

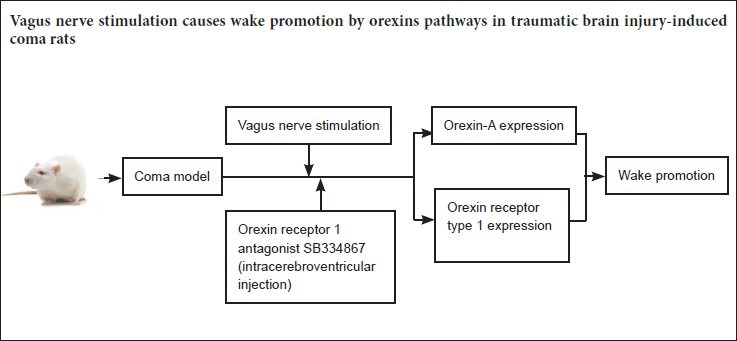
Keywords: nerve regeneration, brain injury, orexin-A, orexin receptor type 1, vagus nerve stimulation, traumatic brain injury, wake-promoting, coma, wakefulness, prefrontal cortex, neurotransmitter, neural regeneration
Abstract
Orexins, produced in the lateral hypothalamus, are important neuropeptides that participate in the sleep/wake cycle, and their expression coincides with the projection area of the vagus nerve in the brain. Vagus nerve stimulation has been shown to decrease the amounts of daytime sleep and rapid eye movement in epilepsy patients with traumatic brain injury. In the present study, we investigated whether vagus nerve stimulation promotes wakefulness and affects orexin expression. A rat model of traumatic brain injury was established using the free fall drop method. In the stimulated group, rats with traumatic brain injury received vagus nerve stimulation (frequency, 30 Hz; current, 1.0 mA; pulse width, 0.5 ms; total stimulation time, 15 minutes). In the antagonist group, rats with traumatic brain injury were intracerebroventricularly injected with the orexin receptor type 1 (OX1R) antagonist SB334867 and received vagus nerve stimulation. Changes in consciousness were observed after stimulation in each group. Enzyme-linked immunosorbent assay, western blot assay and immunohistochemistry were used to assess the levels of orexin-A and OX1R expression in the prefrontal cortex. In the stimulated group, consciousness was substantially improved, orexin-A protein expression gradually increased within 24 hours after injury and OX1R expression reached a peak at 12 hours, compared with rats subjected to traumatic brain injury only. In the antagonist group, the wake-promoting effect of vagus nerve stimulation was diminished, and orexin-A and OX1R expression were decreased, compared with that of the stimulated group. Taken together, our findings suggest that vagus nerve stimulation promotes the recovery of consciousness in comatose rats after traumatic brain injury. The upregulation of orexin-A and OX1R expression in the prefrontal cortex might be involved in the wake-promoting effects of vagus nerve stimulation.
Introduction
Traumatic brain injury (TBI) has a high incidence worldwide, and is associated with high rates of morbidity and mortality. Recent progress in treating traumatic brain injuries has resulted in sharply reduced rates of mortality; however, 14% of TBI patients remain in a long-term coma or vegetative state following treatment. Furthermore, this percentage is gradually increasing, resulting in a heavy burden on society and the affected family members (Harvey, 2013; Durand et al., 2017). The current treatment regimen for TBI-induced coma includes drug therapy, hyperbaric oxygenation, music therapy, and medial nerve stimulation (Kumaria and Tolias, 2012; Cossu, 2014; Kaelber et al., 2016; Gray, 2017; Joseph et al., 2017). However, these treatments do not always result in the patient awakening, and there remains a need for new methods of accelerating the transition from a comatose or vegetative state to arousal. Previous studies have shown that vagus nerve stimulation (VNS) can reduce the amounts of daytime sleep and rapid eye movement, thereby extending the amount of awake time in patients with epilepsy caused by TBI (Malow et al., 2001; Shi et al., 2013; Jain and Glauser, 2014; Neren et al., 2016). VNS is a neurophysiologic method that has been extensively used to treat refractory epilepsy, depression, and cognitive disorders (Yuan and Silberstein, 2015). Recent studies have shown that VNS affects the amounts of time spent awake and asleep, and can decrease sleep duration. The vagus nerve projects to several different brainstem regions, including the locus coeruleus and the parabrachial nucleus. In the parabrachial nucleus, the vagus nerve forms numerous connections with fibers that project to the basal forebrain, thalamus, hypothalamus, and cerebral cortex (Ansari et al., 2007; Frangos and Komisaruk, 2017). Due to this widespread connectivity, VNS has treatment potential for coma. In this study, we evaluated whether VNS could promote grades I–IV consciousness, as determined by observing sensory and motor functions.
Orexin peptides (orexin-A and orexin-B) are produced by the lateral hypothalamus and regulate feeding behavior, energy homeostasis, neuroendocrine activities and the sleep-wake cycle by binding to orexin-1 and orexin-2 receptors (Wu et al., 2007; Boss and Roch, 2015). The orexin receptor type 1 (OX1R) is expressed in many regions of the brain, including the cerebral cortex, prefrontal cortex, ventromedial hypothalamic nucleus, and locus coeruleus. Orexin-A is one of the most important neurotransmitters in the ascending reticular activating system, participating in awareness and the sleep-wake cycle. Therefore, in the present study, we examined whether orexin-A and the OX1R are involved in the wake-promoting effect of VNS.
Materials and Methods
Animals
A total of 120 specific-pathogen-free adult Sprague-Dawley rats (half male and half female), weighing 250−300 g, were obtained from the Institute of Laboratory Animals of Nanchang University of China, and housed in the Laboratory Animal Center of the First Affiliated Hospital of Nanchang University of China. All rats were maintained under controlled temperature and light conditions, and allowed free access to food and water.
The study protocol was approved by the Animal Ethics Committee of the First Affiliated Hospital of Nanchang University (approval number: (2016)(003)). The experimental procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1985).
Establishment of a TBI-induced coma model
The 120 rats were assigned to four different groups (n = 30 each), with 10 rats for each time point per group. In the control group, healthy rats received sham operation and anesthesia. In the TBI group, free-fall drop was used to establish the model of TBI-induced coma (Feeney et al., 1981). In the stimulated group, rats with TBI-induced coma were subjected to VNS. In the antagonist group, comatose rats were intracerebroventricularly injected with the OX1R antagonist SB334867 and received VNS.
Rats in the TBI, stimulated and antagonist groups were anesthetized by inhalation of diethyl ether, and then allowed to breathe air spontaneously. After anesthesia, a 5-mm vertical incision was made to expose the skull. The target for impact was marked with a syringe needle at a spot 2 mm adjacent to the left midline and 1 mm anterior to the coronal suture. Next, a cylindrical impact hammer weighing 400 g and of 2 cm diameter was dropped from a vertical height of 40–44 cm to produce a concave fracture of the skull (Feeney et al., 1981). Following injury, the incision was closed, and each animal was disinfected and placed in a cage.
Evaluation of sensory and motor functions
One hour after the impact, the degree of consciousness was evaluated on a scale of I–IV, based on sensory and motor functions (I–VI consciousness scale) (Stephens and Levy, 1994). The levels of consciousness were as follows: level I: normal activity as seen in the cage; level II: decreased activity; level III: decreased activity accompanied by motor incoordination; level IV: righting reflex could be elicited, and the animal could stand up; level V: the righting reflex was absent; however, the animal could react to pain; level VI: the animal showed no reaction to pain. Rats having consciousness levels of V or VI for at least 30 minutes were deemed to be in a coma state, and were used for the following procedures.
Intracerebroventricular injection of the OX1R antagonist SB334867
Under sterile conditions, an injection catheter was inserted into the left cerebral ventricle of each rat in the antagonist group. Each rat was pretreated with gentamicin (0.1 mL/100 g body weight, intramuscular injection) and anesthetized with 10% chloral hydrate (0.3 mL/100 g body weight, intraperitoneal injection) prior to surgery. The rats were positioned in a stereotaxic frame (ZS-B/S; Beijing Zhongshi Dichuang Science and Technology Development Co., Ltd., Beijing, China). The following coordinates were used to map the guide cannula: 1.0 mm posterior to the bregma, 1.5 mm lateral to the midline, and 4.5 mm ventral to the skull surface, with the incisor bar 3.2 mm below the interauricular line. An injection catheter was inserted into the cerebral ventricle of each rat in the antagonist group under sterile conditions. The OXR1 inhibitor SB334867 (Tocris Bioscience, Ellisville, MO, USA) was dissolved in a 60:40 dimethyl sulfoxide solution and administrated at a dose of 10 mg/kg body weight in a total volume of 5 μL. After awakening from anesthesia, the rats were prepared for VNS.
VNS
After TBI and the first evaluation of consciousness, VNS was performed. Rats in the stimulated and antagonist groups were treated with VNS using a low-frequency electrical stimulator (ES-420; ITO Physiotherapy & Rehabilitation, Tokyo, Japan). After establishment of the TBI-induced coma model and prior to stimulation, the rats were intraperitoneally anesthetized with 10% chloral hydrate (0.3 mL/100 g body weight). Afterwards, the head and neck areas were disinfected with Betadine and then shaved. A small incision was made on the left ventral side of the neck adjoining the midline to approach the left vagus nerve at the cervical level. We performed a blunt dissection of the subcutaneous fat, salivary glands, sternohyoid and sternocleidomastoid, and cut the carotid sheath, including the vagus nerve and carotid artery. A 5-mm segment of the left vagus nerve was separated and attached to an electrode. An ohmmeter was used to ensure that the electrode had good contact with the vagus nerve. VNS was performed with the following parameters: frequency, 30 Hz; current, 1.0 mA; pulse width, 0.5 ms; total stimulation time, 15 minutes. Following surgery, each animal received gentamicin (0.1 mL/100 g body weight) by intramuscular injection. One hour later, behavior and consciousness levels were observed and evaluated based on previously described grading criteria (Stephens and Levy, 1994). Rats in the TBI group underwent a procedure identical to that used for the stimulated groups, but without electrical stimulation.
Tissue extraction
Rats in the stimulated and antagonist groups, as well as rats in the corresponding control and TBI-induced coma groups were simultaneously euthanized with 10% chloral hydrate at 6, 12 and 24 hours after TBI. Prefrontal cortical tissues (within the frontal lobe) were removed and analyzed by enzyme linked immunosorbent assay (ELISA), immunohistochemistry and western blot assay to evaluate orexin-A and OX1R expression.
ELISA
Five rats from each group were sacrificed at 6, 12 and 24 hours after TBI. Tissue samples were tested using an ELISA kit designed for detecting orexin-A protein (cE90607a 96 Tests; Uscn Life Science Inc., Wuhan, Hubei Province, China). Optical density values were measured at 450 nm using a microplate reader (Model 680, Bio-Rad, Hercules, CA, USA). The concentration of orexin-A was calculated using a standard curve.
Western blot assay
At 6, 12 and 24 hours after TBI, five rats from each group were decapitated following intraperitoneal injection of 10% chloral hydrate. Brains were carefully removed, and the prefrontal cortex was quickly dissected on ice. The tissue samples were homogenized using the Tissue Protein Extraction Kit (CW0891; Beijing Kangwei Biotechnology Co., Ltd., Beijing, China). Afterwards, the homogenates were centrifuged at 12,000 × g for 10 minutes at 4°C. The total amount of protein in each supernatant fraction was determined using the Bio-Rad DC protein assay, and an aliquot of each supernatant was removed and stored at −80°C. Equal amounts of total supernatant protein in loading buffer were boiled for 5 minutes, and then separated on a 10% sodium dodecyl sulfate/polyacrylamide gel. The separated proteins were electrophoretically transferred onto polyvinylidene difluoride membranes. The membranes were then blocked for 2 to 3 hours at room temperature with TBST buffer (150 mM NaCl, 20 mM Tris-HCl, pH 7.4, 0.1% Tween-20) containing 5% milk. The blots were then incubated overnight at 4°C with rabbit anti-OX1R polyclonal antibody (1:200, ab68718; Abcam, Hong Kong, China) and rabbit anti-rat β-actin monoclonal antibody (1:400, CW0096; Beijing Kangwei). Following incubation, the membranes were washed extensively with TBST, and then incubated for 1 hour at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgG (H+L) (1:2,000; ZB-2301, Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) diluted in TBST containing 5% milk. After washing, the blots were incubated with a chemiluminescence substrate (32109, ECL Plus; Amersham Biosciences, Piscataway, NJ, USA) and quantified using Image Lab software (Bio-Rad). The blots were then stripped by incubation for 30 minutes at 70°C in a solution containing 2% sodium dodecyl sulfate and 100 mM β-mercaptoethanol in 62.5 mM Tris-HCl, pH 6.8. Subsequently, the blots were re-probed using rabbit anti-β-actin monoclonal antibody (CW0096, Beijing Kangwei) to monitor loading of the gel lanes. Western blot analyses of OX1R in prefrontal cortical tissue were performed at 6, 12 and 24 hours after TBI. The optical density measurements of individual bands were normalized to the optical density of β-actin.
Immunohistochemistry
The rats were anesthetized and decapitated at 6, 12 and 24 hours after TBI, and perfused through the heart with 4% paraformaldehyde. Next, the brains were carefully removed, and 40-µm-thick coronal sections were cut for examination. The sections were rinsed with phosphate-buffered saline and treated with 0.3% hydrogen peroxide (H2O2) for 30 minutes. Afterwards, the sections were rinsed three times for 5 minutes each and then incubated with normal goat serum for 20 minutes. The sections were then incubated overnight at 4°C with rabbit anti-OX1R antibody (1:200; ab68718, Abcam). Following incubation, the tissue sections were extensively rinsed with phosphate-buffered saline and then incubated with a biotinylated goat anti-rabbit antibody. Finally, the sections were reacted with diaminobenzidine and visualized under a light microscope (BX511T-PHD-J11, Olympus, Tokyo, Japan).
Protein expression was quantified as a function of the percentage and staining intensity of immunoreactive cells (Soslow et al., 2000) as follows: A × B, where A represents the percentage of positive cells (0–4, where 0 = 0–1%, 1 = 1–10%, 2 = 10–50%, 3 = 50–80%, 4 = 80–100%) and B represents the intensity of staining (0–3, where 0 = no significant staining, 1 = mild staining, 2 = moderate staining, 3 = dark staining).
Statistical analysis
All data were analyzed with SPSS 17.0 software (SPSS, Chicago, IL, USA). Western blot and ELISA data were expressed as the mean ± SD, and immunohistochemical data as the mean rank. One-way analysis of variance followed by Tukey's test was used for comparison of western blot assay and ELISA data. The Kruskal-Wallis test was used for comparison of immunohistochemical data. A value of P < 0.05 was considered statistically significant.
Results
Evaluation of consciousness after VNS
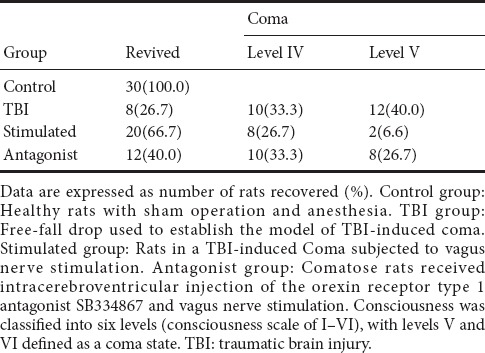
The degree of consciousness was evaluated using a double-blind method 1 hour after the experiment ended. A total of 20 rats died and were excluded from this study in the TBI (3 rats), stimulated (8 rats) and antagonist (9 rats) groups. Only 8 of the 30 rats in the TBI group re-awakened from coma (level IV: 8; level V: 10; level VI: 12), while in the stimulated group, 20 rats re-wakened (level II: 4; level III: 6; level IV: 10; level V: 8; level VI: 2) and 10 rats remained in a comatose state. Twelve rats in the antagonist group re-awakened from coma (level III: 5; level IV: 7; level V: 10; level VI: 8). The number of rats that regained consciousness (levels I–IV) in each group are shown in Table 1, revealing the following order: TBI group < antagonist group < stimulated group < control group.
Table 1.
Effect of vagus nerve stimulation on the recovery of consciousness in rats in a TBI-induced coma
VNS increased orexin-A expression in the prefrontal cortex of rats with TBI-induced coma
ELISA showed that orexin-A expression differed in the various groups at 6, 12 and 24 hours, with the following trend: antagonist group < control group < TBI group < stimulated group (P < 0.05). Orexin-A expression in each of the groups differed temporally as well, with the following trend: 6 hours < 24 hours < 12 hours (P < 0.05; Figure 1).
Figure 1.
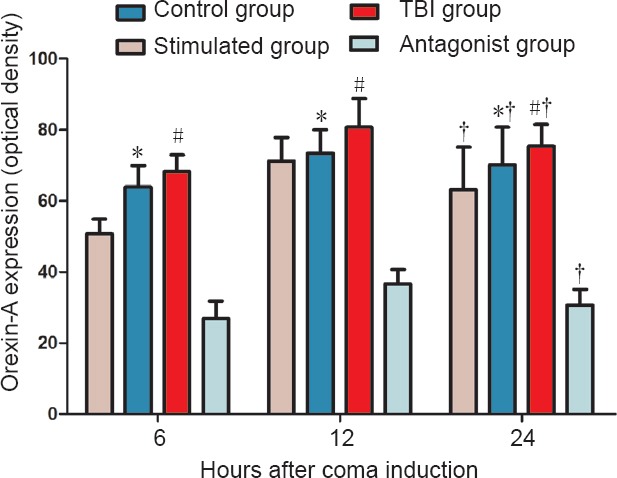
Effect of vagus nerve stimulation on orexin-A expression in the prefrontal cortex of rats in a TBI-induced coma (enzyme-linked immunosorbent assay).
Data are expressed as the mean ± SD (n = 6 per group; one-way analysis of variance followed by Tukey's test); *P < 0.05, vs. control group and stimulated group; #P < 0.05, vs. antagonist group; †P < 0.05, vs. 6 hours after TBI. Control group: Healthy rats with sham operation and anesthesia. TBI group: Free-fall drop was used to establish the model of TBI-induced coma. Stimulated group: TBI-induced comatose rats subjected to vagus nerve stimulation. Antagonist group: Comatose rats received intracerebroventricular injection of the orexin receptor type 1 antagonist SB334867 and vagus nerve stimulation. TBI: Traumatic brain injury.
VNS increased OX1R expression in the prefrontal cortex of rats with TBI-induced coma
OX1R expression levels in the prefrontal cortex were measured by western blot assay. Significant differences in OX1R expression were found among the four groups at 6, 12 and 24 hours. The relative levels of OX1R expression at 6 hours displayed the following trends: control group < antagonist group < TBI group < stimulated group (P < 0.05); at 12 hours: control group < TBI group < antagonist group < stimulated group (P < 0.05); at 24 hours: antagonist group < control group < TBI group < stimulated group (P < 0.05). The mean level of OX1R expression was higher in the TBI group than in the control group, and OX1R expression was substantially higher in the stimulated group than in the TBI group. Furthermore, the levels of OX1R expression after injury were significantly different in the stimulated group from those in the antagonist group. Within-group comparison showed the following trends of OX1R expression at the three time points among the four different groups: 6 hours < 12 hours (P < 0.05); 24 hours < 12 hours (P < 0.05); 6 hours < 24 hours (P > 0.05) (Figure 2).
Figure 2.
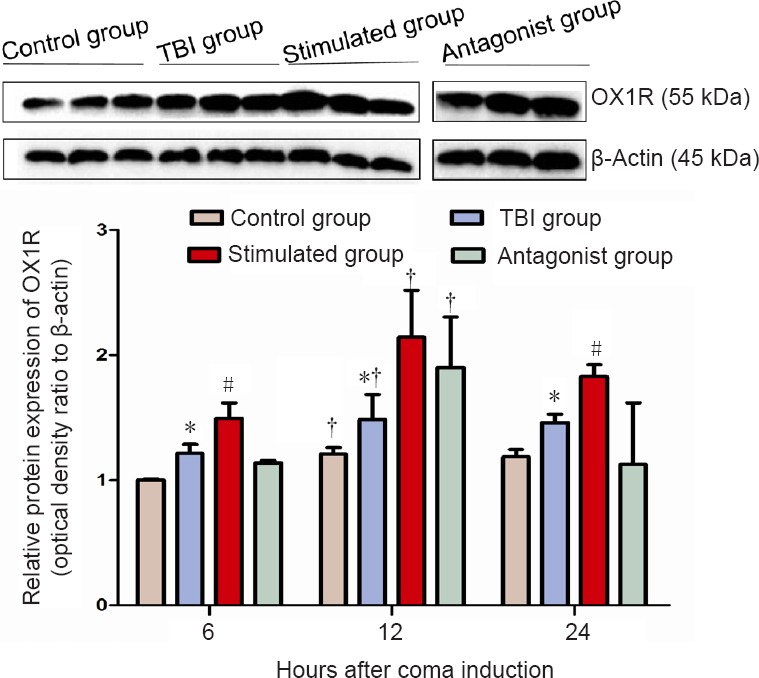
Effect of vagus nerve stimulation on OX1R protein expression in the prefrontal cortex of rats in a TBI-induced coma (western blot assay).
Different expression levels may be associated with the circadian rhythm of hypothalamic orexin secretion. Data are expressed as the mean ± SD (n = 6 per group; one-way analysis of variance). *P < 0.05, vs. control and stimulated groups; #P < 0.05, vs. antagonist group; †P < 0.05, vs. 6 hours after TBI. Control group: Healthy rats with sham operation and anesthesia. TBI group: Free-fall drop was used to establish the model of TBI-induced coma. Stimulated group: Rats in a TBI-induced coma subjected to vagus nerve stimulation. Antagonist group: Comatose rats received intracerebroventricular injection of the OX1R antagonist SB334867 and vagus nerve stimulation. OX1R: Orexin receptor type 1; TBI: traumatic brain injury.
Positive immunostaining for OX1R was found in the cytoplasm and cell membranes of neurons in the prefrontal cortex. OX1R-positive cells were present in all four groups, and the data were analyzed using the Kruskal-Wallis H-test. Samples of prefrontal cortex from the stimulated group showed higher levels of OX1R expression (81.67) compared with prefrontal cortex samples from the control group (40.56), TBI group (59.67) or antagonist group (36.11) (P < 0.001). No significant differences in OX1R expression were observed within each group at the various time points (Figure 3).
Figure 3.

Effect of vagus nerve stimulation on OX1R expression in the prefrontal cortex of rats with TBI-induced coma (immunostaining, light microscope, original magnification, 400×).
Increased OX1R expression was detected within the cytoplasm of neurons in the prefrontal cortex at 12 h after TBI. Control group: Healthy rats with sham operation and anesthesia. TBI group: TBI-induced coma. Stimulated group: Rats in a TBI-induced coma subjected to vagus nerve stimulation. Antagonist group: Comatose rats received intracerebroventricular injection of the OX1R antagonist SB334867 and vagus nerve stimulation. TBI: Traumatic brain injury; OX1R: orexin receptor type 1; h: hours.
Discussion
Previous studies have shown that TBI induces numerous pathophysiological changes, including lipid peroxidation, free radical formation, blood-brain barrier damage, branched chain amino acid release, intracellular Ca2+ overload, oxidative stress, and arachidonic acid decomposition (Singh et al., 2013; Elkind et al., 2015; Hiebert et al., 2015; Lucke-Wold et al., 2015; Hue et al., 2016; Zhu et al., 2017). It is currently believed that there are two main mechanistic causes of TBI-induced coma: (i) impaired reticular activation system and (ii) changes in the levels of important neurotransmitters that regulate the sleep/wake cycle (e.g., orexin-A, norepinephrine, 5-hydroxytryptamine and glutamate). Previous studies showed that orexin levels are decreased in the cerebrospinal fluid of patients with narcolepsy resulting from TBI during the first 2 months after the injury (Ramanjaneya et al., 2009; Jeter et al., 2013). In accordance with the results of our previously published reports, we found in the present study that orexin-A and OX1R expression levels increase acutely after injury and thereafter decrease over time (Feng et al., 2015; Zhong et al., 2015; Feng and Du, 2016). The increase in orexin expression in the first several hours after TBI in our present study might be an acute stress reaction. Alterations in OX1R expression during the first 24 hours after TBI can result from a reduced number of glial cells, low blood pressure, an intracranial pressure change, hypoxia and ischemia, changes in blood glucose levels, or the release of neural specific nucleoprotein (Mihara et al., 2011; Willie et al., 2012). Thus, the orexinergic system appears to play an important role in the pathophysiology of TBI. In this study, we found increased levels of orexin-A and OX1R expression in the prefrontal cortex of rats in the TBI group compared with the control group. These upregulated levels of orexin-A and OX1R during the 24-hour period after TBI might be a response to physiological stress, which is important for protecting neurons during the early stages of TBI.
Orexins (orexin-A and orexin-B) produced in the lateral hypothalamus are hypothalamic peptides involved in food intake, metabolic rate, growth hormone production, autonomic function, and the sleep/wake cycle (Boss and Roch, 2015; Mieda, 2017; Walker and Lawrence, 2017). The orexinergic system comprises many regions of the central nervous system, including the cerebral cortex, thalamus, hypothalamus, brain stem, and limbic system. OX1R is expressed by immunoreactive nerve fiber projections that are extensively distributed throughout the central nervous system, including the hippocampus, dorsal raphe nucleus, arcuate nucleus, anterior pretectal nucleus, tuberomammillary nucleus, and raphe nucleus. Moreover, OX1R is most highly expressed in the prefrontal cortex and locus coeruleus, which play key roles in regulating wakefulness (Xu et al., 2013). A previous report showed that an important function of the orexinergic system is regulating the sleep/wake cycle by directly activating various hypothalamic-cortical pathways (Kampe et al., 2009). The prefrontal cortex has critically important roles in advanced brain activities, including consciousness, integration of information, and cognition. Orexinergic neurons in the hypothalamus project into the prefrontal cortex and excite central nervous system neurons (Xia et al., 2005). Orexin-A-mediated activation of two G-protein coupled receptors, OX1R and OX2R, upregulates Ca2+ levels in the cytoplasm and induces the activation of phosphatases, second messengers, and protein kinases (Kukkonen and Leonard, 2014; Shu et al., 2014; Kukkonen, 2016).
Since its approval for therapeutic use by the Food and Drug Administration in 1997, VNS has been widely used for treating refractory epilepsy, depression, and cognitive disorders (Yuan and Silberstein, 2016, 2017; Ekmekci and Kaptan, 2017; Fulton et al., 2017). While few studies have investigated the relationship between VNS and wakefulness, some evidence suggests that VNS can reduce daytime sleep in patients with epilepsy caused by TBI.
For the following reasons, VNS might be a potentially effective new method for improving the status of patients in a TBI-induced coma: (1) Extensive fibrous projections. The nucleus of the solitary tract receives the majority of vagal afferent fibers and projects into many brainstem regions, including the locus coeruleus, parabrachial nucleus, thalamus, basal forebrain, hypothalamus, and cerebral cortex (Ansari et al., 2007). Electrical stimulation of the vagus nerve should activate the ascending reticular activating system, which plays a key role in promoting arousal, thereby alleviating the comatose condition. (2) Influence of related neurotransmitters. Neurotransmitters such as orexin, noradrenaline, glutamate and dopamine are known to have wake-promoting effects. Recent studies suggest that some of the effects of VNS may involve stimulation of the locus coeruleus to release noradrenaline throughout the central nervous system. Noradrenaline significantly affects recovery from TBI by promoting wakefulness and by inhibiting sleep (Smith et al., 2005). Other reports show that VNS significantly increases extracellular noradrenaline levels in the hippocampus and prefrontal cortex, as well as 5-HT levels in the dorsal raphe nucleus and dopamine levels in the prefrontal cortex and nucleus accumbens (Manta et al., 2013). (3) Anti-inflammatory effects. The cholinergic anti-inflammatory pathway that systemically inhibits pro-inflammatory cytokine release is well characterized. Vagal efferents are thought to regulate systemic inflammation by modulating the release of tumor necrosis factor from macrophages. VNS has been shown to decrease cerebral edema, thereby helping avoid further damage to neurons after TBI (Bonaz et al., 2013; Xiang et al., 2015). (4) Increasing cerebral blood flow. Following VNS, significant increases in blood flow have been reported in the left posterior limb of the internal capsule/medial putamen, right dorsal anterior cingulate, right superior temporal gyrus, left cerebellum, and left dorsolateral prefrontal cortex (Kosel et al., 2011; Conway et al., 2012). The enhanced blood flow should improve neural survival and promote wakefulness. (5) Neurotrophic factors and synaptic plasticity. Neurotrophic factors, such as brain-derived neurotrophic factor and nerve growth factor, are crucial for neuronal survival, development, function and synaptic plasticity, all of which can be affected when previously inactive synapses become functional. VNS rapidly activates the brain-derived neurotrophic factor receptor TrκB and upregulates nerve growth factor levels in the rat brain (Follesa et al., 2007; Cossu, 2014). (6) Electrical activity of the brain. VNS has been reported to increase slow-wave sleep prior to rapid eye movement in rats and upregulate the frequencies of sleep spindles, δ-waves and ponto-geniculo-occipital waves (Valdes-Cruz et al., 2008). (7) Other mechanisms. VNS has been reported to decrease damage to the blood-brain barrier, modulate depolarization activity, upregulate endogenous neurogenesis, and attenuate glutamate-mediated excitotoxicity in models of TBI (Kumaria and Tolias, 2012).
Our previous studies showed that median nerve stimulation promotes wakefulness by upregulating orexin-A and OX1R in the prefrontal cortex and hypothalamus in rats with TBI-induced coma (Feng et al., 2015; Zhong et al., 2015; Feng and Du, 2016). Therefore, our current study was designed to explore the relationship between orexin-A levels in the prefrontal cortex and VNS, and assess the wake-promoting effect of VNS by evaluating the level of consciousness using a I−VI grading scale. To our knowledge, there has been no similar study reported to date.
In this study, only 8 of 30 rats re-awakened in the TBI group, 20 of 30 rats re-awakened in the stimulated group, and 12 of 30 rats recovered from a comatose state in the antagonist group. These results suggest that VNS promotes wakefulness in rats with TBI-induced coma. We also found that orexin-A levels were significantly upregulated in the prefrontal cortex of rats in the stimulated group compared with the other groups. Western blot assay and immunohistochemistry studies revealed trends of increasing OX1R and orexin-A expression in the stimulated group compared with the other groups. We also found lower expression levels of orexin-A and OX1R in the control group compared with the stimulated group after VNS. Moreover, similar results were observed at 6 and 12 hours in the group administered an OX1R antagonist (SB334867). These findings suggest that VNS directly impacts orexin-A and OX1R levels at 6, 12 and 24 hours. Within each group, levels of orexin-A and OX1R were significantly higher at 12 hours than at 6 or 24 hours; this may be associated with the rhythmic patterns of orexin-A neurons. It has been reported that lateral hypothalamic orexin-A neurons are rhythmic and innervated by suprachiasmatic nucleus efferents, which are important components of the arousal system. Orexin neuronal activity is higher during the night than during the day (Belle et al., 2014). Moreover, genes for orexin receptors are expressed in mouse suprachiasmatic nucleus efferents, and OX1R becomes upregulated at dusk (Alo et al., 2017). This circadian rhythmicity of orexin neurons might underlie the observed changes in orexin-A and OX1R expression at the different time points. In the present study, the time during the day that the rats were killed was not constant, and was partially dependent on individual differences in recovery time following TBI or VNS treatment.
Our study has some limitations that should be mentioned. For example, we could have used electroencephalograms, the Glasgow Coma Scale or the evoked potential test to examine the wake-promoting effects of VNS. Also, a larger sample size and a more precise method of measuring TBI could have been used. Additionally, we only examined orexin-A and OX1R expression in the prefrontal cortex, although other regions of the brain, such as the hypothalamus and hippocampus, also participate in promoting wakefulness. Further studies are needed to clarify how expression of orexin-A and its receptor change following TBI-induced coma, how VNS causes increased orexin-A expression, and the mechanisms and pathways underlying these changes. Despite the limitations, our study suggests that orexin-A plays a key role in promoting consciousness, and that VNS helps an animal recover consciousness from TBI-induced coma by upregulating orexin-A and OX1R expression.
Additional wake-promoting mechanisms might be discovered if other neurotransmitters and regions of the brain related to orexin-A and/or wakefulness are studied to identify whether orexin-A acts as an “arousal switch”. Orexin-A knockout animals could be used in future studies to provide additional insight into the effects found in the current study.
In conclusion, upregulation of orexin-A and OX1R expression in the prefrontal cortex might contribute to the wake-promoting effect of VNS in TBI-induced comatose rats. Our findings suggest that VNS is a promising method for awakening patients in a TBI-induced coma. However, further studies are required to test the clinical effects of VNS and its possible complications.
Footnotes
Funding: This study was supported by the Natural Science Foundation of China, No. 81260295 and the Graduate Student Innovation Fund of Jiangxi Province of China, No. YC2015-S090.
Conflicts of interest: None declared.
Financial support: This study was supported by the Natural Science Foundation of China, No. 81260295 and the Graduate Student Innovation Fund of Jiangxi Province of China, No. YC2015-S090. Funders had no involvement in the study design; data collection, management, analysis, and interpretation; paper writing; or decision to submit the paper for publication.
Research ethics: The study protocol was approved by the Animal Ethics Committee of the First Affiliated Hospital of Nanchang University of China (approval number (2016) (003)). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1985).
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
(Copyedited by Patel B, Maxwell R, Wang J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Alo R, Avolio E, Mele M, Fazzari G, Carelli A, Facciolo RM, Canonaco M. Role of leptin and orexin-a within the suprachiasmatic nucleus on anxiety-like behaviors in hamsters. Mol Neurobiol. 2017;54:2674–2684. doi: 10.1007/s12035-016-9847-9. [DOI] [PubMed] [Google Scholar]
- 2.Ansari S, Chaudhri K, Al Moutaery KA. Vagus nerve stimulation: indications and limitations. Acta Neurochir Suppl. 2007;97:281–286. doi: 10.1007/978-3-211-33081-4_31. [DOI] [PubMed] [Google Scholar]
- 3.Belle MD, Hughes AT, Bechtold DA, Cunningham P, Pierucci M, Burdakov D, Piggins HD. Acute suppressive and long-term phase modulation actions of orexin on the mammalian circadian clock. J Neurosci. 2014;34:3607–3621. doi: 10.1523/JNEUROSCI.3388-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonaz B, Picq C, Sinniger V, Mayol JF, Clarencon D. Vagus nerve stimulation: from epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol Motil. 2013;25:208–221. doi: 10.1111/nmo.12076. [DOI] [PubMed] [Google Scholar]
- 5.Boss C, Roch C. Recent trends in orexin research-2010 to 2015. Bioorg Med Chem Lett. 2015;25:2875–2887. doi: 10.1016/j.bmcl.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Conway CR, Sheline YI, Chibnall JT, Bucholz RD, Price JL, Gangwani S, Mintun MA. Brain blood-flow change with acute vagus nerve stimulation in treatment-refractory major depressive disorder. Brain Stimul. 2012;5:163–171. doi: 10.1016/j.brs.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cossu G. Therapeutic options to enhance coma arousal after traumatic brain injury: state of the art of current treatments to improve coma recovery. Br J Neurosurg. 2014;28:187–198. doi: 10.3109/02688697.2013.841845. [DOI] [PubMed] [Google Scholar]
- 8.Durand E, Chevignard M, Ruet A, Dereix A, Jourdan C, Pradat-Diehl P. History of traumatic brain injury in prison populations: a systematic review. Ann Phys Rehabil Med. 2017;60:95–101. doi: 10.1016/j.rehab.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Ekmekci H, Kaptan H. Vagus nerve stimulation. Open Access Maced J Med Sci. 2017;5:391–394. doi: 10.3889/oamjms.2017.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkind JA, Lim MM, Johnson BN, Palmer CP, Putnam BJ, Kirschen MP, Cohen AS. Efficacy, dosage, and duration of action of branched chain amino acid therapy for traumatic brain injury. Front Neurol. 2015;6:73. doi: 10.3389/fneur.2015.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 12.Feng Z, Du Q. Mechanisms responsible for the effect of median nerve electrical stimulation on traumatic brain injury-induced coma: orexin-A-mediated N-methyl-D-aspartate receptor subunit NR1 upregulation. Neural Regen Res. 2016;11:951–956. doi: 10.4103/1673-5374.184494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Z, Zhong YJ, Wang L, Wei TQ. Resuscitation therapy for traumatic brain injury-induced coma in rats: mechanisms of median nerve electrical stimulation. Neural Regen Res. 2015;10:594–598. doi: 10.4103/1673-5374.155433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L, Puligheddu M, Marrosu F, Biggio G. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007;1179:28–34. doi: 10.1016/j.brainres.2007.08.045. [DOI] [PubMed] [Google Scholar]
- 15.Frangos E, Komisaruk BR. Access to vagal projections via cutaneous electrical stimulation of the neck: fMRI evidence in healthy humans. Brain Stimul. 2017;10:19–27. doi: 10.1016/j.brs.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Fulton SP, Van Poppel K, McGregor AL, Mudigoudar B, Wheless JW. Vagus nerve stimulation in intractable epilepsy associated with SCN1A gene abnormalities. J Child Neurol. 2017;32:494–498. doi: 10.1177/0883073816687221. [DOI] [PubMed] [Google Scholar]
- 17.Gray SN. An overview of the use of neurofeedback biofeedback for the treatment of symptoms of traumatic brain injury in military and civilian populations. Med Acupunct. 2017;29:215–219. doi: 10.1089/acu.2017.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey HH. Reducing traumatic brain injuries in youth sports: youth sports traumatic brain injury state laws January 2009-December 2012. Am J Public Health. 2013;103:1249–1254. doi: 10.2105/AJPH.2012.301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350:132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 20.Hue CD, Cho FS, Cao S, Nicholls RE, Vogel Iii EW, Sibindi C, Arancio O, Dale Bass CR, Meaney DF, Morrison Iii B. Time course and size of blood-brain barrier opening in a mouse model of blast-induced traumatic brain injury. J Neurotrauma. 2016;33:1202–1211. doi: 10.1089/neu.2015.4067. [DOI] [PubMed] [Google Scholar]
- 21.Jain SV, Glauser TA. Effects of epilepsy treatments on sleep architecture and daytime sleepiness: an evidence-based review of objective sleep metrics. Epilepsia. 2014;55:26–37. doi: 10.1111/epi.12478. [DOI] [PubMed] [Google Scholar]
- 22.Jeter CB, Hergenroeder GW, Ward NH, 3rd, Moore AN, Dash PK. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30:671–679. doi: 10.1089/neu.2012.2491. [DOI] [PubMed] [Google Scholar]
- 23.Joseph B, Khan M, Rhee P. Non-invasive diagnosis and treatment strategies for traumatic brain injury: an update. J Neurosci Res. 2017 doi: 10.1002/jnr.24132. doi: 10.1002/jnr.24132. [DOI] [PubMed] [Google Scholar]
- 24.Kaelber S, Pantcheva P, Borlongan CV. Drug- and cell-based therapies for targeting neuroinflammation in traumatic brain injury. Neural Regen Res. 2016;11:1575–1576. doi: 10.4103/1673-5374.193231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kampe J, Tschop MH, Hollis JH, Oldfield BJ. An anatomic basis for the communication of hypothalamic, cortical and mesolimbic circuitry in the regulation of energy balance. Eur J Neurosci. 2009;30:415–430. doi: 10.1111/j.1460-9568.2009.06818.x. [DOI] [PubMed] [Google Scholar]
- 26.Kosel M, Brockmann H, Frick C, Zobel A, Schlaepfer TE. Chronic vagus nerve stimulation for treatment-resistant depression increases regional cerebral blood flow in the dorsolateral prefrontal cortex. Psychiatry Res. 2011;191:153–159. doi: 10.1016/j.pscychresns.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Kukkonen JP. OX2 orexin/hypocretin receptor signal transduction in recombinant Chinese hamster ovary cells. Cell Signal. 2016;28:51–60. doi: 10.1016/j.cellsig.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Kukkonen JP, Leonard CS. Orexin/hypocretin receptor signalling cascades. Br J Pharmacol. 2014;171:314–331. doi: 10.1111/bph.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumaria A, Tolias CM. Is there a role for vagus nerve stimulation therapy as a treatment of traumatic brain injury? Br J Neurosurg. 2012;26:316–320. doi: 10.3109/02688697.2012.663517. [DOI] [PubMed] [Google Scholar]
- 30.Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, Bailes JE, Lee JM, Rosen CL, Huber JD. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl Res. 2015;166:509–528. doi: 10.1016/j.trsl.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Malow BA, Edwards J, Marzec M, Sagher O, Ross D, Fromes G. Vagus nerve stimulation reduces daytime sleepiness in epilepsy patients. Neurology. 2001;57:879–884. doi: 10.1212/wnl.57.5.879. [DOI] [PubMed] [Google Scholar]
- 32.Manta S, El Mansari M, Debonnel G, Blier P. Electrophysiological and neurochemical effects of long-term vagus nerve stimulation on the rat monoaminergic systems. Int J Neuropsychopharmacol. 2013;16:459–470. doi: 10.1017/S1461145712000387. [DOI] [PubMed] [Google Scholar]
- 33.Mieda M. The roles of orexins in sleep/wake regulation. Neurosci Res. 2017;118:56–65. doi: 10.1016/j.neures.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Mihara Y, Dohi K, Yofu S, Nakamachi T, Ohtaki H, Shioda S, Aruga T. Expression and localization of the orexin-1 receptor (OX1R) after traumatic brain injury in mice. J Mol Neurosci. 2011;43:162–168. doi: 10.1007/s12031-010-9438-6. [DOI] [PubMed] [Google Scholar]
- 35.Neren D, Johnson MD, Legon W, Bachour SP, Ling G, Divani AA. Vagus nerve stimulation and other neuromodulation methods for treatment of traumatic brain injury. Neurocrit Care. 2016;24:308–319. doi: 10.1007/s12028-015-0203-0. [DOI] [PubMed] [Google Scholar]
- 36.Ramanjaneya M, Conner AC, Chen J, Kumar P, Brown JE, Johren O, Lehnert H, Stanfield PR, Randeva HS. Orexin-stimulated MAP kinase cascades are activated through multiple G-protein signalling pathways in human H295R adrenocortical cells: diverse roles for orexins A and B. J Endocrinol. 2009;202:249–261. doi: 10.1677/JOE-08-0536. [DOI] [PubMed] [Google Scholar]
- 37.Shi C, Flanagan SR, Samadani U. Vagus nerve stimulation to augment recovery from severe traumatic brain injury impeding consciousness: a prospective pilot clinical trial. Neurol Res. 2013;35:263–276. doi: 10.1179/1743132813Y.0000000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu Q, Hu ZL, Huang C, Yu XW, Fan H, Yang JW, Fang P, Ni L, Chen JG, Wang F. Orexin-A promotes cell migration in cultured rat astrocytes via Ca2+-dependent PKCalpha and ERK1/2 signals. PLoS One. 2014;9:e95259. doi: 10.1371/journal.pone.0095259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh IN, Gilmer LK, Miller DM, Cebak JE, Wang JA, Hall ED. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J Cereb Blood Flow Metab. 2013;33:593–599. doi: 10.1038/jcbfm.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith DC, Modglin AA, Roosevelt RW, Neese SL, Jensen RA, Browning RA, Clough RW. Electrical stimulation of the vagus nerve enhances cognitive and motor recovery following moderate fluid percussion injury in the rat. J Neurotrauma. 2005;22:1485–1502. doi: 10.1089/neu.2005.22.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000;89:2637–2645. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 42.Stephens JR, Levy RH. Effects of valproate and citrulline on ammonium-induced encephalopathy. Epilepsia. 1994;35:164–171. doi: 10.1111/j.1528-1157.1994.tb02928.x. [DOI] [PubMed] [Google Scholar]
- 43.Valdes-Cruz A, Magdaleno-Madrigal VM, Martinez-Vargas D, Fernandez-Mas R, Almazan-Alvarado S. Long-term changes in sleep and electroencephalographic activity by chronic vagus nerve stimulation in cats. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:828–834. doi: 10.1016/j.pnpbp.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 44.Walker LC, Lawrence AJ. The role of orexins/hypocretins in alcohol use and abuse. Curr Top Behav Neurosci. 2017;33:221–246. doi: 10.1007/7854_2016_55. [DOI] [PubMed] [Google Scholar]
- 45.Willie JT, Lim MM, Bennett RE, Azarion AA, Schwetye KE, Brody DL. Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. J Neurotrauma. 2012;29:1908–1921. doi: 10.1089/neu.2012.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu HY, Tang CF, He H. Orexin, orexin receptor and sports. Zhongguo Zuzhi Gongcheng Yanjiu. 2007;11:7966–7969. [Google Scholar]
- 47.Xia JX, Chen XW, Cheng SY, Hu ZA. Mechanisms of orexin A-evoked changes of intracellular calcium in primary cultured cortical neurons. Neuroreport. 2005;16:783–786. doi: 10.1097/00001756-200505120-00025. [DOI] [PubMed] [Google Scholar]
- 48.Xiang YX, Wang WX, Xue Z, Zhu L, Wang SB, Sun ZH. Electrical stimulation of the vagus nerve protects against cerebral ischemic injury through an anti-infammatory mechanism. Neural Regen Res. 2015;10:576–582. doi: 10.4103/1673-5374.155430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu TR, Yang Y, Ward R, Gao L, Liu Y. Orexin receptors: multi-functional therapeutic targets for sleeping disorders, eating disorders, drug addiction, cancers and other physiological disorders. Cell Signal. 2013;25:2413–2423. doi: 10.1016/j.cellsig.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 50.Yuan H, Silberstein SD. Vagus nerve stimulation and headache. Headache. 2015 doi: 10.1111/head.12721. doi: 10.1111/head.12721. [DOI] [PubMed] [Google Scholar]
- 51.Yuan H, Silberstein SD. Vagus nerve and vagus nerve stimulation, a comprehensive review: part I. Headache. 2016;56:71–78. doi: 10.1111/head.12647. [DOI] [PubMed] [Google Scholar]
- 52.Yuan H, Silberstein SD. Vagus nerve stimulation and headache. Headache 57 Suppl. 2017;1:29–33. doi: 10.1111/head.12721. [DOI] [PubMed] [Google Scholar]
- 53.Zhong YJ, Feng Z, Wang L, Wei TQ. Wake-promoting actions of median nerve stimulation in TBI-induced coma: an investigation of orexin-A and orexin receptor 1 in the hypothalamic region. Mol Med Rep. 2015;12:4441–4447. doi: 10.3892/mmr.2015.3898. [DOI] [PubMed] [Google Scholar]
- 54.Zhu JQ, Hong J, Cui JZ, Wang KJ. Adipose mesenchymal stem cells for treatment of traumatic brain injury. Zhongguo Zuzhi Gongcheng Yanjiu. 2017;21:71–76. [Google Scholar]