

Abstract
Background
Angiopoietin-like protein 3 (ANGPTL3), a liver-derived protein, plays an important role in the lipid and lipoprotein metabolism. Using data available from the DiOGenes study, we assessed the link with clinical improvements (weight, plasma lipid, and insulin levels) and changes in liver markers, alanine aminotransferase, aspartate aminotransferase (AST), adiponectin, fetuin A and B, and cytokeratin 18 (CK-18), upon low-calorie diet (LCD) intervention. We also examined the role of genetic variation in determining the level of circulating ANGPTL3 and the relation between the identified genetic markers and markers of hepatic steatosis.
Methods
DiOGenes is a multicenter, controlled dietary intervention where obese participants followed an 8-week LCD (800 kcal/day, using a meal replacement product). Plasma ANGPTL3 and liver markers were measured using the SomaLogic (Boulder, CO) platform. Protein quantitative trait locus (pQTL) analyses assessed the link between more than four million common variants and the level of circulating ANGPTL3 at baseline and changes in levels during the LCD intervention.
Results
Changes in ANGPTL3 during weight loss showed only marginal association with changes in triglycerides (nominal p = 0.02) and insulin (p = 0.04); these results did not remain significant after correcting for multiple testing. However, significant association (after multiple-testing correction) were observed between changes in ANGPTL3 and AST during weight loss (p = 0.004) and between ANGPTL3 and CK-18 (baseline p = 1.03 × 10−7, during weight loss p = 1.47 × 10−13). Our pQTL study identified two loci significantly associated with changes in ANGPTL3. One of these loci (the APOA4-APOA5-ZNF259-BUD13 gene cluster) also displayed significant association with changes in CK-18 levels during weight loss (p = 0.007).
Conclusion
We clarify the link between circulating levels of ANGPTL3 and specific markers of liver function. We demonstrate that changes in ANGPLT3 and CK-18 during LCD are under genetic control from trans-acting variants. Our results suggest an extended function of ANGPTL3 in the inflammatory state of liver steatosis and toward liver metabolic processes.
Electronic supplementary material
The online version of this article (10.1186/s12263-018-0597-3) contains supplementary material, which is available to authorized users.
Keywords: Angiopoietin-like protein 3, Liver markers, Liver steatosis, Lipid metabolism, Lipoprotein lipase, Protein quantitative trait locus, Single nucleotide polymorphisms
Background
The metabolic syndrome is a cluster of risk factors that increases the risk of diseases such as type 2 diabetes, hypertension, hyperlipidemia, and non-alcoholic fatty liver disease. The prevalence of the metabolic syndrome increases due to a parallel rise in the occurrence of obesity and insulin resistance [1]. This highlights the need for a more detailed understanding of the underlying molecular mechanisms.
One of the key components in the etiology of the metabolic syndrome is dyslipidemia. Angiopoietin-like proteins (ANGPTLs) have been reported to be involved in the regulation of lipid metabolism [2]. The human gene of angiopoietin-like protein 3 (ANGPTL3) is located on chromosome 1 and encodes one of several structurally similar secreted glycoproteins in the ANGPTL family. The ANGPTLs consists of a signal sequence at the N-terminal followed by an α-helical region forming coiled coil domains, and a fibrinogen-like domain at the C-terminal. ANGPTL8 differs in structure, as it lacks a C-terminal fibrinogen-like domain. ANGPTL3 is found in plasma both as a native protein and in cleaved form [2, 3]. The coiled coil domains at the N-terminal decrease the hydrolysis of plasma triglyceride (TG) through inhibition of lipoprotein lipase (LPL) activity and thereby affect the lipid and lipoprotein metabolism [4]. ANGPTL3 is predominantly expressed in the liver and is secreted by the liver both in mice and in humans [5, 6]. ANGPTL3 deficiency results in a dramatic reduction of the plasma concentration of TG and cholesterol [5, 7], and loss of function mutations in ANGPTL3 are the cause of a recessive form of familial combined hyperlipidemia [8].
In addition to stimulation of lipolysis, ANGPTL3 may be a determining factor in increasing hepatic lipid storage and affecting free fatty acid (FFA)-induced insulin resistance. One study reported a positive association between circulating ANGPTL3 and non-alcoholic steatohepatitis (NASH) [9]. Altogether, ANGPTL3 may be involved in the pathogenesis of the metabolic syndrome and increase the risk of hepatic steatosis.
This study examines the role of ANGPTL3 in lipid metabolism and liver health in the DiOGenes (Diet, Obesity and Genes) study. The DiOGenes study was a randomized, controlled dietary intervention that showed that a reduction in the glycemic index (GI) and an increase in dietary protein content led to an improvement in weight maintenance after an 8-week low-calorie diet (LCD) weight loss in adults [10]. In this study, we first analyze ANGPTL3 concentration in relation to body mass index (BMI), lipid profile, and markers of hepatic steatosis before and during weight loss. Afterwards, we identified genetic variants determining variations of circulating ANGPTL3 level through protein quantitative trait locus (pQTL) analysis and tested their association to ANGPTL3-related covariates.
Methods
Study design
The DiOGenes study (registered at http://www.clinicaltrials.gov, NCT00390637) was an intervention study carried out in eight European centers (Bulgaria, the Czech Republic, Denmark, Germany, Greece, the Netherlands, Spain, and the UK). The primary purpose was to examine the effects of dietary protein and GI on weight regain and metabolic and cardiovascular risk factors in overweight and obese families [10–12]. The study included families with at least one overweight or obese parent less than 65 years of age. The participants aimed to lose ≥ 8% of their initial body weight during 8 weeks of a LCD (800 kcal/day with additional use of 200 g of vegetables/day). Subjects achieving ≥ 8% weight loss were included in a 6-month weight maintenance period. Here, the participants were randomized to one of four ad libitum diets differing in GI and dietary protein content or a control diet following the national dietary guidelines in each of the countries [11].
Ethics
The study was approved by the different local ethical committees. Written informed consent was obtained from all participants, and the study was performed in accordance with the Declaration of Helsinki.
Clinical measurements
In the study, height was measured at the initial screening visit. Body weight was measured on all of the clinical investigation days together with fasting blood sampling. Total cholesterol, high-density lipoprotein cholesterol (HDL-C), TG, fasting glucose, and insulin were analyzed at the Research Laboratory, Department of Clinical Biochemistry, Gentofte University Hospital, Denmark. Low-density lipoprotein cholesterol (LDL-C) was calculated according to Friedewald’s equation [13].
Proteomics analyses
Plasma concentrations of ANGPTL3, alanine aminotransferase (ALT), aspartate aminotransferase (AST), adiponectin, fetuin A, fetuin B, and cytokeratin 18 (CK-18) were quantified before and after the LCD intervention using a multiplexed aptamer-based proteomic technology developed by SomaLogic Inc. (Boulder, CO) and measured as relative fluorescence units (RFU) [14, 15]. Data was normalized and calibrated by SomaLogic™ according to standard operating procedures [16]. This was done to remove systematic biases and correct plate-to-plate variation. Additional post-processing steps removed subjects with potential cell lyses as indicated with high hemoglobin levels (> 9 × 105 RFU) and outliers as detected with principal component analyses. Proteins were also checked for outliers and proportion of missing values before log transformation for analysis [17]. Data were available for 1129 proteins in 512 DiOGenes participants. Protein change during the weight loss intervention was computed as the log2 fold change between the end and the beginning of the intervention.
Genotyping
DNA was extracted from EDTA blood buffy coats with a salting out method. The DNA samples were quality checked, quantified, and normalized to approximately 100 ng/ml and 2.0 mg before genotyping. Genotyping was done using Illumina 660 W-quad according to manufacturer’s protocols (Illumina, San Diego, CA). Detailed information about this dataset can be found in Carayol et al. [17]. Briefly, 498,233 single nucleotide polymorphisms (SNPs) were genotyped; after quality check, additional SNPs were imputed using the Michigan Imputation Server [18] and the European 1000 Genomes set reference panel. SNP information was mapped onto NCBI version 37. Information was available for 4,020,654 SNPs in 494 participants with proteomics data.
QTL mapping
A complete description of the QTL mapping is available in Carayol et al. [17]. In summary, association between SNPs and circulating ANGPTL3 was tested at baseline and during weight loss using linear mixed effect models as implemented in GCTA software adjusting for baseline BMI or change in BMI, center, age, and gender as fixed, and a genetic relationship matrices as random effect [19]. In order to handle the multiple comparisons, p values were corrected using SLIDE (Sliding-window method for Locally Inter-correlated markers with asymptotic Distribution Errors corrected), a method based on a multivariate normal distribution similar to classical permutation but much faster [20]. Considering the large number of tests performed, significance levels were defined at adjusted alpha 10%. Genomic inflation factors (GIF) were estimated for the two pQTL analyses using estlambda function available in the GenABEL R package [21]. Pairwise linkage disequilibrium (LD) was calculated with LDlink, a web-based application using 1000 Genome phase 3 data [22].
Statistical analyses
Association between circulating ANGPTL3 and clinical variables (BMI, fasting glucose and insulin levels, total lipid levels, C-reactive protein (CRP) levels) was performed using a linear model, adjusting for center, age, gender, and baseline BMI. SNP effects were tested as additive effects. In the analyses of data from the weight loss period, models were adjusted for change in BMI. Adjustment for multiple testing was performed applying a Bonferroni correction considering tests performed on data available at baseline and during the LCD intervention separately. Statistical analyses were performed using R version 3.2.3.
Results
Baseline characteristics
In total, 769 participants from the DiOGenes study were included in the analyses. The baseline characteristics are described in Table 1 and have been extensively discussed in previous DiOGenes publications [10, 23, 24]. Briefly, participants were on average 41 years of age, with baseline BMI of 34.5 ± 4.9 kg/m2 (mean ± sd) and were non-diabetics (mean glucose levels = 5.12 ± 0.74 mmol/l and insulin levels = 11.48 ± 8.57 μIU/ml). After the weight loss period, the average BMI was decreased to 30.7 ± 4.5 kg/m2, and glycemic profiles improved to 4.82 ± 0.54 mmol/l for fasting glucose and 8.15 ± 6.12 μIU/ml for insulin.
Table 1.
Participant characteristics
| Variable | Baseline | Change during weight loss | ||
|---|---|---|---|---|
| n | Mean ± sd | n | Mean ± sd | |
| Gender (M/F) | 769 | 263/506 | – | – |
| Age (years) | 769 | 41.28 ± 6.22 | – | – |
| BMI (kg/m2) | 762 | 34.54 ± 4.88 | 638 | 3.80 ± 1.12 |
| Total cholesterol (mmol/l) | 714 | 4.89 ± 1.01 | 620 | 0.66 ± 0.76 |
| HDL-C (mmol/l) | 716 | 1.20 ± 0.33 | 624 | 0.08 ± 0.23 |
| LDL-C (mmol/l) | 711 | 3.07 ± 0.88 | 616 | 0.45 ± 0.64 |
| TG (mmol/l) | 705 | 1.35 ± 0.65 | 611 | 0.31 ± 0.58 |
| FFA (μmol/l) | 630 | 654.9 ± 333.2 | 545 | − 55.3 ± 368.4 |
| Glucose (mmol/l) | 701 | 5.12 ± 0.74 | 606 | 0.29 ± 0.60 |
| Insulin (μIU/ml) | 683 | 11.48 ± 8.57 | 541 | 3.89 ± 5.60 |
| CRP (mg/l) | 698 | 4.30 ± 3.90 | 594 | 1.05 ± 2.55 |
| ANGPTL3 (RFU) | 567 | 349.2 ± 122.0 | 539 | 1.74 ± 85.8 |
| ALT (RFU) | 594 | 5493.6 ± 2399.9 | 543 | 6.55 ± 204.3 |
| AST (RFU) | 594 | 7867.6 ± 2581.6 | 543 | − 0.93 ± 218.5 |
| Adiponectin (RFU) | 594 | 1430.8 ± 555.4 | 543 | − 0.87 ± 196.7 |
| Fetuin A (RFU) | 594 | 1029.2 ± 190.7 | 543 | − 4.66 ± 213.7 |
| Fetuin B (RFU) | 594 | 4026.6 ± 1342.0 | 543 | 2.66 ± 128.9 |
| CK-18 (RFU) | 594 | 222.0 ± 916.1 | 543 | − 4.08 ± 123.2 |
ALT alanine aminotransferase, ANGPTL3 angiopoietin-like protein 3, AST aspartate aminotransferase, BMI body mass index, CK-18 cytokeratin 18, CRP C-reactive protein, FFA free fatty acids, HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, sd standard deviation, TG triglycerides
Circulating ANGPTL3 and clinical measurements
During the weight loss period, ANGPTL3 plasma concentration was marginally associated with weight loss (p = 0.056, see Table 2). Furthermore, ANGPTL3 concentration was positively associated with TG concentration (p = 0.02) and with fasting insulin levels (p = 0.04). For both variables, the associations were independent of weight loss. However, these associations were not significant after adjustment for multiple testing. For other variables (total cholesterol, HDL-C, LDL-C, FFA, glucose, and CRP), there were no significant associations between ANGPTL3 and their concentration at baseline or changes during the weight loss period (Table 2).
Table 2.
Plasma ANGPTL3 and association with BMI and lipid profile
| Variable | Baseline | Weight loss period | ||
|---|---|---|---|---|
| β (95%CI) | p | β (95%CI) | p | |
| BMI (kg/m2) | 0.00 (− 0.44;0.45) | 0.986 | 6.39 (− 0.18;13.0) | 0.056 |
| Total cholesterol (mmol/l) | 0.45 (− 1.75;2.69) | 0.691 | 6.29 (− 3.11;15.7) | 0.189 |
| HDL-C (mmol/l) | − 3.62 (− 10.4;3.67) | 0.322 | − 21.2 (− 55.30;12.8) | 0.221 |
| LDL-C (mmol/l) | − 0.15 (− 2.60;2.37) | 0.906 | 4.81 (− 6.17;15.8) | 0.390 |
| TG (mmol/l) | 3.21 (− 0.34;6.89) | 0.077 | 15.7 (2.15;29.2) | 0.023 |
| FFA (μmol/l) | 0.01 (− 0.00;0.01) | 0.134 | 0.00 (− 0.03;0.02) | 0.728 |
| Glucose (mmol/l) | − 0.35 (− 3.67;3.08) | 0.839 | − 8.13 (− 20.36;4.10) | 0.193 |
| Insulin (μIU/ml) | 0.10 (− 0.18;0.37) | 0.485 | 1.54 (0.06;3.02) | 0.042 |
| CRP (mg/l) | 0.28 (− 0.35;0.91) | 0.383 | 1.89 (− 0.89;4.66) | 0.183 |
Coefficient (β), corresponding 95% confidence intervals, and associated p value from a linear regression are provided. Data are presented as back-transformed β-coefficients in percent with regard to results at baseline. Thus, an increase in ANGPTL3 of 1 RFU results in β (95%CI) percent change of the given variable. The regression models were adjusted for center, age, gender, and BMI. Models with data from the weight loss period were adjusted for the change in BMI due to the weight loss
CI Confidence interval, CRP C-reactive protein, FFA free fatty acids, HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, TG triglycerides
Circulating ANGPTL3 and liver markers
The association between ANGPTL3 and plasma levels of specific liver markers (AST, ALT, adiponectin, fetuin A and B, and CK-18) were tested (Table 3). We observed a strong positive association between circulating ANGPTL3 and CK-18 both at baseline (p = 1.03 × 10−7) and during the weight loss period (p = 1.47 × 10−13). Significant association was also seen between changes in AST and ANGPTL3 levels during weight loss intervention (p = 0.004). All these associations remained significant, even after adjustment for multiple testing. During weight loss, adiponectin displayed marginal association with ANGPTL3 (with nominal p value = 0.03; Bonferroni-adjusted p value = 0.18 and FDR-adjusted p value = 0.06).
Table 3.
Plasma ANGPTL3 and association with liver markers
| Liver markers | Function and association with liver steatosis | Baseline | Weight loss period | ||
|---|---|---|---|---|---|
| β (95%CI) | p | β (95%CI) | p | ||
| ALT (RFU) | Aminotransferase. High levels in liver. Marker of hepatocellular damage (↑). | − 0.78 (− 2.88; 1.31) | 0.463 | 0.02 (− 0.02; 0.05) | 0.405 |
| AST (RFU) | Aminotransferase. High levels in the liver, heart, and muscle (↑). | − 1.00 (− 3.04; 1.04) | 0.336 | − 0.05 (− 0.08; − 0.02) | 0.004 |
| Adiponectin (RFU) | Induce hepatic fatty acid oxidation, inhibits fatty acid synthesis, and suppress TNF-α production in the liver (↓). | 1.51 (− 0.53; 3.56) | 0.150 | 0.04 (0.00; 0.08) | 0.030 |
| Fetuin A (RFU) | Glycoprotein produced predominantly by the liver. Inhibitor of the insulin receptor tyrosine kinase (↑). | − 0.97 (− 3.01; 1.06) | 0.347 | − 0.01 (− 0.04; 0.02) | 0.551 |
| Fetuin B (RFU) | Shares 22% sequence similarity with fetuin A. Linked to inflammation and insulin resistance (↑). | 0.43 (− 1.70; 2.56) | 0.692 | 0.03 (− 0.03; 0.08) | 0.347 |
| CK-18 (RFU) | Activation of caspase 3 (apoptosis) results in cleavage of CK-18, the major intermediate filament in hepatocytes (↑). | 5.90 (3.82; 7.99) | 1.03 × 10 −7 | 0.21 (0.15; 0.26) | 1.47 × 10 −13 |
Coefficient (β), corresponding 95% confidence intervals, and associated p value from a linear regression are provided (in italics, p values passing Bonferroni correction). Data are presented as back-transformed β-coefficients in percent with regard to results at baseline. Thus, an increase in ANGPTL3 of 1 RFU results in β (95%CI) percent change of the given variable. In italics, p values passing Bonferroni correction (p < 0.05/6 = 0.0083). The regression models were adjusted for center, age, gender, and BMI. Models with data from the weight loss period were adjusted for the change in BMI due to the weight loss
ALT alanine aminotransferase, AST aspartate aminotransferase, CI Confidence interval, CK-18 cytokeratin 18, RFU relative fluorescence units, TNF-α tumor necrosis factor α
ANGPTL3 pQTL analyses
Furthermore, we investigated the possible link between circulating ANGPTL3 levels (at baseline and changes during LCD) and genetic markers. We thus performed genome-wide pQTL analyses testing more than 4 million common variants (see the “Methods” section). The results are shown as Manhattan plots in Figs. 1 and 2, respectively for the baseline and LCD pQTLs. Baseline pQTL analysis did not highlight any genome-wide significant signals (at adjusted alpha < 0.10).
Fig. 1.
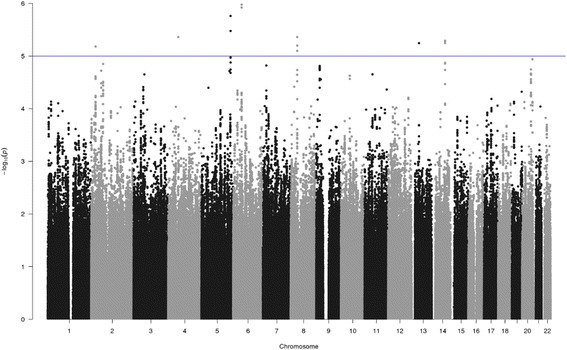
pQTL analysis of SNPs associated with circulating ANGPTL3 at baseline. Manhattan plot of pQTL analysis of SNPs associated with circulating ANGPTL3 at baseline. Each SNP is indicated by a black or a gray dot. They are arranged by chromosomal location (x-axis). The y-axis illustrates the level of statistical significance measured by the negative log of the corresponding p value for each SNP. The blue line represents suggestive association (p < 1 × 10−5)
Fig. 2.
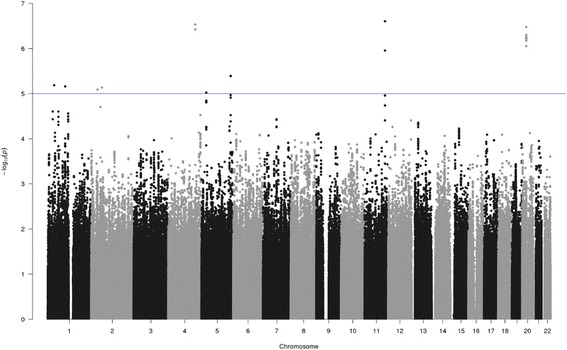
pQTL analysis of SNPs associated with the change in circulating ANGPTL3 during weight loss. Manhattan plot of pQTL analysis of SNPs associated with the change in circulating ANGPTL3 during weight loss. Each SNP is indicated by a black or a gray dot. They are arranged by chromosomal location (x-axis). The y-axis illustrates the level of statistical significance measured by the negative log of the corresponding p value for each SNP. The blue line represents suggestive association (p < 1 × 10−5)
The top SNPs (with nominal p < 1 × 10− 4) are presented in Table 4. However, in the LCD pQTL, three variants were considered genome-wide significant (Table 5). The two first SNPs, rs4360730 (NC_000011.9:g.116488748T>C) and rs74234276 (NC_000011.9:g.116488753G>A) are in perfect LD (R2 = 1) and localized within an intergenic region located 120 kb downstream from BUD13 gene (Fig. 3). This gene belongs to a gene cluster together with APOA4, APOA5, and ZNF259. The third SNP, rs9994520 (NC_000004.11:g.154882844G>C), is located 170 kb upstream from SFRP2 gene (Fig. 4). For both pQTL analyses, no significant p value inflation was observed (GIF were 1.00 and 0.99, respectively for baseline and weight loss pQTL, Additional file 1: Figure S1 and Additional file 2: Figure S2). This indicated no bias due to population substructure.
Table 4.
SNPs associated with circulating ANGPTL3 at baseline (p < 1 × 10−5)
| SNP | Chr | Position (bp) | A1 | A2 | MAF | Coef. | se | p value |
|---|---|---|---|---|---|---|---|---|
| rs36000763 | 6 | 48,687,252 | A | G | 0.064 | 0.202 | 0.041 | 1.06 × 10−6 |
| rs41528149 | 6 | 48,728,020 | T | C | 0.065 | 0.200 | 0.041 | 1.20 × 10−6 |
| rs13185453 | 5 | 166,370,819 | A | G | 0.143 | 0.143 | 0.030 | 1.73 × 10−6 |
| rs35976153 | 5 | 166,365,612 | T | C | 0.146 | 0.136 | 0.029 | 3.34 × 10−6 |
| rs12334611 | 8 | 37,045,986 | C | T | 0.072 | 0.182 | 0.040 | 4.35 × 10−6 |
| rs1277307 | 4 | 57,896,699 | G | T | 0.097 | 0.162 | 0.035 | 4.36 × 10−6 |
| rs76263326 | 14 | 76,627,919 | C | T | 0.100 | 0.151 | 0.033 | 5.12 × 10−6 |
| rs12100883 | 14 | 76,628,814 | T | C | 0.100 | 0.151 | 0.033 | 5.12 × 10−6 |
| rs1900121 | 14 | 76,629,481 | T | C | 0.100 | 0.151 | 0.033 | 5.12 × 10−6 |
| rs3783998 | 14 | 76,632,781 | G | T | 0.101 | 0.151 | 0.033 | 5.26 × 10−6 |
| rs4903381 | 14 | 76,635,675 | C | G | 0.100 | 0.151 | 0.033 | 5.67 × 10−6 |
| rs17521181 | 13 | 42,828,989 | A | T | 0.089 | 0.164 | 0.036 | 5.69 × 10−6 |
| rs4739476 | 8 | 37,046,991 | A | G | 0.070 | 0.184 | 0.041 | 6.33 × 10−6 |
| rs74528305 | 2 | 25,066,379 | G | T | 0.077 | 0.177 | 0.039 | 6.57 × 10−6 |
| rs74571086 | 8 | 37,049,958 | G | A | 0.070 | 0.184 | 0.041 | 7.97 × 10−6 |
Results from the association between SNPs and ANGPTL3 level at baseline
A1 and A2 the minor and major alleles, bp basepair, Chr chromosome, Coef estimated association coefficient, MAF minor allele frequency, se standard error, SNP single nucleotide polymorphism
Table 5.
SNPs associated with change in circulating ANGPTL3 during weight loss intervention (p < 1 × 10−5)
| SNP | Chr | Position (bp) | A1 | A2 | MAF | Coef. | se | p value |
|---|---|---|---|---|---|---|---|---|
| rs4360730* | 11 | 116,488,748 | C | T | 0.057 | 0.166 | 0.032 | 2.48 × 10−7 |
| rs74234276* | 11 | 116,488,753 | A | G | 0.057 | 0.166 | 0.032 | 2.48 × 10−7 |
| rs9994520* | 4 | 154,882,844 | G | C | 0.304 | 0.084 | 0.016 | 2.93 × 10−7 |
| rs113794502 | 20 | 23,631,539 | G | C | 0.237 | − 0.091 | 0.018 | 3.33 × 10−7 |
| rs7661078 | 4 | 154,883,600 | A | G | 0.302 | 0.083 | 0.016 | 3.78 × 10−7 |
| rs55656752 | 20 | 23,631,510 | A | T | 0.236 | − 0.090 | 0.018 | 5.01 × 10−7 |
| rs112213361 | 20 | 23,631,523 | A | G | 0.236 | − 0.090 | 0.018 | 5.01 × 10−7 |
| rs73102376 | 20 | 23,633,232 | T | C | 0.234 | − 0.090 | 0.018 | 5.53 × 10−7 |
| rs73102379 | 20 | 23,633,245 | C | T | 0.234 | − 0.090 | 0.018 | 5.53 × 10−7 |
| rs73102363 | 20 | 23,631,599 | G | C | 0.236 | − 0.089 | 0.018 | 5.78 × 10−7 |
| rs73102364 | 20 | 23,631,602 | G | A | 0.236 | − 0.089 | 0.018 | 5.78 × 10−7 |
| rs73102366 | 20 | 23,631,654 | T | C | 0.236 | − 0.089 | 0.018 | 5.78 × 10−7 |
| rs60143382 | 20 | 23,631,067 | A | G | 0.236 | − 0.090 | 0.018 | 5.84 × 10−7 |
| rs55724037 | 20 | 23,631,068 | C | T | 0.236 | − 0.090 | 0.018 | 5.84 × 10−7 |
| rs58578197 | 20 | 23,631,309 | C | T | 0.236 | − 0.090 | 0.018 | 5.84 × 10−7 |
| rs112950650 | 20 | 23,632,409 | G | A | 0.235 | − 0.089 | 0.018 | 6.05 × 10−7 |
| rs8116240 | 20 | 23,632,730 | T | C | 0.235 | − 0.089 | 0.018 | 6.05 × 10−7 |
| rs8122969 | 20 | 23,632,847 | C | T | 0.235 | − 0.089 | 0.018 | 6.05 × 10−7 |
| rs8124308 | 20 | 23,633,094 | C | T | 0.235 | − 0.089 | 0.018 | 6.17 × 10−7 |
| rs8122922 | 20 | 23,632,776 | C | T | 0.236 | − 0.089 | 0.018 | 6.50 × 10−7 |
Results from the association between SNPs and ANGPTL3 protein level change during weight loss intervention
A1 and A2 the minor and major alleles, bp basepair, Chr chromosome, Coef estimated association coefficient, MAF minor allele frequency, se standard error, SNP Single nucleotide polymorphism
*SNPs with adjusted p value < 0.10 upon the SLIDE (permutation) p value adjustments
Fig. 3.
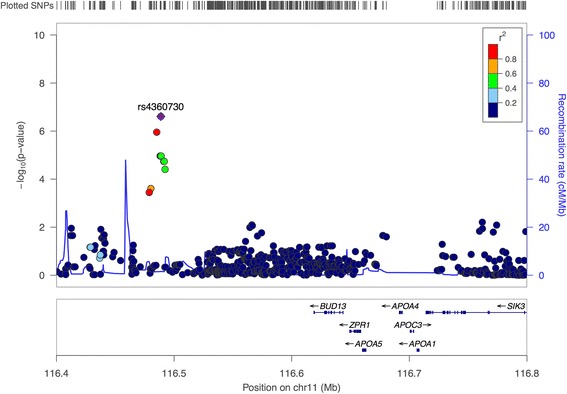
pQTL association signals at baseline in the region of surrounding rs4360730 and rs74234276 and BUD12, APOA4, APOA5, and ZNF259 (ZPR1) genes cluster. Association plot produced using LocusZoom software for SNPs associated to ANGPTL3 protein level at baseline. SNPs’ p values are plotted after −log10 transformation with scale on the y-axis and colors reflect pairwise linkage disequilibrium with the most associated SNP in the region (purple dot) based on the 1000 genomes EUR data set
Fig. 4.
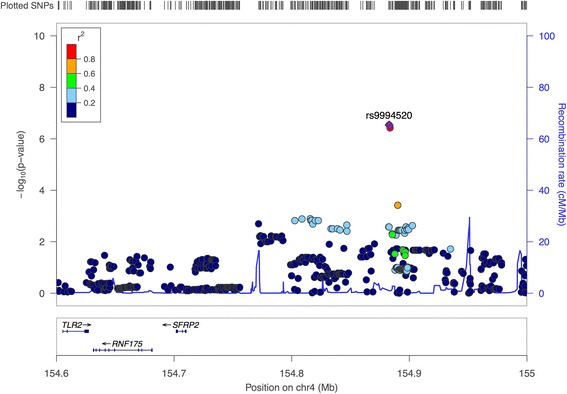
pQTL association signals during weight loss in the region surrounding rs9994520 and SFRP2 gene. Association plot produced using LocusZoom software for SNPs associated to ANGPTL3 protein level change during weight loss intervention. SNPs’ p values are plotted after −log10 transformation with scale on the y-axis and colors reflect pairwise linkage disequilibrium with the most associated SNP in the region (purple dot) based on the 1000 genomes EUR data set
Association between genetic markers and liver markers
Based on the pQTL results, rs4360730 and rs9994520 were chosen for further analysis. Specifically, we assessed whether the two liver markers (CK-18 and AST) associated with ANGPTL3 levels were also under genetic control. rs74234276 was not included due to complete LD with rs4360730. Regarding the rs4360730 SNP, we observed a significant association with CK-18 during weight loss period (with nominal p = 0.007 and Bonferroni adjusted p = 0.028, see Additional file 3: Table S1) and marginal association at baseline (p = 0.086). Effect size per genotype groups are indicated in Additional file 3: Table S1. Association tests with ALT levels did not reveal any significant effect of rs4360730. rs4360730 was not previously identified in published GWAs (EBI GWAs catalog, 01/01/2018 release) nor was it previously identified as an eQTL SNP in GTEX (release 7) [25, 26]. For rs9994520, we did not observe any significant association with CK-18 or ALT levels (at baseline and changes during LCD, see Additional file 3: Table S2).
Discussion
In the current study, we addressed the link between circulating ANGPTL3 levels and clinical improvements (weight, plasma lipid, and insulin profile) during LCD in a large clinical study. We assessed the link between ANGPTL3 and liver markers (released in circulation), and whether ANGPTL3 levels were under genetic control. Finally, we investigated the contribution from genetic markers modulating ANGPTL3 levels on liver markers themselves.
We observed a positive association between circulating ANGPTL3 and TG concentration following weight loss. However, this association was modest and did not remain, when correcting for multiple testing. In general, results on the relationship between circulating ANGPTL3 concentration and plasma lipids in humans are inconsistent [27–32]. In contrast to what could be expected, Robciuc and colleagues reported a negative correlation between ANGPTL3 and TG concentration [31]. This correlation did not remain significant after adjusting for HDL-C and apolipoprotein concentrations. A large study including 1770 participants of European Caucasian ancestry did not observe a correlation between plasma ANGPTL3 and concentration of TG [32]. However, they did report positive correlations between ANGPTL3 concentrations and LDL-C, HDL-C, and total cholesterol. Despite conflicting results concerning the relationship between ANGPTL3 and lipid parameters in humans, there is a consensus about the physiological role of ANGPTL3 regarding inhibition of LPL. But the functional evidence is derived from animal studies [33, 34] and the exact inhibitory mechanisms of ANGPTL3 on LPL in humans are not fully understood. Earlier findings indicate that cleavage is crucial for the function of ANGPTL3. The N-terminal fragment containing the coiled coil domains of the protein is more efficient in inhibiting LPL than the full-length ANGPTL3 [2]. In this study, we used a detection method based on protein binding of aptamers, which are reported to have many advantages, compared to antibodies [35]. However, in this and several other studies, the methods used for detecting ANGPTL3 cannot distinguish between the different fragments of the protein, nor post-translational modification. It is suggested that the functional fraction of ANGPTL3 might not be found in circulation, but exists bound to the endothelial surface of the adipose tissue, cardiac muscle, and skeletal muscle for LPL-mediated lipolysis [36]. This further specifies the need of an improved understanding regarding the LPL inhibitory function of ANGPTL3 and further improvement of the methods to detect and quantify the fragments of the protein.
A study reported that the ANGPTL8 is the rate-limiting protein for the activity of ANGPTL3 [37]. Co-expression of ANGPTL3 and ANGPTL8 in cultured hepatocytes resulted in the appearance of a 33-kDa-sized protein corresponding to the N-terminal domain of ANGPTL3, whereas only full-length ANGPTL3 were detected in cells that did not express ANGTPL8. ANGPTL8 was not assayed on the Somalogic panel, and it was not possible to study the relationship with ANGPTL3 within the DiOGenes study. However, recent in vivo studies have further indicated that ANGPTL3 and ANGPTL8 cooperate in the regulation of plasma TG levels [38, 39]. Davies and colleagues demonstrated that ANGPTL3 and ANGPTL8 as a complex exhibited a greatly enhanced ability to bind LPL compared to either protein alone. This complex was formed more efficiently, when the two proteins were co-expressed [39]. This has led to the suggestion of interplay between ANGPTL3, ANGPTL4, and ANGPTL8 in the regulation of lipid metabolism [40, 41]. ANGPTL8 is induced by feeding and possibly activates the inhibitory effects of ANGPTL3 on LPL in cardiac and skeletal muscles, directing circulating TG to the adipose tissue for storage. In this study, the concentration of circulating ANGPTL3 and lipid parameters were measured in a fasted state, which could explain the lack of significant associations. It is likely that an ANGPTL3 response is only observed post-prandial, and thus, a meal-test challenge would be required to study the dynamics of ANGPTL3. ANGPTL4 is very similar to ANGPTL3 both in structure and in function and is induced by fasting and might inhibit LPL in adipose tissue during energy restriction, directing TG to cardiac and skeletal muscle for oxidation [40, 41].
Consistent with the conflicting results regarding ANGPTL3 and lipid metabolism, the link between ANGPTL3 and glucose metabolism remains unclear [42, 43]. Our results showed a marginal association between circulating ANGPTL3 and fasting insulin concentrations. The mechanisms by which ANGPTL3 influence the insulin remains unclear, but there might be a potential role of the protein to indirectly regulate glucose metabolism.
We found a strong positive association between changes in ANGPTL3 levels and CK-18, together with a negative association between changes in ANGPTL3 and AST, both independently of weight loss. CK-18 is the major intermediate filament protein in the liver. Circulating CK-18 is associated with apoptotic cell death of hepatocytes, and several studies have demonstrated the elevation of CK-18 in the context of NASH and hepatic inflammation [44]. AST is a transaminase enzyme dependent on pyridoxal phosphate and important in the amino acid metabolism. It is present as both cytoplasmic and mitochondrial isoforms. In this study, we measured the cytoplasmic isoform, which independently is a marker of tissue injury. High levels of circulating AST is not exclusively related to the liver steatosis, but could also indicate diseases affecting other organs, as AST is found in high concentrations in the liver, heart, skeletal muscle, and kidney [45, 46]. To our knowledge, only one human study has analyzed circulating ANGPTL3 concentration in relation to liver steatosis. This study found that ANGPTL3 concentration was significantly and independently associated with NASH, but not in patients with simple steatosis [9]. Szalowska et al. induced inflammation in human liver tissues in vitro and identified ANGPTL3 as a biomarker associated with liver diseases [47]. Together with our results regarding CK-18, it could indicate that an increase in plasma ANGPTL3 concentration is the result of liver inflammation or that ANGPTL3 plays a role in the development of the diseased condition. Due to the controversy of non-invasive biomarkers as measurement of liver diseases, additional studies should include actual liver biopsies to further evaluate the role of ANGPTL3 in liver steatosis.
Our pQTL study highlighted SNPs that were modulating changes in circulating ANGPTL3 during the weight loss period, of which one locus also seemed to modulate CK-18 levels. Specifically, these pQTL studies revealed three common genetic variants (rs4360730, rs74234276, and rs9994520) associated with circulating ANGPTL3. SNPs rs4360730 and rs74234276 are located near the APOA4-APOA5-ZNF259-BUD13 gene cluster locus at the chromosome region 11q23.3; and are in perfect LD. Several genetic variants in this region have already been associated to hyperlipidemia [48], serum lipid levels [49], risk of developing metabolic syndrome [50], and plasma TG level [51]. APOA4 and APOA5 encode apolipoproteins involved in lipid metabolism [52]. ZNF259 encodes zinc finger protein, a regulatory protein that is involved in cell proliferation and signal transduction. BUD13 encodes for BUD13 homolog protein, which is a subunit in the retention and splicing (RES) complex that affects nuclear pre-mRNA retention. However, the exact function of ZNF259 and BUD13 in lipid mechanisms is unclear [48]. The region is an interesting target knowing that ANGPTL3 regulates plasma lipid levels and is a potential therapeutic target to treat combined hyperlipidemia [53]. The SNPs in this region, rs4360730 and rs74234276, are trans-acting genetic variants, probably working as distant regulators of ANGPTL3 through mechanisms of the APOA4-APOA5-ZNF259-BUD13 gene cluster. We further demonstrated that CK-18 levels at baseline and during the weight loss period were under genetic control by the rs4360730 SNP.
The rs9994520 SNP is located near the SFRP2 gene. This gene encodes the secreted Frizzled-related protein 2, which operates as soluble modulators of Wnt signaling. The functional relationship between ANGPTL3 and SFRP2 is not known. However, SFRP2 has been associated to adipose tissue mass and may play a role in adipose angiogenesis of which angiopoietin-like proteins are regulation key factors [54–56].
Interestingly, the identified pQTLs affecting circulating ANGPTL3 during the weight loss intervention were not detectable at baseline. This is consistent with our recent large-scale pQTL study on 1129 proteins [17], where the identified pQTL during LCD could not be identified at baseline. This can be explained by effect size consideration (statistical power): very large sample size would be required to identify potential baseline pQTL. By contrast, a clinical intervention (such as LCD) would induce drastic metabolic and physiological changes, thus would lead to very large effect sizes and thereby significantly improve our ability to detect pQTLs associated with such drastic shift in homeostasis [17].
Conclusions
In conclusion, we uncover genetic regulators of circulating ANGPTL3 during LCD and the link with markers of liver function. We report several trans-acting pQTL on changes in circulating ANGPTL3 during LCD. These pQTLs were not detectable at baseline, suggesting a change in the regulation of ANGPTL3 due to calorie restriction. It was not possible to clarify the controversy regarding the function of ANGPTL3 in lipid metabolism as we found a very marginal association with total lipid levels. However, our data suggest strong associations with specific liver markers (CK-18 and AST). These observations are supported by the identification of pQTL signals that affect ANGPTL3 levels during the weight loss period. Our analysis also suggests an extended function of ANGPTL3 in the development of liver steatosis and shows a common genetic regulation for both ANGPTL3 and markers of liver function.
Additional files
{kind=link}
Figure S1. QQ plot of the relationship between expected and observed distribution at baseline. Quantile-quantile plot of baseline data. The relationship between observed (y-axis) and expected (x-axis) distribution. The statistical significance is measured by the negative log of the corresponding p-value for each SNP. (JPEG 92 kb)
{kind=link}
Figure S2. QQ plot of the relationship between expected and observed distribution during weight loss period. Quantile-quantile plot for the analysis of the weight loss period. The relationship between observed (y-axis) and expected (x-axis) distribution. The statistical significance is measured by the negative log of the corresponding p-value for each SNP. (JPEG 94 kb)
Table S1. Effect of rs4360730 on BMI, Lipid Profile and Liver Markers. Table S2 Effect of rs9994520 on BMI, Lipid Profile and Liver Markers. (DOCX 21 kb)
Acknowledgements
We gratefully acknowledge all of the study participants for their contributions to the DiOGenes study. We also wish to thank Mads Vendelbo Lind, Christian Ritz, and Finn Sandø-Pedersen for the advice regarding the statistical analyses.
Funding
The DiOGenes project was funded by a grant from the European Union Food Quality and Safety Priority of the Sixth Framework Programme, contract no. FP6-2005-513946.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALT
Alanine aminotransferase
- ANGPTL3/4/8
Angiopoietin-like protein 3/4/8
- ANGPTLs
Angiopoietin-like proteins
- AST
Aspartate aminotransferase
- BMI
Body mass index
- CK-18
Cytokeratin 18
- CRP
C-reactive protein
- FDR
False discovery rate
- FFA
Free fatty acids
- GI
Glycemic index
- GIF
Genomic inflation factor
- HDL-C
High-density lipoprotein cholesterol
- LCD
Low-calorie diet
- LD
Linkage disequilibrium
- LDL-C
Low-density lipoprotein cholesterol
- LPL
Lipoprotein lipase
- NASH
Non-alcoholic steatohepatitis
- pQTL
Protein quantitative trait locus
- RES complex
Retention and splicing complex
- RFU
Relative fluorescence units
- sd
Standard deviation
- se
Standard error
- SLIDE
Sliding-window method for Locally Inter-correlated markers with asymptotic Distribution Errors corrected
- SNPs
Single nucleotide polymorphisms
- TG
Triglycerides
Authors’ contributions
WS and AA conceived and designed the DiOGenes study; AV, JC, JH, and ADC performed the experiments and data production; AV, JC, LHL, TB, and ALH were responsible for the data analysis, interpretation of the results, and the final version of the manuscript. All authors have read and approved the findings of the study and the final version of the manuscript.
Ethics approval and consent to participate
The study was approved by the local ethical committees in the respective countries, confirming that the study protocol was in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
AA is an advisor to or a member of advisory boards for a number of food and pharmaceutical producers: Basic Research, USA; Beachbody, USA; BioCare Copenhagen, Denmark; Crossfit, USA; Dutch Beer Institute, Netherlands; Feast Kitchen A/S, Denmark; Gelesis, USA; Groupe Éthique et Santé, France; McCain Foods Limited, USA; Nestlé Research Center, Switzerland; Novo Nordisk, Denmark; Pfizer, Germany; Saniona, Denmark; Sanofi-Aventis, Germany; S-Biotek, Denmark; Scandinavian Airlines System, Denmark; TetraPak, Sweden; Weight Watchers, USA; and from Zaluvida, Switzerland. AA does not own stock in, or have other ownership interests in, any of the companies to which he provides scientific advice, or in any nutrition company other than those companies whose stock is held by various mutual fund retirement accounts. Recent research at the University of Copenhagen, Denmark, has been funded by unrestricted grants from or contracts with DC-Ingredients, Denmark; Danish Dairy Foundation; Global Dairy Platform; and Gelesis AS, USA. AA receives payment as associate editor of The American Journal of Clinical Nutrition and as a member of the editorial committee of Annual Review of Nutrition. AA is a recipient of honoraria as speaker for a wide range of Danish and international concerns and of royalties from textbooks and from popular diet and cookery books. AA is a co-inventor of a number of patents, including Methods of inducing weight loss, treating obesity and preventing weight gain (licensee Gelesis, USA) and Biomarkers for predicting degree of weight loss (licensee Nestec SA, CH), owned by the University of Copenhagen, in accordance with Danish law. AA is a co-founder and co-owner of the University of Copenhagen spin-out companies Mobile Fitness A/S, Personalized Weight Management Research Consortium ApS (Gluco-diet.dk), and Flaxslim ApS, where he is also a member of the board. AA is not an advocate or activist for specific diets and is not strongly committed to any specific diet, e.g., veganism, Atkins diet, gluten-free diet, high animal protein diet, or dietary supplements. AV, JC, and JH are full-time employees at Nestlé Institute of Health Sciences. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12263-018-0597-3) contains supplementary material, which is available to authorized users.
Contributor Information
Anne Lundby Hess, Email: alhe@nexs.ku.dk.
Jérôme Carayol, Email: Jerome.carayol@rd.nestle.com.
Trine Blædel, Email: trinebl@nexs.ku.dk.
Jörg Hager, Email: jorg.hager@rd.nestle.com.
Alessandro Di Cara, Email: alessandro.dicara@quartzbio.com.
Arne Astrup, Email: ast@nexs.ku.dk.
Wim H. M. Saris, Email: w.saris@maastrichtuniversity.nl
Lesli Hingstrup Larsen, Email: lehla@nexs.ku.dk.
Armand Valsesia, Email: armand.valsesia@rd.nestle.com.
References
- 1.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28(4):629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 2.Ono M, et al. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J Biol Chem. 2003;278(43):41804–41809. doi: 10.1074/jbc.M302861200. [DOI] [PubMed] [Google Scholar]
- 3.Conklin D, et al. Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics. 1999;62(3):477–482. doi: 10.1006/geno.1999.6041. [DOI] [PubMed] [Google Scholar]
- 4.Shan L, et al. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J Biol Chem. 2009;284(3):1419–1424. doi: 10.1074/jbc.M808477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koishi R, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30(2):151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- 6.Romeo S, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119(1):70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musunuru K, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363(23):2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minicocci I, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97(7):E1266–E1275. doi: 10.1210/jc.2012-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yilmaz Y, et al. Serum concentrations of human angiopoietin-like protein 3 in patients with nonalcoholic fatty liver disease: association with insulin resistance. Eur J Gastroenterol Hepatol. 2009;21(11):1247–1251. doi: 10.1097/MEG.0b013e32832b77ae. [DOI] [PubMed] [Google Scholar]
- 10.Larsen TM, et al. Diets with high or low protein content and glycemic index for weight-loss maintenance. N Engl J Med. 2010;363(22):2102–2113. doi: 10.1056/NEJMoa1007137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen TM, et al. The Diet, Obesity and Genes (Diogenes) Dietary Study in eight European countries—a comprehensive design for long-term intervention. Obes Rev. 2010;11(1):76–91. doi: 10.1111/j.1467-789X.2009.00603.x. [DOI] [PubMed] [Google Scholar]
- 12.Moore CS, et al. Dietary strategy to manipulate ad libitum macronutrient intake, and glycaemic index, across eight European countries in the Diogenes Study. Obes Rev. 2010;11(1):67–75. doi: 10.1111/j.1467-789X.2009.00602.x. [DOI] [PubMed] [Google Scholar]
- 13.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. [PubMed] [Google Scholar]
- 14.Gold L, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5(12):e15004. [DOI] [PMC free article] [PubMed]
- 15.Rohloff JC, et al. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids. 2014;3:e201. doi: 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.SOMAscan Technical White Paper. 2017 [cited 2018 02-02-2018]; Available from: http://www.somalogic.com/somalogic/media/Assets/PDFs/SSM-002-Rev-2-SOMAscan-Technical-White-Paper-3-7-15.pdf.
- 17.Carayol J, et al. Protein quantitative trait locus study in obesity during weight-loss identifies a leptin regulator. Nat Commun. 2017;8(1):2084. doi: 10.1038/s41467-017-02182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han B, Kang HM, Eskin E. Rapid and accurate multiple testing correction and power estimation for millions of correlated markers. PLoS Genet. 2009;5(4):e1000456. doi: 10.1371/journal.pgen.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aulchenko YS, et al. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 22.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valsesia A, et al. Distinct lipid profiles predict improved glycemic control in obese, nondiabetic patients after a low-caloric diet intervention: the Diet, Obesity and Genes randomized trial. Am J Clin Nutr. 2016;104(3):566–575. doi: 10.3945/ajcn.116.137646. [DOI] [PubMed] [Google Scholar]
- 24.Armenise C, et al. Transcriptome profiling from adipose tissue during a low-calorie diet reveals predictors of weight and glycemic outcomes in obese, nondiabetic subjects. Am J Clin Nutr. 2017;106(3):736–746. doi: 10.3945/ajcn.117.156216. [DOI] [PubMed] [Google Scholar]
- 25.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45(D1):D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GT, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatsuda S, et al. Association between plasma angiopoietin-like protein 3 and arterial wall thickness in healthy subjects. J Vasc Res. 2007;44(1):61–66. doi: 10.1159/000098153. [DOI] [PubMed] [Google Scholar]
- 28.Shimamura M, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27(2):366–372. doi: 10.1161/01.ATV.0000252827.51626.89. [DOI] [PubMed] [Google Scholar]
- 29.Stejskal D, et al. Angiopoietin-like protein 3: development, analytical characterization, and clinical testing of a new ELISA. Gen Physiol Biophys. 2007;26(3):230–3. [PubMed] [Google Scholar]
- 30.Shoji T, et al. Plasma angiopoietin-like protein 3 (ANGPTL3) concentration is associated with uremic dyslipidemia. Atherosclerosis. 2009;207(2):579–584. doi: 10.1016/j.atherosclerosis.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 31.Robciuc MR, et al. Quantitation of serum angiopoietin-like proteins 3 and 4 in a Finnish population sample. J Lipid Res. 2010;51(4):824–831. doi: 10.1194/jlr.M002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta N, et al. Differential association of plasma angiopoietin-like proteins 3 and 4 with lipid and metabolic traits. Arterioscler Thromb Vasc Biol. 2014;34(5):1057–1063. doi: 10.1161/ATVBAHA.113.302802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizugawa T, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277(37):33742–33748. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- 34.Koster A, et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology. 2005;146(11):4943–4950. doi: 10.1210/en.2005-0476. [DOI] [PubMed] [Google Scholar]
- 35.Han K, Liang ZQ, Zhou ND. Design strategies for aptamer-based biosensors. Sensors. 2010;10(5):4541–4557. doi: 10.3390/s100504541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonnenburg WK, et al. GPIHBP1 stabilizes lipoprotein lipase and prevents its inhibition by angiopoietin-like 3 and angiopoietin-like 4. J Lipid Res. 2009;50(12):2421–2429. doi: 10.1194/jlr.M900145-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quagliarini F, et al. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci U S A. 2012;109(48):19751–19756. doi: 10.1073/pnas.1217552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haller JF, et al. ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J Lipid Res. 2017;58(6):1166–1173. doi: 10.1194/jlr.M075689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies BSJ, et al. ANGPTL8 promotes the ability of ANGPTL3 to inhibit lipoprotein lipase. FASEB J. 2017;31:1137–1149. [DOI] [PMC free article] [PubMed]
- 40.Zhang R. The ANGPTL3-4-8 model, a molecular mechanism for triglyceride trafficking. Open Biol. 2016;6(4):150272. [DOI] [PMC free article] [PubMed]
- 41.Dijk W, Kersten S. Regulation of lipid metabolism by angiopoietin-like proteins. Curr Opin Lipidol. 2016;27(3):249–256. doi: 10.1097/MOL.0000000000000290. [DOI] [PubMed] [Google Scholar]
- 42.Robciuc MR, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler Thromb Vasc Biol. 2013;33(7):1706–1713. doi: 10.1161/ATVBAHA.113.301397. [DOI] [PubMed] [Google Scholar]
- 43.Haridas PAN, et al. Regulation of angiopoietin-like proteins (ANGPTLs) 3 and 8 by insulin. J Clin Endocrinol Metab. 2015;100(10):E1299–E1307. doi: 10.1210/jc.2015-1254. [DOI] [PubMed] [Google Scholar]
- 44.Diab DL, et al. Cytokeratin 18 fragment levels as a noninvasive biomarker for nonalcoholic steatohepatitis in bariatric surgery patients. Clin Gastroenterol Hepatol. 2008;6(11):1249–1254. doi: 10.1016/j.cgh.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172(3):367–379. doi: 10.1503/cmaj.1040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gowda S, et al. A review on laboratory liver function tests. Pan Afr Med J. 2009;3:17. [PMC free article] [PubMed] [Google Scholar]
- 47.Szalowska E, et al. Comparative analysis of the human hepatic and adipose tissue transcriptomes during LPS-induced inflammation leads to the identification of differential biological pathways and candidate biomarkers. BMC Med Genet. 2011;4:71. doi: 10.1186/1755-8794-4-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aung LHH, et al. Association of the variants in the BUD13-ZNF259 genes and the risk of hyperlipidaemia. J Cell Mol Med. 2014;18(7):1417–1428. doi: 10.1111/jcmm.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LHH A, et al. Association between the MLX interacting protein-like, BUD13 homolog and zinc finger protein 259 gene polymorphisms and serum lipid levels. Sci Rep. 2014;4:5565. [DOI] [PMC free article] [PubMed]
- 50.Lin E, et al. Association and interaction of APOA5, BUD13, CETP, LIPA and health-related behavior with metabolic syndrome in a Taiwanese population. Sci Rep. 2016;6:36830. doi: 10.1038/srep36830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu Q, et al. Effects of polymorphisms in APOA4-APOA5-ZNF259-BUD13 gene cluster on plasma levels of triglycerides and risk of coronary heart disease in a Chinese Han population. PLoS One. 2015;10(9):e0138652. doi: 10.1371/journal.pone.0138652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgado-Lista J, et al. Effects of variations in the APOA1/C3/A4/A5 gene cluster on different parameters of postprandial lipid metabolism in healthy young men. J Lipid Res. 2010;51(1):63–73. doi: 10.1194/jlr.M800527-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52(2):187–193. doi: 10.1007/s12020-015-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowley RK, et al. SFRP2 is associated with increased adiposity and VEGF expression. PLoS One. 2016;11(9):e0163777. [DOI] [PMC free article] [PubMed]
- 55.Courtwright A, et al. Secreted frizzle-related protein 2 stimulates angiogenesis via a Calcineurin/NFAT signaling pathway. Cancer Res. 2009;69(11):4621–4628. doi: 10.1158/0008-5472.CAN-08-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hato T, Tabata M, Oike Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc Med. 2008;18(1):6–14. doi: 10.1016/j.tcm.2007.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. QQ plot of the relationship between expected and observed distribution at baseline. Quantile-quantile plot of baseline data. The relationship between observed (y-axis) and expected (x-axis) distribution. The statistical significance is measured by the negative log of the corresponding p-value for each SNP. (JPEG 92 kb)
Figure S2. QQ plot of the relationship between expected and observed distribution during weight loss period. Quantile-quantile plot for the analysis of the weight loss period. The relationship between observed (y-axis) and expected (x-axis) distribution. The statistical significance is measured by the negative log of the corresponding p-value for each SNP. (JPEG 94 kb)
Table S1. Effect of rs4360730 on BMI, Lipid Profile and Liver Markers. Table S2 Effect of rs9994520 on BMI, Lipid Profile and Liver Markers. (DOCX 21 kb)
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author on reasonable request.