

Abstract
Background
Sheeppox (SPP) and goatpox (GTP) caused by sheeppox virus (SPPV) and goatpox virus (GTPV), respectively of the genus Capripoxvirus in the family Poxviridae, are severely afflicting small ruminants’ production systems in Africa and Asia. In endemic areas, SPP and GTP are controlled using vaccination with live attenuated vaccines derived from SPPV, GTPV or Lumpy skin disease virus (LSDV).
Sometimes outbreaks occur following vaccination. In order to successfully control the spread of the virus, it is essential to identify whether the animals were infected by the field strain and the vaccine did not provide sufficient protection. Alternatively, in some cases the vaccine strain may cause adverse reactions in vaccinated animals or in rare occasions, re-gain virulence. Thus, diagnostic tools for differentiation of virulent strains from attenuated vaccine strains of the virus are needed.
The aim of this study was to identify an appropriate diagnostic target region in the capripoxvirus genome by comparing the genomic sequences of SPPV field isolates with those of the most widely used SPP vaccine strains.
Results
A unique 84 base pair nucleotide deletion located between the DNA ligase gene and the VARV B22R homologue gene was found only in SPPV vaccines derived from the Romanian and Yugoslavian RM/65 strains and absent in SPPV field isolates originated from various geographical locations of Asia and Africa.
In addition, we developed and evaluated a conventional PCR assay, exploiting the targeted intergenic region to differentiate SPPV vaccine virus from field isolates. The assay produced an amplicon size of 218 bp for the vaccine strains, while the SPPV field isolates resulted in a 302 bp PCR fragment. The assay showed good sensitivity and specificity, and the results were in full agreement with the sequencing data of the PCR amplicons.
Conclusion
The developed assay is an improvement of currently existing diagnostic tools and, when combined with a capripox virus species-specific assay, will enhance SPP and GTP diagnosis and surveillance and facilitate epidemiological investigations in countries using live attenuated SPP vaccines. In addition, for laboratories with limited resources, the assay provides a simple and cost-effective alternative for sequencing.
Electronic supplementary material
The online version of this article (10.1186/s12985-018-0969-8) contains supplementary material, which is available to authorized users.
Keywords: CaPV, Sheeppox vaccine, Sheeppox virus, VARV B22R homologue gene, DNA ligase gene
Background
Sheeppox (SPP) and goatpox (GTP) are caused by sheeppox virus (SPPV) and goatpox virus (GTPV) of the genus Capripoxvirus of the family Poxviridae [1]. In endemic areas SPP and GTP have a serious economic impact on small ruminant production systems, causing losses in productivity, mortality, damaging skins and hides, as well as inflicting international trade restrictions [2]. They are listed in the group of economically important animal diseases for which outbreaks have to be notified immediately to the World Organization for Animal Health [3].
The main mode of virus transmission is the direct contact between diseased and non-infected animals, but indirect transmission may also occur [4]. Clinical signs of SPPV and GTPV infections are characterized by ocular and nasal discharge and pock-like lesions in the skin and mucosae of the respiratory and gastrointestinal tracts [2, 4, 5]. Most of the isolates are host specific and cause disease mainly in sheep or in goats, whereas some isolates can cause serious disease in both animal species [6].
SPP and GTP are endemic in many African, Middle Eastern and Asian countries and recurrent epidemics have occurred in Greece and Bulgaria in 2013–2014 [7] and in Greece in 2016 and 2017. SPPV is also circulating in the Russian Federation where it causes sporadic outbreaks of disease.
In endemic regions, control of the disease is through effective immunization using killed or live attenuated vaccines derived from SPPV, GTPV or Lumpy skin disease virus (LSDV). In general, live attenuated vaccines are the better choice as compared to inactivated vaccines, as they confer long-lasting immunity. For instance, the Yugoslavian RM65, the Romanian Fanar and KSGP0240 strains, the most commonly used vaccines strains against SPPV, are reported to provide high levels of protection [7]. The Yugoslavian RM65 is widely used in the Middle East, Asia and in the Horn of Africa, while the Romanian Fanar is used in India and Maghreb countries. The Yugoslavian RM65 was attenuated by 30 serial passages on ovine kidney cells, and the Romanian Fanar by 26 serial passages on lamb testis cells [7].
The KSGP 0240 is widely used in several endemic regions in Africa. Nevertheless, KSGP0240 has been shown, by sequencing, to be a LSDV, thus, it can be differentiated from virulent isolates of SPPV using available capripoxvirus genotyping methods [7].
However, when using live attenuated vaccines, the epidemiological investigation of outbreaks can become quite challenging. When outbreaks occur following vaccination, it is essential to identify whether the animals were infected by the field strain because the vaccine did not provide sufficient protection. Alternatively, in some cases the vaccine strain may cause adverse reactions in vaccinated animals or, in rare occasions, re-gain the virulence as suggested by Lee and co-workers for herpesvirus vaccine [8].
Unfortunately, the current live attenuated capripox (CaP) vaccines do not offer the possibility to differentiate vaccinated animals from infected ones. This creates a need to identify a suitable genomic target and develop molecular tools that would enable the differentiation of SPPV field isolates from vaccine strains to rule out the involvement of SPP vaccines during a CaP outbreak in a vaccinated herd. Such a tool will facilitate the management and control of CaP infections in small ruminants.
The present study describes the use of a suitable diagnostic target of the SPPV genome to develop an assay that can discriminate SPP vaccines derived from the Romanian and the Yugoslavian RM/65 strains from SPPV field isolates, to facilitate the diagnosis and surveillance of capripox virus (CaPV) infections in small ruminants.
Methods
Virus and nucleic acid extraction
The information related to the field isolates and vaccine strains of SPPV as well as other CaPVs used in this study are presented in Table 1. Viral multiplication was performed on embryonic skin cell from sheep (ESH-L cells) grown in Hank’s Minimum Essential Medium (MEM) supplemented with 10% foetal calf serum and 1% antibiotics. DNA was extracted from infected cell culture supernatants and clinical samples using the AllPrep DNA/RNA extraction kit (QIAGEN) following the manufacturer’s instructions. Extracted DNA was eluted with 80 μl elution buffer and stored at − 20 °C until further use.
Table 1.
List of capripoxvirus isolates used in this study
| Sample No. | Isolate/Strain name | Source | Sample type | Country | Collection-date | Host |
|---|---|---|---|---|---|---|
| 1 | SPPV Morocco vaccine | Biopharma/Morocco | Cell culture | Morocco | Unknown | Sheep |
| 2 | SPPV Algeria vaccine Lot/7 | INMV-LCV/Algeria | Cell culture | Algeria | Unknown | Sheep |
| 3 | SPPV Senegal vaccine | LNERV-ISRA/Senegal | Cell culture | Iran | 1966 | Sheep |
| 4 | SPPV PANVAC/6 vaccine | PANVAC/Ethiopia | Cell culture | Kenya | 2010 | Sheep |
| 5 | SPPV Turkey/98 Corum | VCRI-Pendik/Turkey | Cell culture | Turkey | 1998 | Sheep |
| 6 | SPPV Oman/84 | IAH-Pirbright/UK | Cell culture | Oman | 1984 | Sheep |
| 7 | SPPV Turkey/98 Denizli | VCRI-Pendik/Turkey | Cell culture | Turkey | 1998 | Sheep |
| 8 | SPPV Algeria/93 Djelfa | INMV-LCV/Algeria | Cell culture | Algeria | 1993 | Sheep |
| 9 | SPPV Algeria/05 Illizi | INMV-LCV/Algeria | Cell culture | Algeria | 2005 | Sheep |
| 10 | SPPV Turkey/98 Sivas | VCRI-Pendik/Turkey | Cell culture | Turkey | 1998 | Sheep |
| 11 | SPPV MOG/SP/T/2/07 | IVM/Mongolia | Skin scrapping | Mongolia | 2007 | Sheep |
| 12 | SPPV MOG/SP/T/3/07 | IVM/Mongolia | Skin scrapping | Mongolia | 2007 | Sheep |
| 13 | GTPV Iraq/61 Gorgan | IAH-Pirbright/UK | Cell culture | Iraq | 1961 | Goat |
| 14 | GTPV MOG/GP/T/6/08 | IVM/Mongolia | Skin scrapping | Mongolia | 2008 | Goat |
| 15 | GTPV MOG/GP/L/5/08 | IVM/Mongolia | Skin scrapping | Mongolia | 2008 | Goat |
| 16 | GTPV Awi/O13/2011 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2011 | Goat |
| 17 | GTPV Bale/O14/2007 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2007 | Goat |
| 18 | GTPV Giner/O15/2007 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2007 | Goat |
| 19 | LSDV KS-1 | HSL-AGES/Austria | Cell culture | Kenya | 1976 | Sheep |
| 20 | LSDV Egypt/89 Ismalia | HSL-AGES/Austria | Cell culture | Egypt | 1989 | Cattle |
| 21 | LSDV Guder/B5/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 22 | LSDV Humbo/B23/2010 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2010 | Cattle |
| 23 | SPPV Algeria vaccine Lot/10 | INMV-LCV/Algeria | Cell culture | Algeria | Unknown | Sheep |
| 24 | SPPV Mauritania/85 Gorgol | LNERV-ISRA/Senegal | Cell culture | Mauritania | 1985 | Sheep |
| 25 | SPPV Turkey/98 Darica | VCRI-Pendik/Turkey | Cell culture | Turkey | 1998 | Sheep |
| 26 | SPPV MOG/SP/T/1/2006 | IVM/Mongolia | Skin scrapping | Mongolia | 2006 | Sheep |
| 27 | GTPV Saudi Arabia/93 | IAH-Pirbright/UK | Cell culture | Saudi Arabia | 1993 | Goat |
| 28 | SPPV Nigeria/77 | IAH-Pirbright/UK | Cell culture | Nigeria | 1993 | Sheep |
| 29 | GTPV Turkey/98 Denizli | VCRI-Pendik/Turkey | Cell culture | Turkey | 1998 | Goat |
| 30 | GTPV Oman/84 | IAH-Pirbright/UK | Cell culture | Oman | 1984 | Goat |
| 31 | GTPV MOG/GP/T/4/08 | IVM/Mongolia | Skin scrapping | Mongolia | 2008 | Goat |
| 32 | GTPV Yemen/83 | IAH-Pirbright/UK | Cell culture | Yemen | 1983 | Goat |
| 33 | GTPV Towele/O17/2013 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2013 | Goat |
| 34 | GTPV Halasya/G18/2013 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2013 | Goat |
| 35 | LSDV Galesa/B12/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 36 | LSDV Sodo/B24/2010 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2010 | Cattle |
| 37 | LSDV Chilimo/B11/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 38 | LSDV Ambo/B8/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 39 | LSDV Toke/B6/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 40 | LSDV Ginchi/B10/2008 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2008 | Cattle |
| 41 | LSDV Sodo/B22/2010 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2010 | Cattle |
| 42 | LSDV Adama/B4/2011 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2011 | Cattle |
| 43 | LSDV Ziway/B3/2011 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2011 | Cattle |
| 44 | LSDV Asella/B2/2011 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2011 | Cattle |
| 45 | LSDV Arsi/B1/2011 | NAHDIC/Ethiopia | Skin scrapping | Ethiopia | 2011 | Cattle |
| 46 | LSDV Sundus/2012 | CVRL/Sudan | Skin scrapping | Sudan | 2012 | Cattle |
Abbreviations: VCRI Veterinary Control and Research Institute, LNERV-ISRA Laboratoire National de l’Elevage et de Recherches Vétérinaires, Institut Sénégalais de Recherches Agricoles, INMV-LCV Institut National de la Médecine Vétérinaire, Laboratoire Central Vétérinaire, IVM Institute of Veterinary Medicine, IAH Institute for Animal Health, PANVAC Pan African Veterinary Vaccine Centre, NAHDIC National Animal Health Diagnostic and Investigation Center, HSL-AGES High Security Laboratory, Austrian Agency for Health and Food Safety, CVRL Central Veterinary Research Laboratories
Primer design and PCR
The target region was selected based on the alignment of the full genomes of three SPPVs, Sheeppox virus 10,700–99 (AY077832), Sheeppox virus A (AY077833) and Sheeppox virus NISKHI, (AY077834), retrieved from GenBank, and the unpublished genome of the Romanian vaccine strain used in Morocco. PCR primers, flanking a unique nucleotide deletion in the Romanian vaccine strain, were designed using Primer3Plus online tool. The target was an intergenic region located between the DNA ligase gene and the VARV B22R homologue gene of CaPVs corresponding to position 121,500–122,799 of SPPV A (AY077833). The primers (Table 2) were designed to amplify amplicons of 302 bp in SPPV field isolates and 218 bp fragments for SPPV vaccines. The specificity of the primers was checked by using the Basic Local Alignment Search Tool (BLAST). The primers were synthesized and purified by HPLC by Eurofins Genomics (Austria).
Table 2.
Primers used in this study. The length of the predicted amplicons are given
| Primer name | Sequences | Length | Amplicon size |
|---|---|---|---|
| SPPV_DIV_Fow | 5’-ATCTGCTACAAGTTTTAACGAACTTA- 3’ | 26 | 218 bp (SPPV vaccines) |
| SPPV_DIV_Rev | 5’-TGAATGTGATCTCATATCCTTATTG-3’ | 25 | 302 (SPPV field isolates and GTPV) and 336 (LSDV) |
PCR was conducted in a reaction volume of 20 μl, containing 500 nM of each forward and reverse primers, 0.2 mM dNTPs, 2.5 U Taq DNA polymerase (QIAGEN), 1× PCR buffer, and 2 μl template DNA. The cycling conditions were as follow: 95 °C for 4 min followed by 35 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s and a final extension at 72 °C for 2 min. PCR products were cheeked by electrophoresis on a 2% agarose gel for 1 h at 100 V.
Preparation of controls
The PCR amplicons of SPPV Denizli and SPPV Morocco vaccine (Romanian vaccine strain) were selected to prepare positive control plasmids, representing the field isolates and vaccine strains respectively. For plasmid preparation, each amplicon was purified and ligated into pGEM-T Easy Vector Systems (Promega). The ligated products were used to transform DH5α competent cells (Invitrogen). Plasmids containing the inserts were purified from the positive bacteria clones using the PureYield Plasmid Midiprep System (Promega) following the manufacturer’s instructions. The purified plasmids were sequenced to confirm the presence of the correct target region and quantified using the Quant-iTPicoGreen dsDNA Assay Kit (Invitrogen) on a NanoDrop 3300 fluorospectrometer. The concentration of the plasmids was determined following the steps described by Lamien et al., (2011) and the plasmids were kept at − 20 °C until analysis.
Analytical sensitivity and specificity of the assay
The analytical sensitivity of the method was assessed by amplifying 10-fold serial dilutions, from107 to 100 copies/reaction, then follow by 80, 60, 40, 20, 10 and 1 copies/reaction dilutions, of plasmids containing the corresponding target of SPPV Denizli and SPPV Morocco vaccine. The lowest number of viral genome copies that could be detected by the assay was recorded.
The specificity was evaluated by amplifying DNA extracted from all available SPP vaccines and field isolates as well as GTPV and LSDV isolates from various geographical regions (Table 1). The genotype of each sample was confirmed using a capripox species specific PCR (Gelaye et al., 2013). Additionally, the specificity in amplifying only CaPVs was determined by attempting to amplify DNA extracted from Orf virus (ORFV), Mycoplasma capricolum ssp. capripneumonia (Mccp), Bovine herpes virus (BOHV), Bovine popular stomatitis virus (BPSV) and cDNA derived from peste des petits ruminants (PPR) virus (Additional file 1: Table S1).
Sequence analysis
For all CaPVs used in this study, the targeted region of the genome was sequenced to confirm the accuracy of the assay. Thus, all amplified PCR products of the above described PCR reactions were purified using Wizard SV Gel and PCR Clean Up System (Promega) and sequenced commercially by LGC Genomics (Germany). The sequence data were edited and the fragments assembled using Vector NTI 11.5 software (Invitrogen). Multiple sequence alignments were performed using the CLUSTALW algorithm implemented in BioEdit 7.5 software package to compare SPPV field isolates and vaccine strains. All sequences were deposited in GenBank under accession number MG764242 to MG764286.
Results
Assay design and optimization
Primers were designed to amplify a region between the DNA ligase gene and the VARV B22R homologue gene of SPPV. The region was first selected based on the sequence alignment of three SPPV publicly available full genome sequences and a draft genome sequence of the SPP Morocco vaccine derived from the Romanian vaccine strain. The SPP vaccine had an 84 bp nucleotide deletion as compared to SPPV field isolates (Fig. 1). The initial evaluation of the assay showed that the Morocco vaccine strain could be differentiated from SPPV Denizli field isolate based on the size difference of the produced PCR amplicons: 218 bp for the vaccine strain and 302 bp for the SPPV field isolate. In the subsequent steps, the assay was optimized and further evaluated.
Fig. 1.
Multiple sequence alignments showing a 84-nucleotide deletion in SPPV vaccines. The nucleotide sequences of the intergenic region located between the DNA ligase gene and the VARV B22R homologue gene for 45 Capripoxviruses of this study and 7 other retrieved from Genbank were compared
Evaluation of the assay
The optimal assay parameters are presented in Methods. The optimised assay was further evaluated by testing 46 CaPV isolates or clinical samples including 12 field isolates of SPPV, 5 SPP vaccines, 13 field isolates of GTPV and 15 field isolates of LSDV and 1 LSDV vaccine (Table 1). All 5 SPP vaccines produced 218 bp PCR products while the SPPV field isolates produced a 302 bp product (Fig. 2 and Additional file 2: Figure S1). The SPPV field isolates could not be clearly differentiated from GTPVs and LSDVs (Fig. 2 and Additional file 2: Figure S1).
Fig. 2.
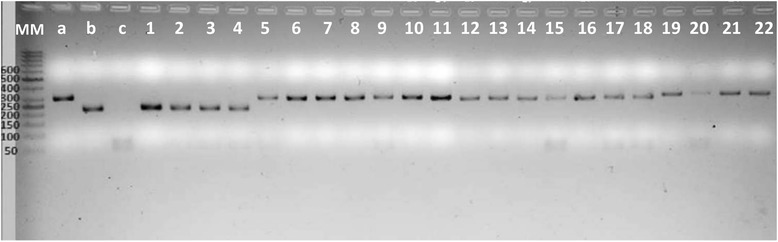
Gel picture of PCR products for selected capripoxvirus samples. The SPPV vaccines appear to be shorter than SPPV field isolates, GTPV and LSDV due to the 84 bp sequence difference. The PCR products of 218 bp, 302 bp and 338 bp represent SPPV vaccine strains, SPPV field isolates/GTPVs, and LSDVs respectively. MM: 50 bp DNA ladder; a: positive control plasmid of the SPPV field isolates; b: positive control plasmid of the SPPV vaccine strain; c: Negative control; Lanes 4–7: SPPV vaccine strains (sample 1 to 4 of Table 1); Lanes 8–15: SPPV field isolates (sample 5 to 12 of Table 1); Lanes 16–21: GTPVs (sample 13 to 18 of Table 1), Lanes 22–25: LSDVs (sample 19 to 22 of Table 1)
Limit of detection and specificity of the assay
The limit of detection of the assay was evaluated by amplifying 10-fold serial dilutions of plasmids as described in Methods. The results showed that the limit of detection for SPPV field isolates and vaccine strains were 80 and 10 copies/reaction (Additional file 3: Figure S2) respectively. The specificity of the assay was tested by attempting to amplify non-capripoxvirus DNA from Orf virus, Mycoplasma capricolum ssp. capripneumonia (Mccp), Bovine herpes virus (BOHV), Bovine popular stomatitis virus (BPSV) and cDNA from peste des petits ruminant virus. No amplification was observed (Additional file 4: Figure S3).
Sequencing of the PCR amplicons
All PCR amplicons were sequenced for further validation of this PCR approach. The results of the multiple sequence alignment showed the absence of the 84-nucleotide deletion in all SPPV field isolates, GTPV and LSDV. Additionally, the targeted region was found to be well conserved among the SPPV field isolates and the SPPV vaccine strains except for the 84 nucleotides deletion. Ten nucleotide variations were observed between SPPV field isolates and GTPVs. LSDV sequences are longer than SPPV field isolates and GTPVs due to an insertion of 34–36 nucleotides. GTPV Saudi Arabia appeared to be a SPPV and SPPV Oman showed the GTPV specific features. The vaccine KS1 (renamed in our paper as LSDV KS-1) presented a 34-nucleotide insertion as most LSDV of this study.
Discussion
In this study, we identified a suitable target in CaPV genome and developed a PCR method to discriminate SPP vaccine strains from SPPV field isolates as well as from other CaPVs.
By aligning the full genome sequences of SPPV field isolates with the unpublished full genome of SPPV Morocco vaccine, a Romanian strain, we identified a region of 84 bp nucleotide deletion in the vaccine strains. Primers were designed to amplify this intergenic region located between the DNA ligase gene and the VARV B22R homologue gene of CaPV. The sequencing of this region in SPPV vaccine strains and SPPV field isolates available for this study, showed this deletion to be unique to the SPP vaccines derived from the Romanian and the Yugoslavian RM/65 strains produced in Algeria, Egypt, Morocco and Senegal. However, the sequence of the NISKHI vaccine, whose full genome has been previously published [9] did not carry such a deletion. Nevertheless, the use of the NISHKI strain seems to be confined to Russia and countries of the former Soviet Union such as Kazakhstan [10]. By using the targeted intergenic region, the aim of this study was to develop an assay to discriminate the live attenuated Romanian and Yugoslavian RM/65 SPP vaccines strains from virulent SPPV field isolates and other CaPVs.
Interestingly, Romanian and Yugoslavian RM/65 SPPV vaccines produced amplicons of shorter lengths, as compared to field isolates of SPPV and other CaPVs, and thus could be easily discriminated from them. The assay was found to be very specific and sensitive, and the accuracy and reliability was confirmed by sequencing the corresponding amplicons of SPP vaccine strains and SPPV, GTPV and LSDV field isolates available for this study.
The high sensitivity of this assay for SPPV vaccine strain, as compared to the field isolates, is likely due to the shortest amplicon size in the vaccine strains. No amplification was detected for any non-CaPV samples by the assay. However, the assay could not differentiate SPPV field isolates from LSDVs and GTPVs. Although an insertion of 34–36 nucleotides was observed in all LSDVs, making them longer than GTPVs and SPPV field isolates, it was not possible to resolve these differences on the agarose gel. Owing to the availability of species-specific PCR [11–13] for CaPV genotype determination, we suggest that the current assay could be used once the genotype of CaPV is established. Alternatively, the exact capripoxvirus genotype can be determined by sequencing the region targeted in this study. Within the SPPV genotype, the test, undoubtedly discriminates between the virulent field isolates and the vaccines derived from the Romanian and the Yugoslavian RM/65 strains.
The availability of an easy-to-use molecular method is needed for the identification of SPPV, to rule out the involvement of SPP vaccines following a CaP outbreak in previously vaccinated flock of small ruminants. In Middle Eastern and Asian countries, small ruminants are protected from CaPV infections using various SPPV, GTPV or LSDV derived vaccines, however, SPP vaccines derived from Romanian and the Yugoslavian RM/65 strains are predominant [14]. In Africa, small ruminants are protected against CaPV using either KSGP O-240 and O-180 vaccines or SPP vaccines derived from the Romanian and the Yugoslavian RM/65 strains. Since KSGP O-240 and O-180 strains are of LSDV genotype, the use of a CaPV species-specific assay [11–13] can allow for determination of their involvement when the disease occurs in a previously vaccinated herd. However, if a herd is vaccinated with a SPP vaccine from the Romanian and the Yugoslavian RM/65 strains, the full genome sequencing of the viral isolate, collected during an outbreak, would be required to rule out vaccine involvement when an outbreak occurs. This is time-consuming and cost prohibitive for most laboratories in limited resourced countries. Thus, the identification of suitable target in the viral genome to differentiate SPPV vaccine strains from SPPV field isolates greatly reduces the costs, by allowing the sequencing of small specific genome fragments. Furthermore, this region can be targeted in a simple molecular method such as the PCR approach presented in the work, thereby avoiding the use of sequencing. The current assay is intended to be used by all veterinary laboratories, including those with limited resources. It can also be used as a front-line tool for the direct screening of pathological samples collected during CaPV outbreaks, especially those occurring in previously vaccinated small ruminant populations. A study was recently conducted in Morocco to rule out the involvement of the vaccine strain in 2010 SPP outbreaks using a PCR based approach [15]. When compared to the assay developed by Haegeman and co-workers [15], our method presents the advantage of using only one primer pair to target both viruses, and thus, is much simpler to conduct and interpret. In addition, more vaccine strains and field isolates, from various geographical locations were included in this study, which broadened the scope of the applicability to all countries where SPP vaccines derived from the Romanian or the Yugoslavian RM/65 strains are used.
Conclusions
The molecular assay described herein is a reliable and rapid method that can easily be implemented for the differentiation of SPP vaccine derived from the Romanian or the Yugoslavian RM/65 strains from virulent SPPV field isolates. The method is applicable as a routine tool for outbreak investigations and disease surveillance in both SPP and GTP enzootic and disease-free countries. It is expected that its adoption by veterinary laboratories in CaPV affected countries, will help facilitate the control and management of CaP disease in small ruminants.
Additional files
Table S1. Non-capripoxvirus samples tested for specificity study. (DOCX 16 kb)
Figure S1. Gel picture of PCR products for the remaining capripoxvirus samples. These sample were tested in this study, but not presented in Fig. 2 of the manuscript. The PCR products of 218 bp, 302 bp and 338 bp represent SPPV vaccine strains, SPPV field isolates/GTPVs, and LSDVs respectively. First row: MM = 50 bp DNA ladder; a = positive control plasmid of the SPPV field isolates; b = positive control plasmid of the SPPV vaccine strain; c = Negative control; Lane 5 to 15 (sample 23 to 33 in Table 1 of the manuscript). Second row: MM = 50 bp; Lane 2 to 14 (sample 34 to 46 in Table 1 of the manuscript). (PDF 104 kb)
Figure S2. Determination of the limits of detection of the PCR assay. Defined amount for the plasmid genotype standard (104, 103, 100, 80, 60, 40, 20, 10, 1 and 0) for SPPV vaccine (A) and SPPV field isolates (B) were tested in parallel reactions and run on agarose gel. (PDF 77 kb)
Figure S3. Gel picture of PCR for the non-capripoxvirus samples tested in this study. MM = 50 bp DNA ladder; a = positive control plasmid of the SPPV field isolates; b = positive control plasmid of the SPPV vaccine strain; c = Negative control; 1–5 (ORF viruses); 6 (BPSV); 7–8 (Mccp); 9 (cDNA, PPRV); 10 (BOHV-1); 11 (BOHV-2). (PDF 45 kb)
Acknowledgements
The authors would like to thank Dr. El Harrak M. (Biopharma, Morocco), Dr. Velý Gulyaz (Pendik-VCRI, Turkey) and Dr. Erdenebaataar Janchivdorj (IVM, Mongolia) and Dr. Adi Steinrigl (AGES, Austria) for providing capripoxvirus and bovine herpes virus samples. We also like to thank, Dr. Esayas Gelaye (NVI, Ethiopia) and Dr. Jenna E. Achenbach (Battelle, VA, USA) for critically assessing the manuscript.
Funding
This study was funded by the International Atomic Energy Agency (IAEA) project “Improvement of Veterinary Laboratory Capacities in Sub-Saharan African Countries”.
Availability of data and materials
All relevant information is provided in this current manuscript.
Authors’ contributions
Conceived and designed the experiments: CEL, TRC. Performed the experiments: TRC, AO. Analyzed the data: TRC, CEL, RG, AL, AO, TBKS. Contributed reagents/materials/analysis tools: AL, NN, KT, HM, MD, ET. Wrote the paper: TRC, CEL, RG, AD, Supervised the study: AD, GC, RG, CEL. Edited the final manuscript: AD, GC, RG, NN, KT, ET, TBKS, HM, MD. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12985-018-0969-8) contains supplementary material, which is available to authorized users.
References
- 1.Buller RM, Arif BM, Black DN, Dumbell KR, Esposito JJ, Lefkowitz EJ, Moss B, Mercer AA, Moyer RW, Skinner MA, Tripathy DN. Family Poxviridae. In: Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. Fauquet CM. Virus taxonomy: VIIIth report of the International Committee on Taxonomy of Viruses. Academic Press; 2005. [Google Scholar]
- 2.Hosamani M, Nandi S, Mondal B, Singh RK, Rasool TJ, Bandyopadhyay SKA. Vero cell-attenuated Goatpox virus provides protection against virulent virus challenge. Acta Virol. 2003;48:15–21. [PubMed] [Google Scholar]
- 3.OIE. Sheep pox and goat pox. In: Manual of diagnostic tests and vaccines for terrestrial animals: 2017. Chapter 27.13. http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.07.13_S_POX_G_POX.pdf
- 4.Balinsky CA, Delhon G, Smoliga G, Prarat M, French RA, Geary SJ, Rock DL, Rodriguez LL. Rapid preclinical detection of sheeppox virus by a real-time PCR assay. J Clin Microbiol. 2008;46:438–442. doi: 10.1128/JCM.01953-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan XM, Chu YF, Wu GH, Zhao ZX, Li J, Zhu HX, Zhang Q. An outbreak of sheep pox associated with goat poxvirus in Gansu province of China. Vet Microbiol. 2012;156:425–428. doi: 10.1016/j.vetmic.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 6.Babiuk S, Bowden TR, Boyle DB, Wallace DB, Kitching RP. Capripoxviruses: an emerging worldwide threat to sheep, goats and cattle. Transbound Emerg Dis. 2008;55:263–272. doi: 10.1111/j.1865-1682.2008.01043.x. [DOI] [PubMed] [Google Scholar]
- 7.EFSA AHAW Panel (EFSA panel on animal health and welfare) Scientific opinion on sheep and goat pox. EFSA J. 2014;12(11):3885. [Google Scholar]
- 8.Lee SW, Markham PF, Coppo MJ, Legione AR, Markham JF, Noormohammadi AH, Browning GF, Ficorilli N, Hartley CA, Devlin JM. Attenuated vaccines can recombine to form virulent field viruses. Science. 2012;337:188. doi: 10.1126/science.1217134. [DOI] [PubMed] [Google Scholar]
- 9.Tulman ER, Afonso CL, Lu Z, Zsak L, Sur JH, Sandybaev NT, Kerembekova UZ, Zaitsev VL, Kutish GF, Rock DL. The genomes of sheeppox and goatpox viruses. J Virol. 2002;76:6054–6061. doi: 10.1128/JVI.76.12.6054-6061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parthiban M, Govindarajan R, Manoharan S, Purushothaman V, Daniel joy Chandran N, Koteeswaran A. Comparative sequence analysis of diagnostic PCR amplicons from Indian sheeppox virus. Veterinarski arhiv. 2005;75:203–209. [Google Scholar]
- 11.Lamien CE, Le GC, Silber R, Wallace DB, Gulyaz V, Tuppurainen E, Madani H, Caufour P, Adam T, El HM, Luckins AG, Albina E, Diallo A. Use of the Capripoxvirus homologue of vaccinia virus 30 kDa RNA polymerase subunit (RPO30) gene as a novel diagnostic and genotyping target: development of a classical PCR method to differentiate goat poxvirus from sheep poxvirus. Vet Microbiol. 2011;149:30–39. doi: 10.1016/j.vetmic.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 12.Lamien CE, Lelenta M, Goger W, Silber R, Tuppurainen E, Matijevic M, Luckins AG, Diallo A. Real time PCR method for simultaneous detection, quantitation and differentiation of capripoxviruses. J Virol Methods. 2011;171:134–140. doi: 10.1016/j.jviromet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Gelaye E, Lamien CE, Silber R, Tuppurainen ES, Grabherr R, Diallo A. Development of a cost-effective method for capripoxvirus genotyping using snapback primer and dsDNA intercalating dye. PLoS One. 2013;8:e75971. doi: 10.1371/journal.pone.0075971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuppurainen ES, Venter EH, Shisler JL, Gari G, Mekonnen GA, Juleff N, Lyons NA, De CK, Upton C, Bowden TR, Babiuk S, Review BLA. Capripoxvirus diseases: current status and opportunities for control. Transbound Emerg Dis. 2017;64(3):729–745. doi: 10.1111/tbed.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haegeman A, Zro K, Sammin D, Vandenbussche F, Ennaji MM, De CK. Investigation of a possible link between vaccination and the 2010 sheep pox epizootic in Morocco. Transbound Emerg Dis. 2016;63:e278–e287. doi: 10.1111/tbed.12342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Non-capripoxvirus samples tested for specificity study. (DOCX 16 kb)
Figure S1. Gel picture of PCR products for the remaining capripoxvirus samples. These sample were tested in this study, but not presented in Fig. 2 of the manuscript. The PCR products of 218 bp, 302 bp and 338 bp represent SPPV vaccine strains, SPPV field isolates/GTPVs, and LSDVs respectively. First row: MM = 50 bp DNA ladder; a = positive control plasmid of the SPPV field isolates; b = positive control plasmid of the SPPV vaccine strain; c = Negative control; Lane 5 to 15 (sample 23 to 33 in Table 1 of the manuscript). Second row: MM = 50 bp; Lane 2 to 14 (sample 34 to 46 in Table 1 of the manuscript). (PDF 104 kb)
Figure S2. Determination of the limits of detection of the PCR assay. Defined amount for the plasmid genotype standard (104, 103, 100, 80, 60, 40, 20, 10, 1 and 0) for SPPV vaccine (A) and SPPV field isolates (B) were tested in parallel reactions and run on agarose gel. (PDF 77 kb)
Figure S3. Gel picture of PCR for the non-capripoxvirus samples tested in this study. MM = 50 bp DNA ladder; a = positive control plasmid of the SPPV field isolates; b = positive control plasmid of the SPPV vaccine strain; c = Negative control; 1–5 (ORF viruses); 6 (BPSV); 7–8 (Mccp); 9 (cDNA, PPRV); 10 (BOHV-1); 11 (BOHV-2). (PDF 45 kb)
Data Availability Statement
All relevant information is provided in this current manuscript.