

Abstract
RNAs are an important class of cellular regulatory elements, and they are well characterized by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy in their folded or bound states. However, the apo or unfolded states are more difficult to characterize by either method. Particularly, effective NMR spectroscopy studies of RNAs in the past were hampered by chemical shift overlap of resonances and associated rapid signal loss due to line broadening for RNAs larger than the median size found in the PDB (∼25 nt); most functional riboswitches are bigger than this median size. Incorporation of selective site-specific 13C/15N-labeled nucleotides into RNAs promises to overcome this NMR size limitation. Unlike previous isotopic enrichment methods such as phosphoramidite, de novo, uniform-labeling and selective biomass approaches, this newer chemical-enzymatic selective method presents a number of advantages for producing labeled nucleotides over these other methods. For example total chemical synthesis of nucleotides, followed by solid-phase synthesis of RNA using phosphoramidite chemistry, while versatile in incorporating isotope labels into RNA at any desired position, faces problems of low yields (<10 %) that drop precipitously for oligonucleotides larger than 50 nt; de novo pyrimidine biosynthesis of NTPs, also a robust technique with modest yields of up to 45%, comes at the cost of using 16 enzymes, expensive substrates, and difficulty in making some needed labeling patterns such as selective labeling of the ribose C1′ and C5′ and the pyrimidine nucleobase C2, C4, C5 or C6; the method of biomass-produced uniformly- or selectively-labeled NTPs suffers from low overall yield per labeled input metabolite and isotopic scrambling with only modest suppression of 13C-13C couplings. In contrast, our current chemo-enzymatic approach overcomes most of these shortcomings and allows for the synthesis of gram quantities of nucleotides with >80% yields while using a limited number of enzymes, six at most. The unavailability of selectively labeled ribose and base precursors had prevented the effective use of this versatile method until now. Recently, we combined an improved organic synthetic approach that selectively places 13C/15N labels in the pyrimidine nucleobase (either 15N1, 15N3, 13C2, 13C4, 13C5, or 13C6 or any combination) with a very efficient enzymatic method to couple ribose with uracil to produce previously unattainable labeling patterns (Alvarado et al 2014). Herein we provide detailed steps of both our chemo-enzymatic synthesis of custom nucleotides and their incorporation into RNAs with sizes ranging from 29 to 155 nt, and showcase the dramatic improvement in spectral quality of reduced crowding and narrow linewidths. Applications of this selective labeling technology should prove valuable in overcoming two major obstacles, chemical shift overlap of resonances and associated rapid signal loss due to line broadening, that have impeded studying the structure and dynamics of large RNAs such as full length riboswitches larger than the ∼25 nt median size of RNA NMR structures found in the PDB.
Keywords: RNA, NMR, TROSY, Stable isotopes, Site-specific labeling, Nucleotide synthesis and purification
1. Theory
Ribonucleic acid (RNA) is central to key biological processes such as signaling, gene regulation, catalysis, and viral infectivity (Breaker, 2009; Lu et al., 2011; Mattick, 2007; Newman & Nagai, 2010; Serganov & Nudler, 2013; Steitz, 2008). This functional diversity is due in part to the elaborate and pliable three-dimensional structures RNAs can adopt. Nuclear magnetic resonance (NMR) spectroscopy is one of the major methods utilized for RNA structure elucidation. However, for RNAs longer than 25 nucleotides, this technique suffers from conformational heterogeneity, extensive chemical shift overlap, and rapid signal decay (Dayie, 2008).
Four approaches were previously proposed to address these limitations: (i) total chemical synthesis of NTP using phosphoramidite chemistry; (ii) de novo biosynthesis of NTPs; (iii) biomass synthesis of NTPs; or (iv) selective-biomass synthesis of NTPs (Batey, Inada, Kujawinski, Puglisi, & Williamson, 1992; Hoffman & Holland, 1995; Johnson, Julien, & Hoogstraten, 2006; Lemaster & Kushlan, 2001; Milecki, 2002; Nikonowicz et al., 1992; Quant et al., 1994; Schultheisz, Szymczyna, Scott, & Williamson, 2011; Thakur & Dayie, 2012; Thakur, Sama, Jackson, Chen, & Dayie, 2010; Wunderlich et al., 2012). NTPs obtained from methods (ii)-(iv) are then used in T7 RNA polymerase based RNA transcription. The biggest advantage of method (i) is the maximum flexibility it affords in positioning the label simply by varying the phosphoramidites used; however, the solid-phase synthesis methodology is extremely inefficient for making RNAs greater than 50 nucleotides as the yield drops significantly with increasing size of the polynucleotide (Wunderlich et al., 2012). Method (ii) proceeds with modest yields of ∼45% for pyrimidine biosysnthesis (Schultheisz, Szymczyna, Scott, & Williamson, 2011), and requires ∼16 enzymes, expensive precursor glucose and aspartic acid substrates, and inaccessibility to some pyrimidine labels. Traditionally large quantities of uniformly labeled nucleotides were cost-effectively produced using biomass growth of bacteria on labeled media containing 15NH4Cl and a variety of uniformly labeled carbon sources such as 13C-acetate, 13C-glucose, 13C-glycerol, 13C-methanol, or 13C-pyruvate (Batey, Inada, Kujawinski, Puglisi, & Williamson, 1992; Hoffman & Holland, 1995; Johnson, Julien, & Hoogstraten, 2006; Lemaster & Kushlan, 2001; Nikonowicz et al., 1992; Thakur & Dayie, 2012; Thakur, Sama, Jackson, Chen, & Dayie, 2010). However, by using site specifically labeled forms of these carbons sources and metabolically modified bacteria, 13C isotopes could be readily incorporated at designated locations (Hoffman & Holland, 1995; Johnson, Julien, & Hoogstraten, 2006; Lemaster & Kushlan, 2001; Thakur & Dayie, 2012; Thakur, Sama, Jackson, Chen, & Dayie, 2010). Nonetheless, both of these methods (iii and iv) suffer from low overall yield per labeled input metabolite, and residual isotopic scrambling invariably leads to inadequate suppression of 13C-13C coupling (Alvarado et al., 2014; Thakur & Dayie, 2012).
A fifth approach, the chemo-enzymatic synthesis of NTPs followed by in vitro RNA transcription, is potentially the most versatile method available. Until recently, lack of commercially available selectively labeled ribose and base precursors, unfortunately, prevented the realization of its full potential (Alvarado et al., 2014). We recently made an important technological advance in combining chemical synthesis of selective 13C/15N labeled pyrimidine nucleobases with enzymatic synthesis to achieve site-specific labeling that overcomes most of these earlier deficiencies (Alvarado et al., 2014).
Here we outline this robust approach that efficiently couples chemically synthesized ribose and nucleobase using enzymes from the pentose phosphate pathway to synthesize NTPs, followed by in vitro RNA transcription (Alvarado et al., 2014; Tolbert & Williamson, 1996). This approach enables labeling pyrimidine nucleobases selectively with any combination of the following: 15N1, 15N3 and 13C2 from 13C-15N labeled urea; 13C4 and 13C5 from 13C-labeled bromoacetic acid; and 13C6 from K13CN. Additionally, it enables labeling ribose at any carbon position to produce previously unattainable labeled NTP patterns on a gram scale with >80% yields (based on input nucleobase). These labels contain isolated two-spin systems in both the ribose and the nucleobase and, thus, are ideal for both structural and dynamic studies for large RNAs such as riboswitches.
As an example, the starting materials 1′,5′-13C2-D-ribose and 6-13C-1,3-15N2-uracil can be enzymatically coupled to synthesize 1′,5′,6-13C3-1,3-15N2-uridine 5′-triphosphate (UTP, Figure 7.1), as showcased in our recent work (Alvarado et al., 2014). Importantly, any labeled combination of either ribose or uracil moieties can be used in our method. In the first step, UTP is synthesized in a one-pot reaction, followed by affinity purification. Cytidine 5′-triphosphate (CTP) is then synthesized in a one-pot reaction from UTP, followed by affinity purification. These newly synthesized nucleotides are then used directly for in vitro RNA transcription (Brunelle & Green, 2013). The labeled RNA is then purified to homogeneity by denaturing purification as detailed by Puglisi and coworkers (Petrov, Wu, Puglisi, & Puglisi, 2013). Herein, we detail the methodology to synthesize these nucleotides.
Figure 7.1.
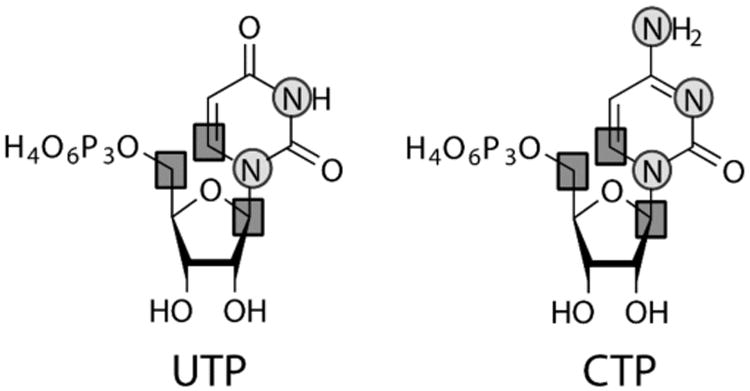
Selective site-specifically 13C/15N-labeled Uridine (left) and Cytidine (right) 5′-triphosphates. This is one of the potential labeling combinations to be synthesized using our methodology. Squares: 13C; circles: 15N.
Finally we demonstrate the versatility of these labels for obtaining structural and dynamic data for small (∼ 20 nt) to large (>150 nt) RNAs. We show representative examples of how NMR spectral resolution and signal-to-noise ratios are enhanced with the incorporated specific isotopic labels in three RNAs of interest: IRE RNA (29 nt), a riboswitch (63 nt), and HIV-1 core encapsidation signal (155 nt). It is anticipated that this methodology should find wide application in probing hitherto “difficult” to characterize RNAs such as full length riboswitches that include both aptamer and expression platform regions.
2. Equipment
0.22-μm cellulose acetate filters (GE Healthcare)
0.5-mL 3K MWCO microcentrifuge spin columns (Millipore, RNase, DNase, pyrogen free)
0.5-mL microcentrifuge tubes (RNase, DNase, pyrogen free)
100 mL round bottom flask
100-mL three necked round bottom flask
2-mL Pasteur pipet, long neck
50-mL conical tubes (RNase, DNase, pyrogen free)
50-mL round bottom flask
600/800 MHz nuclear magnetic resonance instrument equipped with at least 1H/13C/15N probes.
Balloons with a wall thickness of at least 0.3 mm
Bent adapters with NS-stopcocks
C18 reverse-phase Vydac analytical column
Freeze dryer
Freezer (-20 & -80 °C)
High vacuum rotary vane pump
Liquid chromatography system
Low-speed tabletop centrifuge
Low-volume Shigemi tubes
Magnetic stirrer with heating and an oil bath or heat block
Magnetic stirring bar
Microcentrifuge
Micropipettor tips (RNase, DNase, pyrogen free)
Micropipettors
pH meter and electrode
Polyacrylamide gel electrophoresis (PAGE) equipment, preparative size
Razor
Reflux condenser
Refrigerator (4 °C)
Rotary evaporator with a diaphragm pump
Sorbtech® solvent-resistant column
Speedvac
Suction filter
Syringe
UV/Vis spectrophotometer
Water bath
3. Materials
Unless stated otherwise, our chemicals were obtained from Sigma-Aldrich.
13C-potassium cyanide (K13CN)
15N2-urea
2′-deoxy-adenosine 5′-triphosphate (dATP)
2-Bromoacetic acid
4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS)
40% Acrylamide/bis-acrylamide (19:1)
5% Palladium on barium sulfate (5% Pd/BaSO4)
50% aqueous acetic acid
Acetic anhydride
Acid phenol:chloroform 5:1, pH 4.5 (Ambion)
Affi-Gel Boronate Gel (BioRad)
Ammonium persulfate (APS)
Ampicillin
Boric acid (H3BO3)
Bovine Serum Albumin (BSA)
Bromophenol blue
Celite
Concentrated hydrochloric acid (HCl)
Creatine kinase (CK) (see Table 1)
Creatine phosphate
Cytidine triphosphate synthetase (CTPS) (Recombinantly expressed)
Diethylether
Dithiothreitol (DTT)
Dry ice
Ethanol, 100%
Ethylenediaminetetraacetic acid (EDTA)
Formamide
Hydrochloric acid (HCl)
Hydrogen gas
Magnesium chloride (MgCl2)
Methanol, >98% (HPLC grade)
Myokinase (MK)
N,N,N′,N′-Tetramethylethylenediamine (TEMED)
Nucleoside monophosphate kinase (NMPK) (Roche)
pH indicator paper
Phosphoribosylpyrophosphate synthetase (PRPPS) (Recombinantly expressed)
Polyethylene glycol, MW 8000
Potassium chloride (KCl)
Ribokinase (RK) (Recombinantly expressed)
Ribose (unlabeled and in various labeled forms)
RNase/DNase-free water
rNTPs: rATP, rUTP, rCTP, rGTP
Sodium acetate (NaOAc)
Sodium carbonate (Na2CO3)
Sodium phosphate dibasic heptahydrate (Na2HPO4•7H2O)
Sodium phosphate monobasic monohydrate (NaH2PO4•H2O)
Spermidine
T7 RNA polymerase (Processive P266L mutant, recombinantly expressed)
Thermostable pyrophosphatase (TIPP) (New England Biolabs)
Triethylamine bicarbonate (TEABC)
Tris base
Triton X-100
Uracil (unlabeled and in various labeled forms by chemical synthesis, vide infra)
Urea
Uridine phosphoribosyl transferase (UPRT) (Recombinantly expressed)
Xylene cyanol
Table 1. Enzymes utilized in the synthesis of UTP and CTP.
| Enzyme | Abbreviation | E.C. | Source | Vendor |
|---|---|---|---|---|
| Ribokinase | RK | 2.7.1.15 | E. coli* | * |
| Phosphoribosyl pyrophosphate synthetase | PRPPS | 2.7.6.1 | Human* | * |
| Uridine phosphoribosyl transferase | UPRT | 2.4.2.9 | E. coli* | * |
| Cytidine triphosphate synthetase | CTPS | 6.3.4.2 | E. coli* | * |
| Nucleoside monophosphate kinase | NMPK | 2.7.4.4 | Bovine liver | Roche |
| Creatine Kinase | CK | 2.7.3.2 | Rabbit muscle | Sigma |
| Myokinase (Adenylate kinase) | MK | 2.7.4.3 | Rabbit muscle | Sigma |
| Thermostable inorganic pyrophosphatase | TIPP | 3.6.1.1 | T. litoralis | NEB |
3.1 Solutions and buffers
| Step 1: 50% aqueous acetic acid | |||
|
| |||
| Slowly add 100 mL glacial acetic acid to 100 mL distilled water. | |||
|
| |||
| Steps 2-5: 1M Triethylammonium bicarbonate, pH 9.4 | |||
|
| |||
| Dissolve 121 mL triethylamine in 1 L water (final volume) | |||
| Bubble CO2 into the solution until pH 9.4 | |||
|
| |||
| Acidified water, pH 4.6 | |||
|
| |||
| Bubble CO2 into autoclaved water until pH 4.6 | |||
|
| |||
| Vydac column buffer A | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| NaH2PO4•H2O | 12.5 mM | 1 M | 12.5 mL |
|
| |||
| Na2HPO4•7H2O | 12.5 mM | 0.5 M | 25 mL |
|
| |||
| Adjust to pH 2.8 membrane with glacial acetic acid. Add water to 1 L. Filter through 0.22-μm hydrophilic | |||
| Vydac column buffer B | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| NaH2PO4•H2O | 62.5 mM | 1 M | 62.5 mL |
|
| |||
| Na2HPO4•7H2O | 62.5 mM | 0.5 M | 125 mL |
|
| |||
| Adjust to pH 2.8 membrane with glacial acetic acid. Add water to 1 L. Filter through 0.22-μm hydrophilic | |||
| Nucleoside monophosphate kinase (NM PK) solution | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| NMPK | - | - | 60 mg |
|
| |||
| Tris-HCl, pH 6.5 | 50 mM | 1 M | 50 μL |
|
| |||
| Glycerol | 50% (v/v) | 100% (v/v) | 0.5 mL |
|
| |||
| Add water to 1 mL | |||
| Creatine kinase (CK) solution | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| Creatine kinase | - | - | 1 mg |
|
| |||
| Tris-HCl, pH 7.5 | 50 mM | 1 M | 50 μL |
|
| |||
| Glycerol | 50% (v/v) | 100% (v/v) | 0.5 mL |
|
| |||
| Add water to 1 mL | |||
| Step 6: 10X transcription buffer | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| Tris-HCl, pH 8.0 | 400 mM | 1 M | 4 mL |
|
| |||
| DTT | 100 mM | 1 M | 1 mL |
|
| |||
| Triton X-100 | 0.1% (v/v) | 10% | 0.1 mL |
|
| |||
| Spermidine, pH 7.0 | 10 mM | 500 mM | 0.2 mL |
|
| |||
| Add water to 10 mL | |||
| 2X Formamide RNA loading buffer | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| Formamide | 95% (v/v) | 100% (v/v) | 9.5 mL |
|
| |||
| SDS | 0.025% (w/v) | 10% (w/v) | 25 μL |
|
| |||
| EDTA, pH 8.0 | 0.5 mM | 50 mM | 0.1 mL |
|
| |||
| Bromophenol blue | 0.05% (w/v) | - | 5 mg |
|
| |||
| Xylene cyanol | 0.05% (w/v) | - | 5 mg |
|
| |||
| Add water to 10 mL | |||
| 10X Tris-Borate-EDTA buffer | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| Tris | 0.9 M | - | 108 g |
|
| |||
| Boric acid | 0.9 M | - | 240 g |
|
| |||
| EDTA, pH 8.0 | 10 mM | 500 mM | 20 mL |
|
| |||
| Add water to 1 L | |||
| 13% Denaturing polyacrylamide gel (PAGE) solution | |||
|
| |||
| Component | Final concentration | Stock | Amount |
|
| |||
| Acrylamide/bis-acrylamide (19:1) | 13% | 40% | 162.5 mL |
|
| |||
| Urea | 8 M | - | 240 g |
|
| |||
| TBE buffer | 1 X | 10 X | 50 mL |
|
| |||
| Add water to 500 mL. Protect from light | |||
| 3 M Sodium acetate, pH 5.2 | |||
|
| |||
| Dissolve 24.6 g NaOAc in 100 mL water (final volume) | |||
|
| |||
4. Protocol
1. Duration
| Preparation | About 1 week |
|
| |
| Step 1 | 4 – 5 days |
|
| |
| Step 2-5 | About 2 weeks |
|
| |
| Step 6 | 1 week |
|
| |
2. Preparation (See Figure 7.2)
Figure 7.2.
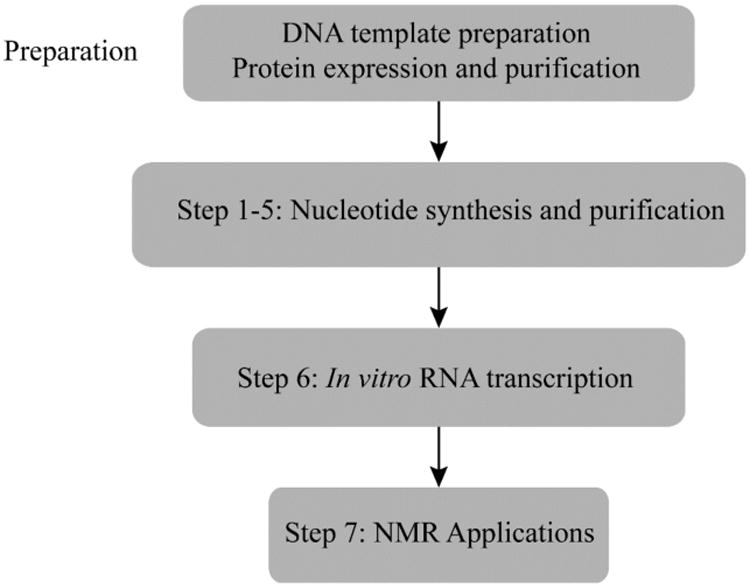
Flowchart of the complete protocol, including preparation.
If the desired RNA is > 70 nt, generate a DNA template for in vitro transcription carrying the T7 promoter sequence (5′ TAATACGACTCACTATAGGG) upstream of the desired RNA by standard PCR techniques. Alternately, if desired RNA is < 70 nt, you may utilize a synthetic DNA template overhang carrying the sequence of interest. Approximately 10 – 50 pmol of DNA is needed for each transcription reaction.
Express and purify RK, PRPPS, UPRT, and CTPS as described previously by Arthur et al. (Arthur et al., 2011). Preferably, stock enzyme solutions concentrations will be ∼ 10 mg/mL.
Purchase the corresponding nucleotide building blocks, e.g. ribose and nucleobase, from commercial sources (Isotec, Cambridge Isotope Laboratories). Alternately, synthesize the nucleobases as described by Kreutz and coworkers (Wunderlich et al., 2012).
3. Caution
The homogeneity of the DNA templates is of utmost importance in RNA preparation. It is critical that all synthetic DNA templates used to produce RNAs < 70 nt are purified by denaturing gel electrophoresis to minimize resulting heterogeneous RNA populations after in vitro transcription. Additionally, including two final 2′ OCH3 nucleotides on the 5′ end of the template strand will reduce N+1 transcript heterogeneity.
The activity of RK, PRPPS, UPRT, and CTPS may decrease > 50% after six months of storage at -20 °C. Therefore, if possible, store the minimum amount needed at -20 °C, and the rest at -80 °C.
RNase-free conditions are of utmost importance to maintaining the integrity of RNA. All reagents, buffers, and solutions should be autoclaved (except for urea-containing solutions). Alternately, they can be sterile filtered. All materials, such as glass plates, should be thoroughly washed and oven-dried before use. Wear gloves at all times.
Some of the buffers used in the protocol are highly volatile. Please prepare these buffers under a chemical fume hood.
5. Step 1: Synthesis of Uracil
5.1 Overview
Uracils with various stable isotope labeling patterns are accessible via chemical synthesis. Starting from potassium cyanide and 2-bromoacetic acid, the cyano acetylurea precursor is obtained in good yields. In the final step, uracil is formed under reductive reaction conditions using palladium on barium sulfate under a hydrogen atmosphere. Various 13C/15N-labeling patterns are amenable using this approach. Here, we exemplify the uracil synthesis for the 6-13C-15N2-uracil derivative.
5.2 Duration
4 – 5 days
1.1 Synthesis of 3-13C-cyanoacetic acid: Dissolve 2-bromoacetic acid (6.99 g, 50.3 mmol) in 20 mL of water in a 100-mL round bottom flask equipped with a reflux condenser and a magnetic stirring bar.
1.2 Add sodium carbonate (Na2CO3, 3.4 g, 32.1 mmol) predissolved in 10 mL of water until pH 9 is reached. Check the pH with a pH indicator paper.
1.3 Dissolve K13CN (3.24 g, 49 mmol) in 10 mL of water and add to the 100-mL round bottom flask. Heat the reaction mixture using an oil bath or a heat block to 80 °C for 3 h while stirring.
1.4 Remove the heating source and continue stirring at room temperature for 20 h.
1.5 Next, add concentrated HCl stepwise until pH 1 is reached. Check the pH after every addition.
1.6 Remove the solvent by evaporation using a rotary evaporator. The residue is then dried in high vacuum for 30 minutes. A yellow-white semi-solid salt cake is obtained.
1.7 Extract the 3-13C-cyanoacetic acid from the semi-solid mass by suspending the salt cake in diethylether (five 100-mL portions). The ether extract is filter through a suction filter and the yellow ethereal solution is evaporated to dryness. The oily residue solidifies upon cooling on ice.
1.8 The solidified yellowish to orange 3-13C-cyanoacetic acid is then dried in high vacuum for 2 h.
1.9 Synthesis of 3-13C-15N2-cyanoacetyl urea: The 3-13C-cyanoacetic acid from the previous step (2.0 g, 23.3 mmol) is placed in a 50 mL round bottom flask equipped with a reflux condenser and a magnetic stirring bar.
1.10 Add 15N2-urea (1.5 g, 25 mmol) and 5 mL of acetic anhydride. Heat this reaction mixture to 90 °C for 30 minutes. After about 5 minutes a white precipitate can be observed.
1.11 Add 1 – 2 mL water and let cool to room temperature. The 3-13C-15N2-cyanoacetyl urea can then be isolated by filtration and is then dried in high vacuum for 5 h.
1.12 Synthesis of 6-13C-15N2-Uracil: Add 5% Pd/BaSO4 (800 mg) and 10 mL 50% aqueous acetic to a 100-mL three-necked round bottom flask equipped with a magnetic stirring bar and bent adapters with NS-stopcocks. The adapters are used to either evaporate the flask or to spill the evacuated flask with hydrogen gas using a balloon.
1.13 The evacuation hydrogen spill procedure is repeated three times. The brown suspension then turns black.
1.14 Simultaneously, 3-13C-15N2-cyanoacetyl urea (1.6 g, 12.5 mmol) is dissolved in 40 mL of boiling aqueous 50% acetic acid and then added to the reduced palladium catalyst.
1.15 The reaction is stirred at room temperature under a hydrogen atmosphere. Refill the hydrogen balloon if necessary.
1.16 Before filtering through a celite pad on a suction filter, the mixture is heated to 70 °C for 1 h.
1.17 The filtrate is concentrated until a white precipitate is observed. Then, 6-13C-15N2-uracil is precipitated by storing the suspension at 4 °C overnight.
1.18 Uracil is obtained by filtration over a suction filter and the white solid is dried in high vacuum. The expected yield is 1.15g (82%).
5.3 Tip
As the labeled compounds are rather expensive, it is advisable to carry out the reactions using unlabeled compounds first to gain familiarity with the procedure.
5.4 Tip
Add the sodium carbonate solution in step 1.2 slowly to avoid frothing due to CO2 evolution. In step 1.5, add the HCl again slowly to avoid frothing due to CO2 evolution.
5.5 Tip
The 1 – 2 mL water added in step 1.11 are needed to dissolve the precipitated urea to facilitate isolation by filtration.
5.6 Tip
The reaction progress starting at step 1.12 should be monitored every 12 h. For that purpose, take a small aliquot (500 μL) of the reaction suspension and centrifuge to clear the suspension. Remove the supernatant and transfer to a 10-mL round bottom flask. Then, the solvent is removed by evaporation and the remaining white solid is dried in high vacuum for 1 h. Then, dissolve the residue in 500 μL deuterated dimethylsulfoxide (DMSO-d6) and acquire a 1D 13C spectrum. The conversion yield to uracil can be qualitatively estimated by comparing the starting material 13C peak at 116 ppm and the product 13C peak at 142 ppm.
6. Step 2: Synthesis of UTP
6.1 Overview
Site-specifically labeled uracil and ribose are combined to produce uridine monophosphate using enzymes from the nucleotide salvage pathway. Uridine monophosphate is then phosphorylated, taking advantage of NMPK and creatine kinase. All aliquots taken at various reaction time points are analyzed on a C18 reverse-phase Vydac analytical column as described in Step 4 below to track the progress of the reaction.
6.2 Duration
11 hours
-
2.1 Prepare the reaction mixture for the synthesis of UMP by adding the following reagents in the order shown (with UPRT added last) to a 50-mL conical tube.
Component Final concentration Stock Amount
Sodium Phosphate Monobasic 9.4 mM 1 M 94 μL
Sodium Phosphate Dibasic 40.6 mM 500 mM 812 μL
MgCl2 10 mM 1 M 100 μL
Ampicillin 2 mg/mL 100 mg/mL 200 μL
DTT 10 mM 1 M 100 μL
dATP 0.5 mL 100 mM 50 μL
Creatine Phosphate 100 mM 500 mM 2000 μL
Uracil 8 mM 50 mM 1600 μL
Bovine serum albumin 0.1 mg/mL 10 mg/mL 100 μL
Creatine Kinase 0.005 mg/mL 1 mg/mL 50 μL
Myokinase 0.010 U/μL 5 U/μL 20 μL
Pyrophosphatase 0.004 U/μL 2 U/μL 20 μL
RK 0.005 U/μL 0.5 U/μL 100 μL
PRPPS 0.0003 U/μL 0.01 U/μL 300 μL
UPRT 0.005 U/μL 0.4 U/μL 125 μL
Add 4.23 mL water to 9.9 mL final volume 2.2 Incubate at 37 °C for 10 minutes to equilibrate temperature.
2.3 Start reaction by adding 100 μL of 1 M ribose to bring the final concentration of ribose to 10 mM.
2.4 Remove a 50 μL aliquot of the reaction (Time 0).
2.5 Place the reaction in a 37 °C water bath and incubate for 5 hours.
2.6 Collect aliquots at 2 hours (Time 2) and 5 hours (Time 5).
-
2.7 Once uracil is completely depleted, as determined by FPLC, add the following components to the reaction mixture.
Component Final concentration Stock Amount
KCl 10 mM 1 M 10 μL
dATP 0.1 mM 100 mM 10 μL
Creatine phosphate 10 mM 500 mM 200 μL
NMPK 0.05 mg/mL 10 mg/mL 50 μL
2.8 Incubate at 37 °C for 4 hours. Alternately, the reaction may run overnight.
2.9 Remove a final aliquot of the reaction (Time Final).
2.10 At this stage, the reaction can either be immediately purified or, alternatively, frozen and purified later.
6.3 Tip
If possible, use a magnetic stirrer. This is to ensure a homogeneous enzymatic reaction throughout the allotted time.
6.4 Tip
In the past we have observed white precipitate. This does not affect the outcome or yield of the reaction.
6.5 Tip
The final concentrations of RK, PRPPS, and UPRT can be decreased or increased if needed without adversely affecting the yield of the reaction. It only affects the completion time.
6.6 Tip
The phosphorylation state of dATP is of utmost importance for the ATP-regeneration system. If possible, aliquot stock dATP solution in small amounts to minimize freeze-thaw cycles that may promote dATP hydrolysis.
6.7 Tip
Commercial myokinase is stored as an ammonium sulfate precipitate. However, due to the low volume needed and the large final reaction volume, it can be used directly without centrifuging it and using the pellet. Thoroughly mix the re-suspension before taking the corresponding aliquot.
7. Step 3: Synthesis of CTP
7.1 Overview
UTP is converted into CTP in a single-step reaction catalyzed by CTP synthetase. The progress of the reaction is monitored by C18 reverse-phase Vydac analytical chromatography as described in Step 4 below to track the progress of the reaction.
7.2 Duration
8 hours
-
3.1 Add the following reagents in the order shown (with CTPS added last).
Component Final concentration Stock Amount
Tris-HCl pH 8.0 50 mM 1 M 500 μL
MgCl2 10 mM 1 M 100 μL
Ampicillin 2 mg/mL 100 mg/mL 200 μL
dATP 4 mM 100 mM 400 μL
UTP 2 mM 50 mM 400 μL
CTPS 0.10 mg/mL 9.6 mL 17.8 mg/mL 56.2 μL
Add 7.94 mL water to final volume 3.2 Incubate at 37 °C for 10 minutes.
3.3 Add 400 μL of 500 mM 15NH4Cl to bring the final concentration to 20 mM.
3.4 Take a 50-μL aliquot (Time 0).
3.5 Incubate at 37 °C for 6 hours.
3.6 Take 50-μL aliquots at 3 and 6 hours (Time 3 & Final).
3.7 At this stage, the reaction can either be immediately purified or, alternatively, frozen at -20 °C and purified later.
7.3 Tip
If possible, use a magnetic stirrer. This is to ensure a homogeneous enzymatic reaction throughout the allotted time.
7.4 Tip
The final concentration of CTPS can be decreased or increased if needed without majorly affecting the yield of the reaction. It would only affect the completion time.
7.5 Tip
The phosphorylation state of dATP is of utmost importance for the ATP-regeneration system. If possible, aliquot stock dATP solution in small amounts to minimize freeze-thaw cycles that may promote dATP hydrolysis.
8. Step 4: Purification & Quantification
8.1 Overview
Synthesized UTP and CTP are purified on a Sorbtech solvent-resistant column packed with approximately 10 g of Affi-Gel Boronate Gel. Purified nucleotides are lyophilized and redissolved at high concentrations for use in in vitro transcription reactions.
8.2 Duration
3 days
4.1 Thaw the UTP or CTP reaction on ice.
4.2 Add 10 mL 1 M TEABC pH 9.4 to the reaction and let sit at room temperature for 15 minutes.
4.3 Centrifuge at 12,800× g for 10 minutes to pellet all precipitated proteins.
4.4 Sterile filter the reaction mixture by passing it through a 0.22-μm syringe filter.
4.5 The reaction is slowly loaded manually onto a Sorbtech column packed with 10 g Affi-Gel Boronate Gel kept at 4 °C.
4.6 The column is washed, at 4 mL/min, with 4 column volumes (CV) of 1 M TEABC to wash-off dATP and any remaining proteins.
4.7 Acidified water pH 4.3 is then used to elute the UTP or CTP off of the column until the UV trace has returned to baseline (Figure 7.4).
4.8 The elution fractions are transferred directly to a lyophilization vessel when the absorbance measured at 254 nm begins to rise.
4.9 The collected flow through is frozen with constant spinning in a dry ice-ethanol bath, ca. -78 °C.
4.10 The sample is lyophilized in vacuo for one and a half days to remove all water and residual TEABC. This step is performed with a large-scale freeze-dryer.
4.11 At the end of the lyophilization, add 2 mL of ddH2O to redissolve the ribonucleotides. Transfer to two 1.5-mL Eppendorf tubes, and wash the lyophilization vessel twice with 2 mL of ddH2O, transferring to Eppendorf tubes as before.
4.12 Reduce the sample volume to dryness (or near dryness) for 2 hours, transfer the contents to 2 tubes, and reduce sample volume for 2 more hours. This step is performed with a Speedvac.
4.13 Finally, consolidate the sample to one tube each for UTP and CTP and calculate the nucleotide concentration by measuring absorbance at 260 nm for UTP (molar extinction coefficient 10,000 M-1 cm-1) or 271 nm or CP (molar extinction coefficient 9,000 M-1 cm-1). The final yield can be calculated based on a theoretical yield of 80 μmol of UTP or 20 μmol of CTP. Adjust the nucleotide concentrations to 50 – 100 mM and store at -20 °C in 10 mM Tris-HCl pH 7.5 and 0.5 mM EDTA.
Figure 7.4.
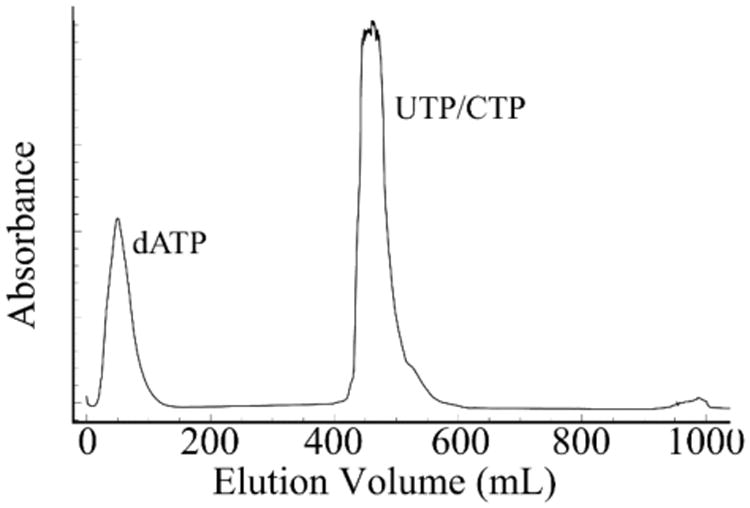
FPLC Chromatogram of boronate purification of UTP or CTP monitored at 254 nm. When purifying CTP, which has an absorbance maximum at 270 nm, the peak will be significantly less intense.
8.3 Tip
The binding capacity of Affi-Gel Boronate gel is 50 mg of ligand (e.g. ribonucleotides) per one gram of resin.
8.4 Tip
The packing of the boronate beads in the column is crucial. Before running any samples through, test the column with commercial standards. Boronate beads double or triple their size in 1 M TEABC. Conversely, they shrink when in acidified water. If the user notes a decrease in purification yields, the column should be re-packed, and washed thoroughly. A stringent regenerating protocol is suggested by the manufacturer in which the beads are washed with 0.1 M glacial acetic acid, rinsed extensively with water, and finally re-equilibrated with 1 M TEABC.
8.5 Tip
The pH for both 1 M TEABC and acidified water may drift over time. Thus, it is recommended that these solution be made fresh prior to the purification.
8.6 Tip
Sample must be manually loaded onto the boronate column due to the high back pressure caused by the volatility of 1 M TEABC.
8.7 Tip
To avoid sample loss during sample concentration, transfers should be made from samples with lowest concentrations to highest concentration.
8.8 Tip
Most FPLC UV detectors are limited to a 254 nm detection wavelength. The molar extinction coefficient for CTP is rather low at such wavelength, thus the purification chromatogram may appear lower than it actually is; always check the final product on a spectrophotometer. In our experience purification yields are > 90%.
9. Step 5: Quality Control
9.1 Overview
During the course of the reaction, aliquots are analyzed to ensure that reactions proceed to completion. The final quality control is performed using a combination of NMR and liquid chromatography.
9.2 Duration
4 Hours
- 5.1 Vydac Analysis of Aliquots:
- 5.1.1 Spin each 50-μL aliquot for 15 minutes at 8,000× g in a 0.5 mL, 3K Molecular Weight Cut-Off centrifugal filtration column to remove protein contaminants.
- 5.1.2 With a syringe, load 10 μL of the filtrate into a 100-μL sample loop.
- 5.1.3 Run the following protocol to separate the nucleotide components in each aliquot. Representative traces for the UTP and CTP reaction are shown in Figure 7.5: 0% Vydac buffer A for 4 CV, linear gradient from 0 – 100% Vydac buffer B for 4 CV, 100% Vydac buffer B for 2 CV, and 0% Vydac buffer B for 4 CV (reequilibration) (Figure 7.5).
- 5.2 NMR Verification:
-
5.2.1 Prepare purified UTP or CTP in the following mixture.
Component Final concentration Stock Amount
UTP/CTP 1 mM 50 mM 13 μL
D2O 10% (v/v) 100% (v/v) 65 μL
DSS 0.1 mM 1 mM 65 μL
Add ddH2O to 650 μL and transfer to a regula r-volume NM R tube - 5.2.2 For both UTP and CTP, run a 2D Heteronuclear Single Quantum Correlation (HSQC) experiment of the C1′ region to verify complete conversion of ribose to UTP. Run other experiments such as 1D 31P and 1D 13C to further validate phosphorylation state and coupling patterns, respectively.
-
Figure 7.5.
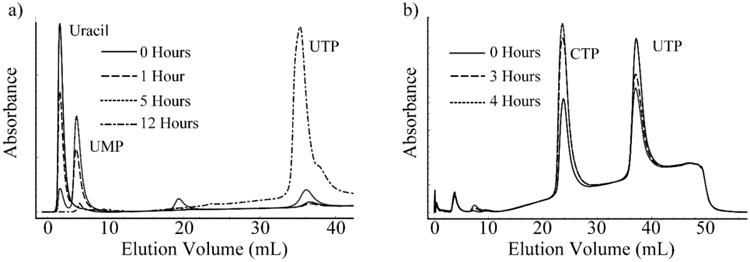
FPLC Chromatograms of the one-pot syntheses of UTP and CTP. (a) The synthesis of UMP is nearly complete at 5 hours. At this point, the components to synthesize UTP are added, completing its synthesis at 12 hours. (b) The synthesis of CTP is nearly complete at 4 hours. Both UTP/dATP and CTP/dADP have identical elution volumes, hence the peaks do not appear to be completely depleted, as dATP and dADP are both in large excess in the reaction mixture.
9.3 Tip
Thorough cleaning of the injection syringe is recommended to avoid cross-sample contamination. Otherwise, sample carry-over from prior time points may appear as incomplete reactions.
9.4 Tip
Typical acquisition parameters for 2D HSQC experiments of the ribose region are: 4.7 ppm 1H carrier, 80 ppm 13C carrier, 13 ppm 1H spectral width, and 50 ppm 13C spectral width.
Typical acquisition parameters for 2D HSQC experiments of the base region are: 4.7 ppm 1H carrier, 130 ppm 13C carrier, 13 ppm 1H spectral width, and 94 ppm 13C spectral width.
Typical acquisition parameters for 1D 31P experiments of the phosphate region are: 0 ppm for the 31P carrier and 60 ppm spectral width.
Typical acquisition parameters for 1D 13C experiments of both the ribose and base regions are: 110 ppm for the 13C carrier and 120 ppm spectral width.
10. Step 6: In vitro RNA transcription
10.1 Overview
Site-specifically labeled UTP and/or CTP are used to transcribe RNA in vitro using T7 RNA polymerase. Optimization of NTP and Mg2+ concentrations at small and mid-scales is extremely important to maximize yields before scaling up to larger volumes. This optimization has been described elsewhere (Milligan, Groebe, Witherell, & Uhlenbeck, 1987). Synthesized RNA is purified by denaturing gel electrophoresis and subsequently electroeluted. Labeled RNA is finally exchanged into an appropriate buffer and used for NMR spectroscopy.
10.2 Duration
3 Days
-
6.1 The following reaction is assembled in the order shown (with T7 RNA polymerase added last).
Component Final concentration Stock Amount
Transcription Buffer 1X 10X 1000 μL
MgCl2 Varies 1 M Varies
PEG 80 mg/mL 400 mg/mL 2000 μL
DNA Template 0.3 μM 10 μM 300 μL
DTT 0.01 M 1M 100 μL
TIPP 2 Units/mL 2000 Units/mL 10 μL
ATP Varies 100 mM Varies
GTP Varies 100 mM Varies
Specifically Labeled UTP Varies 100 mM Varies
Specifically Labeled CTP Varies 100 mM Varies
T7 RNA Polymerase 0.10 mg/mL 10 mg/mL 100 μL
Add water to 10 mL 6.2 Incubate reaction at 37 °C for 3 hours.
6.3 Add 10 mL of acid Phenol:Chloroform to reaction and vortex for 10 seconds.
6.4 Spin reaction at 3,200× g for 10 minutes in a tabletop centrifuge to separate aqueous and organic layers.
6.5 Transfer aqueous layer to two fresh 50 mL falcon tubes.
6.6 Add 5 mL water to the remaining organic layer and vortex for 10 seconds.
6.7 Spin reaction at 3,200× g for 10 minutes in a tabletop centrifuge to separate aqueous and organic layers.
6.8 Pool together all aqueous layers.
6.9 Add 1/10 volume of 3 M sodium acetate pH 5.3 and 3 volumes of cold 100% ethanol to precipitate RNA.
6.10 Store at -20 °C overnight.
6.11 Spin down precipitate at 12,800× g for 45 minutes.
6.12 Remove excess ethanol.
6.13 Wash pellets with 2 mL of cold 70% ethanol.
6.14 Spin down precipitate at 12,800× g for 45 minutes.
6.15 Carefully remove excess ethanol.
6.16 Air dry pellet for 30 minutes.
6.17 Redissolve pellet in minimal volume of 8 M Urea, 1X TBE.
6.18 Purify RNA by denaturing gel electrophoresis as described by Puglisi and coworkers. (Petrov et al., 2013).
10.3 Tip
The homogeneity of the purified RNA is of utmost importance for the subsequent steps. Ensure that the sample is of uniform length and conformation by using denaturing and native PAGE analysis.
10.4 Tip
Optimization of both NTP and Mg2+ concentrations is essential for in vitro RNA transcriptions. In our experience, we have had success in optimizing individual NTP concentrations ranging from 1.25 – 5 mM and Mg2+ concentrations ranging from 5 – 25 mM. Additionally, T7 RNA polymerase concentration should also be optimized. In our experience, we have utilized 0.05 – 1 mg/mL of enzyme.
11. Step 7: NMR Applications
11.1 Overview
With increasing RNA size (>35 nt), the utility of traditional RNA labeling and NMR methodologies becomes more limited (Alvarado et al., 2014). However, RNAs transcribed with our site-specific 13C/15N isotopic labeling patterns can be exploited in NMR spectroscopy to obtain structural and dynamics information hitherto unavailable. Extensive protocols for resonance assignment, structure determination, and dynamics characterization have been published elsewhere (Bothe et al., 2011; T. K. Dayie, 2005, 2011; Pardi, 1995). Here, we present some examples of heteronuclear NMR experiments that show the increased resolution and signal-to-noise ratio of both the Iron Responsive Element (IRE, 29 nt), a riboswitch (63 nt), and HIV-1 core encapsidation signal (155 nt) when transcribed with our custom labels.
11.2 Heteronuclear Single Quantum Coherence (HSQC)
Several resonance assignment experiments utilize HSQC, a through-bond experiment that correlates two active nuclei via their J-coupling constant (1JCH), usually ranging from 147 to 216 Hz in RNA. Unfortunately, resonances in both ribose and nucleobases exhibit narrow chemical shift dispersion, and such a narrow dispersion leads to significant overlap in the two-dimensional correlation map. Here we show how our site-specifically labeled IRE RNA, a 63-nt riboswitch, and HIV-1 RNAs reduce the degree of spectral overlap in two-dimensional HSQC experiments (Figure 7.6) without the need of constant-time experiments. Even though these constant-time experiments remove carbon-carbon couplings in uniformly labeled samples, their implementation leads to rapid signal decay and decreased signal-to-noise levels. With our specific isolated two-spin labels, we do not need to compromise on sensitivity or resolution.
Figure 7.6.
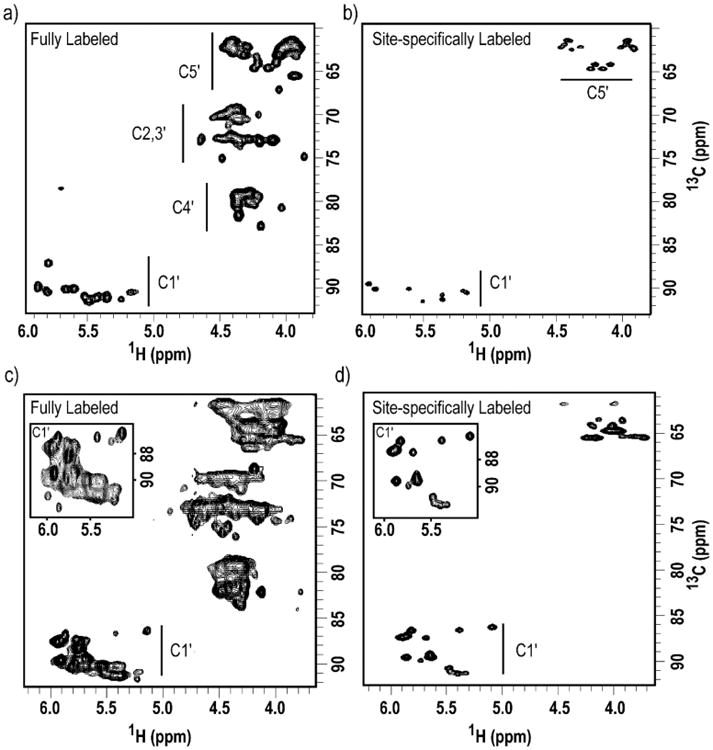
Site-specific labels produced by chemo-enzymatic synthesis provide HSQC spectra with improved resolution and reduced spectral crowding. Two-dimensional HSQC of the ribose region of (a) Fully 13C/15N-labeled and (b) 1′,5′,6-13C3-1,3,4-15N3-CTP labeled IRE RNA; (c) Fully 13C/15N-labeled and (d) 1′,5′,6-13C3-1,3-15N2-UTP labeled 63-nt riboswitch. All experiments were ran with identical parameters and without constant-time intervals. Spectral width: 6009 Hz and 7247 Hz in the 1H and 13C dimensions, respectively. 1024 and 256 complex points were acquired in t2 and t1, respectively, with 64 scans per slice. Insets: Expanded C1′ regions show the degree of resonance overlap in uniform 13C/15N-labeled RNA. Note that the insets are shown at lower level.
11.3 Transverse-Optimized Relaxation Spectroscopy (TROSY)
RNAs synthesized with our selective site-specifically labeled NTPs (prepared using chemo-enzymatic methodology) benefit from TROSY techniques that mitigate problems of crowding, rapid relaxation, low resolution and sensitivity (Miclet et al., 2004; Thakur et al., 2010). Compared to a regular HSQC, TROSY experiments select the slowest relaxing multiplet component of each resonance, leading to enhanced resolution and sensitivity. This approach is particularly important for larger RNAs, such as the HIV-1 RNA used here, in order to observe resonances inaccessible by traditional HSQC experiments (Figure 7.7). Using a C6 methine-optimized TROSY, we obtained a two-fold improvement in signal-to-noise ratio for the HIV-1 RNA (155 nt). A C5′-optimized TROSY showed only marginal improvement for the IRE RNA (29 nt). This highlights the importance of utilizing TROSY-based experiments for structural and dynamics analysis of large RNAs.
Figure 7.7.
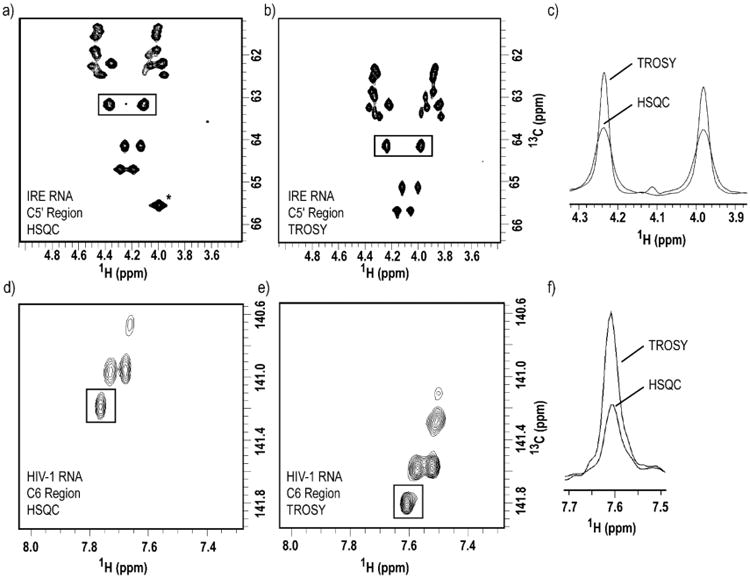
TROSY spectra acquired with RNA samples labeled with site-specific labels produced by chemo-enzymatic synthesis exhibit enhanced resolution and sensitivity than HSQC experiments. Two-dimensional (a) HSQC vs. (b) methylene-optimized TROSY spectra of 1′,5′,6-13C3-1,3-15N2-UTP/1′,5′,6-13C3-1,3,4-15N3-CTP-labeled IRE RNA. (c) One-dimensional slice overlay of the boxed peaks in both (a) and (b), notice the spectral quality enhancement due to the TROSY effect. Two-dimensional (d) HSQC vs. (e) methine-optimized TROSY spectra of 1′,5′,6-13C3-1,3-15N2-UTP-labeled HIV-1 RNA. (f) Overlay of one-dimensional slice of the boxed peak in both (d) and (e), notice the spectral quality enhancement due to the TROSY effect. For IRE RNA, the spectral width used were 3597 Hz and 905 Hz in the 1H and 13C dimensions, respectively. 1024 and 256 complex points were acquired in t2 and t1, respectively, with 8 scans per slice. For HIV-1 RNA, the spectral width used were 3597 Hz and 754 Hz in the 1H and 13C dimensions, respectively. 512 and 128 complex points were acquired in t2 and t1, respectively, with 128 scans per slice. * Peak is not shown in (b) due to the resonance offset in TROSY experiments.
12. Conclusion
We have outlined a fast, efficient, and economical chemo-enzymatic synthetic approach to incorporate site-selectively 13C/15N-labeled pyrimidine nucleotides into any RNA sequence of interest to facilitate structure and dynamics characterization of functional RNAs typically larger than 30 nt. This chemo-enzymatic approach not only provides better yields with less labor, but also new patterns of rNTP labels that are not available with current approaches. Three RNAs— IRE (29 nt), a riboswitch (63 nt), and HIV-1 RNA (155 nt)— were used to illustrate the usefulness of this approach. We hope that this methodology will open up new avenues for multidimensional heteronuclear and homonuclear solution and solid-state NMR methods to study the structure and dynamics of large RNA, such as full length riboswitches, which have till now remain unexplored (Cherepanov, Glaubitz, & Schwalbe, 2010; Marchanka, Simon, & Carlomagno, 2013).
Figure 7.3.
Flowchart of nucleotide synthesis. Notice that it is recommended to perform quality control at all stages to ensure maximal yield.
Acknowledgments
We thank Dr. Michael F. Summers (University of Maryland, Baltimore County) for providing the HIV-1 RNA construct and Dr. Sarah C. Keane (University of Maryland, Baltimore County) for making the HIV-1 core encapsidation signal RNA. Supported by NIH P50 GM103297 (TKD), NSF DBI1040158 (TKD), and the Austrian Sciences Fund (I844 and P26550 to CK).
Footnotes
Disclosures: LJ Alvarado, none; AP Longhini, none; RM LeBlanc, none; B Chen, none; C Kreutz, none; TK Dayie, none.
References
- Alvarado LJ, LeBlanc RM, Longhini AP, Keane SC, Jain N, Yildiz ZF, Tolbert BS, D'Souza VM, Summers MF, Kreutz C, Dayie TK. Regio-selective Chemical-Enzymatic Synthesis of Pyrimidine Nucleotides Facilitates RNA Structure and Dynamics Studies. ChemBioChem. 2014;15(11):1573–1577. doi: 10.1002/cbic.201402130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur PK, Alvarado LJ, Dayie TK. Expression, purification and analysis of the activity of enzymes from the pentose phosphate pathway. Protein Expression and Purification. 2011;76(2):229–237. doi: 10.1016/j.pep.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey RT, Inada M, Kujawinski E, Puglisi JD, Williamson JR. Preparation of isotopically labeled ribonucleotides for multidimensional NMR spectroscopy of RNA. Nucleic Acids Research. 1992;20(17):4515–4523. doi: 10.1093/nar/20.17.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe JR, Nikolova EN, Eichhorn CD, Chugh J, Hansen AL, Al-Hashimi HM. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nature Methods. 2011;8(11):919–931. doi: 10.1038/nmeth.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breaker RR. Riboswitches: from ancient gene-control systems to modern drug targets. Future Microbiology. 2009;4(7):771–773. doi: 10.2217/fmb.09.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JL, Green R. In vitro transcription from plasmid or PCR-amplified DNA. Methods in Enzymology. 2013;530:101–114. doi: 10.1016/B978-0-12-420037-1.00005-1. [DOI] [PubMed] [Google Scholar]
- Cherepanov AV, Glaubitz C, Schwalbe H. High-resolution studies of uniformly 13C,15N-labeled RNA by solid-state NMR spectroscopy. Angewandte Chemie. 2010;49(28):4747–4750. doi: 10.1002/anie.200906885. [DOI] [PubMed] [Google Scholar]
- Dayie KT. Key labeling technologies to tackle sizeable problems in RNA structural biology. International Journal of Molecular Sciences. 2008;9(7):1214–1240. doi: 10.3390/ijms9071214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayie KT. Resolution enhanced homonuclear carbon decoupled triple resonance experiments for unambiguous RNA structural characterization. Journal of Biomolecular NMR. 2005;32(2):129–139. doi: 10.1007/s10858-005-5093-6. [DOI] [PubMed] [Google Scholar]
- Dayie TK. Nucleic Acids: Dynamics Studies by Solution NMR. In: Harris RK, editor. eMagRes. Chichester, UK: John Wiley & Sons, Ltd; 2011. Mar 15, [Google Scholar]
- Hoffman DW, Holland JA. Preparation of carbon-13 labeled ribonucleotides using acetate as an isotope source. Nucleic Acids Research. 1995;23(16):3361–3362. doi: 10.1093/nar/23.16.3361-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Julien KR, Hoogstraten CG. Alternate-site isotopic labeling of ribonucleotides for NMR studies of ribose conformational dynamics in RNA. Journal of Biomolecular NMR. 2006;35(4):261–274. doi: 10.1007/s10858-006-9041-x. [DOI] [PubMed] [Google Scholar]
- Lemaster DM, Kushlan DM. Dynamical Mapping of E. coli Thioredoxin via 13 C NMR Relaxation Analysis. Journal of the American Chemical Society. 2001;118:9255–9264. [Google Scholar]
- Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, Dorjsuren B, Kulandaivel G, Jones S, Atheeth H, Divakaruni SS, LaCotti C, Barton S, Tummillo D, Hosic A, Edme K, Albrecht S, Telesnitsky A, Summers MF. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science. 2011;334(6053):242–245. doi: 10.1126/science.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchanka A, Simon B, Carlomagno T. A suite of solid-state NMR experiments for RNA intranucleotide resonance assignment in a 21 kDa protein-RNA complex. Angewandte Chemie. 2013;52(38):9996–10001. doi: 10.1002/anie.201304779. [DOI] [PubMed] [Google Scholar]
- Mattick JS. A new paradigm for developmental biology. The Journal of Experimental Biology. 2007;210:1526–1547. doi: 10.1242/jeb.005017. [DOI] [PubMed] [Google Scholar]
- Miclet E, Williams DC, Jr, Clore GM, Bryce DL, Boisbouvier J, Bax A. Relaxation-optimized NMR spectroscopy of methylene groups in proteins and nucleic acids. Journal of the American Chemical Society. 2004;126(34):10560–10570. doi: 10.1021/ja047904v. [DOI] [PubMed] [Google Scholar]
- Milecki J. Specific labelling of nucleosides and nucleotides with 13C and 15N. Journal of Labelled Compounds and Radiopharmaceuticals. 2002;45(4):307–337. [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Research. 1987;15(21):8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ, Nagai K. Structural studies of the spliceosome: blind men and an elephant. Current Opinion in Structural Biology. 2010;20(1):82–89. doi: 10.1016/j.sbi.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Nikonowicz EP, Sirr A, Legault P, Jucker FM, Baer LM, Pardi A. Preparation of 13C and 15N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Research. 1992;20(17):4507–4513. doi: 10.1093/nar/20.17.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi A. Multidimensional heteronuclear NMR experiments for structure determination of isotopically labeled RNA. Methods in Enzymology. 1995;261(1991):350–380. doi: 10.1016/s0076-6879(95)61017-0. [DOI] [PubMed] [Google Scholar]
- Petrov A, Wu T, Puglisi EV, Puglisi JD. RNA purification by preparative polyacrylamide gel electrophoresis. Methods in Enzymology. 2013;530:315–330. doi: 10.1016/B978-0-12-420037-1.00017-8. [DOI] [PubMed] [Google Scholar]
- Quant S, Wechselberger RW, Wolter MA, Wörner KH, Schell P, Engels JW, Griesinger C, Schwalbe H. Chemical synthesis of 13C-labelled monomers for the solid-phase and template controlled enzymatic synthesis of DNA and RNA oligomers. Tetrahedron Letters. 1994;35(36):6649–6651. [Google Scholar]
- Schultheisz HL, Szymczyna BR, Scott LG, Williamson JR. Enzymatic de novo pyrimidine nucleotide synthesis. Journal of the American Chemical Society. 2011;133(2):297–304. doi: 10.1021/ja1059685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, Nudler E. A decade of riboswitches. Cell. 2013;152(1-2):17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA. A structural understanding of the dynamic ribosome machine. Nature Reviews Molecular Cell Biology. 2008;9(3):242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- Thakur CS, Dayie TK. Asymmetry of 13C labeled 3-pyruvate affords improved site specific labeling of RNA for NMR spectroscopy. Journal of Biomolecular NMR. 2012;52(1):65–77. doi: 10.1007/s10858-011-9582-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur CS, Sama JN, Jackson ME, Chen B, Dayie TK. Selective 13C labeling of nucleotides for large RNA NMR spectroscopy using an E. coli strain disabled in the TCA cycle. Journal of Biomolecular NMR. 2010;48(4):179–192. doi: 10.1007/s10858-010-9454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert TJ, Williamson JR. Preparation of Specifically Deuterated RNA for NMR Studies Using a Combination of Chemical and Enzymatic Synthesis. Journal of the American Chemical Society. 1996;118:7929–7940. [Google Scholar]
- Wunderlich CH, Spitzer R, Santner T, Fauster K, Tollinger M, Kreutz C. Synthesis of (6-(13)C)pyrimidine nucleotides as spin-labels for RNA dynamics. Journal of the American Chemical Society. 2012;134(17):7558–7569. doi: 10.1021/ja302148g. [DOI] [PubMed] [Google Scholar]