

Abstract
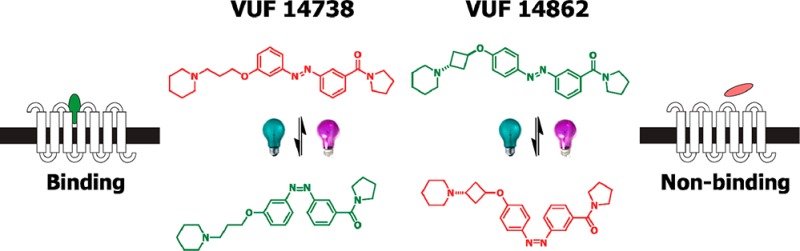
Noninvasive methods to modulate G protein-coupled receptors (GPCRs) with temporal and spatial precision are in great demand. Photopharmacology uses photons to control in situ the biological properties of photoswitchable small-molecule ligands, which bodes well for chemical biological precision approaches. Integrating the light-switchable configurational properties of an azobenzene into the ligand core, we developed a bidirectional antagonist toolbox for an archetypical family A GPCR, the histamine H3 receptor (H3R). From 16 newly synthesized photoswitchable compounds, VUF14738 (28) and VUF14862 (33) were selected as they swiftly and reversibly photoisomerize and show over 10-fold increased or decreased H3R binding affinities, respectively, upon illumination at 360 nm. Both ligands combine long thermal half-lives with fast and high photochemical trans-/cis conversion, allowing their use in real-time electrophysiology experiments with oocytes to confirm dynamic photomodulation of H3R activation in repeated second-scale cycles. VUF14738 and VUF14862 are robust and fatigue-resistant photoswitchable GPCR antagonists suitable for spatiotemporal studies of H3R signaling.
Introduction
Optical control of biological processes is one of the emerging fields in biomedicine due to its high spatiotemporal resolution.1−5 Initially, optogenetics revolutionized the field with major breakthroughs in, e.g., the field of neuroscience.3 Lately, the field of photopharmacology1,2,5,6 has been providing a complementary framework allowing the use of photoswitchable ligand molecules to dynamically modulate native biological targets. In this framework, the photoinduced configuration of the ligand has either lower or higher affinity (directionality) than the “dark” configuration, providing a means to reversibly regulate the biological target with light. It has very recently been applied to, e.g., native membrane-bound ion channels7−9 and G protein-coupled receptors (GPCRs).10−14 Most GPCR examples have so far focused on single ligands typically incorporating a photosensitive group in the periphery of the GPCR ligands,10−14 successfully giving reversible monodirectional photoswitches. In this work, we present a bidirectional photoswitchable GPCR antagonist toolbox, i.e., a set of ligands from the same scaffold amenable to switching in two directions, showing either a light-induced increase or decrease in GPCR affinity, by positioning the photoswitchable moiety in the core of the new ligands.
To this end, we used a prototypic family A GPCR, the histamine H3 receptor (H3R). The H3R is expressed mainly in the central nervous system (CNS) and involved in a variety of CNS processes.15−17 Recently, the first H3R antagonist (pitolisant, Wakix) has been approved for the treatment of narcolepsy.18 The medicinal chemistry of small-molecule H3R ligands is well-developed19,20 and provides interesting chemical opportunities to incorporate photoswitchable groups in order to obtain photoswitchable H3R antagonists. The general H3R antagonist pharmacophore includes a positively charged amine group,21 which is considered to interact with either the amino acid residues D1143.32 or E2065.46 in the transmembrane part of the H3R protein.19,22,23 A linker, usually an alkyl ether, allows the ligand to make a slight kink toward the bottom of the orthosteric H3R binding site. The ether oxygen atom is connected to an aromatic core (e.g., phenyl, naphthyl, and many others19,20), which allows for π–π stacking interactions with aromatic H3R residues, like the Y1153.33, Y3746.51, and F3987.39 residues.24−26 The aromatic core can be further substituted with a variety of groups including lipophilic groups, hydrogen bond acceptors, and basic and acidic moieties.27 Various photoswitchable moieties were evaluated for replacement of the H3R antagonist aromatic core5 and the photoisomerizable azobenzene moiety was deemed the best fit because of the narrow, flat, and elongated aromatic nature of its trans isomer. Although a naphthalene moiety has not been coined as an “azostere”,1 our design started from a naphthalene-containing H3R antagonist (1) developed by Roche et al.,28 which matches the previously mentioned pharmacophore criteria. We postulated that, due to the relatively small nature of the naphthalene core, additional room for π–π stacking interactions is present in the hydrophobic part of the H3R binding site, allowing incorporation of the azobenzene photoswitch in the core of the H3R scaffold (Figure 1) itself rather than in the periphery, as most often observed for GPCR photoligands.
Figure 1.

General design and concept of bidirectional photoswitchable H3R antagonists. In this SAR study, the azobenzene moiety is incorporated in the core of compound 1 to give a series of differently substituted trans isomers (cyan). Illumination leads to their corresponding cis isomers (magenta). Within this study, we aimed to discover photoswitchable H3R antagonists that show at least a 10-fold increase or decrease in GPCR affinity upon illumination.
In this paper, we show how this core-centered strategy led to the new H3R antagonists VUF14738 and VUF14862 that represent a bidirectional set of robust and fatigue-resistant photoswitchable GPCR antagonists and are unique assets to further detail H3R pharmacology. We envision that these ligands are useful molecular tools for, e.g., probing H3R-mediated responses in physiology in a similar fashion as stereoisomers like R- and S-α-methylhistamine,29,30 but having control of H3R affinity using an external trigger. Moreover, in view of the importance of the H3R in various brain functions,17,31 spatiotemporal photopharmacology approaches will bring new experimental options (next to optogenetics) in order to delineate the importance of local neuronal circuits in H3R pharmacology.
Results and Discussion
Synthesis and Photochemical Characterization of Azobenzene-Derived H3 Ligands
Three synthesis routes were developed which allowed for great versatility in the substitution patterns of the azobenzene moiety (Scheme 1). A set of anilines was obtained by an amide coupling from 2–4 to 5–7. The anilines underwent diazotization followed by a quench with phenol to afford 8–11 and Williamson ether synthesis to yield the para-ether-substituted H3R photoswitchable ligands 12–17.
Scheme 1. General Synthetic Scheme for Photoswitchable H3R Antagonists.

Reagents and conditions: (a) pyrrolidine, EDCI·HCl, DIPEA, HOBt·H2O, DMF, rt, 16 h, 57–95%; (b) (I) NaNO2, 1 M aq HCl, 0 °C, 5 min; (II) phenol, aq NaOH, rt, 30 min; (III) 1 M aq HCl, aq satd NH4Cl, rt, 10 min, 30–81%; (c) NaI, K2CO3, Pip-(CH2)n-Cl·HCl, DMF, 130 °C, 16 h, 31–76%; (d) Oxone, H2O/DCM 4:1, rt, 3 h; (e) R-Ar-NH2, AcOH/DCM 1:1, rt, 16 h, 30–76% (over two steps); (f) R-OH, RockPhos, [PdCl(C3H5)]2, Cs2CO3, PhMe, 90 °C, 23 h, 12–47%; (g) R-OH, DEAD, PPh3, THF, 0 °C→ rt, 18 h, 33–73%; (h) 1-methylimidazole, 4-MePhSO2Cl, DCM, rt, 48 h, 43%; (i) (I) Oxone, H2O/DCM 4:1, rt, 6 h; (II) 3-OTBMDS-aniline, AcOH, rt, 16 h; (III) TBAF, THF, 0 °C, 10 min, 13%; (j) (I) NaH, DMF, rt, 30 min; (II) 36, 90 °C, 16 h, 59%; Pip = 1-piperidino.
The second route was designed to overcome the undesired para-selectivity in the diazotization and comprises oxidation of aniline 18 and 19 to a nitroso intermediate, condensation with an aniline to furnish 20–24 and a Pd/RockPhos-catalyzed ether synthesis32 to yield meta-ether substituted ligands 25–30. For the synthesis of constrained ligands, either a Mitsunobu reaction (for 31 and 33) or a Pd/RockPhos-catalyzed ether synthesis32 (for 32) was employed instead of the Williamson ether synthesis. The Mitsunobu approach was unable to yield 34, and instead, a tosylate-displacement strategy through intermediacy of 35–37 was used for this compound.33
We first investigated the photoisomerization of all final compounds as well as their thermal relaxation at 20 °C as determined by the method of Priimagi et al.34 (Table 1, Figure S1). Clear trends in the photochemistry could be deduced. It was observed that para-ether derivatives (scaffold A) have a distinctly higher λmax for the π–π* transition of the trans isomers than meta-ether derivatives (scaffold B). This trend can be explained due to a better “push–pull” profile in the electron density of scaffold A, which is known to red shift the λmax of electronic transitions.35,36 It was found that upon continuous illumination at 10 mM in DMSO-d6 with 360 ± 20 nm light a photostationary state (PSS) containing over 85% cis isomer (LC area at 254 nm) could be obtained for all compounds, with several ligands showing a PSS containing more than 95% cis isomer.
Table 1. Structure–photochemistry Relationship of the Photoswitchable Azobenzene-Derived H3R ligands.


Determined at 25 μM in 50 mM Tris-HCl pH 7.4 buffer + 1% DMSO-d6.
Thermal relaxation half-life times were determined according to the method of Priimagi et al.34 Arrhenius plots are available in Figure S1
Photostationary state area percentages after illumination with 360 ± 20 nm at 1 mM in DMSO-d6 and as determined by LC−MS analysis at 254 nm.
Unsubstituted phenyl moiety.
Various trends can be observed regarding the thermal relaxation half-lives of the compounds. In line with the literature, we observed a 3- to 5-fold difference in thermal relaxation half-life between the para-ethers (12, 13, 15, 17) and their respective meta-ether (25, 26, 28, 30) analogues having a propylpiperidine side chain.37 Such an effect can be attributed to increased conjugation in the para derivatives.38 In the series of ethylpiperidine analogues (14 versus 27, 16 versus 29), this effect of para- versus meta-ether substitution is even more pronounced, increasing the differences in thermal relaxation half-life to 360- and 212-fold, respectively. The propyl, cyclobutyl, and 4-piperidinyl spacers appear to have a comparable influence on the thermal relaxation half-lives (compare 15 versus 31–33 and 15/28 versus 33/34). If the amide group is placed at the ortho (13, 26) or para position (17, 30) the thermal relaxation half-life is comparable to its nonsubstituted counterpart (12, 25). However, having the amide in the meta position as in 15/28 yields a modest increase in thermal relaxation half-life.39
Compounds 28 and 33 were selected for further photochemical characterization based on their favorable pharmacological characteristics (vide infra). The thermal relaxation half-life times for these compounds in 50 mM Tris–HCl pH 7.4 buffer at room temperature were also directly determined, resulting in half-lives of 114 days for compound 28 (Figure S2) and 26 days for compound 33 (Figure S3). These long thermal relaxation half-lives are similar to the values obtained from the Arrhenius plots at 20 °C and uniquely allowed for a thorough analysis of the photoswitching and relaxation process by 1H NMR spectroscopy in conjunction with LC–MS chromatography and UV–vis spectroscopy. We first investigated isomerization of 28 and 33 at 10 mM in DMSO-d6 by monitoring both 1H NMR and LC–MS profiles in time. While multiple 1H NMR signals change upon photoswitching, the signals for the CH or CH2 group adjacent to the ether O atom provided a clearly resolved signal for quantification (Figure 2A,C). For both 28 and 33 the percentages of isomerization as analyzed by 1H NMR spectroscopy and LC–MS analysis at 254 nm (Figure 2B,D) are comparable yet slightly different, reflecting most likely some differences in ε values of the trans and cis isomers (Table S1). Despite these minor differences, LC–MS was subsequently used as a routine analysis in view of the higher throughput.
Figure 2.

(A) Representative part of 1H NMR spectra of 10 mM of compound 33 in DMSO-d6 illuminated at 360 ± 20 nm displayed at various time points (seconds). The presented peak belongs to the hydrogen atom explicitly drawn in the structure shown above the spectrum. Full 1H NMR spectra are available in Figure S18. (B) Representative part of LC–MS chromatograms belonging to the illuminated NMR samples shown in Figure 2A. The full chromatograms are available in Figure S19. (C) Representative part of 1H NMR spectra of 10 mM of compound 28 in DMSO-d6 illuminated at 360 ± 20 nm displayed at various time points (seconds). The presented peak belongs to the hydrogen atom explicitly drawn in the structure shown above the spectrum. Full 1H NMR spectra are available in Figure S20. (D) Representative part of LC–MS chromatograms belonging to the illuminated NMR samples shown in Figure 2C. The full chromatograms are available in Figure S21. (E) UV–vis spectra of 25 μM of compound 33 (trans) in 50 mM Tris–HCl pH 7.4 buffer containing 1% DMSO-d6. PSS cis represents a sample which has been illuminated for 300 s using 360 ± 20 nm light. PSS trans represents subsequent illumination for 300 s using 434 ± 9 nm. (F) UV–vis spectra of 25 μM of compound 28 (trans) in 50 mM Tris–HCl pH 7.4 buffer containing 1% DMSO-d6. PSS cis represents a sample which has been illuminated for 300 s using 360 ± 20 nm. PSS trans represents subsequent illumination for 300 s using 434 ± 9 nm. (G) Absorbance at 344 nm of 25 μM of compound 33 in 50 mM Tris–HCl pH 7.4 buffer +1% DMSO-d6. UV–vis spectra were obtained with 1 s intervals under alternating illumination with 360 ± 20 nm and 434 ± 9 nm perpendicular to the light source of the UV–vis spectrometer. (H) Absorbance at 319 nm of 25 μM of compound 28 in 50 mM Tris–HCl pH 7.4 buffer +1% DMSO-d6. UV–vis spectra were obtained with 1 s intervals under alternating illumination with 360 ± 20 nm and 434 ± 9 nm perpendicular to the light source of the UV–vis spectrometer. (I) Repeated isomerization cycles of 25 μM of compound 33 in a pH 7.4 buffer containing 15 mM HEPES, 64 mM NaCl, 25 mM KCl, 0.4 mM CaCl2, and 0.8 mM MgCl2 containing 1% DMSO-d6 analyzed at 349 nm. PSS cis was obtained by using illuminations for 20 s at 360 ± 20 nm. PSS trans was obtained by using illuminations for 20 s at 434 ± 9 nm. An extended figure is available in Figure S16. (J) Repeated isomerization cycles of 25 μM of compound 28 in a pH 7.4 buffer containing 15 mM HEPES, 64 mM NaCl, 25 mM KCl, 0.4 mM CaCl2, and 0.8 mM MgCl2 containing 1% DMSO-d6 analyzed at 319 nm. PSS cis was obtained by using illuminations for 20 s at 360 ± 20 nm. PSS trans was obtained by using illuminations for 20 s at 434 ± 9 nm. An extended figure is available in Figure S17.
Next, the photoisomerization rate was investigated at 25 μM in 50 mM Tris–HCl pH 7.4 buffer supplemented with 1% DMSO-d6 using UV–vis spectroscopy (Figure 2E,F). Rapid photoisomerization was observed for both compounds during illuminations at both 360 ± 20 nm and 434 ± 9 nm to obtain high amounts of cis isomer or considerable amounts of trans isomer, respectively (Figures S4–S6 and S10–S12). After 30 s of illumination, isomerization of both key compounds had reached isomerization percentages within 5% of their PSS. Thus, illuminating for 300 s was always sufficient to reach PSS. Arguably due to both solvent and concentration effects,36,40,41 the measured isomerization rates are considerably higher in the experiments performed at 25 μM in buffer, compared to the measurements at 10 mM in DMSO-d6 of the two H3R ligands.
We subsequently studied the real-time isomerization behavior of both compounds at a concentration of 25 μM in 50 mM Tris–HCl pH 7.4 buffer supplemented with 1% DMSO-d6 using a setup which allowed the illumination perpendicular to the path of the UV–vis spectrometer light source. Under these conditions, we determined a photoisomerization half-life for 33 of 4 ± 0.2 s under illumination with 360 ± 20 nm and 8 ± 0.6 s under illumination with 434 ± 9 nm (Figure 2G). For 28, the values were found to be 16 ± 0.9 s under illumination at 360 ± 20 nm and 11 ± 0.3 s under illumination at 434 ± 9 nm (Figure 2H).
Both photoswitchable compounds proved to be very photostable. Even after 48 h of illumination at 360 ± 20 nm with an intensity of 0.77 mW/mm2, no apparent photodegradation was visible in both 1H NMR and LC–MS analyses of 28 and 33. To exemplify the resistance to fatigue of 28 and 33, over 1000 isomerization cycles switching between illumination at 360 ± 20 and 434 ± 9 nm with 20 s intervals were executed. The absorbance at λmax of the π–π* transition of the trans isomer did not alter considerably (Figure 2I,J and Figures S16 and S17), indicating that both key photoswitches are highly resistant to photobleaching.
Photopharmacological Characterization
Because of their long thermal relaxation half-lives, the newly synthesized photoswitchable GPCR ligands are uniquely compatible with competition radioligand-binding experiments. All new azobenzene-derived H3R ligands, either illuminated using 360 ± 20 nm to PSS (as determined by LC–MS) or kept in the dark, were evaluated for their affinity for the human H3R by a [3H]-Nα-methylhistamine competition binding experiments performed in the dark. Both histamine and, as previously reported,28 the parent naphthalene compound 1 potently displace [3H]-Nα-methylhistamine binding to membranes overexpressing the H3R (Table 2), and their affinity values were not affected by illumination of the compound solutions at 360 ± 20 nm (data not shown). Replacement of the naphthalene core of 1 with the azobenzene core resulted in a series of moderate to highly potent H3R ligands with pKi values between 6.01 and 8.76 as trans isomers (i.e., without illumination). The direct azobenzene analogue of 1, compound 17, bound the H3R with only a slightly lower affinity (pKi = 7.96 ± 0.06, Table 2) compared to 1 (pKi = 8.29 ± 0.01, Table 2). Furthermore, the affinity for H3R was modulated effectively by shifting the position of the amine side chain, with scaffold A (para-substitution) always resulting in better affinities than scaffold B (meta-substitution) (Table 2). In general, the propyl spacer proved advantageous over the ethyl spacer for H3R affinity. The effects of amide positioning varied among the different subclasses.
Table 2. Affinity for the Human H3R of Photoswitchable Azobenzene-Derived H3R Ligands.


Photostationary state area percentages after illumination with 360 ± 20 nm at 1 mM in DMSO-d6 and as determined by LCMS analysis at 254 nm.
Measured by displacement of [3H]-Nα-methylhistamine on HEK293T cell homogenates transiently expressing the H3R.
Defined as pKi PSS cis – pKitrans.
Unsubstituted phenyl.
Trans sample contained 3.9 area % cis compound.
Rather than absolute affinities of the trans isomers for H3R, the shift in affinity upon illumination was considered the prime parameter for photopharmacology applications and selection of key compounds. An increase in binding affinity upon photoisomerization (illumination at 360 ± 20 nm) was observed for 12, 25, 27, 28, and 30. Interestingly, most of these compounds contain a meta-ether-substituted azobenzene core (scaffold B) with either a 3-carboxamide (27, 28), a 4-carboxamide (30), or no carboxamide substituent (25). The ethyl-spaced 27 has an 8.3-fold decrease in H3R affinity compared to the propyl-spaced analogue 28. Within the set of compounds lacking a carboxamide, meta- and para-ether-substituted azobenzene compounds 25 and 12 (scaffolds B and A) show similar shifts in H3R affinity upon isomerization. The 13.5-fold increase in H3R affinity upon photoswitching of 28 (Figure 3A) led to its identification as one of the key compounds of this study (VUF14738).
Figure 3.

(A, B) Representative curves of [3H]-Nα-methylhistamine binding displacement by increasing concentrations (A) 28 or (B) 33. The curve indicated as PSS cis consists of predominantly (>90%) cis compound. (C, D) Proposed binding modes of (C) 28 and (D) 33 in an H3R homology model based on representative MD snapshots. The cyan and magenta carbon atoms of the ligands correspond to the trans and cis isomer, respectively, and for the purpose of clarity, parts of ECL1/TM3 and ECL2 are not shown.
Compounds that show decreased H3R binding affinity upon isomerization were primarily found to have a para-ether-substituted azobenzene (scaffold A). The trend that ethyl-spaced compounds perform less well than their respective propyl-spaced analogues was also observed for the pairs 14/15 and 16/17. Derivatives with a 2-carboxamide substituents (13, 26) gave smaller shifts in H3R affinity compared to their 3-carboxamide counterparts (15, 28). The observed decreases in H3R binding affinity upon isomerization observed for these compounds (13–17, 26, and 29) were deemed insufficient for proper photopharmacological application. To address this issue, the core azobenzene-substituted scaffold was maintained, but the propyl spacer between the ether and piperidine moiety was constrained using isopropylpiperidinyl and 1,3-cyclobutyl spacers. Such linker rigidification is a known strategy to increase the binding affinity of H3R antagonists.33,42 Importantly, we reasoned that the limited degrees of freedom associated with the linker rigidification would hamper the cis isomer from (partially) readjusting its binding mode in the GPCR protein. Upon linker rigidification, the H3R binding affinities for 31 and 33 were considerably increased compared to their flexible counterpart 15 (Table 2). Additionally, a pronounced increase in the shift in H3R affinity between the trans and cis analogues was observed. Of note is the lower H3R affinity of stereoisomer 32 compared to 33, which is in accordance with previously reported data.33 Compound 33 outperformed 31 based on having a larger shift in H3R affinity (11.2-fold lower, Figure 3B) upon isomerization and higher absolute H3R affinity. Moving the trans-cyclobutyl side chain to the meta-position (34), effectively arriving at a constrained version of key compound 28, virtually abolished the pKi shift and did not increase the binding affinity. All this led to the designation of 33 as the second key compound (VUF14862).
Compounds 28 and 33 either gain or lose at least 10-fold H3R affinity upon illumination (Figure 3A,B). Thus, using the same photoswitchable core and by proper side-chain substitution in the periphery of the scaffold, we developed bidirectional GPCR photoswitchable ligands. Moreover, 28 and 33 show a 10- to 100-fold H3R selectivity over the H1R and no measurable affinity for H2R and H4R (Table S2). With the cautious use of 28 in view of its residual H1R affinity, 28 and 33 offer a bidirectional set of photopharmacological tools for H3R.
Binding Mode Characterization
Molecular modeling studies were performed for tool compounds 28 and 33 in order to gain insights into the molecular mechanism of the observed affinity changes upon isomerization. Molecular docking of 28 and 33 into a series of H3R homology models yielded three potential binding modes in which the ligands form either an ionic interaction with the D1143.32 or E2065.46 residue in transmembrane helices (TM) 3 and 5, respectively.24,43,44 The predicted binding modes share similarity with the binding orientation of ergotamine as observed in crystal structures of serotonin receptors 5-HT1B and 5-HT2B (PDB IDs: 4IAR, 4IB4),45,46 aminergic GPCRs that share 54–62% sequence similarity for the binding site with H3R (Figure S22). Each of the 12 resulting H3R–ligand complexes (2 ligands × 2 isomers × 3 binding modes) was investigated using 50 ns molecular dynamics (MD) simulations. The MD trajectories indicated that only for one binding mode (Figure S22A) (i) a consistent interaction fingerprint (IFP)47 was observed throughout the simulation and (ii) the stable ligand binding mode was in line with the observed SAR data (Table 2). In this binding mode (Figure 3C,D) the ligands form a stable salt bridge with D1143.32 and move upward along TMs 2, 3, and 7 into the extracellular vestibule (ECV). The trans isomer of 33 adopts an elongated binding mode and makes multiple aromatic stacking interactions with Y912.61, Y942.64, Y3947.35, and F3987.39, while the carboxamide substituent is solvent exposed between the extracellular loops 1 and 2 (Figure 3D). The trans isomer of 28, on the other hand, adopts an arched binding mode within the extracellular vestibule (Figure 3C) due to its meta-substitution. The binding modes of the trans isomers of both 28 and 33 are similar to the previously proposed binding modes for bitopic ligands of the aminergic dopamine D2 receptor.48 The cis isomers of both 28 and 33 adopt an almost identical binding mode (Figure 3C,D) in which the carboxamide is buried in the ECV in close proximity to Y3947.35, folded against the azobenzene, and located at the same position as the pyrrolidine moiety of ergotamine bound to 5-HT1B and 5-HT2B crystal structures (Figure S23).
The opposite effects of trans to cis isomerization on the H3R binding affinities of 28 and 33 can be explained by differences in (i) the complementarity of polar or apolar surface areas of the ligand and the protein binding site49 and (ii) the loss of conformational degrees of freedom as the result of ligand binding (conformer focusing50). Both isomers of 33 are expected to be less affected by conformer focusing due to their rigid cyclobutyl-spacer compared to the flexible propyl spacer of 28. The decreased affinity of the trans isomer versus the cis isomer of 28 (Table 2) can be explained by the fact that the apolar surface area of 28 is >4 times more solvent exposed in the trans isomer binding mode than in the cis isomer binding mode in H3R. Conversely, the trans isomer of 33 has a reduced buried polar surface area compared to the folded cis isomer, while the apolar surface area of 33 is able to form a complementary fit with the apolar surface area of the H3R binding site. In addition, the extended trans isomer of 33 allows more degrees of conformational freedom than the folded cis isomer, altogether resulting in an increased H3R affinity for the trans isomer of 33 (Table 2).
Dynamic Photochemical Modulation of H3R
As the new azobenzene-containing H3R photoswitches can be dynamically and reversibly isomerized with light, we applied the two key H3R photoswitches in a dynamic intact cell system with a real-time readout. Two-electrode voltage clamp (TEVC) using Xenopus laevis oocytes co-expressing H3R and a GIRK channel composed of human GIRK1 (Kir3.1) and GIRK4 (Kir3.4) subunits (Figure 4A)51 allows for a real-time H3R readout. In all experiments, H3R-expressing oocytes were used that evoked a high current (>500 nA) upon histamine superfusion in 64 mM NaCl, 25 mM KCl, 15 mM HEPES, 0.8 mM MgCl2, and 0.4 mM CaCl2 pH 7.4 at a membrane potential of −80 mV. Under these conditions, histamine induced GIRK channel activation with a pEC50 value of 7.3 ± 0.06 (Figure 4A). No currents were observed after histamine application to noninjected oocytes (Figure S24). The GIRK channel activation by histamine (1 μM) could be fully inhibited upon simultaneous superfusion with the H3R antagonist clobenpropit (1 μM) or by pretreating the oocytes with the Gαi-protein inhibitor pertussis toxin (1.37 ng/oocyte) (Figures S25 and S26). This shows that the histamine-induced current in Xenopus laevis oocytes coexpressing H3R and a GIRK channel composed of human GIRK1 (Kir3.1) and GIRK4 (Kir3.4) subunits is H3R mediated through Gi-protein coupling to GIRK.
Figure 4.

(A) Schematic drawing of the TEVC setup used for dynamic H3R and GIRK current (in)activation and concentration–response curve of histamine evoked currents in Xenopus oocytes expressing H3R and GIRK. The inset shows the current increase upon continuous histamine (1 μM) perfusion in time. (B) Representative part of a GIRK-mediated current trace during continuous perfusion with 5 μM histamine in competition with 1 μM 33 under illumination of the Xenopus oocyte with alternating 360 ± 20 and 434 ± 9 nm wavelength as measured by TEVC. An extended time trace is available in Figure S34. (C) Representative part of a GIRK-mediated current trace during continuous perfusion with 5 μM histamine in competition with 1 μM 28 and illumination of the oocyte with alternating 360 ± 20 and 434 ± 9 nm wavelength as measured by TEVC. An extended time trace is available in Figure S35.
We first ensured that photoisomerization behavior of 28 and 33 in HEPES pH 7.4 buffer was similar to behavior in 50 mM Tris–HCl pH 7.4 buffer (Figure S4–S15). Next, we superfused Xenopus oocytes expressing H3R and a GIRK channel with histamine and either trans-28 or trans-33 for dynamic receptor inhibition studies. Superfusion of only 28 or 33 on both noninjected and H3R-GIRK expressing oocytes did not result in changes in current, confirming that these H3R ligands act as antagonists (Figure S27–S30). Illumination directly on the oocytes (Figure 4A) in the absence of 28 or 33 did not change the magnitude of histamine evoked currents (Figure S31). Yet, in the presence of either 33 or 28, clear photoswitchable antagonism of the histamine-induced current was observed under alternating illumination. We confirmed that both compounds were able to show this behavior in a concentration-dependent fashion (Figures S32 and S33), yielding Ki values for 33 that nicely match the Ki values obtained in the radioligand binding experiments (TEVC pKi > 99% trans: 8.72 ± 0.11, PSS cis: 7.98 ± 0.15, radioligand binding: pKi > 99% trans: 8.76 ± 0.09, PSS cis: 7.71 ± 0.09). An increase in histamine-induced GIRK activation was observed when trans-33 was illuminated with 360 ± 20 nm, confirming that the H3R antagonistic effect of 33 is reduced upon isomerization to its cis isomer. When subsequently the superfused oocytes were illuminated at 434 ± 9 nm, a decrease in H3R-induced GIRK activation was observed due to a stronger H3R antagonistic effect by the trans isomer of 33 as a consequence of increased H3R binding affinity. This effect could be repeated several times in subsequent illumination cycles in the same experiment (Figure 4B). For 28, the reciprocal light-induced H3R affinity shift compared to 33 translated well to a reciprocal effect upon illumination in the TEVC setup (Figure 4C). That is, 28 reached the highest H3R inhibition, measured as reduced histamine-induced GIRK channel activation, upon illumination with 360 ± 20 nm. Also, 28 could be effectively switched back in the TEVC setup upon illumination with 434 ± 9 nm, and again the GPCR blocking properties could be modulated by alternating illumination (Figure 4C). These experiments demonstrate the rapid bidirectional modulation of the H3R using the two complementary photoswitchable ligands 28 and 33.
Conclusions
A robust bidirectional photochemical toolbox for dynamic antagonism of the H3R has been successfully developed using a core-centered approach replacing a naphthalene moiety in a known scaffold by a properly substituted azobenzene unit. SAR exploration delivered 16 compounds that show a shift in binding affinity for the H3R upon photoisomerization. Two key compounds, 28 and 33, were selected on the basis of their 13.5-fold increase or 11.2-fold decrease in H3R binding affinity upon photoisomerization, respectively. The long thermal relaxation half-lives and resistance to fatigue of these compounds allowed in-depth spectroscopic analysis (NMR, LC–MS, UV–vis) of the photochemical properties. The rapid photochemical isomerization directly translated to dynamic, light-modulated H3R blockade in real-time electrophysiology experiments using H3R and GIRK coexpressing Xenopus oocytes. In this dynamic experimental setup, histamine-induced GPCR activation could be modulated on a time scale of seconds, and we were able to show real-time, light-sensitive blockade of the H3R activation by both new tool compounds. In our view, 28 (VUF14738) and 33 (VUF14862) are highly useful photopharmacological tools for temporal studies to dissect the complex signaling cascade of H3R. Moreover, the presented bidirectional modulation of H3R protein by closely related photosensitive analogs of the same core scaffold further emphasizes the exceptional opportunities of conformational modulation of the GPCR protein family.
Experimental Section
Synthesis and Characterization of Compounds
All starting materials were obtained from commercial suppliers (primarily being Sigma-Aldrich, Acros Organics, Fluorochem, and Strem Chemicals) and used without purification. 4-Hydroxyazobenzene (8) was obtained from Sigma-Aldrich. cis-3-Piperidin-1-ylcyclobutanol (35) was synthesized according to the literature procedure of Wijtmans et al.33 Anhydrous THF and DCM were obtained by passing them through an activated alumina column prior to use. Anhydrous toluene was freshly distilled over CaH2 or dried over activated 4 Å molecular sieves, which were added at least 24 h before use. Anhydrous DMF was purchased from Acros Organics (Geel, Belgium) and used without prior purification. All reactions were carried out under nitrogen atmosphere unless mentioned otherwise. TLC analyses were performed using Merck F254 aluminum-backed silica plates and visualized with 254 nm UV light or a potassium permanganate stain. Flash column chromatography was executed using Silicycle Siliaflash F60 silica gel or by means of a Biotage Isolera equipment using Biotage SNAP columns. All HRMS spectra were recorded on a Bruker micrOTOF mass spectrometer using ESI in positive-ion mode. All NMR spectra were recorded on either a Bruker Avance 250, Bruker Avance 400, Bruker Avance 500, or Bruker Avance 600 spectrometer. The peak multiplicities are defined as follows: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; dd, doublet of doublets; dt, doublet of triplets; td, triplet of doublets; bs, broad singlet; m, multiplet. The spectra were referenced to the internal solvent peak as follows: CDCl3 (1H = 7.26 ppm, 13C = 77.16 ppm), DMSO-d6 (1H = 2.50 ppm, 13C = 39.52 ppm), CD3OD (1H = 3.31 ppm, 13C = 49.00 ppm).53 IUPAC names were adapted from ChemBioDraw Ultra 14.0 (PerkinElmer). Purities were measured with the aid of analytical LC–MS using a Shimadzu LC-20AD liquid chromatography pump system with a Shimadzu SPDM20A diode array detector with the MS detection performed with a Shimadzu LCMS-2010EV mass spectrometer operating in both positive and negative ionization mode. The column used was an Xbridge (C18) 5 μm column (50 mm × 4.6 mm or 100 mm × 4.6 mm). The following solutions are used for the eluents. Solvent A: water/formic acid 999:1 and solvent B: acetonitrile/formic acid 999:1. The eluent program used is as follows: flow rate: 1.0 mL/min, start 95% A in a linear gradient to 10% A over 4.5 min, hold 1.5 min at 10% A, in 0.5 min in a linear gradient to 95% A, hold 1.5 min at 95% A, total run time: 8.0 min. Compound purities were calculated as the percentage peak area of the analyzed compound by UV detection at 254 nm. All chemistry and analyses of photosensitive compounds were carried out under dimmed or red light.
Detailed chemical experimental procedures and chemical analyses are supplied in the Supporting Information.
Photochemistry
UV–vis spectra were obtained using a Thermo-scientific Evolution 201 PC spectrophotometer or a Shimadzu UV-2401 PC spectrophotometer. Fits of UV–vis spectroscopy data were generated using Mathworks Matlab R2014A (8.3.0.532). Illumination was executed using a Sutter instruments Lambda LS with a 300 W full-spectrum lamp connected to a Sutter instruments Lambda 10-3 optical filter changer equipped with 434 ± 9 nm and 360 ± 20 nm filters. For photochemical analyses illuminations were performed in Hellma Suprasil quartz 114-QS cuvettes. Thermal relaxation experiments and Arrhenius extrapolations were performed according to Priimagi et al.34 using a compound concentration of 25 μM in 50 mM Tris–HCl pH 7.4 buffer +1% DMSO-d6 and temperatures of 60, 70, and 80 °C. Illuminations for pharmacological experiments were performed in cylindrical clear glass vials with a volume of 4.5 mL. The typical distance between light source and vial or cuvette was 2 cm. In illuminations during TVEC experiments the light-source was positioned at 5 cm from the chamber containing the oocyte. The focused beam of the light source has a diameter of 1.8 cm and the beam was pointed such that it illuminated the full oocyte. The light intensity on the oocyte is 0.77 mW/mm2 using the 360 ± 20 nm filter and 0.57 mW/mm2 for the 434 ± 9 nm filter as measured using a Thorlabs PM16–401 power meter.
Computational Chemistry
Homology Model Construction
The sequence of H3R was obtained from UniProt (accession code Q9Y5N1) and aligned to the unique sequences of crystallized aminergic GPCRs (PDB accessed at 3 Nov 2015). The sequence similarity and identity were assessed for the full sequence, each TM helix, extracellular loop 2, and the binding pocket as previously defined.23 On the basis of this analysis, the sequence alignment of H3R to H1R, M3R, D3R, 5-HT1BR ,and 5-HT2BR and their crystal structures (PDB codes 3RZE, 4U15, 3PBL, 4IAR, and 4IB4) were used as input for MODELER (version 9.15).54 Five hundred different homology models were constructed from which 36 were selected based on visual inspection. Compounds trans-28, cis-28, trans-33, and cis-33 were subsequently docked into each model using PLANTS55 (settings: speed 1, scoring function ChemPLP, 10-fold docking, 50 proposed poses per fold) into each of the models while treating the side chains of E2065.46 and F3987.39 as flexible. The resulting 26000 docking poses were filtered using IFP47 on either and ionic and H-bond interaction with D1143.32 or E2065.46 from which the top 5 ChemPLP-scored poses were kept for further processing, yielding 396 H3R–ligand complexes. Visual inspection highlighted three clusters of potential binding modes. From each ligand in each binding mode cluster one model was selected for further investigation with molecular dynamics simulations.
Molecular Dynamics Simulations
Each ligand–H3R complex was aligned to the membrane-aligned H1R structure from the OPM database. A sodium ion was added in the sodium binding site based on high-resolution sodium-bound β1-adrenoceptor crystal structure (PDB code 4BVN). Subsequently, the rotamers from the sodium binding site residues were manually optimized.56 The H3R–ligand complexes were embedded in a pre-equilibrated box containing a lipid bilayer (134 POPC lipids), explicit solvent (∼11000 water molecules), and a 0.15 M concentration of NaCl (∼70 ions). Using MOE (v2015.1001; Chemical Computing Group, Inc., Montreal, Quebec, Canada), the ligand–H3R complex was solvated, and waters between the receptor and the membrane and outside the receptor were removed. The ligands were parametrized for GAFF using Amber’s antechamber with AM1-BCC partial charges. The resulting parameters of the ligand were checked, and the azobenzene parameters were optimized in line with the parameters reported by Schäfer et al.57 The AMBER99SB*-ILDN force field was used for the protein and the lipids and waters were described according to the Berger lipid parameters and the TIP3P model, respectively.58 After the system was neutralized, a 1000-step steepest descent minimization of the system was performed after which the complex was equilibrated for 75 and 300 ps using a 1 and 2 fs time step, respectively. A subsequent production run of 50 ns as an NVT ensemble was performed for each combination. The trajectories were analyzed using IFP47 resulting in a time-based interaction profile for each H3R–ligand complex. These MD simulations were performed using GROMACS 5.1.59
Pharmacology
General Data
Within this paper, the 445-isoform of the human H3R is referred to as wild-type receptor. [3H]-Mepyramine (specific activity 20 Ci/mmol–1), [125I]-iodoaminopotentidine (specific activity 2200 Ci/mmol–1), [3H]-Nα-methylhistamine (specific activity 78.3 Ci/mmol–1), and [3H]-histamine (specific activity 17.5 Ci/mmol–1) were purchased from PerkinElmer (Groningen, The Netherlands). All other chemicals were of analytical grade and obtained from commercial sources.
Cell Culture and Transfection
Human embryonic kidney 293T cells (HEK293T cells) were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 IU/mL), and streptomycin (50 μg/mL). HEK293T cells were transiently transfected with H3R cDNA using the polyethylenimine (PEI) method. In brief, 24 h prior to transfection 2 × 106 cells were seeded on a 10 cm dish. Cells were transfected with 5 μg of H3R cDNA and 20 μg of PEI in 500 μL of 150 mM NaCl solution. The DNA–PEI mixture was incubated for 15–30 min at 22 °C before dropwise addition to the cells. Cells were maintained for 48 h at 37 °C with 5% CO2 before harvesting.
Preparation of Cell Homogenates
Forty-eight hours after transfection, cells were detached and collected from the plates by vigorous pipetting using phosphate-buffered saline (PBS). Cells were centrifuged at 3000 rpm for 10 min, and the cell-pellet was stored at −20 °C until the day of the experiment.
Radioligand-Binding Assay
For radioligand binding experiments, samples were pretreated to obtain trans and cis samples. In brief, a 1 mM ligand solution was divided in two samples which were either illuminated at 360 ± 20 nm until their photostationary state was reached as analyzed by LC–MS or kept in the dark for the same duration as the illuminated samples. All subsequent handling was performed in the dark or under near-infrared light. [3H]-Nα-Methylhistamine binding assays were performed in a total volume of 100 μL consisting of cold ligand (increasing concentration ranging from 10–5 to 10–12 M) prepared in assay buffer [50 mM Tris–HCl pH 7.4], 1.6 nM [3H]-Nα-methylhistamine, and HEK293T cell homogenates transiently expressing the human H3R (>25 μg/50 μL). The mixture was incubated for 2 h at 25 °C before termination by rapid filtration over a 96 well GF/C filter plate precoated with 0.5% PEI using PerkinElmer 96-well filtermate-harvester (PerkinElmer). The filter was washed five times with ice-cold wash buffer [50 mM Tris-HCl pH 7.4, 4 °C]. Five hours after addition of Microscint O scintillation liquid, filter-bound radioactivity was measured using a microbeta wallac trilux scintillation counter (PerkinElmer).
Binding Selectivity Screening
H1R radioligand binding assays were performed according to the method of Kuhne et al.60 using an incubation time of 2 h instead of 4 h. H2R radioligand binding assays were performed according to Leurs et al.61 using HEK293T cells instead of CHO cells and a total volume of 400 μL instead of 200 μL. H4R radioligand binding assays were performed according to the method of Nijmeijer et al.62
Data Analysis
All experiments were analyzed using Graphpad prism 6.05 (Graphpad Software, Inc., San Diego, CA). IC50 values obtained from competition binding experiments were converted to Ki values using the Cheng–Prusoff equation.63
Two-Electrode Voltage Clamp Assay
cRNA Synthesis
The Kir3.1 and Kir3.4 were both supplied in the pcDNA3.1 vector and were a kind gift of K. Sahlholm (Karolinska Institute, Stockholm, Sweden). The H3R was cloned into the pCIneo vector. Restriction enzymes BAMHI and NdeI were used to linearize the constructs pCIneo-H3R and pcDNA3.1-Kir3.1 and 3.4, respectively. After the digestion, 0.5% SDS and Proteinase K (200 μg/mL) were added, and the mixture was incubated at 50 °C for 30 min. DNA was extracted by phenol/chloroform and the precipitation method. The quality and digestion were checked on 1% agarose gel. The cRNA synthesis was performed with the T7 mMessage mMachine kit (Ambion, Austin, TX). After the synthesis, the cRNA quality and amount were checked on a denaturating gel for RNA as described by Almeida et al.64
H3R and GIRK Expression in Oocytes
Xenopus laevis oocytes were supplied as “topgrade” oocytes (Ecocyte, Castrop-Rauxel, Germany). Each oocyte was injected with a volume of 46 nL, containing 3 ng cRNA of each GIRK subunit and 50 ng cRNA of the H3R or with 46 nL of RNase free water as control with the Nanoject II (Drummond Scientific Company, Broomall, PA). The oocytes were incubated for 4–6 days at 12 °C in modified Barth solution (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 15 mM HEPES, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 2.5 mg pyruvic acid, 100 μg/mL streptomycin, 50 μg/mL gentamycin, adjusted with Tris to pH 7.4), as described by Sahlholm et al.51
Two-Electrode Voltage Clamp
TEVC was performed with an Axoclamp 900A amplifier and a Digidata 1550 Digitizer. The currents were recorded and analyzed with pClamp 10.6 (Molecular Devices, Sunnyvale, CA). Glass micropipettes were pulled from borosilicate capillaries (GC150-10, Harvard Apparatus, Edenbridge, UK) with the P-1000 micropipette puller (Sutter Instrument, Novato, CA) to have a resistance of 1–3 MΩ when filled with 3 M KCl solution. The oocyte was positioned in the recording chamber (RC-1Z, Warner Instruments, Hamden, CT), and by gravity flow, high potassium solution (64 mM NaCl, 25 mM KCl, 0.8 mM MgCl2, 0.4 mM CaCl2, 15 mM HEPES and adjusted with Bis-Tris Propane to pH 7.4) was perfused through the recording chamber. For the study of H3R ligands, the membrane potential was clamped at −80 mV and the evoked currents were measured at room temperature (∼20 °C). The ligands (histamine, clobenpropit, 28 and 33) were diluted in the high potassium solution to obtain the appropriate concentrations. For the photoswitchable ligands (28 and 33), the TEVC was performed in the dark, and the different wavelengths (360 ± 20 nm and 434 ± 9 nm) were applied directly on the oocyte in the recording chamber for 1 min (with 28) or 2 min (with 33) with a Sutter instruments Lambda LS with a 300 W full spectrum lamp connected to a Sutter instruments Lambda 10–3 optical filter changer equipped with 434 ± 9 nm and 360 ± 20 nm filters to observe the (functional) switch of the ligands. For the pertussis toxin experiment, 46 nL (1.38 ng) of PTX or water as control was injected in H3R/GIRK expressing oocytes (4–6 days after cRNA injection) with the Nanoject II The oocytes were incubated for another 7 h before measuring the currents evoked by 1 μM histamine to the oocytes.
Acknowledgments
We acknowledge The Netherlands Organisation for Scientific Research (NWO) for financial support (TOPPUNT, “7 ways to 7TMR modulation (7-to-7)”, 718.014.002). E.W.E.V. is supported by NWO ECHO project 711.013.014. All authors participate in the European Cooperation in Science and Technology Action CM1207 [GPCR-Ligand Interactions, Structures, and Transmembrane Signaling: A European Research Network (GLISTEN)]. We thank Hans Custers for HRMS analyses, Yara Huppelschoten for determining the selectivity of the key compounds amongst the histamine receptor family, and Andrea Van de Stolpe and Ken Lion for setting up the photochemical equipment. Kristoffer Sahlholm (Karolinska institute) is kindly acknowledged for providing the pcDNA3.1-Kir3.1 and -Kir3.4 plasmids.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11422.
Detailed synthetic procedures, chemical analyses and spectra, and additional data as noted in the text (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Broichhagen J.; Frank J. A.; Trauner D. Acc. Chem. Res. 2015, 48, 1947. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Angew. Chem., Int. Ed. 2016, 55, 10978. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- Szobota S.; Isacoff E. Y. Annu. Rev. Biophys. 2010, 39, 329. 10.1146/annurev.biophys.093008.131400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry A. A.; Woolley G. A. Chem. Soc. Rev. 2011, 40, 4422. 10.1039/c1cs15023e. [DOI] [PubMed] [Google Scholar]
- Szymanski W.; Beierle J. M.; Kistemaker H. A.; Velema W. A.; Feringa B. L. Chem. Rev. 2013, 113, 6114. 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. J. Am. Chem. Soc. 2014, 136, 2178. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Stein M.; Middendorp S. J.; Carta V.; Pejo E.; Raines D. E.; Forman S. A.; Sigel E.; Trauner D. Angew. Chem., Int. Ed. 2012, 51, 10500. 10.1002/anie.201205475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J. A.; Moroni M.; Moshourab R.; Sumser M.; Lewin G. R.; Trauner D. Nat. Commun. 2015, 6, 7118. 10.1038/ncomms8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broichhagen J.; Schonberger M.; Cork S. C.; Frank J. A.; Marchetti P.; Bugliani M.; Shapiro A. M.; Trapp S.; Rutter G. A.; Hodson D. J.; Trauner D. Nat. Commun. 2014, 5, 5116. 10.1038/ncomms6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittolo S.; Gomez-Santacana X.; Eckelt K.; Rovira X.; Dalton J.; Goudet C.; Pin J. P.; Llobet A.; Giraldo J.; Llebaria A.; Gorostiza P. Nat. Chem. Biol. 2014, 10, 813. 10.1038/nchembio.1612. [DOI] [PubMed] [Google Scholar]
- Schönberger M.; Trauner D. Angew. Chem., Int. Ed. 2014, 53, 3264. 10.1002/anie.201309633. [DOI] [PubMed] [Google Scholar]
- Bahamonde M. I.; Taura J.; Paoletta S.; Gakh A. A.; Chakraborty S.; Hernando J.; Fernandez-Duenas V.; Jacobson K. A.; Gorostiza P.; Ciruela F. Bioconjugate Chem. 2014, 25, 1847. 10.1021/bc5003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broichhagen J.; Johnston N. R.; von Ohlen Y.; Meyer-Berg H.; Jones B. J.; Bloom S. R.; Rutter G. A.; Trauner D.; Hodson D. J. Angew. Chem., Int. Ed. 2016, 55, 5865. 10.1002/anie.201600957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M.; Schafroth M. A.; Sarott R.; Imhof M.; Bold C.; Leippe P.; Dhopeshwarkar A.; Grandner J.; Katritch V.; Mackie K.; Trauner D.; Carreira E. M.; Frank J. A. J. Am. Chem. Soc. 2017, 139, 18206. 10.1021/jacs.7b06456. [DOI] [PubMed] [Google Scholar]
- Haas H.; Panula P. Nat. Rev. Neurosci. 2003, 4, 121. 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Leurs R.; Bakker R. A.; Timmerman H.; de Esch I. J. P. Nat. Rev. Drug Discovery 2005, 4, 107. 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- Panula P.; Chazot P. L.; Cowart M.; Gutzmer R.; Leurs R.; Liu W. L.; Stark H.; Thurmond R. L.; Haas H. L. Pharmacol. Rev. 2015, 67, 601. 10.1124/pr.114.010249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J. C. Br. J. Pharmacol. 2011, 163, 713. 10.1111/j.1476-5381.2011.01286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebois E. P.; Jones C. K.; Lindsley C. W. Curr. Top. Med. Chem. 2011, 11, 648. 10.2174/1568026611109060648. [DOI] [PubMed] [Google Scholar]
- Leurs R.; Vischer H. F.; Wijtmans M.; de Esch I. J. P. Trends Pharmacol. Sci. 2011, 32, 250. 10.1016/j.tips.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Celanire S.; Wijtmans M.; Talaga P.; Leurs R.; de Esch I. J. Drug Discovery Today 2005, 10, 1613. 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- Uveges A. J.; Kowal D.; Zhang Y.; Spangler T. B.; Dunlop J.; Semus S.; Jones P. G. J. Pharmacol. Exp. Ther. 2002, 301, 451. 10.1124/jpet.301.2.451. [DOI] [PubMed] [Google Scholar]
- Kooistra A. J.; Kuhne S.; de Esch I. J.; Leurs R.; de Graaf C. Br. J. Pharmacol. 2013, 170, 101. 10.1111/bph.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe F. U.; Bembenek S. D.; Szalma S. J. Mol. Graphics Modell. 2006, 24, 456. 10.1016/j.jmgm.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Lorenzi S.; Mor M.; Bordi F.; Rivara S.; Rivara M.; Morini G.; Bertoni S.; Ballabeni V.; Barocelli E.; Plazzi P. V. Bioorg. Med. Chem. 2005, 13, 5647. 10.1016/j.bmc.2005.05.072. [DOI] [PubMed] [Google Scholar]
- Kim S.-K.; Fristrup P.; Abrol R.; Goddard W. A. J. Chem. Inf. Model. 2011, 51, 3262. 10.1021/ci200435b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander K.; von Coburg Y.; Camelin J. C.; Ligneau X.; Rau O.; Schubert-Zsilavecz M.; Schwartz J. C.; Stark H. Bioorg. Med. Chem. Lett. 2010, 20, 1581. 10.1016/j.bmcl.2010.01.089. [DOI] [PubMed] [Google Scholar]
- Roche O.; Nettekoven M.; Vifian W.; Sarmiento R. M. Bioorg. Med. Chem. Lett. 2008, 18, 4377. 10.1016/j.bmcl.2008.06.062. [DOI] [PubMed] [Google Scholar]
- Arrang J. M.; Garbarg M.; Lancelo J. C.; Lecomte J. M.; Pollard H.; Robba M.; Schunack W.; Schwartz J. C. Nature 1987, 327, 117. 10.1038/327117a0. [DOI] [PubMed] [Google Scholar]
- Alves-Rodrigues A.; Leurs R.; Wu T. S.; Prell G. D.; Foged C.; Timmerman H. Br. J. Pharmacol. 1996, 118, 2045. 10.1111/j.1476-5381.1996.tb15642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Alamilla G.; Marquez-Gomez R.; Garcia-Galvez A. M.; Morales-Figueroa G. E.; Arias-Montano J. A. Mol. Pharmacol. 2016, 90, 649. 10.1124/mol.116.104752. [DOI] [PubMed] [Google Scholar]
- Wu X.; Fors B. P.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 9943. 10.1002/anie.201104361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijtmans M.; Denonne F.; Celanire S.; Gillard M.; Hulscher S.; Delaunoy C.; Van Houtvin N.; Bakker R. A.; Defays S.; Gerard J.; Grooters L.; Hubert D.; Timmerman H.; Leurs R.; Talaga P.; de Esch I. J. P.; Provins L. MedChemComm 2010, 1, 39. 10.1039/c0md00056f. [DOI] [Google Scholar]
- Ahmed Z.; Siiskonen A.; Virkki M.; Priimagi A. Chem. Commun. 2017, 53, 12520. 10.1039/C7CC07308A. [DOI] [PubMed] [Google Scholar]
- Bleger D.; Hecht S. Angew. Chem., Int. Ed. 2015, 54, 11338. 10.1002/anie.201500628. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Hansen M. J.; Kalman L.; Woolley G. A. Chem. Commun. (Cambridge, U. K.) 2015, 51, 12981. 10.1039/C5CC02804C. [DOI] [PubMed] [Google Scholar]
- Talaty E. R.; Fargo J. C. Chem. Commun. 1967, 0, 65. 10.1039/C19670000065. [DOI] [Google Scholar]
- Garcia-Amoros J.; Velasco D. Beilstein J. Org. Chem. 2012, 8, 1003. 10.3762/bjoc.8.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T.; Okada T.; Shinkai S.; Shigematsu K.; Kusano Y.; Manabe O. J. Am. Chem. Soc. 1981, 103, 5161. 10.1021/ja00407a034. [DOI] [Google Scholar]
- Serra F.; Terentjev E. M. Macromolecules 2008, 41, 981. 10.1021/ma702033e. [DOI] [Google Scholar]
- Whitten D. G.; Wildes P. D.; Pacifici J. G.; Irick G. J. Am. Chem. Soc. 1971, 93, 2004. 10.1021/ja00737a027. [DOI] [Google Scholar]
- Dvorak C. A.; Apodaca R.; Barbier A. J.; Berridge C. W.; Wilson S. J.; Boggs J. D.; Xiao W.; Lovenberg T. W.; Carruthers N. I. J. Med. Chem. 2005, 48, 2229. 10.1021/jm049212n. [DOI] [PubMed] [Google Scholar]
- Rai B. K.; Tawa G. J.; Katz A. H.; Humblet C. Proteins: Struct., Funct., Genet. 2010, 78, 457. 10.1002/prot.22571. [DOI] [PubMed] [Google Scholar]
- Levoin N.; Labeeuw O.; Krief S.; Calmels T.; Poupardin-Olivier O.; Berrebi-Bertrand I.; Lecomte J. M.; Schwartz J. C.; Capet M. Bioorg. Med. Chem. 2013, 21, 4526. 10.1016/j.bmc.2013.05.035. [DOI] [PubMed] [Google Scholar]
- Wang C.; Jiang Y.; Ma J.; Wu H.; Wacker D.; Katritch V.; Han G. W.; Liu W.; Huang X. P.; Vardy E.; McCorvy J. D.; Gao X.; Zhou X. E.; Melcher K.; Zhang C.; Bai F.; Yang H.; Yang L.; Jiang H.; Roth B. L.; Cherezov V.; Stevens R. C.; Xu H. E. Science 2013, 340, 610. 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. Science 2013, 340, 615. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcou G.; Rognan D. J. Chem. Inf. Model. 2007, 47, 195. 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- Lane J. R.; Donthamsetti P.; Shonberg J.; Draper-Joyce C. J.; Dentry S.; Michino M.; Shi L.; Lopez L.; Scammells P. J.; Capuano B.; Sexton P. M.; Javitch J. A.; Christopoulos A. Nat. Chem. Biol. 2014, 10, 745. 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke P.; Luque F. J.; Murray J. B.; Barril X. J. Am. Chem. Soc. 2011, 133, 18903. 10.1021/ja207494u. [DOI] [PubMed] [Google Scholar]
- Tirado-Rives J.; Jorgensen W. L. J. Med. Chem. 2006, 49, 5880. 10.1021/jm060763i. [DOI] [PubMed] [Google Scholar]
- Sahlholm K.; Nilsson J.; Marcellino D.; Fuxe K.; Arhem P. Eur. J. Pharmacol. 2007, 567, 206. 10.1016/j.ejphar.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. Organometallics 2010, 29, 2176. 10.1021/om100106e. [DOI] [Google Scholar]
- Sali A.; Blundell T. L. J. Mol. Biol. 1993, 234, 779. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stutzle T.; Exner T. E. J. Chem. Inf. Model. 2009, 49, 84. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Miller-Gallacher J. L.; Nehme R.; Warne T.; Edwards P. C.; Schertler G. F.; Leslie A. G.; Tate C. G. PLoS One 2014, 9, e92727 10.1371/journal.pone.0092727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer L. V.; Muller E. M.; Gaub H. E.; Grubmuller H. Angew. Chem., Int. Ed. 2007, 46, 2232. 10.1002/anie.200604595. [DOI] [PubMed] [Google Scholar]
- Cordomi A.; Caltabiano G.; Pardo L. J. Chem. Theory Comput. 2012, 8, 948. 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. Comput. Phys. Commun. 1995, 91, 43. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Kuhne S.; Kooistra A. J.; Bosma R.; Bortolato A.; Wijtmans M.; Vischer H. F.; Mason J. S.; de Graaf C.; de Esch I. J.; Leurs R. J. Med. Chem. 2016, 59, 9047. 10.1021/acs.jmedchem.6b00981. [DOI] [PubMed] [Google Scholar]
- Leurs R.; Smit M. J.; Menge W. M.; Timmerman H. Br. J. Pharmacol. 1994, 112, 847. 10.1111/j.1476-5381.1994.tb13157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijmeijer S.; Vischer H. F.; Rosethorne E. M.; Charlton S. J.; Leurs R. Mol. Pharmacol. 2012, 82, 1174. 10.1124/mol.112.080911. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Biochem. Pharmacol. 1973, 22, 3099. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Almeida P.; de Boer G.-J.; de Boer A. H. J. Plant Physiol. 2014, 171, 438. 10.1016/j.jplph.2013.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.