

Abstract
Evolving studies in models of transplant rejection, inflammatory bowel disease, and cancer, among others, have implicated purinergic signaling in clinical manifestations of vascular injury and thrombophilia, inflammation, and immune disturbance.
Within the vasculature, spatial and temporal expression of CD39 nucleoside triphosphate diphosphohydrolase (NTPDase) family members together with CD73 ecto-5′-nucleotidase control platelet activation, thrombus size, and stability. This is achieved by closely regulated phosphohydrolytic activities to scavenge extracellular nucleotides, maintain P2-receptor integrity, and coordinate adenosinergic signaling responses. The CD38/CD157 family of extracellular NADases degrades NAD+ and generates Ca2+-active metabolites, including cyclic ADP ribose and ADP ribose. These mediators regulate leukocyte adhesion and chemotaxis. These mechanisms are crucial in vascular homeostasis, hemostasis, thrombogenesis, and during inflammation.
There has been recent interest in ectonucleotidase expression by immune cells. CD39 expression identifies Langerhans-type dendritic cells and efficiently distinguishes T regulatory cells from other resting or activated T cells. CD39, together with CD73 in mice, serves as an integral component of the suppressive machinery of T cells. Purinergic responses also impact generation of T helper-type 17 cells. Further, CD38 and changes in NAD+ availability modulate ADP ribosylation of the cytolytic P2X7 receptor that deletes T regulatory cells.
Expression of CD39, CD73, and CD38 ectonucleotidases on either endothelial or immune cells allows for homeostatic integration and control of vascular inflammatory and immune cell reactions at sites of injury. Ongoing development of therapeutic strategies targeting these and other ectonucleotidases offers promise for the management of vascular thrombosis, disordered inflammation, and aberrant immune reactivity.
I. Introduction
This review addresses novel mechanisms concerning the role of purinergic/pyrimidinergic signaling in hemostasis, vascular thrombosis, and cellular immunity in inflammatory disorders such as in transplantation, sepsis, autoimmune diseases, and cancer (Robson et al., 2005, 2006).
Nucleosides are glycosylamines comprising the nucleobase attached to a pentose sugar ring. Examples of these include cytidine, uridine, adenosine, guanosine, thymidine, and inosine. Nucleosides can be phosphorylated by specific kinases (chiefly within the cell but also feasibly outside). These reactions generate nucleotides, the monomeric structural unit of nucleotide chains that form nucleic acids (RNA and DNA). Nucleotides also play important roles in cellular energy transport and transformation (nucleoside triphosphates such as ATP are the energy-rich end products of the majority of biochemical energy releasing pathways), in enzyme regulation, and as intracellular second messengers.
Clearly, cellular injury processes are detected by release of intracellular constituents that, given the parsimony of nature, could also be used as extracellular “danger” or signaling mediators. Hence, it is possible that specific extracellular messenger systems could have developed to facilitate recognition of extracellular nucleotides that would have served as sensors for environmental stresses. One can also propose that such novel modular systems have been generated and refined with the development of multicellular organisms, different organ systems, and the requirement for a circulatory system and mobile cells that need to leave and reenter the blood stream.
The organization of the purinergic/pyrimidinergic system in health and disease might include requirements for new gene families, with conservation or modification of adaptive genes to facilitate specificity of nucleotide metabolism and responses that we address in the context of the critical host defense mechanisms of hemostasis and blood coagulation, inflammation, and immunity.
II. Purinergic Signaling: A Paradigm Linking Coagulation, Inflammation, and Immunity
Extracellular nucleotides are involved in vascular cell and platelet activation that enhance the generation of fibrin after vascular injury: to cause hemostasis under physiological conditions and provoke thrombosis in pathological situations. These mediators, in a manner analogous to coagulation proteases such as thrombin, have cellular activation effects that are fibrin independent and dictated by the specific purinergic receptors.
Purinergic signaling mechanisms mediated by nucleotides and nucleosides have a complex influence on inflammatory processes. These are characterized by obligatory blood and vessel components that cause extravascular responses.
How purinergic responses dictate and/or modulate inflammation is still incompletely understood. A shared P2-mediated mechanism in several different experimental models involves the boosting of innate immune “danger” signals that are initially generated by bacterial infections with endotoxin or, in the context of transplantation, ischemia–reperfusion, or rejection. The pathways ultimately are switched over to promote adenosinergic-type responses, a process that at least in part involves upregulation of ectonucleotidases and ATP scavenging.
There is increasing evidence that purinergic signaling has dramatic positive and negative effects on cells involved in adaptive immunity, for example, T lymphocytes, and on inflammatory cells that drive chronic inflammation and fibrosis, for example, dendritic cells (DCs), stellate cells, and myofibroblasts. This review will illustrate key paradigms linking coagulation, inflammation, and immunity as integrated purinergic responses. Several inflammatory diseases with thrombophilia have been linked genetically and mechanistically to ectonucleotidases of the CD39 and CD38 families, among others. These ectonucleotidases, as well as principles of purinergic signaling, will be addressed here. We will also describe the importance of dysregulated nucleotide-mediated signaling in cancer and inflammatory bowel disease (IBD) and touch on how pathogens might also subvert these defenses.
III. Extracellular Nucleotides, Nucleosides, and Ectonucleotidases
Extracellular nucleotides (e.g., ATP, UTP, ADP, NAD), and the derivative nucleosides (e.g., adenosine from ATP), are released in a regulated manner by most cells to provide the initiators and primary components for purinergic responses (Luthje, 1989).
These nucleotide/nucleoside mediators bind specific purinergic receptors that after mediator release comprise the second requirement for such a signaling network. Almost all cells express multiple type-2 purinergic/pyrimidinergic (P2) receptors for nucleotides and adenosine or type-1 purinergic (P1) receptors (Burnstock & Knight, 2004). There are seven ionotropic (P2X1–7), at least eight metabotropic (P2Y1,2,4,6,11–14) and four adenosine receptor subtypes (A1, A2A, A2B, A3) that have been identified and characterized to date (Burnstock, 2002). In short, P2X and P2Y11 receptors are chiefly activated by extracellular ATP; P2Y2 by ATP and UTP; P2Y1 and P2Y13 by ADP; P2Y4 by UTP; P2Y6 by UDP; and P2Y14 by UDP-glucose.
The final regulatory component of purinergic systems comprises ectonucleotidases (Robson et al., 2001, 2006) that are the focus of this review. Such ectoenzymes hydrolyze extracellular nucleotides to generate other nucleotides and nucleosides that in turn differentially activate other P2 and ultimately adenosine receptors with often opposing effects to those seen with the initial P2-mediated effects (Beldi et al., 2008a). As an alternative mechanism, these enzymes can also act indirectly by limiting nucleotide (i.e., substrate) availability for other cell surface enzymes, which act on purinergic receptors, thereby indirectly modulating P2 receptor activities (Malavasi et al., 2008).
Within the past decade, ectonucleotidases belonging to several enzyme families have been discovered, cloned, and functionally characterized. In this review, we provide a brief overview of the vascular and immune ectonucleotidases and then focus on CD39, CD73, and CD38 to dissect out their pathophysiological roles (Deaglio & Malavasi, 2006; Di Virgilio et al., 2009; Dwyer et al., 2007; Malavasi et al., 2008; Sitkovsky et al., 2008).
CD39 is the prototype of the ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase) family (EC 3.6.1.5). These proteins comprise a group of ectoenzymes that hydrolyze extracellular nucleoside tri- and diphosphates. The ecto-nucleotidase chain or cascade, as initiated by NTPDases, is terminated by ecto-5′-nucleotidase (CD73; EC 3.1.3.5; Resta et al., 1998; Robson et al., 2006).
Together, ecto-5′-nucleotidase and adenosine deaminase (ADA; EC 3.5.4.4), another ectoenzyme that is involved in purine salvage pathways by converting adenosine to inosine, closely regulate local and pericellular extracellular concentrations of adenosine (Robson et al., 2006).
Most notably, however, in many tissues and cells, there are complex cell surface-located nucleotide hydrolyzing and interconverting machinery (Fig. 1). These mutiple ensembles include the ecto-nucleotide pyrophosphatase phosphodiesterases (E-NPPs; EC 3.1.4.1, EC 3.6.1.9), CD38, NAD-glycohydrolases, alkaline and acid phosphatases, diadenosine polyphosphate hydrolases, adenylate kinases, nucleoside diphosphate kinase, and potentially ecto-F1-Fo ATP synthases (Robson et al., 2006).
FIGURE 1.
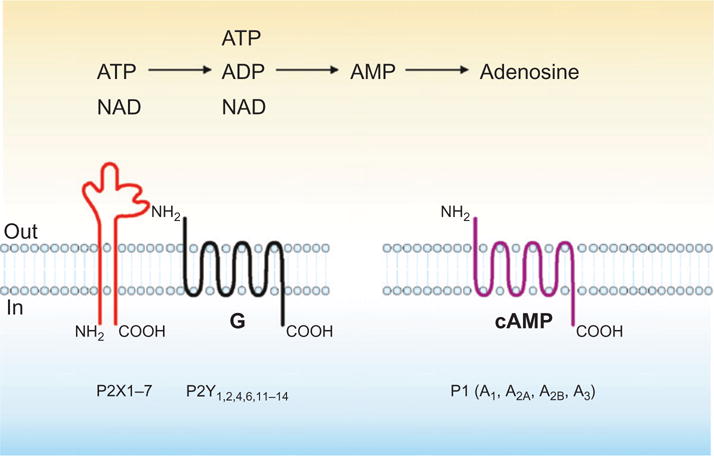
Purinergic mediators that are metabolized by CD39, CD73, and CD38 and respective receptors. Schematic depiction of extracellular nucleotides, for example, ATP, ADP, and NAD with derived adenosine nucleotide products as ligands for the corresponding purinergic receptors—see text for details of NAD catalysis by CD38 and impacts on purinergic signaling.
The CD38/CD157 gene family encodes two extracellular enzymes characterized by a widespread and often complementary cell distribution (Malavasi et al., 2008). Notwithstanding a high structural homology in the extracellular domain, they differ in membrane anchorage in that CD38 is a type II molecule, while CD157 is GPI anchored. These enzymes account for >95% of all NADase activities in mammals and were originally defined as cell surface activation molecules of leukocytes; they became classified as enzymes only later as a consequence of the identification of a sequence similarity between the human lymphocyte antigen CD38 (and later CD157) and the Aplysia ADP ribosyl cyclase (States et al., 1992). The catabolism of NAD+ and NAD(P) mediated by CD38 leads to the generation of potent intracellular Ca2+-mobilizing compounds, including cyclic ADP ribose (cADPR) and ADP-ribose (ADPR); CD38 activity may be also impacted by extracellular ATP (Malavasi et al., 2008). As predicted by the structural homology, soluble CD157 incubated with NAD+ produces cADPR and subsequently ADPR, indicating that this molecule is endowed with both ADPR cyclase and cADPR hydrolase activities. However, the catalytic efficiency of CD157 is several 100-folds lower than that of CD38 (Malavasi et al., 2008).
In addition to binding the TRPM2 membrane Ca2+ channels, ADPR, the main product of the reaction, can be covalently attached to proteins by ADP ribosyl transferases (ARTs). This posttranslational modification of target proteins can have dramatic effects on their functions. Thus, the CD38/CD157 family can modulate purinergic signaling by limiting NAD+ availability to P2Y receptors, primarily P2Y1 and P2Y11 (Malavasi et al., 2008; Moreschi et al., 2008). Alternatively, it is feasible that the enzymatic activity of CD38 may generate products that modulate purinergic signaling responses and also directly impacts on the activity of ARTs, affecting the amount of ADP ribosylated P2X7 (see Fig. 1).
To prevent redundancy and repetition in this issue, extracellular elements of nucleotide interconversion will not be addressed here. The interested reader is referred to Yegutkin et al. (2001, 2003, 2006) and to Chapter 8.
IV. Purinergic Signaling Responses in the Vasculature and Immune Systems
In the blood, extracellular nucleotides (e.g., ATP, ADP, UTP, UDP, and NAD+) are released by leukocytes, lymphocytes, endothelium, and platelets by constituent mechanisms and in an enhanced manner with cell activation inclusive of degranulation and cell death. It is important to note here that many processes of arterial vascular injury are associated with the release of adenine nucleotides that exert a variety of inflammatory effects on the endothelium, platelets, and leukocytes (reviewed in Dubyak & Elmoatassim, 1993; Luthje, 1989). These events result in extracellular signals that are modulated by environmental factors (Luthje, 1989).
As alluded to above, these mediators bind the multiple P2Y and P2X receptors on platelets, endothelium, vascular smooth muscle cells, leukocytes, and immunocytes (Abbracchio & Burnstock, 1994). P2 receptors trigger and mediate short-term (acute) processes affecting cellular metabolism, nitric oxide (NO) release, adhesion, activation, and migration (Dubyak & Elmoatassim, 1993; Luthje, 1989). As an example in the vasculature, ATP released from endothelium during changes in flow (shear stress) or following exposure to hypoxic conditions activates P2Y receptors expressed by these cells and by vascular smooth muscle cells in an autocrine and paracrine manner to release NO, resulting in vessel relaxation as a purinergic event.
The intracellular NAD+ concentration is in the range of 1 mM, whereas the plasma concentration is in the range of 0.1 μM. The latter concentration is below the Km of ARTs (Krebs et al., 2003). It is, however, feasible that high amounts of ecto-NAD+ can be released during tissue injury as a consequence of cell lysis. Further, NAD+ also seems to be released by nonlytic processes under various physiological conditions including hypoxia, inflammation, and mechanical or chemical activation (Bruzzone et al., 2001). Connexin 43 hemichannels, for instance, mediate transmembrane NAD+ fluxes in intact cells (Bruzzone et al., 2001). Hence, high local NAD+ concentrations may be reached under various physiological and pathophysiological situations, which would permit ADP ribosylation of membrane proteins on ART-expressing neighboring cells. The fate of such generated NAD+ is controlled by ecto-NADases, and cellular cross-talk that includes CD38 and select forms of ART, such as ART2.2 involved in immune regulation, has been demonstrated (Adriouch et al., 2008; Haag et al., 2007). Later, responses include those more protracted developmental responses, inclusive of cell proliferation, differentiation, and apoptosis (Burnstock & Knight, 2004; Harden et al., 1997; Weisman et al., 1998).
The dominant ectoenzymes or ectonucleotidases of the vasculature and lymphatics are now more fully characterized as E-NTPDases or E-NTPDases of the CD39 family, CD73/ecto-5′-nucleotidase, and potentially CD157. These important biological properties expressed by the endothelium, and associated cells, are responsible for the regulation of extracellular levels of nucleotides (Kaczmarek et al., 1996; Marcus et al., 1991, 2003; Robson et al., 1997, 2005).
NTPDases, in particular, might be able to serve as molecular switches at the membrane with differing functions according to the context of the signal. Colocalization of NTPDases with P2 and adenosine receptors has been demonstrated by fluorescence resonance energy transfer (FRET) techniques (Schicker et al., 2009); such structural aspects likely correlate with functional integration of responses and may confer added specificity. Such complexities must have been dictated by the ubiquitous nature of the signaling molecules, for example, ATP as mentioned above. However, extracellular nucleotide signaling should still be contrasted with the unique specificity of peptide hormones or vasoactive factors for often single, defined receptors (Goding & Howard, 1998; Sasamura et al., 1994).
Because of its high affinity for NAD+ and its efficient hydrolase activity, CD38 represents the main extracellular NAD+-metabolizing enzyme. When coexpressed with the ARTs, it competes for the substrate and limits their functional activities (Malavasi et al., 2008). This balance directly affects the amount of ADP-ribosylated P2X7 on arginine 125, a process mediated by ART2.2 (Adriouch et al., 2008), which catalyzes the covalent transfer of the ADPR group from NAD+ onto arginine residues of membrane target proteins. Consequently, NAD+ released during inflammation participates in the regulation of T cell homeostasis in vivo through an ART2.2-dependent P2X7-mediated mechanism involving ADP ribosylation (Adriouch et al., 2008; Haag et al., 2007), a process, which is now referred to as NAD-induced cell death (NICD; Grahnert et al., 2010; Hubert et al., 2010).
Besides modulating extracellular NAD+ levels, CD38/CD157 also catalyze the production of Ca2+-regulated metabolites, including cADPR, ADPR, etc. These metabolites bind intracellular receptors and channels that lead to a transient increase in cytosolic Ca2+ concentrations (Malavasi et al., 2008). This activity is believed to potentiate the functions of chemokine receptors, as well as of antigen receptors in B and T lymphocytes, as observed by studying CD38 knockout mice and human disease models (Deaglio & Malavasi, 2006; Deaglio et al., 2001; Morabito et al., 2006).
Many aspects of these ectonucleotidase families and detailed expositions of structure/function relationships have been previously reviewed by us (Beldi et al., 2008a; Deaglio et al., 2006; Di Virgilio et al., 2009; Malavasi et al., 2008; Robson et al., 2006). These topics are fully addressed elsewhere in this issue and are not discussed further here (see Chapter 9).
V. Hemostasis and Thrombosis
A. Pathophysiology
Hemostasis must remain inactive under basal conditions but ready to immediately close off defects, shut off blood loss, and thereby minimize tissue injury. The pathophysiology of coagulation and platelet-activating mechanisms in hemostasis is integral to an understanding of thrombosis and inflammation in the vasculature.
The normal vascular endothelium provides a barrier that separates blood cells and plasma factors from highly reactive elements of the deeper layer of vessel wall and maintains blood fluidity and flow by inhibiting coagulation, platelet activation, and promoting fibrinolysis (Ross, 1995). These properties are governed by important thromboregulatory mechanisms that include the release of prostacyclin (Sinzinger et al., 1991), the generation of NO (Cooke & Dzau, 1997), and heparan sulfate expression (Ihrcke et al., 1993). Other key biological activities of the vasculature include ecto-nucleotide catalysis that generates nucleosides by phosphohydrolysis of the respective nucleotides (Robson et al., 1997b, 2001).
The initiating event of plasma coagulation is the exposure of abluminal or circulating tissue factor (TF) within microparticles to circulating Factor VII. Thus, any disruption in the endothelial barrier between these TF-expressing cells and circulating blood is an initiating event in plasma coagulation. The TF–Factor VIIa complex initiates a chain reaction by activating other zymogen coagulation factors in the blood and initiating feedback loops to enhance clotting activity. TF–Factor VIIa activates two zymogens: Factor IX and Factor X. Factor IX (complexed with Factor VIII which is in turn stabilized by von Willebrand factor (vWF)) serves to activate more Factor X. Activated Factor X in complex with Factor V activates thrombin (Factor II), the major protease responsible for cleavage of fibrinogen to form fibrin. Finally, thrombin accentuates the clotting process with positive feedback loops activating more Factor V, Factor VIII, and Factor XI (increases activated Factor IX) and Factor XIII, which covalently links fibrin molecules in a transglutaminase reaction to form an insoluble mesh. This cascade of activated zymogens and positive feedback loops propagates fibrin formation to impede local hemorrhage. The interested reader is referred to an excellent recent review of this topic by Furie and Furie (2008).
The hemostatic process is also initiated by damage to the endothelium with exposure of circulating platelets to subendothelial surfaces and any associated serine proteases of the coagulation cascade. There appear to be two separate and independent pathways for platelet activation: these involve vascular collagen-dependent activation and thrombin-dependent pathways. Vascular injury exposes circulating platelets to collagen-bound vWF, which binds via the glycoprotein receptor GP1bα and integrins such as GPIIbIIIa (which can also interact with fibrin(ogen)). Thrombin cleaves protease-activated receptors (PAR1 in humans) to initiate platelet activation. This process, in turn, activates other platelets by release of serotonin, thromboxane A2, and nucleotides, which operate in auto- and paracrine manners to amplify platelet activation (Furie & Furie, 2008).
Simultaneous activation of the coagulation cascade and platelets is thought to synergize to propagate thrombus formation (Furie & Furie, 2005). An alternative mechanism is that a form of TF that is initially inactive (or “encrypted”) can be derived from cell-derived circulating microparticles and activated isomerization of a mixed disulfide and a free thiol to an intramolecular disulfide at sites of thrombus formation (Reinhardt et al., 2008). Thrombogenesis is blocked when the extracellular protein disulfide isomerase is inhibited (Reinhardt et al., 2008), perhaps preventing the activation of critical functions in platelet receptors and TF (Cho et al., 2008; Furie, 2009). The hemostatic process can be therefore further facilitated by expression of functionally active TF from cells and/or that expressed by cell-derived microparticles. It is of interest that such thrombus formation promoted by circulating microparticles can develop without implicating direct endothelial damage (Furie, 2009; Furie & Furie, 2008)
B. Impact of Extracellular Nucleotides, Nucleosides, and Ectonucleotidases on Hemostasis and Thrombosis
Over the past decade, extracellular nucleotides have been recognized as important mediators of a variety of processes including vascular inflammation and thrombosis with varying impacts in different systems (Robson et al., 2001). These cellular processes and nucleotide-triggered events are further modulated during angiogenesis and influence the development of atherosclerosis and restenosis following angioplasty (Burnstock & Knight, 2004; Goepfert et al., 2000, 2001; Wang et al., 2003; Wihlborg et al., 2004)
Extracellular nucleotides are continuously released from cells associated with the blood, for example, following the exocytosis of ATP/UTP-containing vesicles, facilitated diffusion by putative ABC transporters or by poorly understood electrodiffusional movements through ATP/nucleotide channels. It has been shown that rates of increase are higher in injured or stressed cells (Abraham et al., 1993; Franceschi et al., 1996; Grierson & Meldolesi, 1995; Luthje, 1989; Tsujimoto, 1997).
Several mechanisms account for the presence of nucleotides or nucleosides in plasma (Traut, 1994). As alluded to above, these include aggregating platelets, degranulating macrophages, excitatory neurons, injured cells, and cells undergoing mechanical or oxidative stress resulting in lysis, selective permeabilization of cellular membranes, and exocytosis of secretory vesicles, such as from platelet dense bodies (Fijnheer et al., 1992; Luthje, 1989). It is important to note here that many processes of arterial vascular injury are associated with the release of adenine nucleotides that exert a variety of inflammatory effects on endothelium, platelets, and leukocytes (reviewed in Dubyak & Elmoatassim, 1993; Luthje, 1989).
Adenosine is recognized as a bioactive agent in vascular inflammatory states, with effects mediated on both vascular cells and leukocytes (Ogura et al., 2006). In addition, adenosine has known antithrombotic effects by blocking induction of TF (Deguchi et al., 1998) via A2A and A3 receptors, particularly during ischemic or atherosclerotic processes, modulates the expression of antiapoptotic genes, and is immunosuppressive (Sitkovsky et al., 2004). Adenosine is constitutively present in the extracellular space at low concentrations, but under metabolically stressful and hypoxic conditions, the levels rise dramatically (Sitkovsky & Lukashev, 2005). Primary release of the mediator could occur ab initio, or this might follow conversion of released nucleotides to adenosine via ectonucleotidases.
The important function of ectonucleotidases in the vasculature is the modulation of P2-receptor-mediated signaling by the removal of extracellular ATP and ADP and related nucleotides. The ultimate generation of extracellular adenosine will abrogate and terminate nucleotide-mediated effects but will also activate adenosine receptors, with often opposing pathophysiological effects. Ectonucleotidases also produce the key molecules for purine salvage and consequent replenishment of ATP stores within multiple cell types. Indeed, although nucleotides appear not to be taken up by cells, their dephosphorylated nucleoside derivatives interact with several specific transporters to enable membrane passage. The regulated dephosphorylation of extracellular nucleotides by ectonucleotidases may be critical for appropriate purinergic/pyrimidinergic signaling and metabolic homeostasis (Enjyoji et al., 1999; Plesner, 1995; Robson et al., 2001, 2005).
The endothelial membrane expressed CD39/NTPDase-1 is the major ecto-nucleotidase in the vasculature (Enjyoji et al., 1999). Other NTPDases associated with the vasculature are the cell-associated ecto-ATPases (CD39L1 or NTPDase-2) and a soluble ecto-ADPases (CD39L2 or NTPDase-6; akin to the monocyte expressed CD39L4 or NTPDase-5; Chadwick & Frischauf, 1998; Mulero et al., 1999; Zimmermann, 1999).
The ectoenzyme CD39/NTPDase1 can be shown to efficiently bind and hydrolyze extracellular ADP (and ATP) to AMP. This phosphohydrolytic reaction limits the platelet activation response that is dependent upon the paracrine release of ADP and activation of specific purinergic receptors (Marcus et al., 1991; Robson et al., 1997b, 2000). CD39L1/NTPDase2, a preferential nucleoside triphosphatase, activates platelets by converting the competitive antagonist (ATP) of platelet ADP receptors to the specific agonist of the P2Y1 and P2Y12 receptors.
In keeping with these biochemical properties, endothelial cells and vascular smooth muscle chiefly express CD39, where it serves as a thromboregulatory factor. In contrast, CD39L1 is associated with the adventitial surfaces of the muscularized vessels, microvascular pericytes, and the stromal cells and would potentially serve as a hemostatic factor (Sevigny et al., 2002).
The ectonucleotidase chain initiated by NTPDases is terminated by ecto-5′-nucleotidase (EC 3.1.3.5; Zimmermann, 1992). The ecto-5′-nucleotidase (CD73) is a glycoprotein member of the family of 5′-nucleotidases that operates in tandem with CD39 in the vasculature. Together, CD73 and ADA (EC 3.5.4.4), another ectoenzyme involved in purine salvage pathways that degrades adenosine to inosine, closely regulate local and pericellular extracellular and plasma concentrations of adenosine (Goding & Howard, 1998; Resta et al., 1998). CD73 exhibits high specificity toward nucleoside monophosphates and is inhibited by nucleoside diphosphates in a manner described as “feed-forward inhibition” that has the effect of promoting a platelet plug and limiting adenosine generation (Gordon et al., 1986) to enhance the process of thrombogenesis. This mechanism also explains, at least in part, why Cd39 deletion might have such pronounced effects on adenosine production and is the dominant functional ectonucleotidase within the vasculature (Gordon et al., 1986; Meghji et al., 1995).
The CD38 family of ectoenzymes is involved in recycling of extracellular nucleotides by metabolizing NAD through the generation of cADPR and ADPR (Howard et al., 1993). The surface expression levels of CD38 are tightly regulated in vivo in response to inflammatory signals and may also impact vascular responsiveness in this process (Malavasi et al., 1994). Specifically, a clear role for CD38 in the interactions taking place between circulating lymphocytes and the vessel wall has been recognized several years ago (Deaglio et al., 1996). The latter finding was the starting point for the search of an endothelial cell-bound ligand, which culminated in the identification of CD31 (platelet endothelial cell adhesion molecule) as a counter receptor for CD38 (Deaglio et al., 1998).
It is plausible that CD31 binding can affect the enzymatic activities of CD38, by influencing the dimerization of the molecule and the opening of the catalytic site (Liu et al., 2005). CD38/CD31 signals have been studied in humans in several models, by mimicking in vitro the conditions that might occur in vivo. The results indicate that this interaction is the starting point of a cascade of events that profoundly affects the gene signature of the cell. The modulated pathways are chiefly those leading to proliferation and chemotaxis (Deaglio & Malavasi, 2006; Deaglio et al., 2001; Morabito et al., 2006).
The CD38 paradigm is also representative of a more general trend involving several nucleotide-metabolizing enzymes (Deaglio et al., 2001). CD38 and CD157 may also serve as receptors, controlling intracellular signaling pathways apparently independently of the ectoenzymatic activity. The two functional activities might represent two separate evolutionary developments resulting in the final multifunctional molecule. Phylogenetic studies support the view that the enzymatic activity is the “older” conserved function, whereas at a later point, CD38 may have acquired membrane anchorage and the ability to function as a receptor (Ferrero & Malavasi, 1999).
These common traits also involve localization in membrane lipid microdomains, and the physical and functional association with partners specialized in signal transduction (Deaglio et al., 2002; Koziak et al., 2000; Pacheco et al., 2005).
C. Purinergic Events in Platelet Thrombus Formation
In vivo, activated platelets appear to contribute to thrombin generation through the exposure of phosphatidylserine, forming a procoagulant catalytic surface, and through platelet–leukocyte–microparticle interactions that result in exposure of TF to blood, thereby promoting clot formation (Falati et al., 2002, 2003). Platelet activation and integrin ligation in response to multiple agonists are known to be dependent upon the release of extracellular nucleotides and therefore regulated by specific antagonists of the P2Y1, P2Y12, and P2X1 receptors (Baurand et al., 2000; Hechler et al., 2003; von Kugelgen & Wetter, 2000).
How platelets and the coagulation systems coordinate the process of thrombogenesis can be directly visualized by in vivo confocal and widefield videomicroscopy visualizing platelet deposition, microparticle accumulation, and fibrin generation after direct, oxidant, or laser-induced vascular injury. Recent work has kinetically plotted the formation of platelet arterial thrombi in the living mouse. This pattern is characterized by rapid accumulation of platelets (accumulation phase), followed by a peak at about 100 s followed by a 50–75% decrease in platelet mass leading to the plateau or stabilization phase (Chou et al., 2004; Falati et al., 2002, 2003), for reasons that are not understood.
Intuitively, one would think that a thrombus would grow indefinitely as platelets accumulate, release ADP, and activate more platelets. But clearly this is not the case as the visualized thrombus decreases in size as bound platelets deaggregate and return to the circulation (Chou et al., 2004).
Plasma microparticles (<1.5 μm) originate from platelet and cell membrane lipid rafts and possibly regulate inflammatory responses and thrombogenesis (Falati et al., 2003; Furie, 2009; Furie & Furie, 2005). These actions are mediated through their phospholipid-rich surfaces and associated cell-derived surface molecules (Falati et al., 2003). Constitutively circulating microparticles are also associated with functional CD39, and accumulation of these at sites of vascular injury appears to influence local thrombus formation and evolution (Banz et al., 2008).
Purified, unstimulated platelets lack or have minimal CD39 functional activity but released CD39 present on microparticles may be a key modulatory influence of thrombosis. In this regard, monocyte-derived microparticles can bind to activated platelets in the developing thrombus in an interaction mediated by platelet P-selectin and microparticle P-selectin glycoprotein ligand 1(PSGL-1; Falati et al., 2002, 2003). We might infer that as the platelet thrombus forms, in parallel, microparticles accumulate in the growing aggregate, which deliver the associated membrane CD39. Hence, CD39, and associated NTPDase activity, is not initially a substantive part of the thrombus but only accumulates after platelet thrombus formation matures. The spatial and temporal expression of NTPDases in the accumulating microparticles may therefore regulate thrombus size by regulating the hydrolysis and hence inactivation of the platelet agonist ADP, ultimately resulting in the observed deaggregation responses (Atkinson et al., 2006).
There is, however, a complicating variable. Although CD39 is a key determinant of extracellular nucleotide fluxes in the vasculature, genetic deletion paradoxically results in disordered hemostasis, secondary to platelet dysfunction. When studied in the CD39-mutant mouse, platelet thrombi were limited after oxidant injury in vivo and purified Cd39-null platelets failed to aggregate to standard agonists in vitro. This pattern of platelet hypofunction was wholly reversible and associated with purinergic type P2Y1 receptor desensitization and membrane integrin dysfunction (Enjyoji et al., 1999).
Cd39 deletion in mice produces a proinflammatory phenotype associated with quantitative and qualitative differences in the microparticle populations, as determined by 2D-gel, Western blot, and flow cytometry. Cd39-null microparticles are also more abundant, have significantly higher proportions of platelet- and endothelial-derived elements and decreased levels of interleukin (IL)-10, tumor necrosis factor receptor-I (TNF-RI), and matrix metalloprotease (MMP-2). Consequently, Cd39-null microparticles boost endothelial activation, as determined by inflammatory cytokine release and upregulation of adhesion molecules in vitro. These findings by Banz and our colleagues suggest a modulatory role for CD39 within microparticles in the exchange of regulatory signals between vascular cells (Banz et al., 2008).
VI. Inflammation
A. Platelets and Leukocytes
Inflammation potentiates thrombosis by both downregulating vascular natural anticoagulant mechanisms and provoking disordered purinergic mechanisms, as alluded to above. Inflammation and thrombosis are closely integrated by close interactions between platelets, circulating leukocytes, endothelial, and other vascular cells.
Platelets are a key element at the interface between thrombosis and inflammation. Activated platelets release extracellular nucleotides and soluble factors that may have both local and systemic effects on blood and vascular cells that compound inflammatory responses.
Polymorphonuclear neutrophils (PMN) are the cardinal cellular component of an acute inflammatory response, and purinergic signaling dictates their functions. Specifically, ATP appears to be released from the leading edge of neutrophils in response to stimulation by chemoattractants, such as the peptide N-formyl-met-leu-phe (fMLP). ATP released by cells among which are PMN activates P2Y2 receptors and consequently forms adenosine that via A3 receptors stimulates neutrophil movement (Chen et al., 2006). CD39 is the critical ectonucleotidase dictating hydrolysis of released ATP and directing cell migration by PMN. Upon stimulation of human PMN or differentiated HL-60 cells in a chemotactic gradient, CD39 tightly associates with the leading edge of polarized cells during their migration in a chemotactic gradient. Inhibition or genetic deletion of CD39 reduces the migration speed of PMN but not the chemotactic ability both in vitro and in vivo (Corriden et al., 2008).
Further, CD39 is the dominant ectonucleotidase expressed by monocyte-macrophages. Upregulation of TF expression by monocytes and major defects in their entry and migration into the substance of Matrigel plugs injected into the subcutaneous tissue of Cd39-null mice have been observed. In parallel, we also evaluated parameters of monocyte transendothelial migration influenced by ATP in vitro and noted failure of Cd39-null cells to migrate in response to exogenous nucleotides. This defect could be overcome by costimulation with serotonin, suggesting a degree of P2Y-receptor desensitization in Cd39-null monocytes, as previously noted in platelet functions (Goepfert et al., 2001; Robson et al., 2001). Ectoenzymes, specifically ectonucleotidases, play a key role in leukocyte trafficking but this complex area will not be dealt with here (for an excellent review on this topic, see Salmi & Jalkanen, 2005).
B. Inflammasome
This recently coined term refers to those multiprotein complexes comprising caspase family members that assemble in the cytoplasm following injury to cells, or exposure to “danger signals.” Activated caspase-1 within the complex cleaves pro-IL-1, and others, leading to cytokine activation and secretion (Latz, 2010). ATP released from damaged cells may serve as a “danger signal” and binds P2X7 which is known to activate the NLRP3/ASC/caspase-1 inflammasome boosting release of active IL-1 (reviewed in Mariathasan et al., 2006; Ogura et al., 2006; and more recently in Aymeric et al., 2010; Stagg & Smyth, 2010).
The autocrine/paracrine release of ATP that generates the inflammasome activation via P2X7 receptor activation can be impacted upon by CD39 in endothelium or by myeloid cells. Crucially, overexpression of CD39 in endothelium efficiently abrogated initial phases of ATP secretion in response to lipopolysaccharide endotoxin and markedly inhibited IL-1 alpha release; comparable results were obtained with soluble NTPDase. These earlier data suggested that CD39 could modulate IL-1 release from endothelium (Imai et al., 2000) and might also impact the inflammasome.
In a more recent study, CD39 was shown to be the dominant ectonucleotidase expressed by murine peritoneal macrophages and to also regulate P2X7-dependent IL-1 secretion responses in these cells (Levesque et al., 2010).
For further details with respect to the impact of ectonucleotidases on inflammation and cytokine elaboration, please refer to Chapter 9.
C. Plasticity of Purinergic Responses in Inflammation
Inflammatory and vascular cells exhibit a certain degree of plasticity to respond to stress, as do adaptive input–output devices. The putative input arises from the extracellular milieu and includes biochemical signals triggered by extracellular nucleotides, and the resulting biomechanical responses are transduced by adhesion receptors in response to P2-mediated signals, such as the affinity changes in integrins and cell adhesion responses. The output is further manifested as alterations in cellular phenotype, such as with activation responses, and includes a number of structural and functional changes implicated in diverse pathophysiological processes.
As an example, acute inflammation and associated oxidative stress, for example, as in graft ischemia–reperfusion injury, have an immediate and direct impact on extracellular nucleotide metabolism secondary to substantive and immediate decreases in the biological activity of CD39. Palmitoylation status of CD39 and cholesterol content of membrane lipid rafts appear to impact changes on NTPDase activity under such episodes of stress. Loss of NTPDase activity can be ameliorated by statins (Kaneider et al., 2002) or by incorporation of saturated fatty acids into cell membranes and limitation of lipid peroxidation responses by antioxidants (Kaczmarek et al., 1996; Robson et al., 1997a).
Recent work has suggested that upregulation of CD39 can be influenced by the select transcription factors CREB (Liao et al., 2010), Sp1, and hypoxia-inducible factor-1 (HIF-1; Eltzschig et al., 2009). CD39 and CD73 as well as certain adenosine receptors are clearly upregulated in the setting of hypoxia (Eltzschig et al., 2003, 2004; Synnestvedt et al., 2002). Downregulation of adenosine transporter expression under such conditions further decreases adenosine uptake (Morote-Garcia et al., 2009). Such hypoxia–adenosinergic mechanisms boost production of extracellular adenosine and receptor signaling. These pathways have different effects in select experimental models when examining leukocyte and lymphocyte infiltration into tissues or peritoneum (Corriden et al., 2008) versus altering egress of cells across epithelial barriers, for example, as in the lung or gut (Eltzschig et al., 2003, 2004; Reutershan et al., 2009).
CD38 expression can be likewise readily modulated, following activation of B and T cells as well as in the context of contacts between lymphocytes and stromal cells (Deaglio et al., 2006). The gene includes a very long first intron that contains regulatory elements for vitamins (A and D) and for interleukins (interferons and IL-6), among others (Malavasi et al., 2008). The presence of a large CpG island suggests epigenetic regulation, as very recently demonstrated in the chronic lymphocytic leukemia (CLL) model. Lastly, CD38 has a well-characterized single-nucleotide polymorphism (SNP) located at the 5′ end of this intron (184 C→G), which leads to the presence (or absence) of a PvuII restriction site. This SNP is located within an E-box and conditions binding of the E2A transcription factor in human B lymphocytes (Saborit-Villarroya et al., 2011).
VII. Immunity
High levels of ATP may be released by CD4+ and CD8+ T cells upon mitogenic or antigenic/TCR stimulation and serve to amplify activation of cells (la Sala et al., 2003). Acute P2 receptor-mediated stimulation of monocytes, lymphocytes, and endothelium causes largely proinflammatory responses, such as the release of IL-1 (or IL-8), as described above (Imai et al., 2000; la Sala et al., 2003; Warny et al., 2001). On DCs, exposure to extracellular ATP induces migration and differentiation to drive cellular immune responses (la Sala et al., 2001).
Multiple P2X and P2Y receptor subtypes are expressed by monocytes and DCs, whereas lymphocytes express only P2Y receptors (Burnstock & Knight, 2004). These various receptors operate in both auto- and paracrine loops and are considered to play a complex, important role in the regulation of vascular and immune cell-mediated responses. We will focus on how CD39 and CD38 impact these functions.
A. CD39
CD39 was first described as a B lymphocyte activation marker (Maliszewski et al., 1994) and has been also shown to be expressed on natural killer (NK) cells, monocytes, DC, and subsets of activated T cells (Koziak et al., 1999). The relevance of the expression of CD39 by these cells is still being explored but there have been several recent advances in understanding brought about, at least in part, by study of mutant mice.
Langerhans DCs have the capacity to recruit, activate, and polarize naive T cells and express high levels of CD39 (Mizumoto et al., 2002). Cd39-null DCs are unresponsive to ATP albeit these are susceptible to cell death, but only after prolonged exposure to nucleotides (Mizumoto et al., 2002). Mutant mice null for Cd39 have amplified inflammatory responses to irritant chemicals as a consequence of the lack of CD39 suppressive properties. However, there are major defects in DC formation, antigen presentation, and T cell responses to haptens in Cd39-null mice. These result in markedly attenuated responses to contact allergens in type IV hypersensitivity cutaneous responses that are also seen in IBD models following haptenic stimulation (Kunzli et al., 2010). These data suggest that Cd39 expression is required for optimal stimulation of hapten-reactive T cells in mice (Mizumoto et al., 2002). Cd39-null DCs are fully functional with respect to homing and phenotypic maturation but are less able to stimulate T cells. These immune findings are relevant to allograft rejection processes (Robson et al., 2001). Sequelae of these putative immune abnormalities include the relative failure of Cd39-null mice to reject allografts under limited costimulation blockade (Li et al., 2003). These data indicate a previously unrecognized role of CD39 and the effects on nucleotide-mediated signaling in immunological responses (Mizumoto et al., 2002) that has been extended in lymphocyte studies.
We have recently shown that absence of CD39 and consequent changes in P2 receptor signaling paradoxically limit interferon gamma (IFNγ) release by NK cells in inflammatory situations. Adoptive transfers of populations of mutant and wild-type immune cells into Rag2/common gamma null mice (deficient in T cells, B cells, and NK/NKT cells) suggest that CD39 deletion on NK cells provides end-organ protection in limited warm ischemia, decreasing tissue damage mediated by these innate immune cells (Beldi et al., 2010).
With Dr. Marilia Cascalho and colleagues, we have noted abnormalities in B cell memory responses to T-dependent antigens in Cd39-null mice with demonstrable abnormalities in postgerminal center terminal B cell differentiation (not shown here).
With Karen Dwyer, Wenda Gao, and colleagues, we have shown that CD39 and CD73 are surface markers of Treg cells (in the mouse) that impart a specific biochemical signature characterized by adenosine generation that has functional relevance for cellular immunoregulation mediated by effector T cell A2A stimulation (Deaglio et al., 2007). CD39, albeit not consistently with CD73, is associated with human CD4–Treg, as defined by high expression of Foxp3 and low levels of CD127 (Dwyer et al., 2010). Comparable work has been also published in human systems (Borsellino et al., 2007; Mandapathil et al., 2010).
In other collaborative work recently published by Dwyer et al., CD39 has been used to characterize blood T cell populations to allow tracking of these cells in health and disease (Dwyer et al., 2010). Differential expression of CD25 and CD39 on circulating CD4+ T cells differentiates between Treg and pathogenic cellular populations associated with secretion of inflammatory cytokines (IFNγ and IL-17). These latter cell populations are increased, with decreases in CD39-expressing Tregs, in patients with renal allograft rejection (Dwyer et al., 2010).
Lastly, work done by Guido Beldi and colleagues has shown an important NKT cell protective phenotype in Cd39-null mice following Concavalin A-induced immune hepatitis. These data indicate a role for modulated purinergic signaling to impact NKT-mediated mechanisms resulting in liver immune injury (Beldi et al., 2008b).
Nucleotides are potent inflammatory factors when suddenly released into the extracellular environment in high concentration, as with platelet degranulation. Hence, depending on the P2 or adenosine receptor subtype, the cell types, and signaling pathway involved, these receptors might preferentially trigger and mediate short-term (acute) processes that affect metabolism, adhesion, activation, or migration. Moreover, purinergic signaling also has profound impacts upon other more protracted reactions, including cell proliferation, differentiation, and apoptosis, such as seen in several chronic inflammatory states (Burnstock, 2002). These mechanisms could be also implicated in immune memory (Koshiba et al., 1997; Sitkovsky et al., 2004). Immune outcomes appear to differ somewhat when there are slow, gradual, and persistent increases in nucleotide fluxes where P2 receptor desensitization might occur. Under these conditions, purinergic effects deviate immune responses more toward nonresponsiveness (tolerance) rather than heightened reactivity (reviewed in Di Virgilio & Robson, 2009; Di Virgilio et al., 2009).
B. CD38
Expression of CD38 by activated T and B lymphocytes was the starting point for functional studies aimed at dissecting the role of the molecule in the immune compartment. The emerging picture is highly complex and mostly dependent on the lineage considered and the activation status of the cell. However, a few common trends emerge that are expanded upon below (reviewed in Malavasi et al., 2008).
It seems apparent that CD38 engagement by agonistic mAbs is followed by a signaling cascade typical of canonical receptors, including tyrosine phosphorylation of a sequential number of intracellular enzymes, nuclear events, and more long-term effects dependent on active protein synthesis. An increase of the cytoplasmic levels of calcium ions is also a common theme upon activation of CD38. The calcium wave is typically slow in rising as compared to the spikes obtained after signaling through the antigen receptors in T and B lymphocytes. Unlike the antigen receptors, a CD38-induced calcium wave may last several minutes before declining, indicating that the molecular mechanisms responsible for calcium mobilization might be different and possibly relying on the enzymatic activities of the molecule, which generate several Ca2+ active compounds.
It is still not clear what initiates the signal. A hypothesis is that this commences with provision of substrate, that is, NAD+, and might be further regulated through protein–protein interactions. In the human, these would include CD31, the only nonsubstrate ligand so far identified that might focus biochemical activity at vascular sites, as alluded to above. The structural requirements for signaling include localization in critical areas of the plasma membrane, in close physical proximity with more typical signaling receptors, such as the B cell receptor complex in B cells (Deaglio et al., 2006), CD16 in NK cells (Deaglio et al., 2002), and also inclusive of the T cell receptor in T lymphocytes, MHC Class II and CD9 in monocytes, and the CCR7 and CXCR4 chemokine receptors, CD83 and CD11b in mature DCs (reviewed in Deaglio & Malavasi, 2006; Malavasi et al., 2008).
The long-term events described as a consequence of the interaction of CD38 with the CD31 ligand, as mimicked by agonistic mAbs, vary according to cell lineage and differentiation status. Two common themes as to the regulation of cellular responses by CD38 are evident. The first is linked to the amplification of the signal mediated by the antigen receptors in T and B lymphocytes and by CD16 in NK cells. The second is associated with the amplification of chemotaxis signals mediated by different chemokine receptors. Both avenues have the potential to modulate immune responsiveness and inflammation in health and disease, as addressed next.
VIII. Disease Processes
A. Cancer
Inflammation and coagulation have important roles in the biology of cancer, and effects mediated by nucleotides and other mediators are often dualistic.
First, patients with cancer might exhibit thrombophilia with coagulation activated toward a prothrombotic state and heightened activation of inflammation. Second, such a procoagulant environment may promote growth and dissemination by several mechanisms involving fibrin deposition, angiogenesis, and platelet activation. Dysregulation of coagulation with heightened levels of inflammatory mediators, such as extracellular nucleotides in cancer, impacts clotting and vascular homeostasis to provoke venous thrombosis. These mediators also stimulate cell signaling in the extra vascular compartment, through interaction with cell receptors like PARs and P2 receptors, to drive tumor growth. Platelet activation and associations with leukocytes may further promote inflammatory reactions to tumors.
Proteases like thrombin may drive malignant cell proliferation and are opposed by anticoagulant and anti-inflammatory actions, inclusive of the protein C-thrombomodulin mechanism. Likewise, nucleotide-mediated effects are abrogated by ectonucleotidases that generate adenosine. In such situations, cancer may promote growth and protect against host responses by subverting protective effects of inflammatory responses.
Experimental approaches that promote ATP-mediated activation of immune responses and tumor cytotoxicity, or inhibit generation of extracellular adenosine have been tested. We have demonstrated that metastatic tumors from a melanoma cell line and other cancers were strongly inhibited in mice with Cd39-null vasculature as a consequence of disordered angiogenesis (Beldi et al., 2008c; Jackson et al., 2007). Tumors likewise were decreased in size in wild-type mice with circulating Cd39-null bone marrow-derived cells (Sun et al., 2010). Our models suggest that functional CD39 expression on CD4(+)Foxp3(+) Tregs suppressed antitumor immunity mediated by NK cells in vitro and in vivo. Inhibition of CD39 activity by polyoxometalate-1, a pharmacologic inhibitor of NTPDase activity, significantly inhibited tumor growth in the experimental system validated by us (Sun et al., 2010).
Several primary tumors contain CD39-expressing Treg. There is another intriguing possibility in that tumor cells express ectonucleotidases that may primarily generate high levels of extracellular adenosine; cancer cells may, therefore, blunt host responses while promoting tumor growth without any need for Treg recruitment (Buffon et al., 2007; Stagg & Smyth 2010; Stagg et al., 2010). To address these possibilities, we and others have proposed that pharmacologic or targeted inhibition of CD39 and/or CD73 enzymatic activity may find utility as an adjunct therapy for primary and secondary malignancies (Hilchey et al., 2009; Mandapathil et al., 2010; Sun et al., 2010; Zhang, 2010).
The role of CD38 in human tumor models is intimately linked to CLL, a relatively common leukemia of adults (Chiorazzi et al., 2005a, 2005b). The disease is invariably characterized by the expansion of a population of mature B cells expressing CD5 and accumulating in the blood and lymphoid organs. The clinical behavior is highly heterogeneous with patients who survive decades without any need for therapy, along with patients who need to be treated in the first 12 months after diagnosis, become rapidly resistant to therapy, and ultimately die because of the leukemia. CD38 is expressed by the aggressive variant of the disease and has become a widely used and trusted marker (Deaglio et al., 2006).
The current view is that CD38 is intimately linked to disease progression and that expression conditions tumor growth and expansion (Deaglio et al., 2010a, 2010b). This view is linked to the role of CD38 in collaborating with the B cell receptor complex in delivering proliferation signals to the neoplastic cell. More recently, CD38 has also been recognized as critical in determining chemotactic responses to the CXCL12 chemokine, by working in physical and functional association with the CXCR4 receptor (Vaisitti et al., 2010).
These overall observations suggest that CD38 is a potential therapeutic target in CLL. It is reasonable to surmise that CD38-blocking reagents of antibody origin or based on modified substrate ligands will perturb growth circuits of the neoplastic clone, probably rendering it more susceptible to conventional chemotherapy. Alternatively, nonsubstrate ligands can be used to block CD38 and consequent biochemical interactions with the surrounding deleterious receptors (Deaglio & Malavasi, 2006; Malavasi et al., 2008). Human antihuman-CD38 mAbs are already being tested in clinical trials, providing further support to this proposal.
B. Inflammatory Bowel Disease
IBD is an often devastating disease and is associated with excessive inflammation in the bowel and extra intestinal tissues in genetically susceptible individuals (Podolsky, 2002). Dynamic balances of Treg to Th17 cells in the gut, alterations in bacterial flora, and other environmental factors may regulate inflammatory responses both locally and systematically (Boden & Snapper, 2008; Paust & Cantor, 2005). Associations between IBD and thrombophilia have been recognized for decades and are of unclear etiology lacking a clear molecular basis (Yoshida & Granger, 2009).
As noted above, purinergic signaling pathways are involved in thrombosis and vascular inflammation (Robson et al., 2005), as well as immune suppressive cellular responses (Deaglio et al., 2007). Importantly, genetic polymorphisms of CD39 are linked to Crohn’s disease, and mice null for Cd39 exhibit severe colitis in several experimental models (Friedman et al., 2009; Kunzli et al., 2010). The purinergic linkages between inflammation and clotting listed above might also indicate, at least in part, why this disease has such a high incidence of venous thrombosis, blood clotting issues, and platelet activation (Yoshida & Granger, 2009).
It may seem relevant to ask here as to the functions CD39 could be serving when expressed by Treg and/or DCs that are relevant to IBD. A population of CD4+ lymphocytes noted to selectively express CD39 suggests a functional role for ectonucleotidases. Without such biochemical activity, Treg and vascular endothelium are not able to rapidly convert nucleotides into adenosine (Deaglio et al., 2007; Robson et al., 2005). Therefore, Cd39-null cells lack an endogenous “stop” or “brake” mechanism that might allow unfettered intestinal inflammation and correlate with the tendency of low expression of CD39 to be genetically associated with Crohn’s disease (Friedman et al., 2009). In a reciprocal manner, heightened levels of CD39 likely promote generation of adenosine to effectively block Th17 functions (Dwyer et al., 2010).
Recent work by Atarashi et al. (2008) indicates that commensal bacteria release ATP that specifically generates Th17 cells following the activation of a unique subset of lamina propria cells. Importantly, the administration of exogenous ATP driving purinergic responses exacerbates an adoptive T transfer colitis model and boosts Th17 differentiation (Atarashi et al., 2008).
C. Infections and Pathogens
Certain parasitic pathogens of humans express surface-located NTPDases that have been linked to virulence (chiefly protozoans and helminths). For those parasites that are purine auxotrophs, these enzymes may play an important role in purine scavenging, although they may also influence the host response to infection.
Although E-NTPDases are rare in bacteria, expression of a secreted NTPDase in Legionella pneumophila has been noted (Sansom et al., 2008a, 2008b). This ectoenzyme enhances intracellular growth of the bacterium and potentially affects virulence.
Staphylococcus aureus and other bacterial pathogens exploit the immunomodulatory attributes of adenosine to escape host immune responses (Thammavongsa et al., 2009). Purine metabolism and ectoenzymes (CD73) influence the host response to infection to Lyme borreliosis, a common arthropod-borne infection caused by Borrelia burgdorferi (Yegutkin et al., 2010).
Those interested in reading more on possible effects of microbial ectonucleotidases on host–pathogen interactions should examine the review (Sansom et al., 2008b).
IX. Conclusion
Extracellular nucleotide-mediated vascular endothelial and accessory cell stimulation have clear consequences for platelet activation, thrombogenesis, angiogenesis, vascular remodeling, and the metabolic milieu of the vasculature, in response to inflammatory stress and/or immune reactions. These mechanisms not only impact hemostasis but also dictate processes of vascular injury, thromboregulatory disturbances, and defective angiogenesis with vascular remodeling. This review has attempted to summarize those specific components of purinergic signaling as pertaining to vascular injury, inflammation, and immunity with a focus on the CD39 and CD38 family of ectonucleotidases (Fig. 1).
We have suggested that modulated, distinctive ectonucleotidase expression (by endothelium, leukocytes, and platelets (and potentially by derived microparticles; Banz et al., 2008)) regulates nucleotide-mediated signaling in the vasculature in a temporal and spatial manner with consequences for thrombogenesis and platelet activation. We have also addressed components of extracellular nucleotide-mediated signaling pathway, chiefly in T cells that are impacted upon largely by CD39, the prototypic member of the E-NTPDase family of ectonucleotidases and those effects on T cell homeostasis pertinent to CD38 (as shown in Fig. 2). Modulated, distinct NTPDase expression appears to regulate nucleotide- and nucleoside-mediated signaling within the vasculature and in the immune system. Hence, expression of ectonucleotidases on either endothelial or immune cells integrates vascular inflammatory and immune cell reactions at sites of injury.
FIGURE 2. Schematic representation of regulation of T regulatory cell homeostasis by extracellular nucleotides.
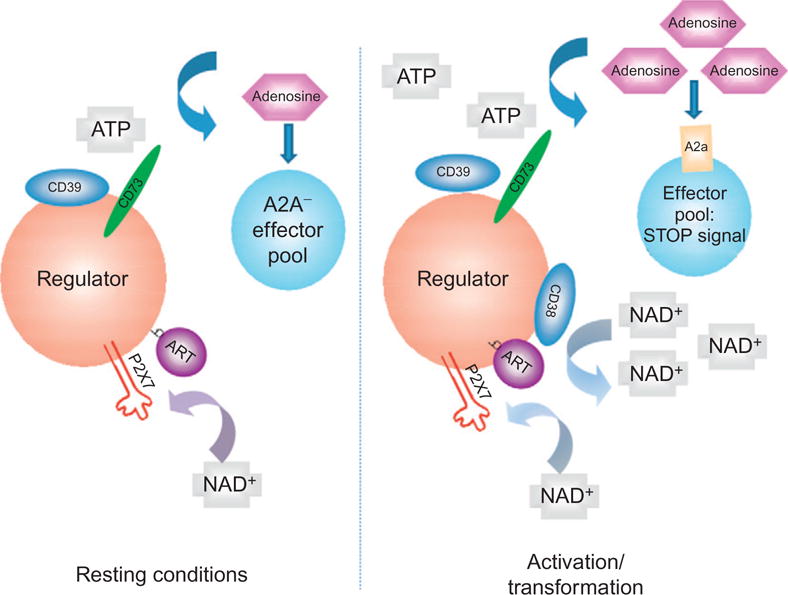
Extracellular nucleotide metabolism may be integrated into a homeostatic mechanism for the T cell compartment. Resting conditions are associated with limited ATP or NAD flux, low levels of CD39, CD38, and CD73. Basal mechanisms involve ADP ribosylation of P2X7, NICD, and maintenance of Treg compartment in quiescent state by low-level A2A stimulation (left panel). With acute inflammation and cellular stress, there are increases of pericellular nucleotides that induce cellular activation with resulting abrogation of select Treg responses (secondary to P2X7 activation). Those surviving cells exhibit marked upregulation of CD39, CD38, and CD73. Ultimately, NAD is converted and effectively removed by CD38, while ATP is subjected to hydrolysis by CD39 and CD73 (right panel). As these surviving immune suppressive cells are “protected” by ectonucleotidases and also have potent internal mechanisms to modulate cAMP, such cells, for example, Treg initially expand, and adenosine accumulates to then activate A2A receptors on T effector cells and NK cells (amongst others). This effect has the result of limiting effector cell expansion and blocking immune responses.
Increasing interest in this field will open up several new avenues for investigation. These developments might result in new treatment modalities for thrombotic disorders, vascular inflammation, and disordered immunity.
Acknowledgments
We apologize in advance if, because of space constraints, we have not adequately referenced the work of others and have cited review articles. We thank our colleagues and reviewers of chapter for their invaluable inputs. Support of NIH is recognized.
Abbreviations
- ART
ADP ribosyl transferase
- cADPR
cyclic ADP ribsose
- DC
dendritic cell
- EC
endothelial cell
- E-NTPDase
ecto-nucleoside triphosphate diphosphohydrolase
- IBD
inflammatory bowel disease
- IL
interleukin
- LC
Langerhans cells
- NAD
nicotinamide dinucleotide
- NICD
NAD-induced cell death
- NPP
nucleotide pyrophosphatase/phosphodiesterase
- TF
tissue factor
- Treg
T regulatory cells
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Abbracchio MP, Burnstock G. Purinoceptors—Are there families of P2X and P2Y purinoceptors. Pharmacology & Therapeutics. 1994;64(3):445–475. doi: 10.1016/0163-7258(94)00048-4. Review. [DOI] [PubMed] [Google Scholar]
- Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, et al. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(1):312–316. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriouch S, Bannas P, Schwarz N, Fliegert R, Guse AH, Seman M, et al. ADP-ribosylation at R125 gates the P2X7 ion channel by presenting a covalent ligand to its nucleotide binding site. The FASEB Journal. 2008;22(3):861–869. doi: 10.1096/fj.07-9294com. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- Atkinson B, Dwyer K, Enjyoji K, Robson SC. Ecto-nucleotidases of the CD39/NTPDase family modulate platelet activation and thrombus formation: Potential as therapeutic targets. Blood Cells, Molecules & Diseases. 2006;10:10. doi: 10.1016/j.bcmd.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Research. 2010;70(3):855–858. doi: 10.1158/0008-5472.CAN-09-3566. [DOI] [PubMed] [Google Scholar]
- Banz Y, Beldi G, Wu Y, Atkinson B, Usheva A, Robson SC. CD39 is incorporated into plasma microparticles where it maintains functional properties and impacts endothelial activation. British Journal Haematology. 2008;142(4):627–637. doi: 10.1111/j.1365-2141.2008.07230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurand A, Eckly A, Bari N, Leon C, Hechler B, Cazenave JP, et al. Desensitization of the platelet aggregation response to ADP: Differential down-regulation of the P2Y1 and P2cyc receptors. Thrombosis and Haemostasis. 2000;84(3):484–491. [PubMed] [Google Scholar]
- Beldi G, Banz Y, Kroemer A, Sun X, Wu Y, Graubardt N, et al. Deletion of CD39 on natural killer cells attenuates hepatic ischemia/reperfusion injury in mice. Hepatology. 2010;51(5):1702–1711. doi: 10.1002/hep.23510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldi G, Enjyoji K, Wu Y, Miller L, Banz Y, Sun X, et al. The role of purinergic signaling in the liver and in transplantation: Effects of extracellular nucleotides on hepatic graft vascular injury, rejection and metabolism. Frontiers in Bioscience. 2008;13:2588–2603. doi: 10.2741/2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldi G, Wu Y, Banz Y, Nowak M, Miller L, Enjyoji K, et al. Natural killer T cell dysfunction in CD39-null mice protects against concanavalin A-induced hepatitis. Hepatology. 2008;48(3):841–852. doi: 10.1002/hep.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldi G, Wu Y, Sun X, Imai M, Enjyoji K, Csizmadia E, et al. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology. 2008;135(5):1751–1760. doi: 10.1053/j.gastro.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden EK, Snapper SB. Regulatory T cells in inflammatory bowel disease. Current Opinion in Gastroenterology. 2008;24(6):733–741. doi: 10.1097/mog.0b013e328311f26e. [DOI] [PubMed] [Google Scholar]
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- Bruzzone S, Franco L, Guida L, Zocchi E, Contini P, Bisso A, et al. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. The Journal of Biological Chemistry. 2001;276(51):48300–48308. doi: 10.1074/jbc.M107308200. [DOI] [PubMed] [Google Scholar]
- Buffon A, Wink MR, Ribeiro BV, Casali EA, Libermann TA, Zerbini LF, et al. NTPDase and 5′ ecto-nucleotidase expression profiles and the pattern of extracellular ATP metabolism in the Walker 256 tumor. Biochimica et Biophysica Acta. 2007;1770(8):1259–1265. doi: 10.1016/j.bbagen.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signaling and vascular cell proliferation and death. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(3):364–373. doi: 10.1161/hq0302.105360. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Knight G. Cellular distribution and functions of P2 receptor subtypes in different systems. International Review of Cytology. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- Chadwick BP, Frischauf AM. The CD39-like gene family—Identification of three new human members (CD39l2, CD39l3, and CD39l4), their murine homologues, and a member of the gene family from Drosophila melanogaster. Genomics. 1998;50(3):357–367. doi: 10.1006/geno.1998.5317. [DOI] [PubMed] [Google Scholar]
- Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314(5806):1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- Chiorazzi N, Hatzi K, Albesiano E. B-cell chronic lymphocytic leukemia, a clonal disease of B lymphocytes with receptors that vary in specificity for (auto)antigens. Annals of the New York Academy of Sciences. 2005;1062:1–12. doi: 10.1196/annals.1358.002. [DOI] [PubMed] [Google Scholar]
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. The New England Journal of Medicine. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. Journal of Clinical Investigation. 2008;118(3):1123–1131. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Mackman N, Merrill-Skoloff G, Pedersen B, Furie BC, Furie B. Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood. 2004;104:3190–3197. doi: 10.1182/blood-2004-03-0935. [DOI] [PubMed] [Google Scholar]
- Cooke JP, Dzau VJ. Nitric oxide synthase—Role in the genesis of vascular disease. Annual Review of Medicine. 1997;48(489):489–509. doi: 10.1146/annurev.med.48.1.489. Review. [DOI] [PubMed] [Google Scholar]
- Corriden R, Chen Y, Inoue Y, Beldi G, Robson SC, Insel PA, et al. Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. The Journal of Biological Chemistry. 2008;283(42):28480–28486. doi: 10.1074/jbc.M800039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D’Arena G, et al. CD38/ CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Molecular Medicine. 2010;16(3–4):87–91. doi: 10.2119/molmed.2009.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Dianzani U, Horenstein AL, Fernandez JE, van Kooten C, Bragardo M, et al. Human CD38 ligand. A 120-KDA protein predominantly expressed on endothelial cells. Journal of Immunology. 1996;156(2):727–734. [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. The Journal of Experimental Medicine. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Malavasi F. The CD38/CD157 mammalian gene family: An evolutionary paradigm for other leukocyte surface enzymes. Purinergic Signalling. 2006;2(2):431–441. doi: 10.1007/s11302-006-9002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Mehta K, Malavasi F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leukemia Research. 2001;25(1):1–12. doi: 10.1016/s0145-2126(00)00093-x. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. Journal of Immunology. 1998;160(1):395–402. [PubMed] [Google Scholar]
- Deaglio S, Vaisitti T, Aydin S, Ferrero E, Malavasi F. In-tandem insight from basic science combined with clinical research: CD38 as both marker and key component of the pathogenetic network underlying chronic lymphocytic leukemia. Blood. 2006;108(4):1135–1144. doi: 10.1182/blood-2006-01-013003. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Vaisitti T, Zucchetto A, Gattei V, Malavasi F. CD38 as a molecular compass guiding topographical decisions of chronic lymphocytic leukemia cells. Seminars in Cancer Biology. 2010;20(6):416–423. doi: 10.1016/j.semcancer.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. 2002;99(7):2490–2498. doi: 10.1182/blood.v99.7.2490. [DOI] [PubMed] [Google Scholar]
- Deguchi H, Takeya H, Urano H, Gabazza EC, Zhou H, Suzuki K. Adenosine regulates tissue factor expression on endothelial cells. Thrombosis Research. 1998;91(2):57–64. doi: 10.1016/s0049-3848(98)00045-0. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Boeynaems JM, Robson SC. Extracellular nucleotides as negative modulators of immunity. Current Opinion in Pharmacology. 2009;9(4):507–513. doi: 10.1016/j.coph.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F, Robson SC. Identification of novel immunosuppressive pathways paves the way for drug discovery. Current Opinion in Pharmacology. 2009;9(4):445–446. doi: 10.1016/j.coph.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubyak GR, Elmoatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. The American Journal of Physiology. 1993;265(3 Part 1):C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. Review. [DOI] [PubMed] [Google Scholar]
- Dwyer K, Deaglio S, Crikis S, Gao W, Enjyoji K, Strom TB, et al. Salutary roles of CD39 in transplantation. Transplantation Reviews. 2007;21:54–63. [Google Scholar]
- Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, et al. Expression of CD39 by human peripheral blood CD4(+) CD25(+) T cells denotes a regulatory memory phenotype. American Journal of Transplantation. 2010;10(11):2410–2420. doi: 10.1111/j.1600-6143.2010.03291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. The Journal of Experimental Medicine. 2003;198(5):783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113(1):224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: Coordination by extracellular nucleotide metabolism. Blood. 2004;104(13):3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JSA, et al. Targeted disruption of Cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nature Medicine. 1999;5(9):1010–1017. doi: 10.1038/12447. [DOI] [PubMed] [Google Scholar]
- Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nature Medicine. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. The Journal of Experimental Medicine. 2003;197(11):1585–1598. doi: 10.1084/jem.20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero E, Malavasi F. The metamorphosis of a molecule: From soluble enzyme to the leukocyte receptor CD38. Journal of Leukocyte Biology. 1999;65(2):151–161. doi: 10.1002/jlb.65.2.151. [DOI] [PubMed] [Google Scholar]
- Fijnheer R, Boomgaard MN, van den Eertwegh AJ, Homburg CH, Gouwerok CW, Veldman HA, et al. Stored platelets release nucleotides as inhibitors of platelet function. Thrombosis and Haemostasis. 1992;68(5):595–599. [PubMed] [Google Scholar]
- Franceschi C, Abbracchio MP, Barbieri D, Ceruti S, Ferrari D, Iliou JP, et al. Purines and cell death. Drug Development Research. 1996;39(3–4):442–449. [Google Scholar]
- Friedman DJ, Kunzli BM, YI AR, Sevigny J, Berberat PO, Enjyoji K, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(39):16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie B. Pathogenesis of thrombosis. Hematology/the Education Program of the American Society of Hematology. 2009:255–258. doi: 10.1182/asheducation-2009.1.255. [DOI] [PubMed] [Google Scholar]
- Furie B, Furie BC. Thrombus formation in vivo. Journal of Clinical Investigation. 2005;115(12):3355–3362. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie B, Furie BC. Mechanisms of thrombus formation. The New England Journal of Medicine. 2008;359(9):938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- Goding JW, Howard MC. Ecto-enzymes of lymphoid cells. Immunological Reviews. 1998;161:5–10. doi: 10.1111/j.1600-065x.1998.tb01567.x. [DOI] [PubMed] [Google Scholar]
- Goepfert C, Imai M, Brouard S, Csizmadia E, Kaczmarek E, Robson SC. CD39 modulates endothelial cell activation and apoptosis. Molecular Medicine. 2000;6(7):591–603. [PMC free article] [PubMed] [Google Scholar]
- Goepfert C, Sundberg C, Sevigny J, Enjyoji K, Hoshi T, Csizmadia E, et al. Disordered cellular migration and angiogenesis in cd39-null mice. Circulation. 2001;104(25):3109–3115. doi: 10.1161/hc5001.100663. [DOI] [PubMed] [Google Scholar]
- Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta. Feed-forward inhibition of adenosine production at the cell surface. Journal of Biological Chemistry. 1986;261(33):15496–15507. [PubMed] [Google Scholar]
- Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. NAD+: A modulator of immune functions. Innate Immunity. 2010 doi: 10.1177/1753425910361989. ePub. [DOI] [PubMed] [Google Scholar]
- Grierson JP, Meldolesi J. Shear stress-induced [Ca2+]i transients and oscillations in mouse fibroblasts are mediated by endogenously released ATP. The Journal of Biological Chemistry. 1995;270(9):4451–4456. doi: 10.1074/jbc.270.9.4451. [DOI] [PubMed] [Google Scholar]
- Haag F, Adriouch S, Brass A, Jung C, Moller S, Scheuplein F, et al. Extracellular NAD and ATP: Partners in immune cell modulation. Purinergic Signalling. 2007;3(1–2):71–81. doi: 10.1007/s11302-006-9038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden TK, Lazarowski ER, Boucher RC. Release, metabolism and interconversion of adenine and uridine nucleotides: Implications for G protein-coupled P2 receptor agonist selectivity. Trends in Pharmacological Sciences. 1997;18(2):43–46. [PubMed] [Google Scholar]
- Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, et al. A role of the fast ATP-gated P2X1 cation channel in thrombosis of small arteries in vivo. The Journal of Experimental Medicine. 2003;198(4):661–667. doi: 10.1084/jem.20030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. Journal of Immunology. 2009;183(10):6157–6166. doi: 10.4049/jimmunol.0900475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262(5136):1056–1059. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, et al. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. The Journal of Experimental Medicine. 2010;207(12):2561–2568. doi: 10.1084/jem.20091154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrcke NS, Wrenshall LE, Lindman BJ, Platt JL. Role of heparan sulfate in immune system-blood vessel interactions. Immunology Today. 1993;14(500):500–505. doi: 10.1016/0167-5699(93)90265-M. [DOI] [PubMed] [Google Scholar]
- Imai M, Goepfert C, Kaczmarek E, Robson SC. CD39 modulates IL-1 release from activated endothelial cells. Biochemical and Biophysical Research Communications. 2000;270(1):272–278. doi: 10.1006/bbrc.2000.2410. [DOI] [PubMed] [Google Scholar]
- Jackson SW, Hoshi T, Wu Y, Sun X, Enjyoji K, Cszimadia E, et al. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1-null mice. The American Journal of Pathology. 2007;171(4):1395–1404. doi: 10.2353/ajpath.2007.070190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39 vascular ATP diphosphohydrolase. The Journal of Biological Chemistry. 1996;271(51):33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Egger P, Dunzendorfer S, Noris P, Balduini CL, Gritti D, et al. Reversal of thrombin-induced deactivation of CD39/ATPDase in endothelial cells by HMG-CoA reductase inhibition: Effects on Rho-GTPase and adenosine nucleotide metabolism. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(6):894–900. doi: 10.1161/01.atv.0000018305.95943.f7. [DOI] [PubMed] [Google Scholar]
- Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. The Journal of Biological Chemistry. 1997;272(41):25881–25889. doi: 10.1074/jbc.272.41.25881. [DOI] [PubMed] [Google Scholar]
- Koziak K, Kaczmarek E, Kittel A, Sevigny J, Blusztajn JK, Esch JSA, et al. Palmitoylation targets CD39/endothelial ATP diphosphohydrolase to caveolae. The Journal of Biological Chemistry. 2000;275(3):2057–2062. doi: 10.1074/jbc.275.3.2057. [DOI] [PubMed] [Google Scholar]
- Koziak E, Sevigny J, Robson SC, Siegel JB, Kaczmarek K. Analysis of CD39/ ATP diphosphohydrolase expression in endothelial cells, platelets and leukocytes. Thrombosis and Haemostasis. 1999;82:1538–1544. [PubMed] [Google Scholar]
- Krebs C, Koestner W, Nissen M, Welge V, Parusel I, Malavasi F, et al. Flow cytometric and immunoblot assays for cell surface ADP-ribosylation using a monoclonal antibody specific for ethenoadenosine. Analytical Biochemistry. 2003;314(1):108–115. doi: 10.1016/s0003-2697(02)00640-1. [DOI] [PubMed] [Google Scholar]
- Kunzli BM, Berberat PO, Dwyer K, Deaglio S, Csizmadia E, Cowan P, et al. Variable impact of CD39 in experimental murine colitis. Digestive Diseases and Sciences. 2010 doi: 10.1007/s10620-010-1425-9. ePub. [DOI] [PubMed] [Google Scholar]
- la Sala A, Ferrari D, Corinti S, Cavani A, Di Virgilio F, Girolomoni G. Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. Journal of Immunology. 2001;166(3):1611–1617. doi: 10.4049/jimmunol.166.3.1611. [DOI] [PubMed] [Google Scholar]
- la Sala A, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G, et al. Alerting and tuning the immune response by extracellular nucleotides. Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. Journal of Leukocyte Biology. 2003;73(3):339–343. doi: 10.1189/jlb.0802418. [DOI] [PubMed] [Google Scholar]
- Latz E. The inflammasomes: Mechanisms of activation and function. Current Opinion in Immunology. 2010;22(1):28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque SA, Kukulski F, Enjyoji K, Robson SC, Sevigny J. NTPDase1 governs P2X7-dependent functions in murine macrophages. European Journal of Immunology. 2010;40(5):1473–1485. doi: 10.1002/eji.200939741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Csizmadia E, Sevigny J, Enjyoji K, Robson SC. CD39/nucleoside triphosphate diphosphohydrolase-1 (NTPDase-1) modulates allograft rejection and cellular immune responses. American Journal of Transplantation. 2003;3(Suppl. 5):545. [Google Scholar]
- Liao H, Hyman MC, Baek AE, Fukase K, Pinsky DJ. cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. The Journal of Biological Chemistry. 2010;285(19):14791–14805. doi: 10.1074/jbc.M110.116905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13(9):1331–1339. doi: 10.1016/j.str.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Luthje J. Origin, metabolism and function of extracellular adenine nucleotides in the blood. Klinische Wochenschrift. 1989;67(6):317–327. doi: 10.1007/BF01741386. [Review] [DOI] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiological Reviews. 2008;88(3):841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- Malavasi F, Funaro A, Roggero S, Horenstein A, Calosso L, Mehta K. Human CD38: A glycoprotein in search of a function. Immunology Today. 1994;15(3):95–97. doi: 10.1016/0167-5699(94)90148-1. [DOI] [PubMed] [Google Scholar]
- Maliszewski CR, Delespesse GJ, Schoenborn MA, Armitage RJ, Fanslow WC, Nakajima T, et al. The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. Journal of Immunology. 1994;153(8):3574–3583. [PubMed] [Google Scholar]
- Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. The Journal of Biological Chemistry. 2010;285(10):7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus AJ, Broekman MJ, Drosopoulos JH, Islam N, Pinsky DJ, Sesti C, et al. Metabolic control of excessive extracellular nucleotide accumulation by CD39/ ecto-nucleotidase-1: Implications for ischemic vascular diseases. The Journal of Pharmacology and Experimental Therapeutics. 2003;305(1):9–16. doi: 10.1124/jpet.102.043729. [DOI] [PubMed] [Google Scholar]
- Marcus AJ, Safier LB, Hajjar KA, Ullman HL, Islam N, Broekman MJ, et al. Inhibition of platelet function by an aspirin-insensitive endothelial cell ADPase. Thromboregulation by endothelial cells. Journal of Clinical Investigation. 1991;88(5):1690–1696. doi: 10.1172/JCI115485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Meghji P, Pearson JD, Slakey LL. Kinetics of extracellular ATP hydrolysis by microvascular endothelial cells from rat heart. The Biochemical Journal. 1995;308(Pt 3):725–731. doi: 10.1042/bj3080725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumoto N, Kumamoto T, Robson SC, Sevigny J, Matsue H, Enjyoji K, et al. CD39 is the dominant Langerhans cell associated ecto-NTPDase: Modulatory roles in inflammation and immune responsiveness. Nature Medicine. 2002;8(4):358–365. doi: 10.1038/nm0402-358. [DOI] [PubMed] [Google Scholar]
- Morabito F, Damle RN, Deaglio S, Keating M, Ferrarini M, Chiorazzi N. The CD38 ectoenzyme family: Advances in basic science and clinical practice. Molecular Medicine. 2006;12(11–12):342–344. doi: 10.2119/2006-00110.Morabito. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreschi I, Bruzzone S, Bodrato N, Usai C, Guida L, Nicholas RA, et al. NAADP+ is an agonist of the human P2Y11 purinergic receptor. Cell Calcium. 2008;43(4):344–355. doi: 10.1016/j.ceca.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136(2):607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- Mulero JJ, Yeung G, Nelken ST, Ford JE. CD39-L4 is a secreted human apyrase, specific for the hydrolysis of nucleoside diphosphates. The Journal of Biological Chemistry. 1999;274(29):20064–20067. doi: 10.1074/jbc.274.29.20064. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: First line of the immune response to cell stress. Cell. 2006;126(4):659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Pacheco R, Martinez-Navio JM, Lejeune M, Climent N, Oliva H, Gatell JM, et al. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(27):9583–9588. doi: 10.1073/pnas.0501050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paust S, Cantor H. Regulatory T cells and autoimmune disease. Immunological Reviews. 2005;204:195–207. doi: 10.1111/j.0105-2896.2005.00247.x. [DOI] [PubMed] [Google Scholar]
- Plesner L. Ecto-ATPases: Identities and functions. International Review of Cytology. 1995;158:141–214. doi: 10.1016/s0074-7696(08)62487-0. [DOI] [PubMed] [Google Scholar]
- Podolsky DK. Inflammatory bowel disease. The New England Journal of Medicine. 2002;347(6):417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. Journal of Clinical Investigation. 2008;118(3):1110–1122. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunological Reviews. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. The FASEB Journal. 2009;23(2):473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- Robson SC, Daoud S, Begin M, Cote YP, Siegel JB, Bach FH, et al. Modulation of vascular ATP diphosphohydrolase by fatty acids. Blood Coagulation & Fibrinolysis. 1997;8(1):21–27. doi: 10.1097/00001721-199701000-00005. [DOI] [PubMed] [Google Scholar]
- Robson SC, Enjyoji K, Goepfert C, Imai M, Kaczmarek E, Lin Y, et al. Modulation of extracellular nucleotide-mediated signaling by CD39/nucleoside triphosphate diphosphohydrolase-1. Drug Development Research. 2001;53(2–3):193–207. [Google Scholar]
- Robson SC, Kaczmarek E, Siegel JB, Candinas D, Koziak K, Millan M, et al. Loss of ATP diphosphohydrolase activity with endothelial cell activation. The Journal of Experimental Medicine. 1997;185(1):153–163. doi: 10.1084/jem.185.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson SC, Sevigny J, Imai M, Enjyoji K. Thromboregulatory potential of endothelial CD39/nucleoside triphosphate diphosphohydrolase: Modulation of purinergic signalling in platelets. Emerging Therapeutic Targets. 2000;4:155–171. [Google Scholar]
- Robson SC, Sevigny J, Zimmermann H. The E-NTPDase family of ecto-nucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signalling. 2006;2:409–430. doi: 10.1007/s11302-006-9003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson SC, Wu Y, Sun X, Knosalla C, Dwyer K, Enjyoji K. Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Seminars in Thrombosis and Hemostasis. 2005;31(2):217–233. doi: 10.1055/s-2005-869527. [DOI] [PubMed] [Google Scholar]
- Ross R. Cell biology of atherosclerosis. Annual Review of Physiology. 1995;57(791):791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- Saborit-Villarroya I, Vaisitti T, Rossi D, D’Arena G, Gaidano G, Malavasi F, et al. E2A is a transcriptional regulator of CD38 expression in chronic lymphocytic leukemia. Leukemia. 2011;25:479–488. doi: 10.1038/leu.2010.291. [DOI] [PubMed] [Google Scholar]
- Salmi M, Jalkanen S. Cell-surface enzymes in control of leukocyte trafficking. Nature Reviews. Immunology. 2005;5(10):760–771. doi: 10.1038/nri1705. [DOI] [PubMed] [Google Scholar]
- Sansom FM, Riedmaier P, Newton HJ, Dunstone MA, Muller CE, Stephan H, et al. Enzymatic properties of an ecto-nucleoside triphosphate diphosphohydrolase from Legionella pneumophila: Substrate specificity and requirement for virulence. The Journal of Biological Chemistry. 2008;283(19):12909–12918. doi: 10.1074/jbc.M801006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom FM, Robson SC, Hartland EL. Possible effects of microbial ecto-nucleoside triphosphate diphosphohydrolases on host-pathogen interactions. Microbiology and Molecular Biology Reviews. 2008;72(4):765–781. doi: 10.1128/MMBR.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasamura H, Dzau VJ, Pratt RE. Desensitization of angiotensin receptor function. Kidney International. 1994;46(6):1499–1501. doi: 10.1038/ki.1994.429. [DOI] [PubMed] [Google Scholar]
- Schicker K, Hussl S, Chandaka GK, Kosenburger K, Yang JW, Waldhoer M, et al. A membrane network of receptors and enzymes for adenine nucleotides and nucleosides. Biochimica et Biophysica Acta. 2009;1793(2):325–334. doi: 10.1016/j.bbamcr.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Sundberg C, Braun N, Guckelberger O, Csizmadia E, Qawi I, et al. Differential catalytic properties and vascular topography of murine nucleoside triphosphate diphosphohydrolase 1 (NTPDase1) and NTPDase2 have implications for thromboregulation. Blood. 2002;99(8):2801–2809. doi: 10.1182/blood.v99.8.2801. [DOI] [PubMed] [Google Scholar]
- Sinzinger H, Virgolini I, Gazso A, Ogrady J. Review—Eicosanoids in atherosclerosis. Experimental Pathology. 1991;43(2):2–19. doi: 10.1016/s0232-1513(11)80133-7. [DOI] [PubMed] [Google Scholar]
- Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nature Reviews. Immunology. 2005;5(9):712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annual Review of Immunology. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A. Adenosine A2A receptor antagonists: Blockade of adenosinergic effects and T regulatory cells. British Journal of Pharmacology. 2008;153(Suppl. 1):S457–S464. doi: 10.1038/bjp.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29(39):5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- States DJ, Walseth TF, Lee HC. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends in Biochemical Sciences. 1992;17(12):495. doi: 10.1016/0968-0004(92)90337-9. [DOI] [PubMed] [Google Scholar]
- Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010;139(3):1030–1040. doi: 10.1053/j.gastro.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. The Journal of Clinical Investigation. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thammavongsa V, Kern JW, Missiakas DM, Schneewind O. Staphylococcus aureus synthesizes adenosine to escape host immune responses. The Journal of Experimental Medicine. 2009;206(11):2417–2427. doi: 10.1084/jem.20090097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. Physiological concentrations of purines and pyrimidines. Molecular and Cellular Biochemistry. 1994;140(1):1–22. doi: 10.1007/BF00928361. Review. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Apoptosis and necrosis—Intracellular ATP level as a determinant for cell death modes. Cell Death and Differentiation. 1997;4(6):429–434. doi: 10.1038/sj.cdd.4400262. Review. [DOI] [PubMed] [Google Scholar]
- Vaisitti T, Aydin S, Rossi D, Cottino F, Bergui L, D’Arena G, et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia. 2010;24(5):958–969. doi: 10.1038/leu.2010.36. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I, Wetter A. Molecular pharmacology of P2Y-receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2000;362(4–5):310–323. doi: 10.1007/s002100000310. Review. [DOI] [PubMed] [Google Scholar]
- Wang L, Andersson M, Karlsson L, Watson MA, Cousens DJ, Jern S, et al. Increased mitogenic and decreased contractile P2 receptors in smooth muscle cells by shear stress in human vessels with intact endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(8):1370–1376. doi: 10.1161/01.ATV.0000080350.37408.5A. [DOI] [PubMed] [Google Scholar]
- Warny M, Aboudola S, Robson SC, Sevigny J, Communi D, Soltoff SP, et al. P2Y(6) nucleotide receptor mediates monocyte interleukin-8 production in response to UDP or lipopolysaccharide. The Journal of Biological Chemistry. 2001;276(28):26051–26056. doi: 10.1074/jbc.M102568200. [DOI] [PubMed] [Google Scholar]
- Weisman GA, Erb L, Garrad RC, Theiss PM, Santiago-Perez LI, Flores RV, et al. P2Y nucleotide receptors in the immune system: Signaling by a P2Y(2) receptor in U937 monocytes. Drug Development Research. 1998;45(3–4):222–228. [Google Scholar]
- Wihlborg AK, Wang L, Braun OO, Eyjolfsson A, Gustafsson R, Gudbjartsson T, et al. ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(10):1810–1815. doi: 10.1161/01.ATV.0000142376.30582.ed. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, Henttinen T, Jalkanen S. Extracellular ATP formation on vascular endothelial cells is mediated by ecto-nucleotide kinase activities via phosphotransfer reactions. The FASEB Journal. 2001;15(1):251–260. doi: 10.1096/fj.00-0268com. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, Hytonen J, Samburski SS, Yrjanainen H, Jalkanen S, Viljanen MK. Disordered lymphoid purine metabolism contributes to the pathogenesis of persistent Borrelia garinii infection in mice. Journal of Immunology. 2010;184(9):5112–5120. doi: 10.4049/jimmunol.0902760. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, Samburski SS, Jalkanen S. Soluble purine-converting enzymes circulate in human blood and regulate extracellular ATP level via counteracting pyrophosphatase and phosphotransfer reactions. The FASEB Journal. 2003;17(10):1328–1330. doi: 10.1096/fj.02-1136fje. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, Samburski SS, Jalkanen S, Novak I. ATP-consuming and ATP-generating enzymes secreted by pancreas. The Journal of Biological Chemistry. 2006;281(40):29441–29447. doi: 10.1074/jbc.M602480200. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Granger DN. Inflammatory bowel disease: A paradigm for the link between coagulation and inflammation. Inflammatory Bowel Diseases. 2009;15(8):1245–1255. doi: 10.1002/ibd.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. CD73: A novel target for cancer immunotherapy. Cancer Research. 2010;70(16):6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H. 5′-nucleotidase: Molecular structure and functional aspects. The Biochemical Journal. 1992;285:345–365. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H. Two novel families of ectonucleotidases: Molecular structure, catalytic properties and a search for function. Trends in Pharmacological Sciences. 1999;20:231–236. doi: 10.1016/s0165-6147(99)01293-6. [DOI] [PubMed] [Google Scholar]