

Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative disorder that is thought to be mediated by autoreactive T lymphocytes that find their way into the central nervous system (CNS). The pathological mechanism of MS is still being elucidated but it involves complex interactions between infiltrating immune cells and resident glial cells within the CNS that culminate into strong neuroinflammation and axonal damage. Most of the current knowledge on the immunopathology of MS has been generated using the rodent model of experimental autoimmune encephalomyelitis (EAE). Among CD4+ T helper cell subsets, interferon gamma (IFN-γ)-producing Th1 cells as well as interleukin 17 (IL-17)-producing Th17 cells are crucial in driving the pathology of EAE. Th1 and Th17 cell differentiation is guided by distinct transcriptional programs induced by polarizing factors. Presumably, following a trigger, some local factors and effector molecules produced by Th1 and Th17 cells facilitate their entry into the CNS and induce a pathological neuroinflammatory response by activating resident glial cells, which further assist massive infiltration of a second wave of immune cells into the CNS. This scenario was evident from the fact that adoptive transfer of in vitro activated myelin oligodendrocyte glycoprotein (MOG)-specific Th1 and Th17 cells into naïve rodent hosts was sufficient to induce EAE (Codarri et al., 2011). Nevertheless, disease severity and clinical manifestation of EAE induced after adoptive transfer of Th1 and Th17 cells were highly variable. Transfer of Th1 cells induced classical paralytic EAE, whereas Th17 cell transfer drove atypical ataxic EAE, an indication that mechanisms used by effector Th1 and Th17 cells in driving neuroinflammation might be different. A number of parameters could account for these differences. First, Th1 and Th17 cells might have different capacities to directly target neurons (Siffrin et al., 2010). Second, by virtue of distinct sets of effector molecules they target and recruit different cells within and towards the CNS and altogether induce a different neuroinflammatory profile. Third, they might have different capacities to regulate repair mechanisms following initial neuroinflammatory damage. Here we focus on the current knowledge of T cell-glial interactions and discuss how effector molecules of Th1 and Th17 cells influence the phenotype and function of resident glial cells within the CNS. There is a great body of evidence describing IFN-γ and IL-17 as major effector molecules of Th1 and Th17 cells, respectively. However, induction of EAE by IFN-γ-/- and IL-17-/- T cells has demonstrated that these factors are dispensable for neuropathology (Codarri et al., 2011). Further studies have identified granulocyte macrophage colony stimulating factor (GM-CSF), largely associated with Th17 cells but also produced by Th1 cells, as an indispensable effector molecule whose overexpression in CD4+ T cells alone was sufficient for driving neuropathology similar to EAE (Codarri et al., 2011; Spath et al., 2017). Following infiltration into the CNS, autoreactive Th1 and Th17 cells are involved in constant crosstalk with microglia and astrocytes and their effector molecules profoundly influence the phenotype and function of these major glial cell types. Microglia are the sentinels of the CNS that rapidly respond to invading pathogens, CNS injury and inflammation. Depending on external cues they can attain pro- (M1-like) or anti-inflammatory (M2-like) phenotypes. Although this dichotomy vastly oversimplifies the plasticity of microglia, the original thought is that an M1-like phenotype is attained by sensing invading pathogens or inflammatory mediators and is considered to be neurotoxic, whereas M2-like microglia are involved in repair mechanisms and are considered to play a neuroprotective role by providing anti-inflammatory mediators and growth factors (Aguzzi et al., 2013). Microglial responses need to be tightly balanced between these phenotypes to maintain the integrity of neural tissue. A sustained pro-inflammatory milieu during MS favors M1-like microglia to populate the lesions triggering demyelination and axonal damage. Similarly, reactive astrogliosis is also a characteristic feature of neurodegenerative disorders like MS. Astrocytes are the most abundant cell type in the CNS with a multitude of functions including support of neural homeostasis. Anatomically astrocytes are active components of the blood-brain barrier (BBB) and are also found in close association with neurons. Therefore they were believed to be less reactive than microglia to avoid any imminent damage to the neural tissue. However, astrocytes do respond to injury by releasing diverse molecules. Primarily, they are a major source of neurotrophic growth factors (nerve growth factor, glial cell-derived neurotrophic factor, ciliary neurotrophic factor, etc.) which drive neurogenesis and assist tissue repair mechanisms. Additionally, astrocytes produce anti-inflammatory factors that dampen any minor inflammation in the CNS and avoid potential damage. Under pathological conditions they also respond to pathogens and infiltrating leukocytes and release a large array of pro-inflammatory cytokines and chemokines, thereby directly contributing to exacerbation of neuroinflammation.
Previously, it was unclear how distinct sets of effector molecules produced by Th1 and Th17 cells influence the neuroinflammatory properties of microglia and astrocytes. In the past, few studies dealing with antigen-specific Th1 and Th17 cells have shown that both subsets can activate microglia and astrocytes (McQuillan et al., 2010; Murphy et al., 2010). However, it is not known to what extent effector molecules released by Th1 and Th17 cells contribute to this process. It is noteworthy that during MS there is massive infiltration of both antigen-specific and non-specific T cells into the CNS. Therefore, cells within the CNS are exposed to both antigen-specific signals due to major histocompatibility complex (MHC) and T-cell receptor ligation as well as to signals delivered by cytokine binding to their respective receptors. Our recent findings indicate that distinct effector molecules produced by Th1 and Th17 cells have different capacities to influence the phenotype and function of microglia and astrocytes (Prajeeth et al., 2014, 2017). Th1 cells and their effector molecules can activate microglia and augment their pro-inflammatory properties, thus rendering them a neurotoxic M1-like phenotype. Furthermore, Th1-derived effector molecules enhanced expression of MHC class II and co-stimulatory molecules (CD40, CD86) on the surface of microglia making them potent antigen-presenting cells (APC) and providing a platform for further T cell activation and differentiation (Prajeeth et al., 2014). So far, Th17 cells are considered to be far more efficient than Th1 cells in driving the CNS pathology of EAE. Intriguingly, Th17-derived effector molecules were found to be ineffective in microglial activation (Prajeeth et al., 2014). This is in contrast to earlier reports where it was shown that amyloid-beta (Aβ) and MOG-specific Th17 cells, when co-cultured with microglia, induced the production of pro-inflammatory cytokines and co-stimulatory molecules (McQuillan et al., 2010; Murphy et al., 2010). This again is an indication that effects observed in response to direct microglia and T cell interaction mediated by antigens is different from that triggered by sensing secreted factors. Unlike microglia, astrocytes react to both Th1- and Th17-derived effector molecules (Prajeeth et al., 2017). In response to these effector molecules astrocytes downregulate the expression of neurotrophic factors that are crucial for repair process and upregulate pro-inflammatory cytokines and chemokines that cause tissue damage (Prajeeth et al., 2017). The ability of effector molecules secreted by Th17 cells to act on astrocytes but not on microglia is quite puzzling, and the molecular explanation for this phenomenon remains enigmatic. One possibility might be that astrocytes are better equipped with receptors and corresponding signaling machinery needed to respond to Th17-derived effector molecules than microglia. An indication for this speculation comes from the work of Kang et al. (2010) who demonstrated that ablation of IL-17 induced Act1-signaling on astrocytes ameliorates EAE, whereas ablation of this signaling in microglia and macrophages has no influence on the disease course. We know that IL-17A alone has minimal effects on astrocytes and is more effective when combined with other factors such as tumor necrosis factor (TNF) (Prajeeth et al., 2017). Therefore, key effector molecules of Th17 cells and the signaling pathways induced by these factors in target cells are the major issues that need thorough investigation.
Enhanced chemokine expression by microglia and astrocytes in response to Th1- and Th17-derived effector molecules is of great significance for understanding the pathology of MS. Chemokines orchestrates the communication between cells that are located at larger distances within the body. Cells expressing a particular chemokine receptor sense and migrate towards the cells producing their respective chemokine ligand. Under physiological conditions microglia, astrocytes, neurons, and vascular endothelial cells constitutively express both chemokines and their receptors. However, their expression is increased under pathological conditions. During MS, chemokines and their receptors act as amplifiers of neuroinflammation by assisting recruitment of microglia, monocytes and other immune cells to the focal lesions (Le Thuc et al., 2015). Several chemokine receptor:ligand pairs have been associated with MS pathology. The most prominent among them are CCL2:CCR2, CCL20:CCR6 and CXCR3:CXCL10 that have been associated with recruitment of T cells, monocytes and microglia (Le Thuc et al., 2015). Our recent findings suggest that inducing a strong chemokine response in microglia and astrocytes is a key mechanism, by which Th1- and Th17-derived effector molecules amplify neuroinflammation and drive pathology during neuroinflammation (Prajeeth et al., 2014, 2017). We have provided evidence how factors released by activated astrocytes guided the recruitment of microglia and transendothelial migration of Th17 cells (Skripuletz et al., 2013; Prajeeth et al., 2017). Furthermore, we show that in the absence of astrocytes microglial recruitment and Th17 infiltration into the CNS is also affected.
Previously it was believed that a small number of autoreactive T cells by unknown mechanism have to cross the BBB and activate glial cells to initiate the neuropathology of MS. Our argument is that this might essentially not be the case. We believe that this can also be achieved by Th1- and Th17-derived effector molecules acting on glial cells (Figure 1). Chemokines such as CCL2, CCL20 and CXCL10 produced by microglia and astrocytes as a result of this activation might assist the infiltration of Th1 and Th17 cells into the CNS. Subsequently, these Th1 and Th17 cells interact with glial cells either in an antigen-specific manner or through effector cytokines and further facilitate bulk recruitment of a second wave of immune cells into the CNS that amplify the neuroinflammatory response. This may finally trigger or amplify demyelination and axonal damage. Our data suggest that the quality of the inflammatory response is highly dependent on the effector molecules that microglia and astrocytes sense early during the initial phase of the disease. An earlier study has reported that Th1 cells reach the CNS first and facilitates the entry of Th17 during EAE (O'Connor et al., 2008). This finding is in accordance to our own results demonstrating that Th1-derived effector molecules induce a robust inflammatory response by activating both microglia and astrocytes unlike Th17-derived effector molecules, which only act on astrocytes. In other words, Th1-derived effector molecules function as a key to open the floodgates for the entry of other leukocytes into the CNS. In our view, understanding the regulation of glial function by T cells is essential to develop strategies that limit damage and support regeneration and repair process. Therapies should target particular effector molecules or subsets of T cells that are dominant at the given stage of disease. It is also noteworthy that not all T cells are destructive and their effectors are also needed for regeneration and repair processes.
Figure 1.
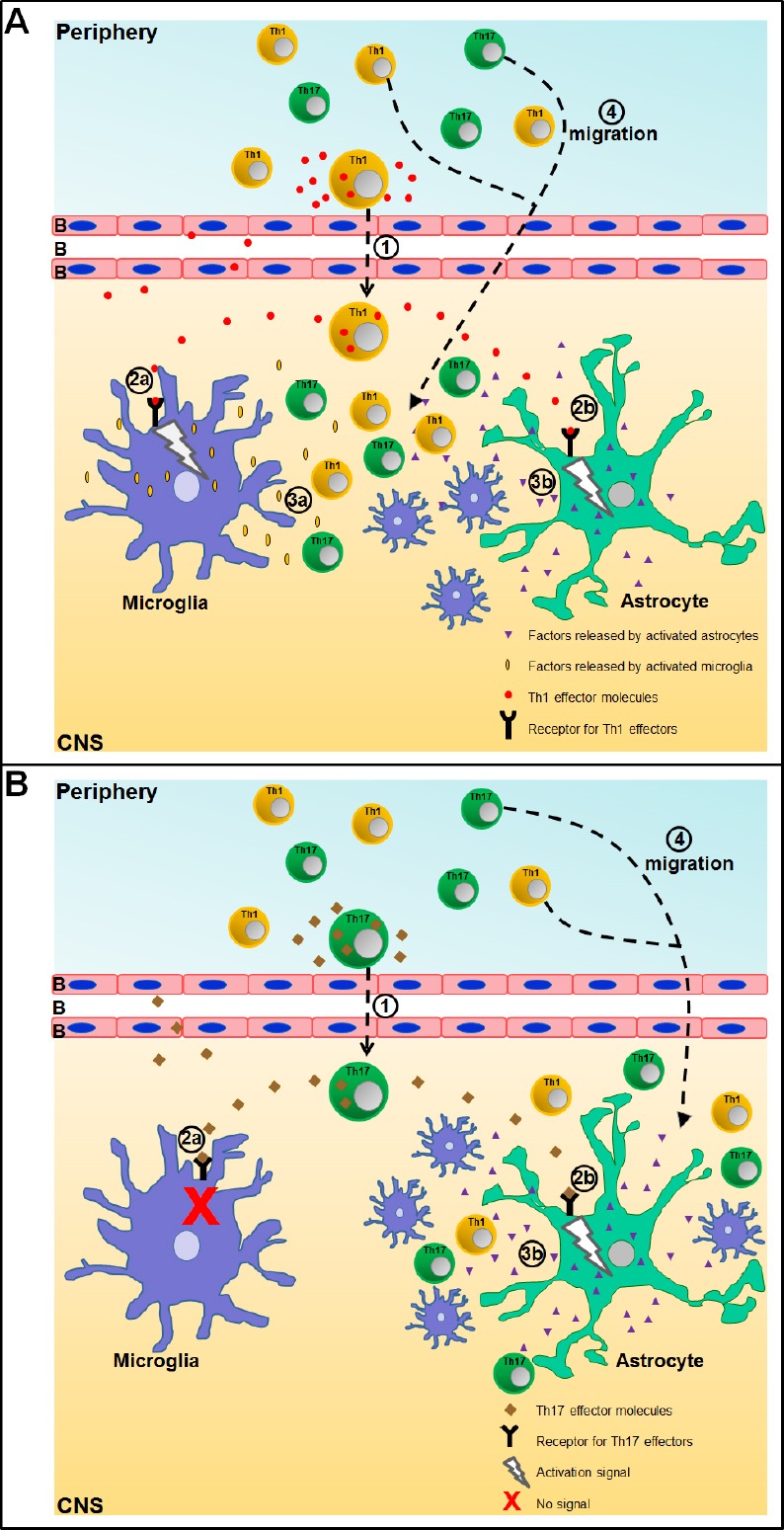
Schematic representation of microglia and astrocyte activation by effector molecules from Th1 and Th17 cells within the central nervous system (CNS) and subsequent neuroinflammatory response.
Here we use an oversimplified scheme to explain the sequence of events that lead to neuroinflammation following infiltration of (A) Th1 cells or (B) Th17 cells into the CNS.
Step 1: Th1 and Th17 cells cross the blood brain barrier (BBB).
Step 2: Their effector molecules bind to receptors expressed on (a) microglia and/or (b) astrocytes and induce a signaling cascade. In the case of Th17 cells, their effector molecules only act on astrocytes.
Step 3: (a) microglia and (b) astrocytes respond and release factors that trigger recruitment of cells to the site of inflammation.
Step 4: Activated microglia and astrocytes express high amounts of chemokines that assist infiltration of a second wave of Th1 and Th17 cells into the CNS.
JH is supported by the Helmholtz-Gemeinschaft, “Zukunftsthema” Immunology and inflammation” (ZT-0027). MS is supported by the Pertermax-Müller-Stiftung and the Niedersachsen Research Network on Neuroinfectiology (N-RENNT) of the Ministry of Science and Culture of Lower Saxony.
Footnotes
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Jigar Pravinchandra Modi, Florida Atlantic University, USA.
References
- 1.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 3.Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Thuc O, Blondeau N, Nahon JL, Rovere C. The complex contribution of chemokines to neuroinflammation: switching from beneficial to detrimental effects. Ann N Y Acad Sci. 2015;1351:127–140. doi: 10.1111/nyas.12855. [DOI] [PubMed] [Google Scholar]
- 5.McQuillan K, Lynch MA, Mills KH. Activation of mixed glia by Abeta-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav Immun. 2010;24:598–607. doi: 10.1016/j.bbi.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 7.O'Connor RA, Prendergast CT, Sabatos CA, Lau CW, Leech MD, Wraith DC, Anderton SM. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prajeeth CK, Lohr K, Floess S, Zimmermann J, Ulrich R, Gudi V, Beineke A, Baumgartner W, Muller M, Huehn J, Stangel M. Effector molecules released by Th1 but not Th17 cells drive an M1 response in microglia. Brain Behav Immun. 2014;37:248–259. doi: 10.1016/j.bbi.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Prajeeth CK, Kronisch J, Khorooshi R, Knier B, Toft-Hansen H, Gudi V, Floess S, Huehn J, Owens T, Korn T, Stangel M. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J Neuroinflammation. 2017;14:204. doi: 10.1186/s12974-017-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, Leuenberger T, Lehmann SM, Luenstedt S, Rinnenthal JL, Laube G, Luche H, Lehnardt S, Fehling HJ, Griesbeck O, Zipp F. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. 2010;33:424–436. doi: 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M, Baumgartner W, Stangel M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 2013;136:147–167. doi: 10.1093/brain/aws262. [DOI] [PubMed] [Google Scholar]
- 12.Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, Becher B. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity. 2017;46:245–260. doi: 10.1016/j.immuni.2017.01.007. [DOI] [PubMed] [Google Scholar]