

Abstract
Human herpesviruses (HVs) have developed ingenious mechanisms that enable them to traverse the defenses of the central nervous system (CNS). The ability of HVs to enter a state of latency, a defining characteristic of this viral family, allows them to persist in the human host indefinitely. As such, HVs represent the most frequently detected pathogens in the brain. Under constant immune pressure, these infections are largely asymptomatic in healthy hosts. However, many neurotropic HVs have been directly connected with CNS pathology in the context of other stressors and genetic risk factors. In this review, we discuss the potential mechanisms by which neurotropic HVs contribute to neurodegenerative disease (NDD) pathology by highlighting two prominent members of the HV family, herpes simplex virus 1 (HSV-1) and human herpesvirus 6 (HHV-6). We (i) introduce the infectious pathways and replicative cycles of HSV-1 and HHV-6 and then (ii) review the clinical evidence supporting associations between these viruses and the NDDs Alzheimer's disease (AD) and multiple sclerosis (MS), respectively. We then (iii) highlight and discuss potential mechanisms by which these viruses exert negative effects on neurons and glia. Finally, we (iv) discuss how these viruses could interact with other disease-modifying factors to contribute to the initiation and/or progression of NDDs.
Keywords: herpes simplex virus 1, human herpesvirus 6, central nervous system, neurodegeneration, demyelination, Alzheimer's disease, multiple sclerosis, viral latency, viral reactivation
Introduction
The human brain is, despite its immune-privileged status, susceptible to infection by a vast number of neurotropic viruses both during development and adulthood. This review discusses the impact of persistent viral infections on the central nervous system (CNS) with a focus on two prevalent members of the human herpesvirus (HV) family, herpes simplex virus 1 (HSV-1), and human herpesvirus-6 (HHV-6). These viruses have been clinically associated with Alzheimer's disease (AD) and multiple sclerosis (MS), respectively, but causal relationships have not been firmly established. We propose that the persistent presence of these viruses establishes a state of vulnerability that can contribute to disease progression and neurodegeneration in AD and MS.
HSV-1, HHV-6A, and HHV-6B (two distinct HHV-6 viruses with HHV-6A being more neurotropic) (De Bolle et al., 2005)), together with six other members of the human HV family (herpes simplex virus 2 (HSV-2), varicella-zoster virus (VZV), Epstein-Barr virus (EBV), cytomegalovirus (CMV), human herpesvirus-7 (HHV-7), and human herpesvirus-8 (HHV-8 or Kaposi's sarcoma-associated herpesvirus)), are known to be associated with human neurological pathologies (reviewed in Soares and Provenzale, 2016). They all share several structural and genomic characteristics, each consisting of a large (125–235 kb), linear, double-stranded DNA genome enclosed within an icosahedral capsid (Boehmer and Nimonkar, 2003). HVs also carry a number of viral and cellular proteins surrounding their nucleocapsids (Curanovic and Enquist, 2009). With the exception of HSV-2 and HHV-8, these viruses are ubiquitous in the population and are mostly acquired in the first years of life.
Neurotropism of HVs
One notable feature of many HVs is their capacity to enter the CNS via peripheral axons and the bloodstream. The CNS parenchyma is separated from the bloodstream and adjacent tissues by the blood-brain barrier (BBB), a highly specialized structure comprised of endothelial cells interconnected by tight junctions and interacting with pericytes and astrocytic end-feet (Abbott et al., 2006). In addition to its function in regulating oxygen, ion, and nutrient flux between the brain and vasculature (Abbott et al., 2010), the BBB plays a significant role in protecting the brain from the non-selective diffusion of macromolecules and, importantly, viruses (Salinas et al., 2010).
Like other microbes, HVs have evolved methods to both avoid and traverse the BBB in order to enter and infect the CNS. Two primary routes of HV infection of the CNS are hematogenous dissemination, or direct transport across the BBB, and retrograde dissemination, in which viruses infect peripheral nerve endings and utilize axonal transport networks to invade the brain (Zhou et al., 2013)
Viral routes to the CNS
While many HVs can directly infect neuronal and glial cells, most establish their initial infections in epithelial cells at the respiratory and oropharyngeal surfaces (Mori et al., 2005). The direct connections between peripheral nervous system (PNS) and CNS neurons provide a direct route of CNS entry for HVs (reviewed in Salinas et al., 2010). CNS projections of the olfactory system are also uniquely susceptible to viral infection. Olfactory receptor neurons are directly exposed to the external environment, and as such provide a direct route for viruses to invade the olfactory and limbic systems of the CNS. Indeed, both HHV-6 and HSV-1 have been identified in the olfactory bulb (Harberts et al., 2011; Menendez and Carr, 2017).
HHV-6 has also been proposed to enter the brain via a ‘trojan horse’ mechanism that exploits the enhanced permeability of endothelial and parenchymal basement membranes in cases of increased inflammation (Kristensson, 2011). The high tropism of HHV-6 for activated CD4+ T lymphocytes (Takahashi et al., 1989) would allow viral entry into the CNS via these cells, although such a route of entry has not yet been has not directly demonstrated.
Viral dissemination and detection within the CNS
The presence of HV DNA and antigen in various brain regions has been well established and suggests that HVs are capable of disseminating throughout the brain. A PCR-based study investigating the prevalence of HVs in healthy CNS tissue identified the viral genomes of HSV, VZV, EBV, CMV, and HHV-6 in 28%, 32%, 38%, 22%, and 43% of samples, respectively (Sanders et al., 1996). HSV-1 viral antigen has been identified in the fronto- and mediotemporal regions of the brain in patients with HSV encephalitis, including the olfactory cortex, amygdala, hippocampus, insula, and cingulate gyrus (Esiri, 1982). This detection is, however, not limited to brains of patients with neurological diseases. A PCR-based detection study by Baringer and Pisani identified HSV-1 DNA in similar regions in healthy human brain samples (Baringer and Pisani, 1994). A recent transcriptomics analysis of the adult brain demonstrated heightened expression of HSV-1 receptors in the hippocampus, which may explain the unusually high tropism the virus has for this limbic structure (Lathe and Haas, 2017).
The presence of HHV-6 genome and antigen have likewise been detected in both healthy and diseased CNS samples. In patients with mesial temporal lobe epilepsy, HHV-6 DNA was detected in the hippocampus and temporal lobe (Donati et al., 2003; Fotheringham et al., 2007), while another study found the viral genome in regions as dispersed as the hindbrain and spinal cord (Harberts et al., 2011).
The mechanism of viral dissemination in the brain is not well understood, but it has been suggested that, at least in the case of HSV-1 release, neurosecretion proteins such as synaptosomal-associated protein 25 (SNAP-25), Rab3A, and growth associated protein 43 (GAP-43), may be involved (Miranda-Saksena et al., 2009).
Stages of the Human Herpesvirus Replicative Cycle
The ability of many HVs to persist in the CNS regions described above depends on unique aspects of their replicative cycle. As viruses rely on their hosts for replication, the evolution of these pathogens has favored infections that allow both virus and host to survive for extended periods of time. HVs have thus developed the ability to silence and reactivate their infectious cycle in response to immune pressure. After primary infection, HVs enter a quasi-quiescent state, termed latency, in which the viral genome is generally silenced but subject to sporadic periods of reactivation (Figure 1).
Figure 1.
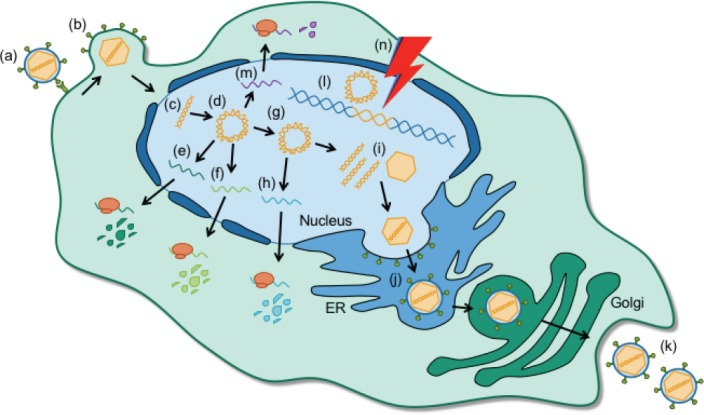
Stages of the human herpesvirus (HV) infectious cycle.
HVs enter cells by (a) binding cell surface receptors and (b) fusing their viral envelope with the host cell membrane. (c) Viral DNA is released into the nucleus where it (d) circularizes and sequentially transcribes (e) immediate early and (f) early viral transcripts and proteins. (g) Upon replication of the viral genome, (h) late transcripts and proteins are expressed. (i) Newly synthesized viral DNA is packaged into capsids, (j) trafficked through the endoplasmic reticulum (ER) and trans golgi network, and (k) released into the extracellular space to infect neighboring cells. (l) Latent viral DNA remains as a circular episome (HSV-1) or integrates into the host genome (HHV-6) and does not actively replicate. (m) Maintenance of HHV latency depends on the expression of latency associated transcripts (LAT in HSV-1) and/or proteins (U94 in HHV-6). (n) External and internal stressors, such as UV exposure and psychological stress, can cause the latent virus to reactivate and produce new viral molecules.
Productive infection
Initial infection of CNS cells depends primarily on cell-specific expression of target receptors (McGavern and Kang, 2011). HSV-1 infection of dorsal root ganglia neurons, for example, is mediated by an interaction between viral glycoprotein D and the cell adhesion molecule nectin-1 (Richart et al., 2003). HHV-6 has a less confined cell tropism. The ubiquitous type-I glycoprotein CD46, a protein expressed on the surface of all nucleated cells, mediates cellular entry of HHV-6 by interacting with viral glycoproteins G, H, and L (Santoro et al., 1999; Mori et al., 2003). Following receptor binding, incorporation of HVs into target cells is accomplished by envelope glycoproteins that promote fusion between viral and cellular membranes (Figure 1a, b) (Radtke et al., 2006).
Upon fusion with the host cell, the de-enveloped HV releases its viral DNA into the nucleus where it circularizes before transcription and replication of viral DNA occur (Figure 1c, d). Productive HV infection is characterized by the sequential expression of three subsets of lytic genes via cellular RNA polymerase II (Figure 1e, f, h) (reviewed in Boehmer and Nimonkar, 2003).
Transcription of the HSV-1 immediate early (IE) gene set is initially regulated by the viral transactivator VP16, a component of the HSV-1 tegument (Boehmer and Nimonkar, 2003), while HHV-6 IE expression has been proposed to rely on the interaction between cellular transcription factors and viral DNA sequences (Mirandola et al., 1998; De Bolle et al., 2005). IE gene products function primarily to initiate viral reproduction by activating expression of early (E) and late (L) gene sets, while E genes promote nucleic acid metabolism, facilitate viral DNA synthesis, and can serve immunomodulatory functions to promote viral survival (Fox et al., 2017; Nishimura et al., 2017). In turn, the structural units of new HV virions encoded by L genes are expressed only following replication of the viral genome (Figure 1g, h). Newly synthesized viral DNA and structural proteins are then assembled into capsids which traverse the trans-Golgi network to acquire viral glycoproteins before they are released from the cell by exocytosis (Figure 1i–k) (Smith, 2012; Agut et al., 2015). Although productive infection by many viruses causes direct cell lysis, HVs are capable of budding from the host cell without rupturing the plasma membrane. Highly productive infections, however, ultimately cause apoptosis or necrosis of the host cell by affecting the DNA replication and protein synthesis machinery (Grinde, 2013; Agut et al., 2015).
Viral latency
Viral latency, a stage in which viral DNA replication is silenced and viral DNA persists without producing new, infectious virions, is a common feature after primary HV infection. Many HVs can persist in the CNS without causing any apparent symptoms (Pantry et al., 2013; Menendez et al., 2016). Latent HV DNA is canonically known to take the form of a circular episome within the nucleus (Figure 1l). This is true for HSV-1, the genome of which associates with core histones (Deshmane and Fraser, 1989). HHV-6, however, is unique in that it establishes latency by direct integration into host DNA (Figure 1l). This is likely due to homologous recombination events between host telomeres and the perfect telomeric repeats found in the periphery of the HHV-6 genome (Arbuckle et al., 2010). Germline transmission of chromosomally integrated HHV-6 (ciHHV-6) has also been demonstrated and results in affected progeny harboring the full HHV-6 genome in every nucleated cell (Tanaka-Taya et al., 2004).
Irrespective of how latency is established, a hallmark of latency in both HSV-1 and HHV-6 infections is the expression of latency-associated gene products (Figure 1m). The HSV-1 latency-associated transcript (LAT) and several viral miRNAs are involved in promoting host cell survival and targeting viral transactivators for degradation to preserve latency (Thompson and Sawtell, 2001; Umbach et al., 2008). The HHV-6 latency product U94 is likewise suggested to play a major role in the establishment and maintenance of latency, although it is dispensable for chromosomal integration (Wallaschek et al., 2016). The state of latency is also maintained by a number of CNS immune factors that suppress viral replication (St Leger and Hendricks, 2011; Rosato and Leib, 2015).
While a longstanding viewpoint poses latency as a benign and “silent” state, it appears that latent infections do not only represent a potential reservoir for viral reactivation (see below), but that the latent virus itself might contribute to disease-modifying events, as discussed later.
Viral reactivation
While latency preserves dormant HVs in the host throughout life, both HSV- and HHV-6 maintain the ability to “wake up” and produce infectious virions (Figure 1n) (Owens et al., 2011; Bennett et al., 2012). This process, termed reactivation, can occur when the host immune system is weakened or impaired. Transplant patients receiving immunosuppressants often present with drastic increases in viral activity (Yamane et al., 2007) and the internal stress of aging has also been seen to weaken the immune system, allowing for the reactivation of HVs (Bennett et al., 2012). Stressors like fever, UV exposure, hormonal fluctuation, and cranial trauma, have all been shown to trigger HSV-1 reactivation, while superinfection with other HVs can reactivate the latent HHV-6 genome (Katsafanas et al., 1996; Wilson and Mohr, 2012). Reactivation of HSV-1 and HHV-6 often leads to cold sores and skin rashes, respectively, but can in rare cases also result in encephalitis and cognitive dysfunction (Steiner, 2011; Zerr et al., 2011; Ogata et al., 2013).
Links between stress and reactivation may originate on either side of the delicate balance between HVs and the host immune response; stressors are known to influence CD8+ T cell behavior and have recently been shown to directly modulate the activity of HSV-1 initiator proteins (Tanguy Le Gac and Boehmer, 2002). Specific pathways triggering transactivation of HHV-6 lytic proteins during reactivation, however, are still undetermined (Agut et al., 2015).
Human Herpesviruses and Neurodegeneration
We have thus far discussed that ability of HVs to enter the brain, disseminate in CNS tissue and enter a stage of latency that allows for long-term persistence. A number of HV proteins have been demonstrated to enhance this persistence by influencing the biology of host cells in their favor. It is now also clear that the latency proteins encoded by some HVs are capable of interfering with host cell function.
The relevance of such interactions is highlighted by growing epidemiological data suggesting a link between HV infections and chronic neurodegenerative diseases (NDDs) of the CNS. NDDs affect 37 million people worldwide and are the fourth leading cause of death in developed countries (Zhou et al., 2013). As average lifespan increases and these diseases become more prevalent, it becomes increasingly important to identify the factors that contribute to disease incidence and progression. Although advances in genome-wide association methodologies have allowed for the identification of several disease-related risk alleles (Brouwers et al., 2008; Baranzini and Oksenberg, 2017), other etiological and/or modulatory factors for NDDs are still largely unknown.
Studies on the impact of viral proteins on neurons and glia in vitro and in vivo illustrate their effects on processes including autophagy and progenitor cell proliferation, migration and maturation. These process are often affected in NDDs, and it is thus timely to consider viral proteins as biologically active agents that have the potential to initiate or exacerbate disease onset and progression. HSV-1 and HHV-6 provide compelling examples of the potential role of resident human viruses in NDDs.
HSV-1 and Alzheimer's disease (AD)
AD is considered one of the most common NDDs and is the leading cause of dementia in the elderly. AD is characterized by the accumulation of extra- and intracellular amyloid plaques consisting of amyloid β (Aβ), a cleavage product of amyloid precursor protein (APP) (Figure 2a) (Baranello et al., 2015). In healthy brains, Aβ oligomers can be trafficked to and degraded within the lysosome (Figure 2b) (Li et al., 2012), and while its normal function is not fully understood, soluble Aβ oligomers may serve important antimicrobial protective functions by binding microbial cell wall carbohydrates and inhibiting pathogen adhesion to host cells (Soscia et al., 2010; Kumar et al., 2016).
Figure 2.
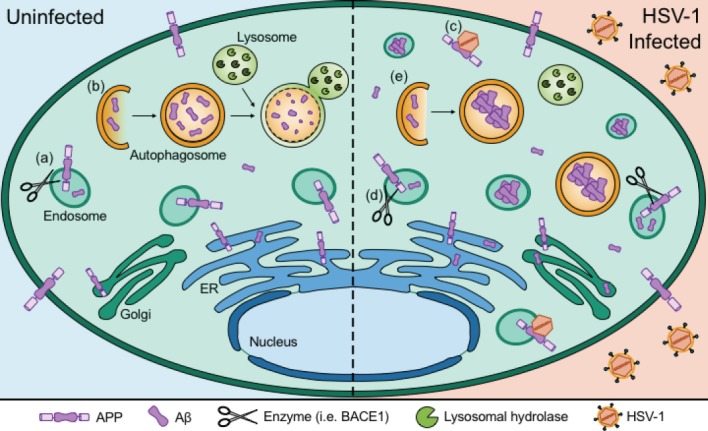
Herpes simplex virus 1 (HSV-1) infection alters production and degradation of amyloid.
Amyloid precursor protein (APP) is produced within the endoplasmic reticulum (ER) and Golgi system and is present on the surfaces of both the plasma membrane and endosomes. (a) APP is cleaved by enzymes such as β-site APP cleaving enzyme (BACE1) to produce amyloid β (Aβ). (b) Aβ proteins and aggregates can be broken down by the process of autophagy, in which it is recruited to autophagosomes that fuse with the lysosomal compartment to be degraded by lysosomal hydrolases. Intracellular Aβ can also accumulate within endosomes and the ER/Golgi system. (c) HSV-1 capsids associate with APP to alter its distribution. (d) HSV-1 infection increases expression of BACE1, which promotes the production of Aβ. (e) HSV-1 inhibits autophagic processing of Aβ, causing intracellular Aβ oligomers to accumulate within autophagosomes and endosomes.
In the case of AD, however, Aβ accumulates due to an imbalance between production and clearance of the protein (Baranello et al., 2015). Defects in autophagic processing of these aggregates has been demonstrated, as autophagosomes accumulate in affected neurites (Nixon et al., 2005) and induce neuronal apoptosis, which in turn can drive degeneration of CNS tissue (Loo et al., 1993).
Clinical evidence for a role of HSV-1 in AD
Jamieson et al. (1991) first suggested a link between HSV-1 infection and neurodegeneration due to the presence of HSV-1 DNA in a large proportion of AD-affected brain samples. However, HSV-1 detection was not specific to AD samples, as a considerable proportion of non-affected elderly people also showed HSV-1 burden. Given its ubiquitous nature and the fact that HSV-1 replication appears to increase in the elderly brain regardless of AD (Wozniak et al., 2005), further studies were conducted to prove a pathogenic role. Investigation of the localization of HSV-1 DNA revealed greater co-localization between HSV-1 DNA and Aβ in AD brains compared to age-matched, non-affected controls (72% vs. 24%), lending credence to the hypothesis that HSV-1 directly impacts the pathological hallmarks of AD (Wozniak et al., 2009). Further support for a link between HSV-1 and AD has been provided by studies on the humoral responses to the virus; the presence of anti-HSV-1 IgM antibodies (indicative of primary or reactivated infection) significantly increased the risk of developing AD in a longitudinal cohort study, while the presence of IgG antibodies (indicative of a life-long infection) did not (Lovheim et al., 2015). These results suggest that the presence of persistent HSV-1 alone does not contribute to the development of AD, but that reactivated virus plays a significant role. It has long been suggested that this viral reactivation occurs in the elderly as a result of natural weakening of the immune response, as is discussed below.
As stated earlier, HSV-1 has been detected in limbic structures of both normal post mortem samples and those derived from patients with HSV encephalitis (Esiri, 1982; Baringer and Pisani, 1994). Parallels have been drawn between these patterns and the distribution of neurodegeneration in AD patients, indirectly supporting the possibility of a link between HSV-1 and AD. Interestingly, recent findings have shown that anti-HSV-1 IgG titers correlate significantly with gray matter volume in temporal and orbitofrontal cortices of AD patients, as measured by magnetic resonance imaging. In an attempt to justify these seemingly counterintuitive findings, the authors suggest that antibodies against HSV-1 may serve a protective role against virus-induced neurodegeneration (Mancuso and Cao, 2014).
More recent studies have expanded the link between HSV-1 and AD to include other forms of cognitive dysfunction across ages and disease states. In children, for example, HSV-1 seropositivity correlates with cognitive impairments, while middle-aged adults show a correlation between seropositivity and impaired reading and visuospatial processing (Tarter et al., 2014). Another study examining schizophrenic patients and their non-psychotic relatives found that exposure to the virus, measured by antibody titers against an HSV-1 glycoprotein, was significantly associated with decreased neurocognitive performance regardless of disease state (Watson et al., 2013). Taken together, these studies suggest that not only does active HSV-1 infection confer greater risk for development of AD specifically, but that infections lead to neuronal death and result in cognitive impairments regardless of age or other comorbidities.
Potential mechanisms of HSV-1 involvement in AD
The past decade has seen a surge of studies identifying potential mechanisms by which HSV-1 infection could produce the pathological hallmarks of AD. The imbalance between the production and degradation of Aβ oligomers likely appears, at least in part, as a consequence of HSV-1's methods of cellular invasion and adaptive response to host cell immunity (Figure 2).
In vitro infection with HSV-1 has been shown to affect processing and distribution of APP, the precursor to neurotoxic Aβ, by multiple mechanisms. Even the earliest event in HSV-1 infection, binding of the virus to neuronal membranes, has been shown to enhance APP phosphorylation and Aβ accumulation (Piacentini et al., 2011). Cheng and colleagues have also shown that HSV-1 itself interacts with APP, hypothesizing that the virus binds APP to promote its own transport (APP is a component of the anterograde transport machinery) at the expense of normal APP transport and distribution (Figure 2c) (Cheng et al., 2011). A more indirect mechanism of HSV-1-induced Aβ accumulation has also been postulated; RNA-activated protein kinase (PKR), a defensive viral RNA sensor, becomes activated in response to HSV-1 and promotes translation of β-site APP cleaving enzyme (BACE1) to enhance Aβ formation (Figure 2d) (Ill-Raga et al., 2011).
While characteristic AD protein aggregation appears to indirectly result from mechanisms that facilitate viral entry and transport, HSV-1 also directly inhibits autophagic processing to promote its own survival through the actions of the viral protein ICP34.5 (Talloczy et al., 2006). Orvedahl et al. (2007) found that the viral protein ICP34.5 can bind Beclin-1, a critical protein in the initiation of autophagy. Subsequent in vivo studies showed that recombinant virus expressing ICP34.5 lacking the N terminal Beclin-1 binding domain was less neurovirulent, replicated to lower viral titers (Orvedahl et al., 2007), and elicited a larger CD4+ T cell response against HSV-1 (Leib et al., 2009). In addition, ICP34.5 has been shown to target the PKR defense mechanism described above. Elongation initiation factor 2α (eIF2α) is normally phosphorylated by PKR in response to detection of virus-derived double-stranded RNA, and mediates translational shut-off, initiation of autophagic degradation of pathogens, and activation of immune sensors (Talloczy et al., 2002). Through activity of its C terminal domain, ICP34.5 has been demonstrated to recruit host cell phosphatases to inactivate eIF2α and inhibit autophagy (He et al., 1997; Talloczy et al., 2002). A more recent study linked HSV-1 infection to the inhibition of Aβ breakdown by showing that HSV-1 infection induces accumulation of intracellular Aβ in autophagosomes that fail to fuse with the lysosomal compartment (Figure 2e) (Santana et al., 2012).
Interactions between HSV-1 and other AD risk factors
Attempts to prove causal links between infection with a ubiquitous virus and development of NDDs are almost always met with skepticism, given the discrepancies between infection prevalence and epidemiological data on neurodegeneration. However, such studies are inherently flawed if one considers HSV-1 as a “secondary insult” that needs to occur in conjunction with other known risk factors. Such a concept has been well established in other neurological disorders, such as autism spectrum disorder (ASD). Many hundreds of genetic risk loci have been identified that alone do not cause ASD, but clearly increase risk of ASD development if the brain is exposed to another genetic or environmental insult (Schmidt et al., 2014). AD is likewise a multifactorial disease, with identified risk factors including gene mutations, traumatic brain injury, and cardiovascular conditions.
The probability of HSV-1 acting as a “secondary insult” in the context of genetic risk factors was first tested by Itzhaki et al. (1997) who showed in a seminal paper that only in patients carrying the APOE-ε4 allele did CNS infection with HSV-1 confer a significant risk for the development of AD. This work led to the current hypothesis that the specific APOE-ε4 isoform renders the CNS more susceptible to primary infection, persistence, and reactivation of HSV-1. Subsequent in vivo studies demonstrated significant increases in HSV-1 IE gene expression and viral titers in the brains of APOE-ε4 transgenic mice as compared to APOE knockout animals and carriers of other APOE alleles (Burgos et al., 2006; Miller and Federoff, 2008), thus supporting this hypothesis. A study by Guzman-Sanchez et al. (2012) further demonstrated that the increase in HSV-1 infection due to APOE expression also leads to impaired cognitive performance in mice.
In recent years, large-scale genetic approaches have identified other possible links between HSV-1 and AD. Datasets from GWAS studies of AD susceptibility suggest enrichment in a number of genes that are associated with viral transport, entry into neurons, and the regulation of host immune defenses (Porcellini et al., 2010). More recent studies found a notable overlap between susceptibility genes for a number of NDDs and HSV-1 associated genes, proposing that HSV-1 binding interactions with proteins encoded by these susceptibility genes might enhance their ability to influence disease risk (Carter, 2013).
The greatest known risk factor for AD is aging. Given the increased prevalence of HSV-1 DNA in elderly compared to young brains, it has been hypothesized that the side effects of aging, namely, weakening of the immune system, directly contribute to the effects of HSV-1 on age-related CNS pathology (Wozniak et al., 2005, 2009). Indeed, immune suppression has already been shown to enhance HSV-1 reactivation within the human brain (Saldanha et al., 1986).
AD, along with other NDDs, is associated with chronic CNS inflammation mediated by glial cells and various inflammatory mediators. While neuroinflammation in AD is initiated to protect against damaging protein aggregates, the unresolved activation of immune cells actually enhances development of AD pathology (Mrak and Griffin, 2005; Heneka et al., 2015). Combined with the presence of latent HSV-1, this presents a scenario of inflammatory feedback; inflammation can reactivate latent HSV-1, which in turn drives further neuro-immune activation (Rock et al., 2004). Even at the level of single neurons, in vitro HSV-1 infection results in upregulation of the proinflammatory cytokine interleukin-1β, sustained release of which is commonly seen in AD (Hill et al., 2009).
A direct link between HSV-1 infection and other AD risk factors such as traumatic brain injury and cardiovascular conditions is less clear. Trauma and cardiovascular diseases such as hypertension are associated with a loss of cerebrovascular control. Cerebrovascular dysregulation is believed to be both caused by AD and to be a driving mechanism in disease progression (Burgmans et al., 2013). In the senescence-accelerated mouse model of AD, animals show increasing permeability of the BBB with age that specifically localizes to the limbic structures affected by AD and HSV-1 infection (Pelegri et al., 2007; Del Valle et al., 2009). While any direct relationship between BBB alterations and HSV-1 infection is unclear, it is possible that BBB permeabilization could enhance HSV-1 entry into the brain to drive further CNS inflammation and neurodegeneration. However, although HSV-1 is capable of infecting peripheral immune cells (Kruse et al., 2000; Paludan et al., 2001), passage across the BBB via these cells in humans has not been explicitly documented.
Taken together, there is growing evidence suggesting that the response to the endemic HSV-1 virus can drive development of AD pathology in the context of other disease-modifying factors. By interacting with alleles that confer greater susceptibility to infection, persistence, and reactivation of HSV-1 in the CNS, this virus appears to play a direct role in AD. While no precise mechanism has been confirmed to be the cause of neurodegeneration in AD patients, considerable evidence has been provided to suggest that HSV-1 gene products enhance the accumulation and impair the autophagic degradation of AD protein aggregates. Other driving forces in AD development, aging, chronic inflammation, and cerebrovascular dysregulation, are also likely to enhance HSV-1 infection of the CNS and contribute to neuronal death. Effects of HSV-1 on other CNS cell types (Bello-Morales et al., 2005; Chucair-Elliott et al., 2014) in the context of other genetic risk factors may link this virus to a variety of NDDs (Carter, 2013) and identify it as a valuable target in NDD therapeutic development.
HHV-6 and multiple sclerosis (MS)
MS is an autoimmune, demyelinating NDD of the human CNS predominantly affecting the white matter of the brain, spinal cord, and optic nerves (reviewed in Garg and Smith, 2015). The disease disproportionally affects young adults with a preponderance in women, in whom it is one of the leading causes of disability in North America and northern Europe (Browne et al., 2014).
The pathology of MS is well documented. Classic MS lesions consist of focal sclerotic demyelinated plaques often accompanied, in early disease, by areas of incomplete remyelination. These areas, called “shadow plaques” contain thinly myelinated axons (Barkhof et al., 2003). The presence of gray matter demyelination and axonal damage has also been recognized in early MS (Trapp et al., 1998). Although a majority of patients present with relapsing-remitting neurological deficits, a subset of patients develop a secondary progressive disease course with few remissions. Primary progressive MS is defined by progressive cumulative disability from the outset (Garg and Smith, 2015).
Overall, the course of MS disease progression seems to be defined by the balance between the severity of demyelination and the success of remyelination and repair processes that are carried out by the endogenous oligodendrocyte progenitor cell (OPC) pool (Tognatta and Miller, 2016). Impairment of the two critical functions of these cells, migration to sites of myelin damage and maturation into myelinating oligodendrocytes, has been suggested to contribute to disease severity and progression in MS (Kuhlmann et al., 2008; Boyd et al., 2013).
Clinical evidence for a role of HHV-6 in MS
Since its first description in the mid-nineteenth century, a wide range of viral and bacterial pathogens have been suggested to play a role in MS pathology. Nearly all potential candidates, however, have subsequently been dismissed due to inconsistent data and a overall lack of reproducibility. Exceptions are EBV (reviewed in Pakpoor et al., 2013) and HHV-6 (reviewed in Leibovitch and Jacobson, 2014), which remain prominent disease-altering candidates.
A proposed association between HHV-6 and MS was first made in the early 1990s with the observation of increased anti-HHV-6 antibody titers in sera of some MS patients in comparison to a cohort of normal controls (Sola et al., 1993). A subsequent study demonstrated increased detection of HHV-6 DNA in the cerebrospinal fluid (CSF) in MS patients (Wilborn et al., 1994). More compelling, however, was the first histopathological study demonstrating increased detection of HHV-6 DNA and proteins within MS plaques and, specifically, within oligodendrocytes when compared to control tissue (Challoner et al., 1995). These findings were supported by studies using a variety of techniques that demonstrated increased HHV-6 DNA in MS plaques versus adjacent normal appearing white matter (Blumberg et al., 2000; Goodman et al., 2003).
Involvement of HHV-6 in MS has also been proposed using immunological readouts. A study by Soldan et al. (1997) reported increased anti-HHV-6 IgM responses to HHV-6 early antigen (p41/38) in patients with relapsing-remitting MS when compared to healthy controls. Importantly, there was no difference in HHV-6 IgG titers, suggesting that differences in IgM production resulted from reactivation of the virus rather than primary infection. A more recent serological study found both an increased prevalence and increased average IgG titer against the predominant HHV-6 latency-associated protein, U94, in MS patients versus controls (Ben-Fredj et al., 2013), suggesting a possible role for latent HHV-6 in MS.
These reports are contrasted by a number of studies failing to show an association between HHV-6 and MS. Merelli et al. (1997) detected HHV-6 DNA more frequently in the cerebral tissue of normal adults compared to MS patients, while another study reported HHV-6 DNA in as many MS as control brains (Gordon et al., 1996). These findings might be explained, in part, by the highly variable levels of latent and reactivated virus in the CNS. However, given the ubiquitous nature of this virus, further studies are needed to discern contributions of latent and reactivated HHV-6 to MS occurrence and progression.
Potential mechanisms of HHV-6 involvement in MS
Given the discrepancies in clinical reports on the link between HHV-6 and MS, it remains important to investigate whether this virus can mechanistically replicate characteristics of disease in relevant cell types. Recent results suggest that active and latent HHV-6 infection of immune and glial cells can alter the sensitive balance between de- and remyelination that defines MS progression.
The inflammatory component of MS has led to the suggestion that HHV-6 infection might have an immunomodulatory effect that indirectly affects surrounding cells (Arena et al., 1997). This possibility was explored by Kong et al. (2003) who demonstrated that supernatant from HHV-6-infected SupT1 cells inhibited the proliferation and viability of an oligodendroglial cell line. This indirect cytotoxicity, however, could theoretically only occur in cases where T cell influx to the CNS has already occurred, and thus cannot be considered a disease-initiating event in MS.
HHV-6 has also been suggested to enhance the development of autoimmunity. Tejada-Simon et al. (2003) made the intriguing observation that the HHV-6 protein U24 shares a seven amino acid sequence with myelin basic protein (MBP), a main component of the myelin sheath. The authors suggested that this shared sequence could trigger cross-reactivity between the viral and host proteins to cause aberrant autoimmune targeting of the myelin sheath (a phenomenon know as “molecular mimicry”). Indeed, their study showed that MS patients exhibited significantly higher frequencies of CD4+ T-cells cross-reactive to the same amino acid sequence in both U24 and MBP. However, antibody titers specific to the shared sequence were not significantly different in MS patients versus controls. More recently, a group identified CD8+ cytotoxic T-cells that were cross-reactive to U24 and MBP, further suggesting that molecular mimicry could result in the direct targeting of oligodendrocytes (Cheng et al., 2012). It thus appears that while HHV-6 infection alone cannot trigger MS onset, it could act to enhance the inflammatory state of the CNS to exacerbate demyelination in MS patients.
As a persistent resident of the CNS, it is also possible that HHV-6 plays a role in the failure of myelin repair that invariably occurs as MS progresses through the lifespan (Tognatta and Miller, 2016). Even in the absence of inflammatory lesion formation, HHV-6 infection of the OPC population could impair remyelination to exacerbate disease progression (Figure 3). We examined this possibility in a 2004 study where we infected proliferating human OPCs with fluorescently labeled HHV-6 virions. Infection led to G1/S cell cycle arrest and premature differentiation of these cells into oligodendrocytes but did not result in increased cell death (Figure 3e–d) (Dietrich et al., 2004). Thus, acute CNS infection or re-activation of HHV-6 later in life could conceivably cause localized depletion and premature differentiation of the OPC pool.
Figure 3.
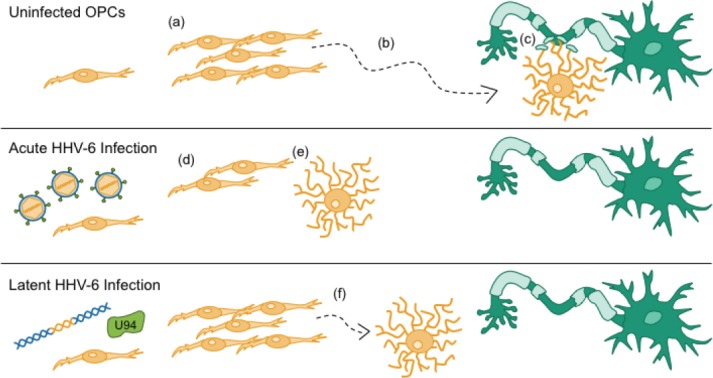
Acute and latent human herpesvirus 6 (HHV-6) infections alter components of the myelin repair response.
In response to demyelinating injury, resident oligodendrocyte precursor cells (OPCs) (a) proliferate and (b) migrate to lesion sites, where they (c) differentiate into mature, myelinating oligodendrocytes (OLs) and wrap denuded axons with new myelin sheaths. Acute infection of OPCs with HHV-6 leads to (d) cell cycle arrest and (e) premature differentiation. Latent HHV-6 infection, as modeled by expression of the latency gene U94, (f) inhibits migration of OPCs. Collectively, the effects of both acute and latent infections of OPCs with HHV-6 alter critical components of the myelin repair response, which may impair remyelination and functional recovery in patients with demyelinating diseases.
We also considered the possibility that latent infection with HHV-6 may play a role, as no studies have shown evidence for actively replicating viral particles in the demyelinative lesions of MS (Opsahl and Kennedy, 2005). As noted earlier, a recent serological study detected significantly higher antibody titers against HHV-6 latency gene U94 in MS patients vs. controls (Ben-Fredj et al., 2013), providing a viral candidate protein that could be active in MS relevant cells during latency and could affect their function. To test this possibility, human OPCs expressing U94 by a lentiviral vector were compared to control OPCs in analyses of cell viability, division, migration, and differentiation, all of which are critical OPC functions during remyelination (Figure 3a–c) (Tognatta and Miller, 2016). Although we found no differences in cell viability, division, or differentiation, we observed a significant migratory impairment in U94+ OPCs both in vitro and in a murine model of demyelination (Figure 3d) (Campbell et al., 2017). These findings suggest that the expression of viral transcripts and proteins during HHV-6 latency may contribute to the progression of chronic demyelinating diseases by direct inhibition of OPC migration and subsequent myelin repair. These data also strongly contradict the dogma that latent HHV-6 in the human CNS is merely commensal, although further research is necessary to investigate whether these effects on OPC migration manifest as impaired remyelination.
Interactions between HHV-6 and other MS risk factors
There exists compelling evidence to suggest that the presence of HHV-6 in the human CNS represents a vulnerability that could disrupt the balance between demyelination and remyelination in patients with MS. Although clinical data are conflicting, it is not likely that a ubiquitous pathogen such as HHV-6 can act as the sole trigger for the development of demyelinating diseases. Rather, HHV-6 may exacerbate oligodendrocyte death and demyelination only in the context of other genetic and environmental risk factors for MS. Even in the absence of reactivated virus, recent evidence suggests that the HHV-6 latency gene product may cause profound impediments to normal myelin repair processes (Campbell et al., 2017).
The risk for acquisition of MS is known to have a strong genetic component with an inheritance pattern characteristic of polygenic diseases. Genome wide association studies have identified multiple single-nucleotide polymorphisms (SNPs) associated with this disease, most of which involve HLA loci (Baranzini and Oksenberg, 2017). In contrast to HSV-1 and AD, however, there have been no studies focused on identifying gene-virus interactions. The novel observation of latency genes being potentially involved in MS progression opens a new avenue for genome wide studies and might well lead to new, thus far unrecognized, interaction between susceptibility genes and viral components.
MS, like AD, exhibits many of the hallmarks of inflammatory CNS disease. Transendothelial migration of leukocytes from the periphery into the CNS is enhanced in MS due to recurrent weakening of the BBB, even in the earliest stages of disease (Ortiz et al., 2014). BBB permeabilization is likely to enhance the entry of HHV-6 into the brain, as HHV-6 has a high tropism for activated CD4+ T lymphocytes (Takahashi et al., 1989). Enhanced CNS entry of HHV-6 in MS patients could exacerbate demyelination by any of the potential mechanisms described above. In parallel, an immune response against HHV-6 could cause further inflammatory damage within the CNS. Strong intrathecal T-cell responses to HHV-6 have been documented in MS patients, along with significantly increased T-cell release of the proinflammatory cytokine tumor necrosis factor α (TNF-α) (Wuest et al., 2014), suggesting that HHV-6-specific immune responses may contribute to inflammatory damage.
To effectively parse out the contributions of HHV-6 to MS disease occurrence and outcome, future studies are encouraged to consider HHV-6 infection, both active and latent, in conjunction with established genetic risk factors and inflammatory hallmarks of disease.
Concluding Remarks
Neurotropic HVs have coexisted with humans and their ancestors for approximately 200 million years (McGeoch et al., 1995), relying on the delicate balance between viral replication and immune pressure to maintain survival in the brains of relatively healthy hosts. As a result, these viruses have traditionally been considered a benign presence in the immunocompetent CNS.
Although clinical associations between HVs and NDs have been demonstrated for years, the ubiquitous nature of HV infection has made it difficult to draw causative connections. In this review, we have addressed a growing body of data considering the possibility that HVs are indeed capable of exacerbating pathogenesis when considered in the context of other genetic and environmental insults.
NDs are often defined by the targeted death of mature cells of the CNS, followed by repair from a limited pool of progenitor cells. It therefore remains important to consider the effects of viruses on both mature and progenitor cell types, as has been done for HHV-6. In the context of HSV-1 infection, viral gene products not only impact Aβ breakdown in neurons, but also drive apoptosis in neuronal progenitor cells (Chucair-Elliott et al., 2014). As neurogenesis appears to be a compensatory mechanism to replace lost neurons in AD brains (Jin et al., 2004), targeting HSV-1 may not only preserve mature neurons, but also protect valuable progenitors.
While most research on latency proteins is focused on determining how such proteins maintain the latency state of the virus and/or allow the virus to be reactivated, the impact of these proteins on cell specific functions of the host are not well understood. It is therefore of critical importance that we investigate latency gene products in the context of CNS disease (Campbell et al., 2017). It is intriguing that the state of latency seems to have profoundly different effects on host cells in HSV-1 compared to HHV-6. For example, HSV-1 LAT enhances neuronal survival (Thompson and Sawtell, 2001), while the HHV-6 latency product U94 inhibits migratory processes in OPCs. It remains to be seen, however, whether these effects are cell type specific.
We propose that it will be necessary to view a latent infection not merely as a benign state of a viral infections, but as a potential state of heighted vulnerability. In respect to HHV-6, it would be beneficial to focus epidemiological studies not only on a potential association of HHV-6 with MS occurrence, but also on specific associations between HHV-6 and different courses of disease progression. It will also be necessary to develop new animal models that recapitulate the potential role of human viral proteins in disease.
Given the fact that many latent viral proteins, like U94, suppress viral replication, targeting such proteins directly may lead to viral reactivation and perhaps severe neurological side effects. Therefore, identification of the central mechanism of proteins like U94 may provide novel avenues for intervention in MS patients with latent HHV-6 infection. This would be particularly exciting, considering that no therapies are yet approved to target endogenous remyelination in MS patients. Finally, if we consider the advance of cell therapy using transplantation approaches, it will be critical to screen human graft cells for the presence of viral proteins before transplantation into human recipients.
Taken together, the studies discussed in this review demonstrate the effects of HSV-1 infection on amyloid-beta accumulation in AD and of HHV-6 infection on oligodendrocyte and OPC function in MS. Together they raise the possibility that these viruses contribute to disease and thus serve as potential targets for therapeutic intervention.
Acknowledgments:
We thank all members of the Mayer-Proschel, Mark Noble and Christoph Proschel laboratories for continuous discussion and input into the ongoing work that is related to the review. We also thank the HHV-6 Foundation for their tireless efforts to educate the scientific community of the role of viruses in human disease. We sincerely apologize for all the important contributions that we were unable to cite here due to the limitation on the number of references allowed and the instruction to focus on the most recent publications.
Footnotes
Conflicts of interest: None declared.
Financial support: None.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review report:
Reviewer: Siyu Zhang, Shanghai Jiao Tong University, China.
Comments to authors: The authors chose a good topic about HVs' effect on neurodegenerative, but they did not limit the scope to make a comprehensive discussion. First, the authors presented characteristics of the HVs, including family members, structure, neurotropism and replicative cycle properties. Then, the authors reviewed the relationship between HSV-1 and AD, HHV-6 and MS. Finally, the authors summarized the potential mechanisms about HVS-1 and HHV-6 involved in these two neurodegenerative diseases.
References
- 1.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 2.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Agut H, Bonnafous P, Gautheret-Dejean A. Laboratory and clinical aspects of human herpesvirus 6 infections. Clin Microbiol Rev. 2015;28:313–335. doi: 10.1128/CMR.00122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, Komaroff AL, Ambros PF, Medveczky PG. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci U S A. 2010;107:5563–5568. doi: 10.1073/pnas.0913586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arena A, Liberto MC, Capozza AB, Foca A. Productive HHV-6 infection in differentiated U937 cells: role of TNF alpha in regulation of HHV-6. New Microbiol. 1997;20:13–20. [PubMed] [Google Scholar]
- 6.Baranello RJ, Bharani KL, Padmaraju V, Chopra N, Lahiri DK, Greig NH, Pappolla MA, Sambamurti K. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer's disease. Curr Alzheimer Res. 2015;12:32–46. doi: 10.2174/1567205012666141218140953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baranzini SE, Oksenberg JR. The genetics of multiple sclerosis: from 0 to 200 in 50 years. Trends Genet. 2017;33:960–970. doi: 10.1016/j.tig.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol. 1994;36:823–829. doi: 10.1002/ana.410360605. [DOI] [PubMed] [Google Scholar]
- 9.Barkhof F, Bruck W, De Groot CJ, Bergers E, Hulshof S, Geurts J, Polman CH, van der Valk P. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol. 2003;60:1073–1081. doi: 10.1001/archneur.60.8.1073. [DOI] [PubMed] [Google Scholar]
- 10.Bello-Morales R, Fedetz M, Alcina A, Tabares E, Lopez-Guerrero JA. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J Neurovirol. 2005;11:190–198. doi: 10.1080/13550280590924179. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Fredj N, Ben-Selma W, Rotola A, Nefzi F, Benedetti S, Frih-Ayed M, Di Luca D, Aouni M, Caselli E. Prevalence of human herpesvirus U94/REP antibodies and DNA in Tunisian multiple sclerosis patients. J Neurovirol. 2013;19:42–47. doi: 10.1007/s13365-012-0138-6. [DOI] [PubMed] [Google Scholar]
- 12.Bennett JM, Glaser R, Malarkey WB, Beversdorf DQ, Peng J, Kiecolt-Glaser JK. Inflammation and reactivation of latent herpesviruses in older adults. Brain Behav Immun. 2012;26:739–746. doi: 10.1016/j.bbi.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blumberg BM, Mock DJ, Powers JM, Ito M, Assouline JG, Baker JV, Chen B, Goodman AD. The HHV6 paradox: ubiquitous commensal or insidious pathogen? A two-step in situ PCR approach. J Clin Virol. 2000;16:159–178. doi: 10.1016/s1386-6532(99)00084-0. [DOI] [PubMed] [Google Scholar]
- 14.Boehmer PE, Nimonkar AV. Herpes virus replication. IUBMB Life. 2003;55:13–22. doi: 10.1080/1521654031000070645. [DOI] [PubMed] [Google Scholar]
- 15.Boyd A, Zhang H, Williams A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013;125:841–859. doi: 10.1007/s00401-013-1112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer's disease: an update. Ann Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- 17.Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, Thompson AJ. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83:1022–1024. doi: 10.1212/WNL.0000000000000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgmans S, van de Haar HJ, Verhey FR, Backes WH. Amyloid-β interacts with blood-brain barrier function in dementia: a systematic review. J Alzheimers Dis. 2013;35:859–873. doi: 10.3233/JAD-122155. [DOI] [PubMed] [Google Scholar]
- 19.Burgos JS, Ramirez C, Sastre I, Valdivieso F. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;80:5383–5387. doi: 10.1128/JVI.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell A, Hogestyn JM, Folts CJ, Lopez B, Proschel C, Mock D, Mayer-Proschel M. Expression of the human herpesvirus 6A latency-associated transcript U94A disrupts human oligodendrocyte progenitor migration. Sci Rep. 2017;7:3978. doi: 10.1038/s41598-017-04432-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter CJ. Susceptibility genes are enriched in those of the herpes simplex virus 1/host interactome in psychiatric and neurological disorders. Pathog Dis. 2013;69:240–261. doi: 10.1111/2049-632X.12077. [DOI] [PubMed] [Google Scholar]
- 22.Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng SB, Ferland P, Webster P, Bearer EL. Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS One. 2011;6:e17966. doi: 10.1371/journal.pone.0017966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng W, Ma Y, Gong F, Hu C, Qian L, Huang Q, Yu Q, Zhang J, Chen S, Liu Z, Chen X, Zhou T, Zhang D. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front Biosci (Landmark Ed) 2012;17:1648–1658. doi: 10.2741/4010. [DOI] [PubMed] [Google Scholar]
- 25.Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. 2014;62:1418–1434. doi: 10.1002/glia.22689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curanovic D, Enquist L. Directional transneuronal spread of alpha-herpesvirus infection. Future Virol. 2009;4:591. doi: 10.2217/fvl.09.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Bolle L, Van Loon J, De Clercq E, Naesens L. Quantitative analysis of human herpesvirus 6 cell tropism. J Med Virol. 2005;75:76–85. doi: 10.1002/jmv.20240. [DOI] [PubMed] [Google Scholar]
- 28.Del Valle J, Duran-Vilaregut J, Manich G, Camins A, Pallas M, Vilaplana J, Pelegri C. Time-course of blood-brain barrier disruption in senescence-accelerated mouse prone 8 (SAMP8) mice. Int J Dev Neurosci. 2009;27:47–52. doi: 10.1016/j.ijdevneu.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Deshmane SL, Fraser NW. During latency herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989;63:943–947. doi: 10.1128/jvi.63.2.943-947.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dietrich J, Blumberg BM, Roshal M, Baker JV, Hurley SD, Mayer-Proschel M, Mock DJ. Infection with an endemic human herpesvirus disrupts critical glial precursor cell properties. J Neurosci. 2004;24:4875–4883. doi: 10.1523/JNEUROSCI.5584-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, Heiss JD, Cogen P, Gaillard WD, Sato S, Theodore WH, Jacobson S. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology. 2003;61:1405–1411. doi: 10.1212/01.wnl.0000094357.10782.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esiri MM. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci. 1982;54:209–226. doi: 10.1016/0022-510x(82)90183-6. [DOI] [PubMed] [Google Scholar]
- 33.Fotheringham J, Donati D, Akhyani N, Fogdell-Hahn A, Vortmeyer A, Heiss JD, Williams E, Weinstein S, Bruce DA, Gaillard WD, Sato S, Theodore WH, Jacobson S. Association of human herpesvirus-6B with mesial temporal lobe epilepsy. PLoS Med. 2007;4:e180. doi: 10.1371/journal.pmed.0040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox HL, Dembowski JA, DeLuca NA. A herpesviral immediate early protein promotes transcription elongation of viral transcripts. MBio. 2017;8:e00745–17. doi: 10.1128/mBio.00745-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garg N, Smith TW. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015;5:e00362. doi: 10.1002/brb3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis. 2003;187:1365–1376. doi: 10.1086/368172. [DOI] [PubMed] [Google Scholar]
- 37.Gordon L, McQuaid S, Cosby SL. Detection of herpes simplex virus (types 1 and 2) and human herpesvirus 6 DNA in human brain tissue by polymerase chain reaction. Clin Diagn Virol. 1996;6:33–40. doi: 10.1016/0928-0197(95)00203-0. [DOI] [PubMed] [Google Scholar]
- 38.Grinde B. Herpesviruses: latency and reactivation - viral strategies and host response. J Oral Microbiol. 2013;5 doi: 10.3402/jom.v5i0.22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guzman-Sanchez F, Valdivieso F, Burgos JS. Aging-related neurostructural, neuropathological, and behavioral changes associated with herpes simplex virus type 1 brain infection in mice. J Alzheimers Dis. 2012;30:779–790. doi: 10.3233/JAD-2012-120070. [DOI] [PubMed] [Google Scholar]
- 40.Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A. 2011;108:13734–13739. doi: 10.1073/pnas.1105143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015;16:229–236. doi: 10.1038/ni.3102. [DOI] [PubMed] [Google Scholar]
- 43.Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport. 2009;20:1500–1505. doi: 10.1097/WNR.0b013e3283329c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ill-Raga G, Palomer E, Wozniak MA, Ramos-Fernandez E, Bosch-Morato M, Tajes M, Guix FX, Galan JJ, Clarimon J, Antunez C, Real LM, Boada M, Itzhaki RF, Fandos C, Munoz FJ. Activation of PKR causes amyloid ss-peptide accumulation via de-repression of BACE1 expression. PLoS One. 2011;6:e21456. doi: 10.1371/journal.pone.0021456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. Lancet. 1997;349:241–244. doi: 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- 46.Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer's disease brains. J Med Virol. 1991;33:224–227. doi: 10.1002/jmv.1890330403. [DOI] [PubMed] [Google Scholar]
- 47.Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katsafanas GC, Schirmer EC, Wyatt LS, Frenkel N. In vitro activation of human herpesviruses 6 and 7 from latency. Proc Natl Acad Sci U S A. 1996;93:9788–9792. doi: 10.1073/pnas.93.18.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong H, Baerbig Q, Duncan L, Shepel N, Mayne M. Human herpesvirus type 6 indirectly enhances oligodendrocyte cell death. J Neurovirol. 2003;9:539–550. doi: 10.1080/13550280390241241. [DOI] [PubMed] [Google Scholar]
- 50.Kristensson K. Microbes' roadmap to neurons. Nat Rev Neurosci. 2011;12:345–357. doi: 10.1038/nrn3029. [DOI] [PubMed] [Google Scholar]
- 51.Kruse M, Rosorius O, Kratzer F, Stelz G, Kuhnt C, Schuler G, Hauber J, Steinkasserer A. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J Virol. 2000;74:7127–7136. doi: 10.1128/jvi.74.15.7127-7136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 53.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016;8:340ra372. doi: 10.1126/scitranslmed.aaf1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lathe R, Haas JG. Distribution of cellular HSV-1 receptor expression in human brain. J Neurovirol. 2017;23:376–384. doi: 10.1007/s13365-016-0504-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol. 2009;83:12164–12171. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leibovitch EC, Jacobson S. Evidence linking HHV-6 with multiple sclerosis: an update. Curr Opin Virol. 2014;9:127–133. doi: 10.1016/j.coviro.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–44601. doi: 10.1074/jbc.M112.420224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lovheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer's disease. Alzheimers Dement. 2015;11:593–599. doi: 10.1016/j.jalz.2014.04.522. [DOI] [PubMed] [Google Scholar]
- 60.Mancuso L, Cao G. Acute toxicity test of CuO nanoparticles using human mesenchymal stem cells. Toxicol Mech Methods. 2014;24:449–454. doi: 10.3109/15376516.2014.928920. [DOI] [PubMed] [Google Scholar]
- 61.McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nat Rev Immunol. 2011;11:318–329. doi: 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 63.Menendez CM, Carr DJJ. Herpes simplex virus-1 infects the olfactory bulb shortly following ocular infection and exhibits a long-term inflammatory profile in the form of effector and HSV-1-specific T cells. J Neuroinflammation. 2017;14:124. doi: 10.1186/s12974-017-0903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menendez CM, Jinkins JK, Carr DJ. Resident T cells are unable to control herpes simplex virus-1 activity in the brain ependymal region during latency. J Immunol. 2016;197:1262–1275. doi: 10.4049/jimmunol.1600207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merelli E, Bedin R, Sola P, Barozzi P, Mancardi GL, Ficarra G, Franchini G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J Neurol. 1997;244:450–454. doi: 10.1007/s004150050121. [DOI] [PubMed] [Google Scholar]
- 66.Miller RM, Federoff HJ. Isoform-specific effects of ApoE on HSV immediate early gene expression and establishment of latency. Neurobiol Aging. 2008;29:71–77. doi: 10.1016/j.neurobiolaging.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 67.Miranda-Saksena M, Boadle RA, Aggarwal A, Tijono B, Rixon FJ, Diefenbach RJ, Cunningham AL. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J Virol. 2009;83:3187–3199. doi: 10.1128/JVI.01579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mirandola P, Menegazzi P, Merighi S, Ravaioli T, Cassai E, Di Luca D. Temporal mapping of transcripts in herpesvirus 6 variants. J Virol. 1998;72:3837–3844. doi: 10.1128/jvi.72.5.3837-3844.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mori I, Nishiyama Y, Yokochi T, Kimura Y. Olfactory transmission of neurotropic viruses. J Neurovirol. 2005;11:129–137. doi: 10.1080/13550280590922793. [DOI] [PubMed] [Google Scholar]
- 70.Mori Y, Yang X, Akkapaiboon P, Okuno T, Yamanishi K. Human herpesvirus 6 variant A glycoprotein H-glycoprotein L-glycoprotein Q complex associates with human CD46. J Virol. 2003;77:4992–4999. doi: 10.1128/JVI.77.8.4992-4999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mrak RE, Griffin WS. Potential inflammatory biomarkers in Alzheimer's disease. J Alzheimers Dis. 2005;8:369–375. doi: 10.3233/jad-2005-8406. [DOI] [PubMed] [Google Scholar]
- 72.Nishimura M, Wang J, Wakata A, Sakamoto K, Mori Y. Crystal structure of the DNA-binding domain of human herpesvirus 6A immediate early protein 2. J Virol. 2017;91:e01121–17. doi: 10.1128/JVI.01121-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 74.Ogata M, Satou T, Kadota J, Saito N, Yoshida T, Okumura H, Ueki T, Nagafuji K, Kako S, Uoshima N, Tsudo M, Itamura H, Fukuda T. Human herpesvirus 6 (HHV-6) reactivation and HHV-6 encephalitis after allogeneic hematopoietic cell transplantation: a multicenter, prospective study. Clin Infect Dis. 2013;57:671–681. doi: 10.1093/cid/cit358. [DOI] [PubMed] [Google Scholar]
- 75.Opsahl ML, Kennedy PG. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain. 2005;128:516–527. doi: 10.1093/brain/awh390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ortiz GG, Pacheco-Moises FP, Macias-Islas MA, Flores-Alvarado LJ, Mireles-Ramirez MA, Gonzalez-Renovato ED, Hernandez-Navarro VE, Sanchez-Lopez AL, Alatorre-Jimenez MA. Role of the blood-brain barrier in multiple sclerosis. Arch Med Res. 2014;45:687–697. doi: 10.1016/j.arcmed.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 77.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 78.Owens GP, Gilden D, Burgoon MP, Yu X, Bennett JL. Viruses and multiple sclerosis. Neuroscientist. 2011;17:659–676. doi: 10.1177/1073858411386615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pakpoor J, Giovannoni G, Ramagopalan SV. Epstein-Barr virus and multiple sclerosis: association or causation? Expert Rev Neurother. 2013;13:287–297. doi: 10.1586/ern.13.6. [DOI] [PubMed] [Google Scholar]
- 80.Paludan SR, Ellermann-Eriksen S, Kruys V, Mogensen SC. Expression of TNF-alpha by herpes simplex virus-infected macrophages is regulated by a dual mechanism: transcriptional regulation by NF-kappa B and activating transcription factor 2/Jun and translational regulation through the AU-rich region of the 3' untranslated region. J Immunol. 2001;167:2202–2208. doi: 10.4049/jimmunol.167.4.2202. [DOI] [PubMed] [Google Scholar]
- 81.Pantry SN, Medveczky MM, Arbuckle JH, Luka J, Montoya JG, Hu J, Renne R, Peterson D, Pritchett JC, Ablashi DV, Medveczky PG. Persistent human herpesvirus-6 infection in patients with an inherited form of the virus. J Med Virol. 2013;85:1940–1946. doi: 10.1002/jmv.23685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pelegri C, Canudas AM, del Valle J, Casadesus G, Smith MA, Camins A, Pallas M, Vilaplana J. Increased permeability of blood-brain barrier on the hippocampus of a murine model of senescence. Mech Ageing Dev. 2007;128:522–528. doi: 10.1016/j.mad.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 83.Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, Azzena GB, Palamara AT, Grassi C. HSV-1 promotes Ca2+-mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol Aging. 2011;32:2323.e13–26. doi: 10.1016/j.neurobiolaging.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 84.Porcellini E, Carbone I, Ianni M, Licastro F. Alzheimer's disease gene signature says: beware of brain viral infections. Immun Ageing. 2010;7:16. doi: 10.1186/1742-4933-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Radtke K, Dohner K, Sodeik B. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell Microbiol. 2006;8:387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 86.Richart SM, Simpson SA, Krummenacher C, Whitbeck JC, Pizer LI, Cohen GH, Eisenberg RJ, Wilcox CL. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J Virol. 2003;77:3307–3311. doi: 10.1128/JVI.77.5.3307-3311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosato PC, Leib DA. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015;11:e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saldanha J, Sutton RN, Gannicliffe A, Farragher B, Itzhaki RF. Detection of HSV1 DNA by in situ hybridisation in human brain after immunosuppression. J Neurol Neurosurg Psychiatry. 1986;49:613–619. doi: 10.1136/jnnp.49.6.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salinas S, Schiavo G, Kremer EJ. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol. 2010;8:645–655. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- 91.Sanders VJ, Felisan S, Waddell A, Tourtellotte WW. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol. 1996;2:249–258. doi: 10.3109/13550289609146888. [DOI] [PubMed] [Google Scholar]
- 92.Santana S, Recuero M, Bullido MJ, Valdivieso F, Aldudo J. Herpes simplex virus type I induces the accumulation of intracellular beta-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol Aging. 2012;33:430 e419–433. doi: 10.1016/j.neurobiolaging.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell. 1999;99:817–827. doi: 10.1016/s0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 94.Schmidt RJ, Tancredi DJ, Krakowiak P, Hansen RL, Ozonoff S. Maternal intake of supplemental iron and risk of autism spectrum disorder. Am J Epidemiol. 2014;180:890–900. doi: 10.1093/aje/kwu208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith G. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol. 2012;66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soares BP, Provenzale JM. Imaging of herpesvirus infections of the CNS. AJR Am J Roentgenol. 2016;206:39–48. doi: 10.2214/AJR.15.15314. [DOI] [PubMed] [Google Scholar]
- 97.Sola P, Merelli E, Marasca R, Poggi M, Luppi M, Montorsi M, Torelli G. Human herpesvirus 6 and multiple sclerosis: survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J Neurol Neurosurg Psychiatry. 1993;56:917–919. doi: 10.1136/jnnp.56.8.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, Brennan MB, Maloni HW, McFarland HF, Lin HC, Patnaik M, Jacobson S. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nat Med. 1997;3:1394–1397. doi: 10.1038/nm1297-1394. [DOI] [PubMed] [Google Scholar]
- 99.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.St Leger AJ, Hendricks RL. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol. 2011;17:528–534. doi: 10.1007/s13365-011-0062-1. [DOI] [PubMed] [Google Scholar]
- 101.Steiner I. Herpes simplex virus encephalitis: new infection or reactivation? Curr Opin Neurol. 2011;24:268–274. doi: 10.1097/WCO.0b013e328346be6f. [DOI] [PubMed] [Google Scholar]
- 102.Takahashi K, Sonoda S, Higashi K, Kondo T, Takahashi H, Takahashi M, Yamanishi K. Predominant CD4 T-lymphocyte tropism of human herpesvirus 6-related virus. J Virol. 1989;63:3161–3163. doi: 10.1128/jvi.63.7.3161-3163.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Talloczy Z, Virgin HW, 4th, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 104.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tanaka-Taya K, Sashihara J, Kurahashi H, Amo K, Miyagawa H, Kondo K, Okada S, Yamanishi K. Human herpesvirus 6 (HHV-6) is transmitted from parent to child in an integrated form and characterization of cases with chromosomally integrated HHV-6 DNA. J Med Virol. 2004;73:465–473. doi: 10.1002/jmv.20113. [DOI] [PubMed] [Google Scholar]
- 106.Tanguy Le Gac N, Boehmer PE. Activation of the herpes simplex virus type-1 origin-binding protein (UL9) by heat shock proteins. J Biol Chem. 2002;277:5660–5666. doi: 10.1074/jbc.M108316200. [DOI] [PubMed] [Google Scholar]
- 107.Tarter KD, Simanek AM, Dowd JB, Aiello AE. Persistent viral pathogens and cognitive impairment across the life course in the third national health and nutrition examination survey. J Infect Dis. 2014;209:837–844. doi: 10.1093/infdis/jit616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. 2003;53:189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- 109.Thompson RL, Sawtell NM. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol. 2001;75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tognatta R, Miller RH. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology. 2016;110:539–547. doi: 10.1016/j.neuropharm.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 112.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wallaschek N, Gravel A, Flamand L, Kaufer BB. The putative U94 integrase is dispensable for human herpesvirus 6 (HHV-6) chromosomal integration. J Gen Virol. 2016;97:1899–1903. doi: 10.1099/jgv.0.000502. [DOI] [PubMed] [Google Scholar]
- 114.Watson AM, Prasad KM, Klei L, Wood JA, Yolken RH, Gur RC, Bradford LD, Calkins ME, Richard J, Edwards N, Savage RM, Allen TB, Kwentus J, McEvoy JP, Santos AB, Wiener HW, Go RC, Perry RT, Nasrallah HA, Gur RE, et al. Persistent infection with neurotropic herpes viruses and cognitive impairment. Psychol Med. 2013;43:1023–1031. doi: 10.1017/S003329171200195X. [DOI] [PubMed] [Google Scholar]
- 115.Wilborn F, Schmidt CA, Brinkmann V, Jendroska K, Oettle H, Siegert W. A potential role for human herpesvirus type 6 in nervous system disease. J Neuroimmunol. 1994;49:213–214. doi: 10.1016/0165-5728(94)90198-8. [DOI] [PubMed] [Google Scholar]
- 116.Wilson AC, Mohr I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012;20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. 2009;217:131–138. doi: 10.1002/path.2449. [DOI] [PubMed] [Google Scholar]
- 118.Wozniak MA, Shipley SJ, Combrinck M, Wilcock GK, Itzhaki RF. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer's disease patients. J Med Virol. 2005;75:300–306. doi: 10.1002/jmv.20271. [DOI] [PubMed] [Google Scholar]
- 119.Wuest SC, Mexhitaj I, Chai NR, Romm E, Scheffel J, Xu B, Lane K, Wu T, Bielekova B. A complex role of herpes viruses in the disease process of multiple sclerosis. PLoS One. 2014;9:e105434. doi: 10.1371/journal.pone.0105434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yamane A, Mori T, Suzuki S, Mihara A, Yamazaki R, Aisa Y, Nakazato T, Shimizu T, Ikeda Y, Okamoto S. Risk factors for developing human herpesvirus 6 (HHV-6) reactivation after allogeneic hematopoietic stem cell transplantation and its association with central nervous system disorders. Biol Blood Marrow Transplant. 2007;13:100–106. doi: 10.1016/j.bbmt.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 121.Zerr DM, Fann JR, Breiger D, Boeckh M, Adler AL, Xie H, Delaney C, Huang ML, Corey L, Leisenring WM. HHV-6 reactivation and its effect on delirium and cognitive functioning in hematopoietic cell transplantation recipients. Blood. 2011;117:5243–5249. doi: 10.1182/blood-2010-10-316083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou L, Miranda-Saksena M, Saksena NK. Viruses and neurodegeneration. Virol J. 2013;10:172. doi: 10.1186/1743-422X-10-172. [DOI] [PMC free article] [PubMed] [Google Scholar]