbell of BBR·8PF6 confers, however, two distinct properties upon this rotaxane: (1) the rate of shuttling is reduced significantly relative to those of its predecessors, resulting in marked electrochemical hysteresis observed by cyclic voltammetry for switching between the BBR8+/BBR4(+•) states, and (2) the formally tetraradical form of the rotaxane, BBR4(+•), exhibits a diamagnetic ground state, which, as a result of the slow shuttling motions within BBR4(+•), has a long enough lifetime to be characterized by 1H NMR spectroscopy.
Short abstract
A rotaxane was designed using a redox-active ring-in-ring recognition motif to create a nanoswitch that exhibits hysteresis for switching between states with strikingly different electronic features.
Introduction
Mechanically interlocked molecules1−17 (MIMs) are of interest on account of their unique chemical,18−22 physical,23−27 and electronic properties.28−36 Their distinct characteristics have been employed in a variety of advanced nanomaterials, including nanoswitches,28−32,36−42 molecular muscles,43−48 molecular motors,49−51 and nanoscopic stabilized radicals.33,34,52,53 The features of these nanomaterials are realized, at least in part, by the control over the individual component’s relative motion,6 or lack thereof, that is a consequence of the mechanical link. Thus, it is of interest to develop even more precise methods for controlling the movement and spatial arrangement of the mechanically interlocked components, while paradoxically, the practical demands of possible applications54 require that this control be achieved using minimal synthetic effort.
The use55−57 of radical-pairing interactions58−62 to template the formation of catenanes and rotaxanes has been a significant recent development55−57 in the field of functional MIMs, not only offering a facile means to their synthesis but also imbuing the products with distinct electronic properties33,34 and stimulus responsive motion.32,33,48 This research began with the discovery63,64 that the dicationic diradical state of cyclobis(paraquat-p-phenylene), CBPQT2(+•), and the monocationic monoradical state of methyl viologen, MV+•, form a strongly associated tricationic trisradical complex [CBPQT⊂MV]3(+•). Variations of this host–guest complex have been used (Scheme 1) to template55−57 the formation of a variety of MIMs, such as the [2]rotaxane57CBPQT-RV-[2]R6+ and the homo[2]catenane34bis-CBPQT-[2]C(7+)(1•). The restricted motion of the interlocked cyclophanes in bis-CBPQT-[2]C(7+)(1•) results in this MIM exhibiting an air-stable radical state,34 while reduction and oxidation of CBPQT-RV-[2]R6+ toggle between strong attraction and repulsion of its viologen components.57 These features are promising for applications in molecular electronics32,36/muscles47,48/machines,65,66 but there are currently limitations. For example, bis-CBPQT-[2]C(7+)(1•) exhibits only one translational state,34 while CBPQT-RV-[2]R6+ exhibits33 very rapid redox-stimulated shuttling behavior. As a consequence, both MIMs have limited potential for hysteretic28,30,32 electrochemical behavior that is desired for electronic applications. Additionally, rotaxanes based on the trisradical complex [CBPQT⊂MV]3(+•) are inherently paramagnetic in their reduced states, a property which prevents characterization of these states by the easily accessible and well-developed techniques of NMR spectroscopy.67
Scheme 1. Structural Formulas and Space-Filling Representations of Examples of Radical Host–Guest Complexes and Their Mechanically Interlocked Derivatives.
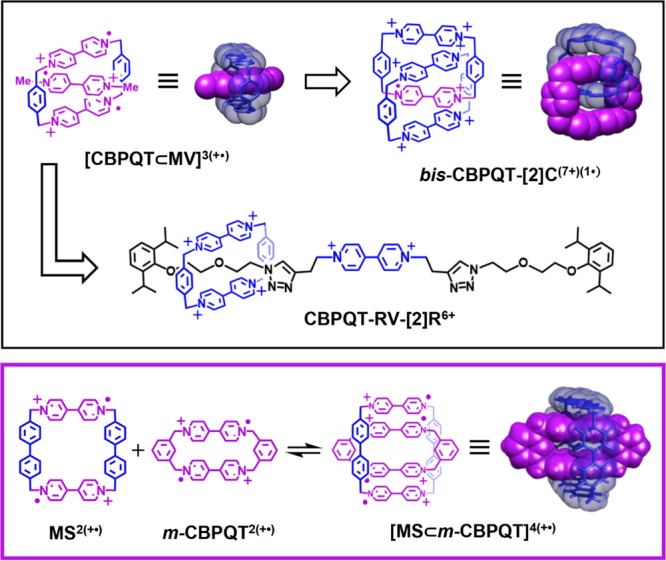
The radical ring-in-ring complex68[MS⊂m-CBPQT]4(+•)—formed between a small dicationic diradical cyclophane59m-CBPQT2(+•) and a dicationic diradical molecular square69MS2(+•)—offers a possible solution to the shortcomings of the first generation of radical MIMs. Although [MS⊂m-CBPQT]4(+•) resembles (Scheme 1) an expanded version of [CBPQT⊂MV]3(+•), the former was found68 to exhibit much slower rates of association/dissociation. This second generation complex is also comparable to bis-CBPQT-[2]C(7+)(1•) insofar as both are composed of two bis-viologen cyclophanes, but unlike the catenane, MIMs based on [MS⊂m-CBPQT]4(+•) have the potential to display bistability. Lastly, [MS⊂m-CBPQT]4(+•) is a ground-state singlet, suggesting that NMR spectroscopy might be used to characterize MIMs designed around this complex. Herein, we describe a ring-in-ring rotaxane—a simple but as yet uncommon16,70,71 type of MIM—which incorporates a derivative of m-CBPQT2(+•) in its dumbbell component as a recognition unit for MS2(+•).
Results and Discussion
Synthesis of a Ring-In-Ring Rotaxane BBR·8PF6
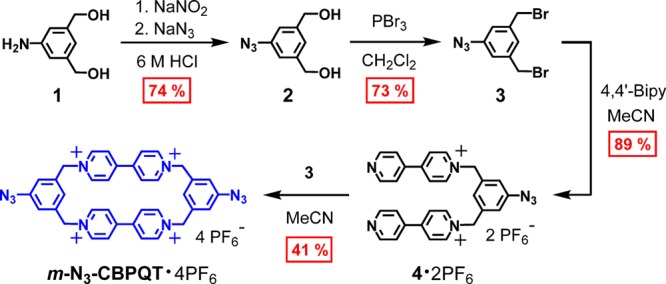
In order to synthesize a rotaxane utilizing the host–guest complex [MS⊂m-CBPQT]4(+•) as a template, it was, first of all, necessary to prepare a derivative of the m-CBPQT2(+•) guest that would be amenable to further functionalization. Since copper-catalyzed alkyne–azide cycloaddition (CuAAC) has recently been shown57 to be useful for synthesizing radically templated MIMs, a bis-azide substituted cyclophane m-N3-CBPQT·4PF6 was targeted.
The synthesis (Scheme 2) of m-N3-CBPQT·4PF6 was accomplished in four steps, starting from the known72 3,5-bis(hydroxymethyl)aniline (1). This starting material can be obtained commercially, or by employing a published procedure72 involving the reduction of inexpensive dimethyl 5-aminoisophthalate. The intermediate compounds 2, 3, and 4, were obtained in good yields and, moreover, could be purified by crystallization or by washing with an appropriate solvent. The target cyclophane, m-N3-CBPQT·4PF6, was also obtained pure without the need for chromatography. A mixed Br–/PF6– salt of the product precipitated from the reaction mixture in nearly pure form; additional crops were obtained by concentrating the supernatant. Pure m-N3-CBPQT·4PF6 was obtained in 41% yield after washing the precipitate sparingly with MeCN, followed by salt metathesis with aqueous NH4PF6. This relatively easy synthesis is notable since, at the outset, it seemed possible that incorporating a cyclophane component into the dumbbell of a rotaxane would introduce a considerable synthetic burden at an early stage.
Scheme 2. Synthesis of m-N3-CBPQT·4PF6.
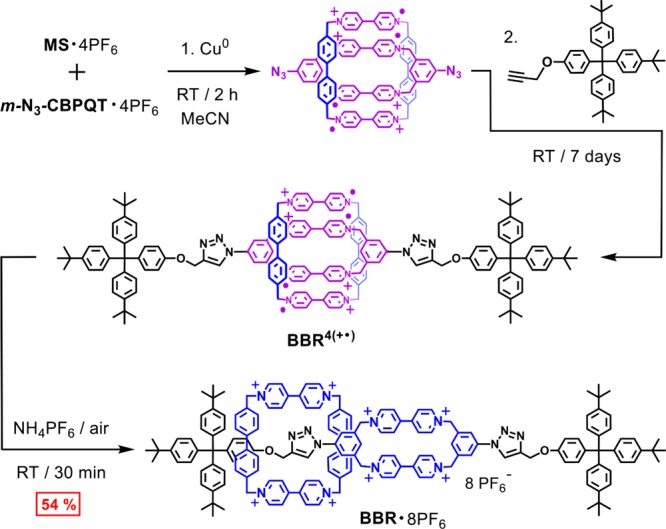
Scheme 3 depicts the use of m-N3-CBPQT·4PF6 in the synthesis of the Box-in-Box-Rotaxane·8PF6 (BBR·8PF6), a name that recognizes the common use73 of the term “box” to refer to rigid viologen-based cyclophanes. Copper powder was used as a reducing agent in order to generate the host–guest complex [MS⊂m-N3-CBPQT]4(+•) from a 1:1 ratio mixture of m-N3-CBPQT·4PF6 and MS·4PF6 in MeCN. As reported previously in the synthesis57 of CBPQT-RV-[2]R·6PF6, this reduction generates (MeCN)4Cu+ as a byproduct, which mediates the cycloadditions between [MS⊂m-N3-CBPQT]4(+•) and a bulky alkyne. The tetracationic tetraradical rotaxane BBR4(+•), which is formed initially, is converted into its octacationic state, BBR8+, upon oxidation with air. Examination of the crude product by 1H NMR spectroscopy indicated that 70–80% of m-N3-CBPQT·4PF6 was converted to the rotaxane, while the remainder was converted to the m-Box-Dumbbell4+ (m-BDB4+) component that was not encircled by the molecular square. The rotaxane was isolated in 54% yield after a series of solvent washes, salt metathesis steps, crystallization of the tetracationic tetraradical form, and reoxidation with air. The purification of BBR·8PF6 is, thus, more complicated than that for m-N3-CBPQT·4PF6, but the individual steps are operationally simple and the need for chromatography is once again avoided.
Scheme 3. Synthesis of the Box-In-Box Rotaxane BBR·8PF6.
Spectroscopic Characterization of BBR8+ and BBR4(+•)
The mechanically interlocked nature of the rotaxane was confirmed by ESI-HRMS, DOSY NMR spectroscopy, and UV–vis–NIR dilution measurements of its reduced BBR4(+•) state. See the synthetic details and Figures S3 and S10. The 1H NMR spectrum (Figure 1) also reveals the mechanically interlocked nature of BBR8+. All the resonances of the dumbbell component of BBR8+ exhibit anisochronicity because the molecular square is confined, at least on the NMR exchange time scale, to one end of the dumbbell. The asymmetry is observed most clearly in (i) the −CH2O– proton resonances and (ii) the ortho Ar–H signals of the phenolic group. These signals are shifted upfield considerably for the side of the dumbbell that is encircled by the molecular square, indicating that this electron deficient cyclophane resides over the electron rich ether unit.
Figure 1.
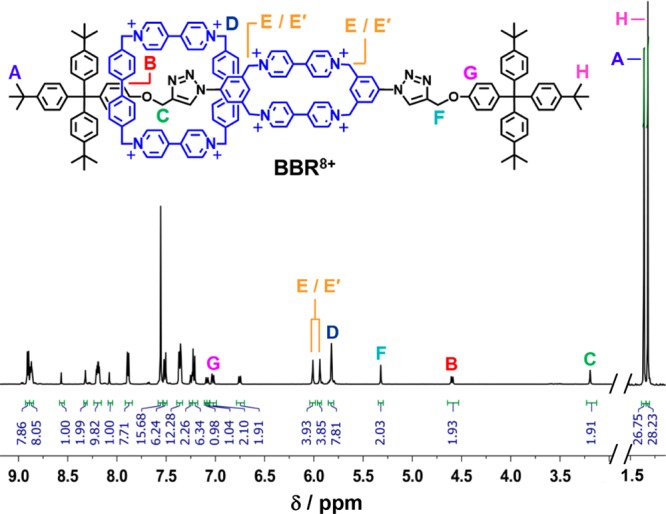
1H NMR spectrum of a CD3CN solution of BBR·8PF6. The spectrum is abridged in the region of the residual CD2HCN and HOD resonances, while all of the signals for the BBR8+ cation are displayed. Selected resonances are labeled in order to illustrate the coconstitutional asymmetry of the rotaxane. See Figure S1 for full 1H NMR spectroscopic assignments.
The proton resonances of BBR8+ exhibit only slight broadening of ≤2 Hz at half-height in 1H NMR spectra that were collected at 75 °C compared with those recorded at 25 °C in CD3CN. See Figures S11 and S12. Significant broadening, let alone coalescence, was not observed at 75 °C, even for the pair of potentially exchangeable resonances that exhibited the smallest separation (ca. 6.5 Hz at 500 MHz) from each other. From this latter observation, it can be inferred74 that the molecular square translates across the cyclophane portion of the dumbbell at a rate, kex, that is ≪29 s–1 at 75 °C. Furthermore, it can be inferred that the degenerate coconformations of BBR8+ have lifetimes of >1 s at 25 °C. In contrast, the 1H NMR spectrum of CBPQT-RV-R·6PF6 in CD3CN exhibits significant broadening/coalescence for several of the signals at 25 °C. Although the rates33,57 of shuttling of rings along dumbbells in hexacationic rotaxanes of this type are slower in CD3COCD3 solutions, fast shuttling is observed33,57 for these rotaxanes, even in this solvent, at elevated temperatures. The differences in the shuttling rates observed for hexacationic rotaxanes and BBR8+ can be attributed to the greater charge repulsion present in the latter rotaxane, as well as to the comparatively tight fit68 between the cyclophane components of BBR8+.
The conversion of BBR8+ to BBR4(+•) was probed (Figure 2a) by UV–vis–NIR spectroscopic monitoring of the sequential addition of 1–4 equiv of the 1 e– reductant75 Cp2Co to a solution of BBR·8PF4 in MeCN. The resulting spectra exhibit linear increases with each equivalent that is added, indicating that BBR8+ is converted portionwise directly to BBR4(+•). Intermediate oxidation states must, therefore, not be thermodynamically accessible to any significant extent. The spectrum obtained following the addition of 4 equiv of Cp2Co corresponds to full conversion of BBR8+ to BBR4(+•), which exhibits an intense NIR band (λmax = 950 nm) that is very similar to that16 (λmax = 941 nm) observed for the host–guest complex [MS⊂m-CBPQT]4(+•). This comparison indicates that this radical ring-in-ring complex provides an accurate coconformational model for the associated cyclophane components of BBR4(+•).
Figure 2.
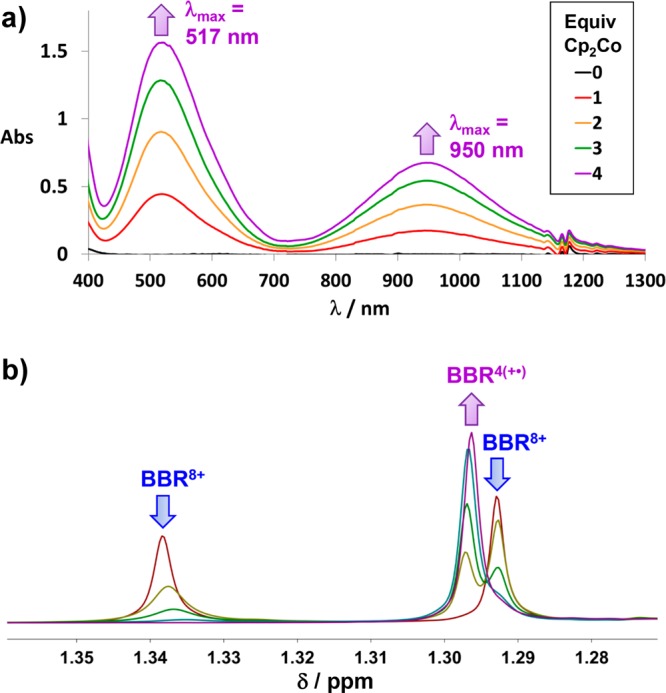
Titrations of solutions of BBR·8PF6 with 1 equiv at a time of Cp2Co. (a) UV–vis–NIR spectra of a 0.050 mM solution of BBR·8PF6 in MeCN after the addition of 0–4 equiv of Cp2Co. (b) Partial 1H NMR spectra displaying the tBu resonances of a 1 mM solution of BBR·8PF6 in CD3CN, after the addition of 0–4 equiv of Cp2Co.
1H NMR spectroscopy was also employed to monitor the titration of BBR·8PF6 with Cp2Co in CD3CN. The two tBu proton resonances of BBR8+ are observed (Figure 2b) to undergo stepwise decreases in intensities upon sequential addition of Cp2Co, while the single tBu resonance of BBR4(+•) grows in strength concomitantly. Other 1H resonances of BBR4(+•) are also observed (Figure 3a) in the spectrum after the addition of 4 equiv of the reductant. The signals for both BBR4(+•) and BBR8+ are observed in the 1H NMR spectra collected during intermediate stages of the titration. See Figures S24–S26. Selective broadening of some resonances of each oxidation state are, however, evident at the midpoint of the titration. Since this broadening does not affect all of the signals, it can be concluded that the two oxidation states do not rapidly undergo degenerate interconversion—a 4-electron based process—on the 1H NMR time scale. Instead, there may be some degree of single-electron transfer to provide BBR(7+)(1•) and BBR(5+)(3•) transiently, a process which would affect primarily the NMR signals arising from protons that are on or near the cyclophane components.
Figure 3.
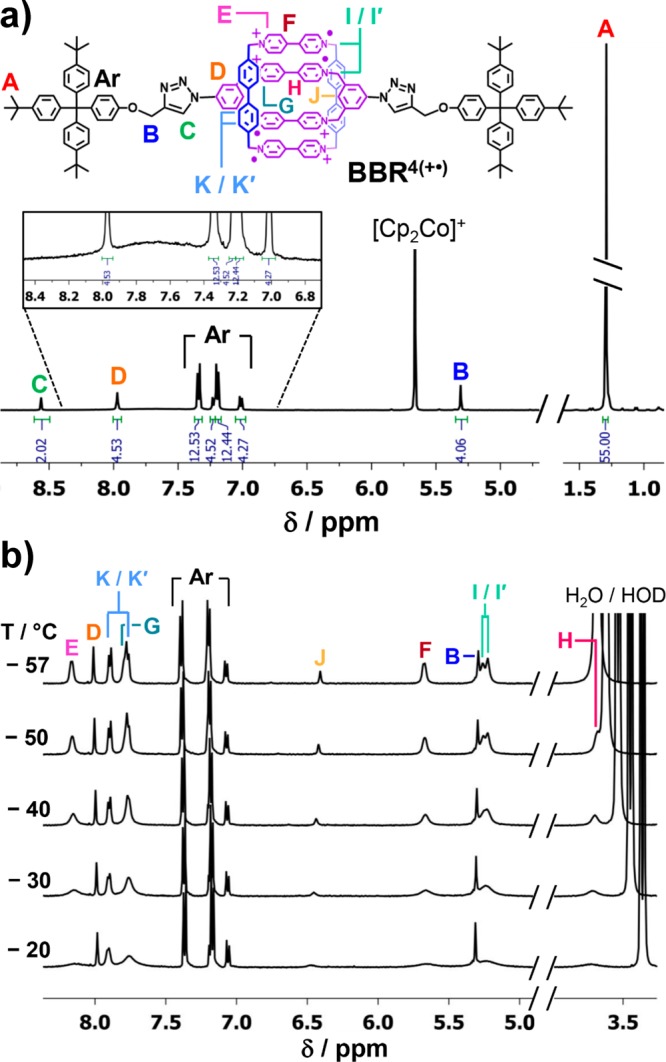
(a) 1H NMR spectrum of a CD3CN solution of BBR4(+•) that was prepared in situ by the addition of 4 equiv of Cp2Co to a solution of BBR·8PF6. The spectrum is abridged in the region of the residual CD2HCN and HOD signals, and the height of the tBu resonance is truncated. See Figure S4 for the full spectrum. (b) Partial 1H NMR spectra, collected over a temperature range of −20 to −57 °C, of a solution of BBR4(+•) in CD3COCD3 that was prepared in situ by stirring a solution of BBR·8PF6 over Zn dust. Although the resonances arising from the BBR4(+•) cation are best resolved in the spectrum collected at −57 °C, signal H is obscured by the H2O/HOD signal at this temperature. In the interest of preserving space, the tBu resonance of BBR4(+•) is not displayed. See Figure S14 for the full 1H NMR spectrum recorded at −57 °C.
The 1H NMR spectrum of BBR4(+•) exhibits several well-resolved signals, including those for the four outward facing protons of the m-phenylene linker, which are located very close to the viologen radical cations. The ability to observe sharp signals from these protons can be attributed to the strong radical-pairing interactions between the two cyclophanes, a phenomenon which provides a diamagnetic ground state. In contrast, the closest observable protons in the 1H NMR spectrum (Figure S6) of the free dicationic diradical dumbbell, m-DBD2(+•), are the −CH2O– methylene protons, a full seven bonds away from the m-phenylene positions that can be observed in the spectrum of BBR4(+•). Other 1H NMR resonances arising from the cyclophane components of BBR4(+•) could not be observed individually in the spectra collected at 25 °C, but broad signals, such as that observed in the inset of Figure 3a, can be attributed to these groups. These broad signals are most well-resolved when 3–4 equiv of Cp2Co has been added, or when Zn dust was used as the reducing agent, but disappear upon the addition of even a small excess (4.25 equiv) of Cp2Co. See Figures S26 and S27. Further characterization of lower (<4+) oxidation states of this rotaxane is described below in the section “Lower Oxidation States of BBRx(+•)”.
1H NMR spectra of BBR4(+•) were collected at reduced temperatures in CD3COCD3 in the hopes of resolving the broad resonances observed in the spectra collected at 25 °C. These resonances sharpened and resolved into individual signals in the spectra collected at temperatures between +25 and −57 °C. Resonances for all the possible protons of BBR4(+•) can be observed (Figure 3b) clearly at −50 °C. At −57 °C, the proton resonances of the cyclophane units are even better resolved, although one of them becomes obscured by the H2O/HOD residual signal. The resonances of BBR4(+•) integrate appropriately in the spectrum collected at −57 °C, and J-coupling (J = 5.4 Hz) can even be discerned for the two viologen Ar–H doublets that do not overlap with other signals. See Figure S15 for a depiction of the integrated spectrum.
The four aromatic resonances of the viologen units can be assigned based on the shielding of the protons that is expected by considering their proximity to other aromatic units. The innermost viologen protons of the smaller cyclophane are, thus, assigned to the most upfield signal since these protons are surrounded on both sides by other viologen units. The 2-positions of the pyridinium groups, in contrast, give rise to 1H NMR signals with normal pyridinum chemical shifts. These protons are not in the proximity of other viologen units because of the nearly perpendicular geometry that is enforced between these units on the smaller cyclophane compared with the larger one. The assumptions of this analysis were validated by 1H NMR spectroscopic studies of the cyclophane59o-CBPQT2(+•), which also exhibits diamagnetic NMR behavior. See Figure S8. The o-phenylene linkers of this small cyclophane enforce an eclipsed, rather than perpendicular, alignment of the viologen units, such that the 2- and 3-positions on the pyridinum groups are shielded to a similar extent by the adjacent viologen unit. This geometric arrangement is reflected in the 1H NMR chemical shifts of o-CBPQT2(+•); the viologen units give rise to more tightly clustered 1H NMR chemical shifts than those observed in the spectra of BBR4(+•).
It is remarkable that well-resolved 1H NMR spectra could be obtained for BBR4(+•) and o-CBPQT2(+•). The latter species was first reported59 ca. 30 years ago, but it was not investigated by 1H NMR spectroscopy even though EPR spectroscopy indicated that it is diamagnetic. To our knowledge,67,76−80 well-resolved 1H NMR spectra have never been observed for any other diamagnetic viologen radical dimers, which in most cases can be attributed to rapid equilibration between the dimers and a significant concentration of the paramagnetic monomers. It is conceivable that similar processes—equilibration between BBR4(+•) and a metastable coconformation (MSCC), BBR4(+•)D, in which the two cyclophane components are dissociated from each other—might be responsible for the broadening of the cyclophane resonances in the 1H NMR spectrum of BBR4(+•) at 25 °C. This possibility can, however, be discounted based on variable scan-rate cyclic voltammetry described in the next section.
Another possible explanation for the selective broadening of the 1H NMR signals of BBR4(+•) is that rapid electron transfer between BBR4(+•) and BBR8+ could transiently form BBR(7+)(1•) and BBR(5+)(3•), as is invoked (vide supra) to account for the broadening of signals of BBR8+ when a large concentration of BBR4(+•) is present. This explanation is, however, inadequate when it comes to explaining the NMR spectroscopic data of BBR4(+•) for two reasons: (1) Reduction of BBR8+ with an excess of Zn dust is expected to provide nearly complete and selective conversion to BBR4(+•) (see Figure S35 for the relevant EPR spectrum), and (2) o-CBPQT2(+•) consistently exhibits sharp 1H NMR resonances at a much higher temperature (+25 °C) than does BBR4(+•) (−57 °C). This latter observation is particularly significant because the rigid covalent linkers in o-CBPQT2(+•) must greatly reduce the rearrangement energy needed for electron transfer between o-CBPQT2(+•) and its higher oxidation states, such that the 1H NMR spectrum of o-CBPQT2(+•) should be more sensitive to electron transfer processes. Consistent with this assessment, 1H NMR signals could not be observed for a 1:1 ratio mixture of o-CBPQT2(+•) and o-CBPQT4+ in CD3CN. See Figure S31.
The best explanation for the broadened 1H NMR signals in the case of BBR4(+•) is that there is a thermally accessible triplet electronic state. Previous DFT calculations68 on [MS⊂m-CBPQT]4(+•) revealed that its HOMO consists of a bonding combination of the viologen SOMO orbitals, while the LUMO corresponds to an antibonding combination. The lowest energy triplet state of BBR4(+•) would involve the thermal excitation of an electron across this HOMO–LUMO gap, thus disrupting one of the radical-pairing interactions. The o-CBPQT2(+•) cyclophane presumably has a larger HOMO–LUMO separation because the eclipsed arrangement of its viologen units provides greater orbital overlap.62 The relative HOMO–LUMO gaps for o-CBPQT2(+•) and BBR4(+•) were assessed by comparing their NIR absorption bands, which arise60,61 from the photoinduced promotion of an electron from the HOMO to the LUMO level within radical-paired dimers. It should be noted that the energies of these photoexcitations do not provide a direct measure of the energies of thermally promoted singlet/triplet interconversion because the optical absorption is a singlet-to-singlet transition and is subject to the Franck–Condon principle. Since, however, these effects apply to both o-CBPQT2(+•) and BBR4(+•), it can be concluded that the HOMO–LUMO gap is larger in this smaller cyclophane (λmax = 850 nm)59 relative to the rotaxane (λmax = 950 nm, Figure 2a), as expected based on the 1H NMR spectra for each compound. Despite its influence on the 1H NMR spectrum of BBR4(+•), the triplet state of this rotaxane could not be observed by EPR spectroscopy (see Figure S35) even for samples that were warmed to 90 °C.
Electrochemical Characterization of BBR·8PF6
Cyclic voltammetry was used to probe the interconversion between BBR8+, BBR4(+•), and a neutral form of this rotaxane BBR0. These are the three oxidation states of the rotaxane that appear to be accessible based on the CV data presented in Figure 4a. The more negative BBR4(+•)/BBR0 redox couple exhibits completely reversible behavior at scan rates between 50 and 500 mV/s, while, in contrast, the BBR8+/BBR4(+•) redox couple displays a large peak separation (ΔEp ≥ 130 mV) even at the lowest scan rates examined, i.e., 50 mV/s in Figure 4a and 10 mV/s in Figure S38. This large peak separation can be explained by a slow rate of dissociation for the two cyclophanes from the radical-paired state of BBR4(+•), resulting in an overpotential for oxidation back to the BBR8+ state. Similar electrochemical behavior was observed68 previously for the corresponding redox couple of a 1:1 molar ratio mixture of MS·4PF6 and m-CBPQT·4PF6, though, in contrast with BBR8+, reversible behavior was observed for the host–guest complex at sufficiently low scan rates (≤25 mV/s). Thus, the two cyclophane components of BBR4(+•) come apart even more slowly than the two cyclophane components of [MS⊂m-CBPQT]4(+•), a difference that might be attributed to steric factors associated with the dumbbell component of BBR4(+•).
Figure 4.
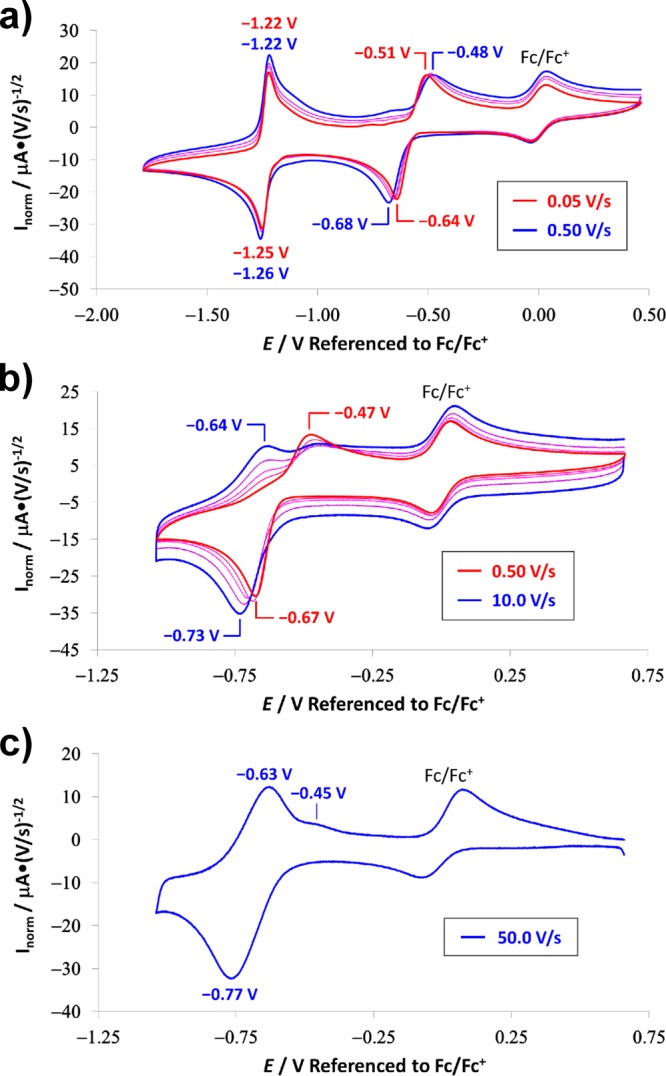
Cyclic voltammograms (CVs) of a 0.125 mM solution of BBR·8PF6 in MeCN containing a 0.1 M concentration of [Bu4N][PF6] electrolyte and ferrocene as an internal redox standard. In all the CVs, the current is normalized relative to the square root of the scan rate. (a) Scan rates of 0.05, 0.1, 0.2, and 0.5 V/s with a potential window that includes all accessible oxidation states of the rotaxane. (b) Scan rates of 0.5, 1, 2.5, 5.0, and 10.0 V/s over a potential window that includes only the BBR8+ and BBR4(+•) oxidation states of the rotaxane. (c) Scan rate of 50.0 V/s over a potential window that includes only the BBR8+ and BBR4(+•) oxidation states of the rotaxane. The capacitive current at 50.0 V/s was measured independently and subtracted from this CV.
The more positive redox couple of BBR8+ was examined in more detail to determine if the slow kinetics of association observed68 previously between m-CBPQT2(+•) and MS2(+•) would be preserved within the context of the rotaxane. As the scan rate was increased beyond 500 mV/s, the CVs of BBR·8PF6 revealed the appearance of a new reoxidation wave at potentials that were negative relative to those of the wave that is assigned to the radical-paired state of this rotaxane. See Figure 4b. The appearance of this wave can be attributed to the oxidation of a metastable coconformation (MSCC) BBR4(+•)D in which the two rings are dissociated from each other. Such a wave would appear at higher scan rates if, after reduction of BBR8+ to BBR4(+•)D, the two cyclophane components came together too slowly to form the associated BBR4(+•)A ground state coconformation (GSCC). The wave corresponding to the BBR4(+•)A state disappears almost completely at a scan rate of 50 V/s, such that the more negative BBR4(+•)D wave is part of a nearly reversible redox couple. The E1/2 value (−0.70 V) is similar to those observed68 for the individual MS4+/MS2(+•) and m-CBPQT4+/m-CBPQT2(+•) redox processes and is consistent with the assignment of the wave to a reversible BBR8+/BBR4(+•)D couple.
The electrochemical mechanism in Scheme 4 was used to simulate the CVs of the BBR8+/BBR4(+•) redox couple. It is assumed that, like the individual cyclophanes, this rotaxane undergoes two-electron redox processes. Furthermore, the rate constant, k1, for the conversion of BBR(6+)(2•)D to BBR(6+)(2•)A can be assumed to be lower than that (k2) for the conversion of BBR4(+•)D to BBR4(+•)A simply because the latter association process occurs with less charge repulsion. This consideration implies that the pathway associated with k2 will be the primary route that leads to the formation of the BBR4(+•)A state. From the CV data, it is also evident that k–1 > k2 because the metastable coconformation BBR(6+)(2•)A of the 6+ oxidation state does not have a long enough lifetime to permit observation of a BBR(6+)(2•)A/BBR8+A oxidation wave. When these considerations were taken into account, CVs simulated with a rate constant of k2 = 10–25 s–1 provided good agreement81 with the experimental data relating to the appearance and disappearance of the BBR4(+•)D/BBR8+D and BBR4(+•)A/BBR(6+)(2•)A oxidation waves. See Figure S41 for simulated CVs obtained with k2 = 15 s–1. The rate constant, k–2, must be orders of magnitude smaller since the BBR4(+•)D/BBR4(+•)A equilibrium lies far toward the associated state.
Scheme 4. Proposed Mechanisms of BBR8+/BBR4(+•) Interconversion.
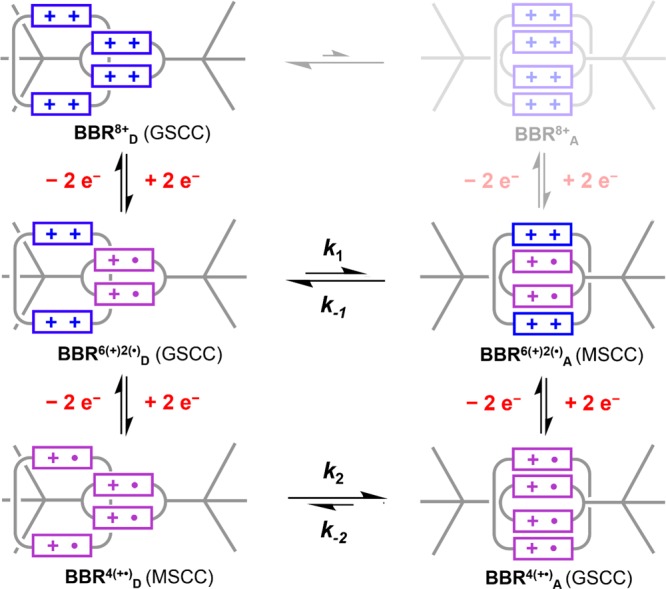
CV studies demonstrate that electrochemical hysteresis is observed during the switching between BBR8+ and BBR4(+•). Thus, this rotaxane preserves the comparatively slow kinetics of association/dissociation observed for the ring-in-ring complex68 upon which it is based. In contrast, related hexacationic rotaxanes, e.g., CBPQT-RV-[2]R6+, exhibit33 2–3 reversible redox couples associated with the electrochemical switching between the dissociated 6+ state and the associated trisradical 3+ state, even though the corresponding host–guest complexes can exhibit63,64 some degree of electrochemical hysteresis involving their association/dissociation at elevated scan rates (1–10 V/s). The slow rate of switching between BBR4(+•)D and BBR4(+•)A is particularly notable, given the distinct electronic nature, i.e., open- versus closed-shell electron configurations, of these two states of this rotaxane. Owing to their odd electron count, the trisradical rotaxanes cannot exhibit this same distinction between their associated and dissociated states since all coconformations of CBPQT-RV-[2]R3(+•) are paramagnetic.
Lower Oxidation States of BBRx(+•)
CV studies indicate that only three thermodynamically stable oxidation states—namely, BBR8+, BBR4(+•), and BBR0—exist for this ring-in-ring rotaxane. These results are consistent with data from Cp2Co titration experiments on the conversion of BBR8+ to BBR4(+•). In contrast, similar studies on the conversion of BBR4(+•) to BBR0 reveal that at least two intermediate oxidation states are accessible. This result was evident from changes in the UV–vis–NIR spectrum (Figure 5a) of a solution of BBR·8PF6 in Me2CO82 as 4–14 equiv of Cp2Co were added. Nearly identical results are obtained (Figure S19) when employing MeCN as the solvent.
Figure 5.
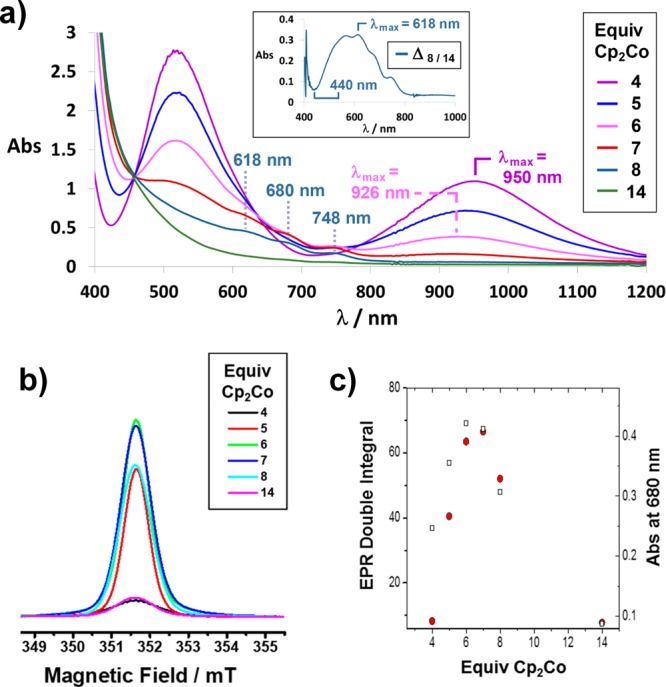
(a) UV–vis–NIR spectra of a 0.33 mM (initial concentration) solution of BBR·8PF6 in Me2CO following the additions of 4, 5, 6, 7, 8, and 14 equiv of Cp2Co. The inset depicts the difference between the spectra obtained after adding 8 and 14 equiv of Cp2Co, with the latter spectrum scaled appropriately to account for different concentrations of the fully reduced viologen units in each sample. (b) Relative integrated EPR signal intensities of 0.25 mM solutions of BBR·8PF6 in Me2CO after the additions of 4, 5, 6, 7, 8, and 14 equiv of Cp2Co. See Figure S34 for the full EPR spectra. (c) Comparison of the relative integrated EPR signal intensities (red circles) with the visible light absorption of these samples at 680 nm (squares) that was measured by UV–vis–NIR spectroscopy.
Upon the addition of 5 and 6 equiv of the reductant, the NIR absorption band of BBR4(+•) decreases in step sizes of >30%, and this decrease is accompanied by a shift in wavelength from λmax(4equiv) = 950 nm to λmax(6equiv) = 926 nm. Both observations indicate that BBR0 is not the only product formed upon partial reduction of BBR4(+•), particularly because BBR0 does not absorb in the NIR region. The blue shift of the NIR band implies that there is a significant quantity of an intermediate oxidation state of this rotaxane that also features radical-pairing interactions. This new radical-paired state is assigned (Scheme 5) to BBR2(+•)A, which features two of the viologen units in a paired radical state and two in their fully reduced state. Owing to the mixed valency of the viologen units in BBR2(+•)A, its electronic absorption spectrum is very similar to that of a 1:1 mixture of BBR4(+•) and BBR0, such that the individual concentrations of these three states of this rotaxane could not be determined in the sample mixture.
Scheme 5. Electrochemically Hidden Oxidation States of BBRx(+•).
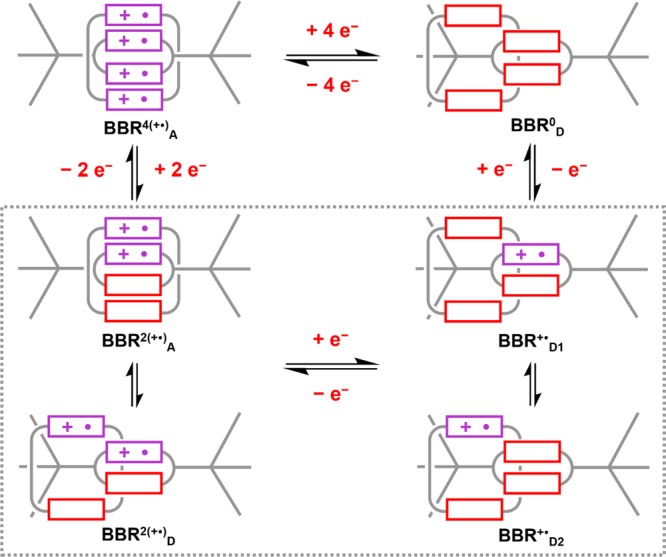
In addition to changes in the NIR region, the addition of 6 equiv of Cp2Co resulted in the observation of subtle features in the visible region of the spectrum that suggested that unpaired viologen radicals are present. These spectroscopic features became more evident after 7 and 8 equiv of Cp2Co had been added, while the NIR band disappeared almost completely. Note that full reduction of BBR8+ to the neutral BBR0 state is not achieved upon the addition of 8 equiv of Cp2Co because the Cp2Co/Cp2Co+ redox couple has a potential75ECp2Co/Co+ = −1.3 V which is not sufficiently negative compared to that of the BBR4(+•)/BBR0 couple (EBBR4+/BBR0 = −1.24 V). This limitation was overcome by the addition of a large excess of Cp2Co (14 equiv) to BBR8+, resulting in complete reduction to BBR0 as indicated by a UV–vis spectrum consistent with the presence of only fully reduced viologen cyclophanes.
The inset in Figure 5a displays a spectrum corresponding only to the radical viologen units of BBR+•. This spectrum was obtained by subtraction of the BBR0 spectrum, with appropriate scaling, from that obtained after the addition of 8 equiv of Cp2Co to the solution of BBR·PF6. The correct scaling of the BBR0 spectrum was assessed using the assumption that, in the resulting viologen radical spectrum, the absorption maximum at 618 nm and minimum at 440 nm would have a similar ratio of intensities to those observed for other unpaired viologen radicals. From this scaling, it was determined that 85% of the viologen units were in the fully reduced state after the addition of 8 equiv of Cp2Co, leaving 15% in the radical state. A slightly higher estimate (18%) of the radical concentration was determined by comparing the integrated EPR signal intensity arising from BBR+• with that of a MV+• standard. The spectrum of the BBR+• viologen radical resembles that observed68 previously for a 1:1 mixture of MS2(+•) and m-CBPQT2(+•) under conditions in which there is little association between the two cyclophanes. Thus, it can be concluded that two mixed-valence isomers of BBR+• are present, which are distinguished by whether the unpaired electron resides on the smaller or larger cyclophane.
The Cp2Co titration was also monitored by EPR spectroscopy in order to provide the data displayed in Figure 5b,c. The integrated EPR signal intensity increased in approximately equal steps after addition of the fifth and sixth equivalents of the reductant, suggesting that BBR3(+•) is not a major component of the resulting mixture of BBRx(+•) oxidation states. The addition of a seventh equivalent of Cp2Co resulted in a slight increase in the EPR signal intensity, corresponding to the maximum value that was observed during these experiments. This result is consistent with the assignment of the EPR signal primarily to the BBR+• state of this rotaxane, which is formed upon the addition of 7 electrons to the initial BBR8+ state. The change in the EPR signal intensity between adding the sixth and seventh equivalents was, however, relatively small. This observation indicates that there might be some contribution to the EPR signal from a dissociated BBR2(+•)D state, as illustrated in Scheme 5, although this possibility can be concluded with less certainty than the existence of the BBR2(+•)A and BBR+• states of this rotaxane.
Further information was sought using 1H NMR spectroscopy, but in contrast to the higher oxidation states, only a single set of resonances, corresponding to groups remote from the viologen cyclophanes, could be observed in each of the spectra obtained after adding >4 equiv of Cp2Co to a solution of BBR8+. See Figures S28–S30. It appears that electrons exchange rapidly between the lower oxidation states of this rotaxane, resulting in the observation of 1H NMR signals that are the average of multiple states, including some with paramagnetic character. Since some of these states involve association between the two rings, while others do not, it can be inferred that the rings associate and dissociate more rapidly in the lower BBRx(+•) (x < 4) oxidation states of this rotaxane than in the higher ones.
Conclusions
A recently characterized68 tetracationic tetraradical ring-in-ring complex has now been incorporated into the design of a new rotaxane, BBR8+, which features a cyclophane unit within its dumbbell component. This rotaxane is similar in its design to those of hexacationic rotaxanes based on the tricationic trisradical complex [CBPQT⊂MV]3(+•). The properties of BBR8+ are, however, influenced to a great extent by the rigid preorganization of two redox-active viologen units within the cyclophane unit of its dumbbell. The tetracationic charge and physical size of this unit create a significant barrier to the movement of the larger, square-shaped cyclophane from one end of the rotaxane to the other in its BBR8+ oxidation state. Relatively slow kinetics of translational motion are also observed in the case of the reduced BBR4(+•) state of the rotaxane: strong radical-pairing between the two cyclophanes causes slow dissociation of the rings from this favorable state, while charge repulsion and steric features create a significant barrier to the reverse process. Only in the lower oxidation states of the rotaxane, which feature reduced charge repulsion and weakened radical-pairing, does the larger cyclophane move rapidly along the dumbbell component.
The slow shuttling processes exhibited by BBR8+ and BBR4(+•) result in easily observable hysteresis for switching between these states, whereas closely related tris-viologen rotaxanes exhibit reversible electrochemical behavior. Furthermore, the associated and dissociated states of BBR4(+•) have strikingly different electronic properties. The transient BBR4(+•)D coconformation exhibits, for example, redox processes comparable to those of individual open-shell viologen cyclophanes, whereas radical-pairing interactions provide diamagnetic character to the BBR4(+•)A state while also stabilizing it against oxidation. The distinct electronic features of BBR4(+•)A are underscored by the ability to observe well-resolved 1H NMR spectra for this form of the rotaxane.
The striking differences in the electronic properties of the associated and dissociated states of the ring-in-ring rotaxane—as well as marked hysteresis in switching between them—make this rotaxane design promising for the development of molecular electronic materials. More broadly, the ability to identify the reduced BBR4(+•)A state of this rotaxane by 1H NMR spectroscopy represents a major step forward in the investigation of complex functional MIMs using radical-pairing interactions. This realization provides a looking glass for peering at the formally radical states of these MIMs, which, in turn, will enable much more detailed understanding of their complex structural features than is available using UV–vis–NIR and EPR spectroscopic methods.
Acknowledgments
This research is part of the Joint Center of Excellence in Integrated Nano-Systems (JCIN) at King Abdulaziz City for Science and Technology (KACST) and Northwestern University (NU). The authors would like to thank both KACST and NU for their continued support of this research. Electron paramagnetic resonance studies were supported by the U.S. National Science Foundation under Grant No. CHE-1565925 (M.R.W.). Y. Wu thanks the Fulbright Scholar Program for a Fellowship and the NU International Institute of Nanotechnology for a Ryan Fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00535.
Experimental details, including full synthetic procedures, routine characterization data, cyclic voltammograms, UV–vis–NIR spectra, EPR and 1H NMR spectra, and details of data processing (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dietrich-Buchecker C.; Sauvage J.-P. Interlocking of molecular threads: from the statistical approach to the templated synthesis of catenands. Chem. Rev. 1987, 87, 795–810. 10.1021/cr00080a007. [DOI] [Google Scholar]
- Amabilino D. B.; Stoddart J. F. Interlocked and Intertwined Structures and Superstructures. Chem. Rev. 1995, 95, 2725–2829. 10.1021/cr00040a005. [DOI] [Google Scholar]
- Sauvage J.-P., Dietrich-Buchecker C., Eds. Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology; Wiley: Weinheim, Germany, 1999. [Google Scholar]
- Stoddart J. F.; Colquhoun H. M. Big and Little Meccano. Tetrahedron 2008, 64, 8231–8263. 10.1016/j.tet.2008.06.035. [DOI] [Google Scholar]
- Forgan R. S.; Sauvage J.-P.; Stoddart J. F. Chemical topology: complex molecular knots, links, and entanglements. Chem. Rev. 2011, 111, 5434–5464. 10.1021/cr200034u. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Bruns C. J.; Li H.; Trabolsi A.; Coskun A.; Stoddart J. F. Ground-State Kinetics of Bistable Redox-Active Donor–Acceptor Mechanically Interlocked Molecules. Acc. Chem. Res. 2014, 47, 482–493. 10.1021/ar400161z. [DOI] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F.. The Nature of the Mechanical Bond: From Molecules to Machines; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Wasserman E. The Preparation of Interlocking Rings: A Catenane. J. Am. Chem. Soc. 1960, 82, 4433–4434. 10.1021/ja01501a082. [DOI] [Google Scholar]
- Schill G.; Lüttringhaus A. The Preparation of Catena Compounds by Directed Synthesis. Angew. Chem., Int. Ed. Engl. 1964, 3, 546–547. 10.1002/anie.196405461. [DOI] [Google Scholar]
- Harrison I. T.; Harrison S. Synthesis of a stable complex of a macrocycle and a threaded chain. J. Am. Chem. Soc. 1967, 89, 5723–5724. 10.1021/ja00998a052. [DOI] [Google Scholar]
- Ashton P. R.; Goodnow T. T.; Kaifer A. E.; Reddington M. V.; Slawin A. M. Z.; Spencer N.; Stoddart J. F.; Vicent C.; Williams D. J. A [2]Catenane Made to Order. Angew. Chem., Int. Ed. Engl. 1989, 28, 1396–1399. 10.1002/anie.198913961. [DOI] [Google Scholar]
- Harrison I. T. The effect of ring size on threading reactions of macrocycles. J. Chem. Soc., Chem. Commun. 1972, 231–232. 10.1039/c39720000231. [DOI] [Google Scholar]
- Ashton P. R.; Bĕlohradský M.; Philp D.; Spencer N.; Stoddart J. F. The self assembly of [2]- and [3]-rotaxanes by slippage. J. Chem. Soc., Chem. Commun. 1993, 0, 1274–1277. 10.1039/C39930001274. [DOI] [Google Scholar]
- Chiu C.-W.; Lai C.-C.; Chiu S.-H. Threading-Followed-by-Swelling”: A New Protocol for Rotaxane Synthesis. J. Am. Chem. Soc. 2007, 129, 3500–3501. 10.1021/ja069362i. [DOI] [PubMed] [Google Scholar]
- Barin G.; Coskun A.; Friedman D. C.; Olson M. A.; Colvin M. T.; Carmielli R.; Dey S. K.; Bozdemir O. A.; Wasielewski M. R.; Stoddart J. F. A Multistate Switchable [3]Rotacatenane. Chem. - Eur. J. 2011, 17, 213–222. 10.1002/chem.201002152. [DOI] [PubMed] [Google Scholar]
- Vajpayee V.; Song Y. H.; Cook T. R.; Kim H.; Lee Y.; Stang P. J.; Chi K.-W. A unique non-catenane interlocked self-assembled supramolecular architecture and its photophysical properties. J. Am. Chem. Soc. 2011, 133, 19646–19649. 10.1021/ja208495u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R.; Wakatsuki K.; Yamasaki R.; Mutoh Y.; Kasama T.; Saito S. Synthesis of rotacatenanes by the combination of Cu-mediated threading reaction and the template method: the dual role of one ligand. Chem. Commun. 2014, 50, 204–206. 10.1039/C3CC47425A. [DOI] [PubMed] [Google Scholar]
- Leigh D. A.; Marcos V.; Wilson M. R. Rotaxane Catalysts. ACS Catal. 2014, 4, 4490–4497. 10.1021/cs5013415. [DOI] [Google Scholar]
- Beswick J.; Blanco V.; Bo G. D.; Leigh D. A.; Lewandowska U.; Lewandowska B.; Mishiro K. Selecting reactions and reactants using a switchable rotaxane organocatalyst with two different active sites. Chem. Sci. 2015, 6, 140–143. 10.1039/C4SC03279A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli M.; Lewis J. E. M.; Goldup S. M. A Stimuli-Responsive Rotaxane–Gold Catalyst: Regulation of Activity and Diastereoselectivity. Angew. Chem., Int. Ed. 2015, 54, 13545–13549. 10.1002/anie.201505464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.-L. Supramolecular catalysis: A rotaxane with the golden touch. Nat. Chem. 2016, 8, 8–9. 10.1038/nchem.2403. [DOI] [PubMed] [Google Scholar]
- Movsisyan L. D.; Franz M.; Hampel F.; Thompson A. L.; Tykwinski R. R.; Anderson H. L. Polyyne Rotaxanes: Stabilization by Encapsulation. J. Am. Chem. Soc. 2016, 138, 1366–1376. 10.1021/jacs.5b12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K. Novel Cross-Linking Concept of Polymer Network: Synthesis, Structure, and Properties of Slide-Ring Gels with Freely Movable Junctions. Polym. J. 2007, 39, 489–499. 10.1295/polymj.PJ2006239. [DOI] [Google Scholar]
- Sawada J.; Aoki D.; Uchida S.; Otsuka H.; Takata T. Synthesis of Vinylic Macromolecular Rotaxane Cross-Linkers Endowing Network Polymers with Toughness. ACS Macro Lett. 2015, 4, 598–601. 10.1021/acsmacrolett.5b00242. [DOI] [PubMed] [Google Scholar]
- Murakami T.; Schmidt B. V. K. J.; Brown H. R.; Hawker C. J. One-Pot “Click” Fabrication of Slide-Ring Gels. Macromolecules 2015, 48, 7774–7781. 10.1021/acs.macromol.5b01713. [DOI] [Google Scholar]
- Nakahata M.; Mori S.; Takashima Y.; Yamaguchi H.; Harada A. Self-Healing Materials Formed by Cross-Linked Polyrotaxanes with Reversible Bonds. Chem. 2016, 1, 766–775. 10.1016/j.chempr.2016.09.013. [DOI] [Google Scholar]
- Aoki D.; Aibara G.; Uchida S.; Takata T. A Rational Entry to Cyclic Polymers via Selective Cyclization by Self-Assembly and Topology Transformation of Linear Polymers. J. Am. Chem. Soc. 2017, 139, 6791–6794. 10.1021/jacs.7b01151. [DOI] [PubMed] [Google Scholar]
- Collier C. P.; Mattersteig G.; Wong E. W.; Luo Y.; Beverly K.; Sampaio J.; Raymo F. M.; Stoddart J. F.; Heath J. R. A [2]Catenane-Based Solid State Electronically Reconfigurable Switch. Science 2000, 289, 1172–1175. 10.1126/science.289.5482.1172. [DOI] [PubMed] [Google Scholar]
- Steuerman D. W.; Tseng H.-R.; Peters A. J.; Flood A. H.; Jeppesen J. O.; Nielsen K. A.; Stoddart J. F.; Heath J. R. Molecular-Mechanical Switch-Based Solid-State Electrochromic Devices. Angew. Chem., Int. Ed. 2004, 43, 6486–6491. 10.1002/anie.200461723. [DOI] [PubMed] [Google Scholar]
- Green J. E.; Choi J. W.; Boukai A.; Bunimovich Y.; Johnston-Halperin E.; DeIonno E.; Luo Y.; Sheriff B. A.; Xu K.; Shin Y. S.; Tseng H.-R.; Stoddart J. F.; Heath J. R. A 160-kilobit molecular electronic memory patterned at 1011 bits per square centimeter. Nature 2007, 445, 414–417. 10.1038/nature05462. [DOI] [PubMed] [Google Scholar]
- Cao D.; Amelia M.; Klivansky L. M.; Koshkakaryan G.; Khan S. I.; Semeraro M.; Silvi S.; Venturi M.; Credi A.; Liu Y. Probing Donor–Acceptor Interactions and Co-Conformational Changes in Redox Active Desymmetrized [2]Catenanes. J. Am. Chem. Soc. 2010, 132, 1110–1122. 10.1021/ja909041g. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Zhu Z.; Cao D.; Liu W.-G.; Li H.; Dey S. K.; Basu S.; Trabolsi A.; Botros Y. Y.; Goddard W. A. III; Stoddart J. F. Radically Enhanced Molecular Switches. J. Am. Chem. Soc. 2012, 134, 16275–16288. 10.1021/ja306044r. [DOI] [PubMed] [Google Scholar]
- Li H.; Zhu Z.; Fahrenbach A. C.; Savoie B. M.; Ke C.; Barnes J. C.; Lei J.; Zhao Y.-L.; Lilley L. M.; Marks T. J.; Ratner M. A.; Stoddart J. F. Mechanical Bond-Induced Radical Stabilization. J. Am. Chem. Soc. 2013, 135, 456–467. 10.1021/ja310060n. [DOI] [PubMed] [Google Scholar]
- Barnes J. C.; et al. A Radically Configurable Six-State Compound. Science 2013, 339, 429–433. 10.1126/science.1228429. [DOI] [PubMed] [Google Scholar]
- Götz G.; Zhu X.; Mishra A.; Segura J.-L.; Mena-Osteritz E.; Bäuerle P. π-Conjugated [2]Catenanes Based on Oligothiophenes and Phenanthrolines: Efficient Synthesis and Electronic Properties. Chem. - Eur. J. 2015, 21, 7193–7210. 10.1002/chem.201406039. [DOI] [PubMed] [Google Scholar]
- Sun J.; Wu Y.; Wang Y.; Liu Z.; Cheng C.; Hartlieb K. J.; Wasielewski M. R.; Stoddart J. F. An Electrochromic Tristable Molecular Switch. J. Am. Chem. Soc. 2015, 137, 13484–13487. 10.1021/jacs.5b09274. [DOI] [PubMed] [Google Scholar]
- Bissell R. A.; Cordova E.; Kaifer A. E.; Stoddart J. F. A chemically and electrochemically switchable molecular shuttle. Nature 1994, 369, 133–137. 10.1038/369133a0. [DOI] [Google Scholar]
- Linke M.; Chambron J.-C.; Heitz V.; Sauvage J.-P.; Semetey V. Complete rearrangement of a multi-porphyrinic rotaxane by metallation–demetallation of the central coordination site. Chem. Commun. 1998, 2469–2470. 10.1039/a805746j. [DOI] [Google Scholar]
- Armaroli N.; Balzani V.; Collin J.-P.; Gaviña P.; Sauvage J.-P.; Ventura B. Rotaxanes Incorporating Two Different Coordinating Units in Their Thread: Synthesis and Electrochemically and Photochemically Induced Molecular Motions. J. Am. Chem. Soc. 1999, 121, 4397–4408. 10.1021/ja984051w. [DOI] [Google Scholar]
- Kihara N.; Hashimoto M.; Takata T. Redox Behavior of Ferrocene-Containing Rotaxane: Transposition of the Rotaxane Wheel by Redox Reaction of a Ferrocene Moiety Tethered at the End of the Axle. Org. Lett. 2004, 6, 1693–1696. 10.1021/ol049817d. [DOI] [PubMed] [Google Scholar]
- Murakami H.; Kawabuchi A.; Matsumoto R.; Ido T.; Nakashima N. A Multi-Mode-Driven Molecular Shuttle: Photochemically and Thermally Reactive Azobenzene Rotaxanes. J. Am. Chem. Soc. 2005, 127, 15891–15899. 10.1021/ja053690l. [DOI] [PubMed] [Google Scholar]
- Davidson G. J. E.; Sharma S.; Loeb S. J. A [2]Rotaxane Flip Switch Driven by Coordination Geometry. Angew. Chem., Int. Ed. 2010, 49, 4938–4942. 10.1002/anie.201001486. [DOI] [PubMed] [Google Scholar]
- Jiménez M. C.; Dietrich-Buchecker C.; Sauvage J.-P. Towards Synthetic Molecular Muscles: Contraction and Stretching of a Linear Rotaxane Dimer. Angew. Chem., Int. Ed. 2000, 39, 3284–3287. . [DOI] [PubMed] [Google Scholar]
- Tsukagoshi S.; Miyawaki A.; Takashima Y.; Yamaguchi H.; Harada A. Contraction of Supramolecular Double-Threaded Dimer Formed by α-Cyclodextrin with a Long Alkyl Chain. Org. Lett. 2007, 9, 1053–1055. 10.1021/ol063078e. [DOI] [PubMed] [Google Scholar]
- Romuald C.; Busseron E.; Coutrot F. Very Contracted to Extended co-Conformations with or without Oscillations in Two- and Three-Station [c2]Daisy Chains. J. Org. Chem. 2010, 75, 6516–6531. 10.1021/jo101234u. [DOI] [PubMed] [Google Scholar]
- Bruns C. J.; Li J.; Frasconi M.; Schneebeli S. T.; Iehl J.; Jacquot de Rouville H.-P.; Stupp S. I.; Voth G. A.; Stoddart J. F. An Electrochemically and Thermally Switchable Donor–Acceptor [c2]Daisy Chain Rotaxane. Angew. Chem., Int. Ed. 2014, 53, 1953–1958. 10.1002/anie.201308498. [DOI] [PubMed] [Google Scholar]
- Bruns C. J.; Frasconi M.; Iehl J.; Hartlieb K. J.; Schneebeli S. T.; Cheng C.; Stupp S. I.; Stoddart J. F. Redox Switchable Daisy Chain Rotaxanes Driven by Radical–Radical Interactions. J. Am. Chem. Soc. 2014, 136, 4714–4723. 10.1021/ja500675y. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Frasconi M.; Liu W.-G.; Sun J.; Wu Y.; Nassar M. S.; Botros Y. Y.; Goddard W. A. III; Wasielewski M. R.; Stoddart J. F. Oligorotaxane Radicals under Orders. ACS Cent. Sci. 2016, 2, 89–98. 10.1021/acscentsci.5b00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh D. A.; Wong J. K. Y.; Dehez F.; Zerbetto F. Unidirectional rotation in a mechanically interlocked molecular rotor. Nature 2003, 424, 174–179. 10.1038/nature01758. [DOI] [PubMed] [Google Scholar]
- Wilson M. R.; Solá J.; Carlone A.; Goldup S. M.; Lebrasseur N.; Leigh D. A. An autonomous chemically fuelled small-molecule motor. Nature 2016, 534, 235–240. 10.1038/nature18013. [DOI] [PubMed] [Google Scholar]
- Kassem S.; van Leeuwen T.; Lubbe A. S.; Wilson M. R.; Feringa B. L.; Leigh D. A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46, 2592–2621. 10.1039/C7CS00245A. [DOI] [PubMed] [Google Scholar]
- Spruell J. M.; et al. Highly stable tetrathiafulvalene radical dimers in [3]catenanes. Nat. Chem. 2010, 2, 870–879. 10.1038/nchem.749. [DOI] [PubMed] [Google Scholar]
- Coskun A.; Spruell J. M.; Barin G.; Fahrenbach A. C.; Forgan R. S.; Colvin M. T.; Carmieli R.; Benítez D.; Tkatchouk E.; Friedman D. C.; Sarjeant A. A.; Wasielewski M. R.; Goddard W. A. III; Stoddart J. F. Mechanically Stabilized Tetrathiafulvalene Radical Dimers. J. Am. Chem. Soc. 2011, 133, 4538–4547. 10.1021/ja110584c. [DOI] [PubMed] [Google Scholar]
- Stoddart J. F. Putting Mechanically Interlocked Molecules (MIMs) to Work in Tomorrow’s World. Angew. Chem., Int. Ed. 2014, 53, 11102–11104. 10.1002/anie.201408043. [DOI] [PubMed] [Google Scholar]
- Li H.; Fahrenbach A. C.; Dey S. K.; Basu S.; Trabolsi A.; Zhu Z.; Botros Y. Y.; Stoddart J. F. Mechanical Bond Formation by Radical Templation. Angew. Chem., Int. Ed. 2010, 49, 8260–8265. 10.1002/anie.201004488. [DOI] [PubMed] [Google Scholar]
- Gibbs-Hall I. C.; Vermeulen N. A.; Dale E. J.; Henkelis J. J.; Blackburn A. K.; Barnes J. C.; Stoddart J. F. Catenation through a Combination of Radical Templation and Ring-Closing Metathesis. J. Am. Chem. Soc. 2015, 137, 15640–15643. 10.1021/jacs.5b10623. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Sun J.; Liu Z.; Nassar M. S.; Botros Y. Y.; Stoddart J. F. Symbiotic Control in Mechanical Bond Formation. Angew. Chem., Int. Ed. 2016, 55, 12387–12392. 10.1002/anie.201605454. [DOI] [PubMed] [Google Scholar]
- Kosower E. M.; Cotter J. L. Stable Free Radicals. II. The Reduction of 1-Methyl-4-cyanopyridinium Ion to Methylviologen Cation Radical. J. Am. Chem. Soc. 1964, 86, 5524–5527. 10.1021/ja01078a026. [DOI] [Google Scholar]
- Geuder W.; Hunig S.; Suchy A. Single and double bridged viologenes and intramolecular pimerization of their cation radicals. Tetrahedron 1986, 42, 1665–1677. 10.1016/S0040-4020(01)87583-9. [DOI] [Google Scholar]
- Lü J.-M.; Rosokha S. V.; Kochi J. K. Stable (Long-Bonded) Dimers via the Quantitative Self-Association of Different Cationic, Anionic, and Uncharged π-Radicals: Structures, Energetics, and Optical Transitions. J. Am. Chem. Soc. 2003, 125, 12161–12171. 10.1021/ja0364928. [DOI] [PubMed] [Google Scholar]
- Fumanal M.; Capdevila-Cortada M.; Ribas-Arino J.; Novoa J. J. Electronic Excitation Energies in Dimers between Radical Ions Presenting Long, Multicenter Bonding. J. Chem. Theory Comput. 2015, 11, 2651–2660. 10.1021/acs.jctc.5b00381. [DOI] [PubMed] [Google Scholar]
- Geraskina M. R.; Dutton A. S.; Juetten M. J.; Wood S. A.; Winter A. H. The Viologen Cation Radical Pimer: A Case of Dispersion-Driven Bonding. Angew. Chem., Int. Ed. 2017, 56, 9435–9439. 10.1002/anie.201704959. [DOI] [PubMed] [Google Scholar]
- Trabolsi A.; Khashab N.; Fahrenbach A. C.; Friedman D. C.; Colvin M. T.; Cotí K. K.; Benítez D.; Tkatchouk E.; Olsen J.-C.; Belowich M. E.; Carmielli R.; Khatib H. A.; Goddard W. A. III; Wasielewski M. R.; Stoddart J. F. Radically enhanced molecular recognition. Nat. Chem. 2010, 2, 42–49. 10.1038/nchem.479. [DOI] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Barnes J. C.; Lanfranchi D. A.; Li H.; Coskun A.; Gassensmith J. J.; Liu Z.; Benítez D.; Trabolsi A.; Goddard W. A. III; Elhabiri M.; Stoddart J. F. Solution-Phase Mechanistic Study and Solid-State Structure of a Tris(bipyridinium radical cation) Inclusion Complex. J. Am. Chem. Soc. 2012, 134, 3061–3072. 10.1021/ja2089603. [DOI] [PubMed] [Google Scholar]
- Cheng C.; McGonigal P. R.; Liu W.-G.; Li H.; Vermeulen N. A.; Ke C.; Frasconi M.; Stern C. L.; Goddard W. A. III; Stoddart J. F. Energetically Demanding Transport in a Supramolecular Assembly. J. Am. Chem. Soc. 2014, 136, 14702–14705. 10.1021/ja508615f. [DOI] [PubMed] [Google Scholar]
- Cheng C.; McGonigal P. R.; Schneebeli S. T.; Li H.; Vermeulen N. A.; Ke C.; Stoddart J. F. An artificial molecular pump. Nat. Nanotechnol. 2015, 10, 547–553. 10.1038/nnano.2015.96. [DOI] [PubMed] [Google Scholar]
- To our knowledge, NMR investigations of viologen radical cations have never been reported. We have found that well-resolved 1H NMR signals are not discernible for these organic radicals in solution. Researchers outside of our laboratory have informed us, in private discussions, that they also have been unable to obtain NMR spectra for these compounds, even when radical-pairing ostensibly provides a diamagnetic ground state.
- Lipke M. C.; Cheng T.; Wu Y.; Arslan H.; Xiao H.; Wasielewski M. R.; Goddard W. A. III; Stoddart J. F. Size-Matched Radical Multivalency. J. Am. Chem. Soc. 2017, 139, 3986–3998. 10.1021/jacs.6b09892. [DOI] [PubMed] [Google Scholar]
- Barin G.; Frasconi M.; Dyar S. M.; Iehl J.; Buyukcakir O.; Sarjeant A. A.; Carmieli R.; Coskun A.; Wasielewski M. R.; Stoddart J. F. Redox-Controlled Selective Docking in a [2]Catenane Host. J. Am. Chem. Soc. 2013, 135, 2466–2469. 10.1021/ja3125004. [DOI] [PubMed] [Google Scholar]
- To our knowledge, ref (16) describes the only other example of a rotaxane in which the dumbbell component includes a macrocyclic site on which the outer ring can reside. For the related concept of a catenane that features a macrocyclic component embedded within one of its rings as a recognition unit for the other ring, see ref (71).
- Juric̆ek M.; Barnes J. C.; Strutt N. L.; Vermeulen N. A.; Ghooray K. C.; Dale E. J.; McGonigal P. R.; Blackburn A. K.; Avestro A.-J.; Stoddart J. F. An ExBox [2]catenane. Chem. Sci. 2014, 5, 2724–2731. 10.1039/c4sc00488d. [DOI] [Google Scholar]
- Neelarapu R.; Holzle D. L.; Velaparthi S.; Bai H.; Brunsteiner M.; Blond S. Y.; Petukhov P. A. Design, Synthesis, Docking, and Biological Evaluation of Novel Diazide-Containing Isoxazole- and Pyrazole-Based Histone Deacetylase Probes. J. Med. Chem. 2011, 54, 4350–4364. 10.1021/jm2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale E. J.; Vermeulen N. A.; Juríc̆ek M.; Barnes J. C.; Young R. M.; Wasielewski M. R.; Stoddart J. F. Supramolecular Explorations: Exhibiting the Extent of Extended Cationic Cyclophanes. Acc. Chem. Res. 2016, 49, 262–273. 10.1021/acs.accounts.5b00495. [DOI] [PubMed] [Google Scholar]
- Bryant R. G. The NMR time scale. J. Chem. Educ. 1983, 60, 933–935. 10.1021/ed060p933. [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- Though NMR spectroscopic studies have not, to our knowledge, been reported for viologen radical-cation dimers, solid-state 13C NMR spectra of tetracyanoethylene radical-anion dimers, tetrathiafulvalene radical-cation dimers, and mixed-valency tricyanobenzene trimers have been described. See refs (77) – (80).
- Strohmeier M.; Barich D. H.; Grant D. M.; Miller J. S.; Pugmire R. J.; Simons J. Solid-State NMR Spectra and Long Intradimer Bonds in the π-[TCNE]22- Dianion. J. Phys. Chem. A 2006, 110, 7962–7969. 10.1021/jp061920s. [DOI] [PubMed] [Google Scholar]
- Bagnato J. D.; Shum W. W.; Strohmeier M.; Grant D. M.; Arif A. M.; Miller J. S. The Structure of Fractionally Charged Tetracyanobenzenen– Present in [TCNB]32–. Angew. Chem., Int. Ed. 2006, 45, 5322–5326. 10.1002/anie.200601070. [DOI] [PubMed] [Google Scholar]
- Miller J. S.; Novoa J. J. Four-Center Carbon–Carbon Bonding. Acc. Chem. Res. 2007, 40, 189–196. 10.1021/ar068175m. [DOI] [PubMed] [Google Scholar]
- Halling M. D.; Bell J. D.; Pugmire R. J.; Grant D. M.; Miller J. S. Solid-State NMR Spectra and Long, Intra-Dimer Bonding in the π-[TTF]22+ (TTF = Tetrathiafulvalene) Dication. J. Phys. Chem. A 2010, 114, 6622–6629. 10.1021/jp910509f. [DOI] [PubMed] [Google Scholar]
- For a given scan rate in the simulated CVs, the ratios of the reoxidation waves for the BBR4(+•)D and BBR4(+•)A coconformations are most sensitive to variation of the rate constant k2, as expected, since this rate constant corresponds to the conversion of BBR4(+•)D to BBR4(+•)A. There is, however, a smaller dependence of the peak current of the simulated BBR4(+•)A oxidation wave on the rate constant k–1. As a consequence of these considerations, the simulations allow the value of k2 to be estimated within a narrow range, but not determined exactly.
- Nearly identical results are obtained (Figure S19) when employing MeCN as the solvent. The low solubility of BBR0 in MeCN, however, leads to precipitation of this oxidation state of this rotaxane from solution, making MeCN a less suitable solvent for some measurements. See Figures S20 and S33.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.