

Abstract
Background
Skeletal muscle responds to eccentric contractions (ECC) with an anabolic response that involves the induction of protein synthesis through the mechanistic target of rapamycin complex 1. While we have reported that repeated ECC bouts after cachexia initiation attenuated muscle mass loss and inflammatory signalling, cachectic muscle's capacity to induce protein synthesis in response to ECC has not been determined. Therefore, we examined cachectic muscle's ability to induce mechano‐sensitive pathways and protein synthesis in response to an anabolic stimulus involving ECC and determined the role of muscle signal transducer and activator of transcription 3 (STAT3)/nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB) signalling on ECC‐induced anabolic signalling.
Methods
Mechano‐sensitive pathways and anabolic signalling were examined immediately post or 3 h after a single ECC bout in cachectic male Apc Min/+ mice (n = 17; 16 ± 1% body weight loss). Muscle STAT3/NFκB regulation of basal and ECC‐induced anabolic signalling was also examined in an additional cohort of Apc Min/+ mice (n = 10; 16 ± 1% body weight loss) that received pyrrolidine dithiocarbamate 24 h prior to a single ECC bout. In all experiments, the left tibialis anterior performed ECC while the right tibialis anterior served as intra‐animal control. Data were analysed by Student's t‐test or two‐way repeated measures analysis of variance with Student‐Newman‐Keuls post‐hoc when appropriate. The accepted level of significance was set at P < 0.05 for all analysis.
Results
Apc Min/+ mice exhibited a cachectic muscle signature demonstrated by perturbed proteostasis (Ribosomal Protein S6 (RPS6), P70S6K, Atrogin‐1, and Muscle RING‐finger protein‐1 (MuRF1)), metabolic (adenosine monophosphate‐activated protein kinase, Peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α), and Cytochrome c oxidase subunit IV (COXIV)), and inflammatory (STAT3, NFκB, extracellular signal‐regulated kinases 1 and 2, and P38) signalling pathway regulation. Nonetheless, mechano‐sensitive signalling pathways (P38, extracellular signal‐regulated kinases 1 and 2, and Protein kinase B (AKT)) were activated immediately post‐ECC irrespective of cachexia. While cachexia did not attenuate ECC‐induced P70S6K activation, the protein synthesis induction remained suppressed compared with healthy controls. However, muscle STAT3/NFκB inhibition increased basal and ECC‐induced protein synthesis in cachectic Apc Min/+ mice.
Conclusions
These studies demonstrate that mechano‐sensitive signalling is maintained in cachectic skeletal muscle, but chronic STAT3/NFκB signalling serves to attenuate basal and ECC‐induced protein synthesis.
Keywords: ApcMin/+, Cancer cachexia, Eccentric contractions, Interleukin‐6, Muscle protein synthesis
Introduction
Skeletal muscle mass depletion associated with cancer cachexia contributes to increased patient morbidity and mortality.1, 2 Skeletal muscle size is influenced by the dynamic balance between the rates of protein synthesis and breakdown,3, 4 and disrupted protein turnover accompanies cancer cachexia.5, 6, 7 While our understanding of suppressed basal protein synthesis and activated breakdown during cachexia has increased dramatically,7, 8, 9 we have a more limited understanding of how the cachectic environment affects skeletal muscle responsiveness to anabolic stimuli, which is clinically relevant for the treatment of the cachectic cancer patient. Resistance exercise is a potent anabolic stimulus that stimulates muscle hypertrophy through the activation of mechanistic target of rapamycin complex 1 (mTORC1) in healthy adults10, 11, 12 and can attenuate skeletal muscle mass loss in several muscle wasting conditions.13, 14, 15 Despite the clinical significance of maintaining or improving muscle mass during cancer, limited information currently exists on the cachectic muscle's anabolic response to resistance exercise.
Eccentric contractions (ECC) induced by high‐frequency electrical stimulation have been used to examine signalling associated with muscle hypertrophy in rodents16, 17, 18, 19 and have demonstrated great utility for improving our mechanistic understanding of contraction‐induced protein synthesis and mTORC1 signalling.18, 20, 21, 22 Related to cancer cachexia, evidence suggests that increased loading by synergist ablation or ECC can maintain muscle mass in tumour‐bearing mice.23, 24, 25, 26 Indeed, we have reported that repeated ECC bouts after the initiation of cachexia can attenuate muscle mass loss through reduced inflammatory signalling24; however, cachectic muscle's capacity to induce protein synthesis in response to ECC has not been determined and warrants further investigation. We have found that mechano‐activation of protein synthesis in stretched myotubes can be disrupted by conditioned media from Lewis lung carcinoma cells,27 suggesting that tumour‐derived cachectic factors can interfere with mechanical signalling inducing protein synthesis in vitro. While these studies demonstrate that skeletal muscle from tumour‐bearing animals may be responsive to exercise training or loading, the regulation of protein synthesis by muscle contraction in the presence of a systemic cachectic environment requires further investigation.
Suppressed muscle protein synthesis and mTORC1 signalling are associated with interleukin‐6 (IL‐6) induction of signal transducer and activator of transcription 3 (STAT3), nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB), and 5'‐adenosine monophosphate‐activated protein kinase (AMPK) in tumour‐bearing mice.28, 29 Indeed, we have previously demonstrated an inverse relationship between plasma IL‐6 and muscle protein synthesis during cachexia progression, and systemic IL‐6 overexpression can suppress mTORC1 signalling in tumour‐bearing mice.28 In contrast, blocking muscle IL‐6 signalling through STAT3 inhibition or glycoprotein 130 (gp130) receptor loss attenuated wasting in tumour‐bearing mice.30, 31 Additionally, acute and chronic muscle STAT3/NFκB inhibition improved mTORC1 signalling in cachectic mice.29, 32 Lastly, extracellular signal‐regulated kinases 1 and 2 (ERK1/2) and P38 mitogen‐activated protein kinase (MAPK) inhibition restored myotube stretch‐induced protein synthesis in the presence of Lewis lung carcinoma‐derived cachectic factors.27 Thus, there is a clear rationale that muscle inflammatory signalling involving STAT3/NFκB can disrupt basal and mechanical‐induced regulation of protein synthesis during cancer cachexia. However, it is currently unknown if suppressed muscle protein synthesis and mTORC1 signalling can be activated in the cachectic environment. Therefore, we examined cachectic muscle's ability to induce protein synthesis in response to an anabolic stimulus involving ECC and determined the role of muscle STAT3/NFκB signalling on ECC‐induced anabolic signalling. Interestingly, we report that mechano‐sensitive signalling is maintained in cachectic skeletal muscle, but STAT3/NFκB signalling attenuates basal and ECC‐induced protein synthesis.
Methods
Animals
Male Apc Min/+ mice on a C57BL/6 background were originally purchased from Jackson Laboratories and bred at the University of South Carolina's Animal Resource Facility. Mice used in the current study were obtained from the investigators breeding colony in the Center for Colon Cancer Research Mouse Core. Mice were individually housed, kept on a 12:12 h light‐dark cycle, and had access to standard rodent chow (cat#8604 Rodent Diet; Harlan Teklad) and water ad libitum. Body weight and food measurements were taken weekly, and the percentage body weight loss from peak body weight was calculated. Mice lacking the Apc Min/+ mutation (C57BL/6) served as controls for all experiments. The University of South Carolina's Institutional Animal Care and Use Committee approved all animal experimentation in this study.
Experimental designs
Male C57BL/6 (n = 15) and Apc Min/+ (n = 27) mice (20 weeks of age) were used to determine cachectic muscle's ability to induce mechano‐sensitive pathways and protein synthesis in response to a single ECC bout. In the first experiment, C57BL/6 (n = 6) and Apc Min/+ (n = 7) mice were sacrificed immediately following a single ECC bout. In the second experiment, C57BL/6 (n = 9) and Apc Min/+ (n = 10) mice were sacrificed 3 h after a single ECC bout. In the third experiment, an additional cohort of cachectic Apc Min/+ mice (n = 10; 16 ± 1% body weight loss) received a single pyrrolidine dithiocarbamate (PDTC) treatment (10 mg/kg body weight; cat#: P8765; Sigma Aldrich) 24 h prior to a single ECC bout. We have previously found that this treatment paradigm can sufficiently lower muscle inflammatory signalling prior to muscle contraction.32 In the current study, a single PDTC treatment did not alter total tumour number (86 ± 4; P = 0.91) or plasma IL‐6 levels (44 ± 5; P = 0.71), as we have previously observed following short‐term treatment after the initiation of cachexia.29 In all experiments, mice were fasted 2 h prior to contraction and remained fasted until sacrifice (immediately or 3 h post). There were no differences in cachexia indices (e.g. body weight, muscle mass, and fat loss) between Apc Min/+ mice in all experiments; therefore, general animal characteristics from each cohort are summarized in Table 1. Additionally, protein expression in the non‐contracted, control leg from Apc Min/+ mice in Experiments 1 and 2 was used to determine the cachectic muscle phenotype (Figure 1).
Table 1.
C57BL/6 and Apc Min/+ mice that performed a single eccentric contraction bout
| C57BL/6 | Apc Min/+ | |
|---|---|---|
| No. of mice | 15 | 27 |
| Body weight, g | ||
| Peak | 26.7 ± 0.7 | 24.7 ± 0.4 |
| ECC | 26.7 ± 0.7 | 20.7 ± 0.4a , b |
| % change | 0 ± 0 | −16 ± 0.6a |
| Tibialis anterior, mg | 49 ± 0.7 | 28 ± 0.9a |
| Epididymal fat, mg | 361 ± 44 | 3 ± 2a |
| Spleen, mg | 71 ± 4 | 506 ± 21a |
| Testes, mg | 201 ± 7 | 103 ± 8a |
| LABC, mg | 88 ± 2 | 31 ± 2a |
| Seminal vesicle, mg | 261 ± 8 | 32 ± 3a |
| Plasma IL‐6, pg/mL | 0 ± 0 | 43 ± 3a |
| Tumour number | 0 ± 0 | 87 ± 4a |
| Tibia length, mm | 16.9 ± 0.1 | 16.8 ± 0.1 |
Data are means ± standard error. There were no differences between control and stimulated TA muscles; therefore, the average is presented. A two‐way repeated measures analysis of variance was used to determine differences between body weight at peak and immediately prior to ECC. Post‐hoc analyses were performed with Student‐Knewman‐Keuls methods when appropriate. Student's t‐test was used to determine differences in all other variables. Statistical significance was set at P < 0.05. ECC, eccentric contraction; g, grams; IL‐6, interleukin‐6; LABC, levator ani/bulbocavernosus muscle; mg, milligrams; mL, millilitre; mm, millimetre; No., number; pg, picogram.
Significantly different from C57BL/6.
Significantly different from difference from peak body weight.
Figure 1.
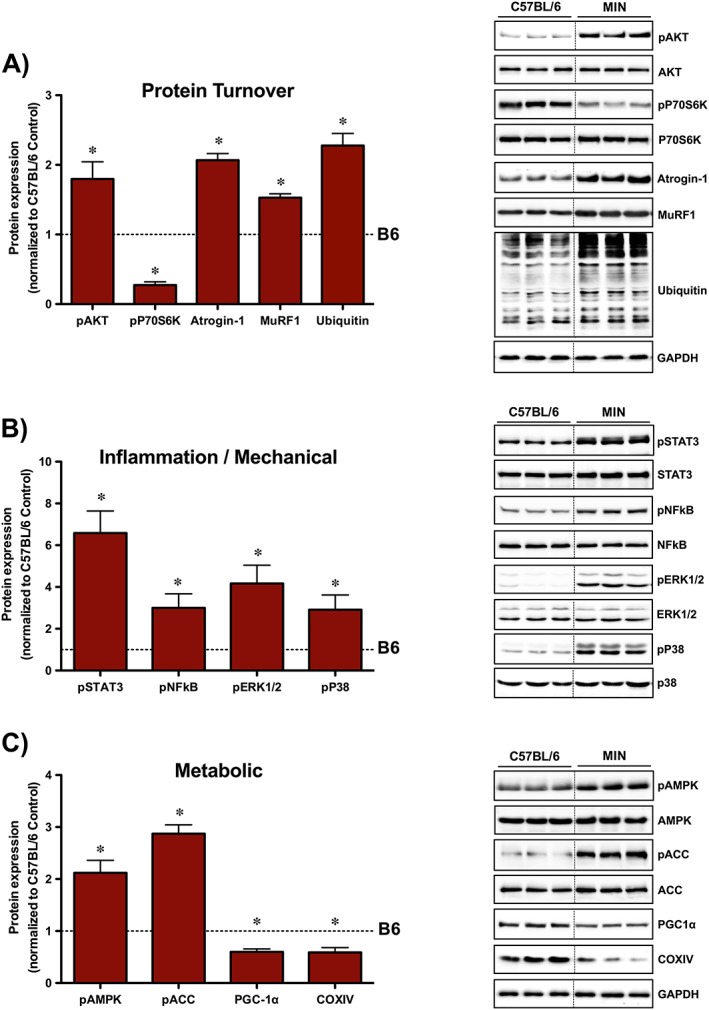
Cachectic muscle phenotype. (A) Protein turnover regulation in C57BL/6 and Apc Min/+ mice. (B) Inflammatory and mechano‐sensitive pathways in C57BL/6 and Apc Min/+ mice. (C) Metabolic signalling regulation in C57BL/6 and Apc Min/+ mice. Tibialis anterior protein expression was examined in the non‐contracted, control muscle. The activation of signalling molecules was determined by the phosphorylated and total ratio when appropriate. For protein expression, values were corrected for equal protein loading using GAPDH. All samples were run on the same gel and normalized to C57BL/6 Control values. Dotted lines indicate that images were cropped for representative purposes. Data are means ± standard error; n = 15; C57BL/6, n = 17; Apc Min/+. Student's t‐test was used to determine differences between C57BL/6 and Apc Min/+ mice. Statistical significance was set at P < 0.05. *, Significantly different from C57BL/6; ACC, Acetyl‐CoA carboxylase; AMPK, adenosine monophosphate‐activated protein kinase; ERK1/2, extracellular signal‐regulated kinases 1 and 2; NFκB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3.
Eccentric contractions
Eccentric contractions of the left tibialis anterior (TA) muscle was induced by high‐frequency electrical stimulation of the sciatic nerve as previously described with slight modifications.16, 24 Mice were anaesthetized via isoflurane (2% in O2 with 1.5% maintenance), the left leg/hip region was shaved, and two needle electrodes were placed subcutaneously on the left leg to stimulate the sciatic nerve. Tetanic muscle contractions were generated using a Grass Stimulator (Model S88, Grass Instruments, Quincy, MA, USA) for 10 sets of six repetitions (100 Hz, 6–12 V, 1 ms duration). Ten seconds of rest was given between repetitions, and 50 s of rest was given between sets. The stimulation protocol recruits all motor units and results in net plantar flexion of the ankle.33, 34 The dorsiflexors (TA and EDL) undergo ECCs while the plantar flexors (gastrocnemius, soleus, and plantaris) perform concentric muscle contractions. In all experiments, the left TA performed ECC while the right TA served as intra‐animal, non‐contracted control. Our laboratory and others have demonstrated repeated ECC bouts, but not concentric contractions, can induce muscle and myofiber growth in rodents.16, 23, 24, 33 Thus, the TA was examined in all experiments. Mice were given an intraperitoneal injection of warm saline following the stimulation procedure and returned to cages upon complete recovery.
Tissue collection
Mice received an intraperitoneal injection of puromycin (0.04 μmol/kg body weight) 30 min prior to sacrifice.29, 35 Mice were anaesthetized with a subcutaneous injection of ketamine/xylazine/acepromazine cocktail (1.4 mL/kg body weight) at the time of sacrifice. Muscles and organs were rapidly excised, cleared of excess connective tissue, rinsed in phosphate‐buffered saline, dried on blotting paper, weighed, and snap frozen in liquid nitrogen. Immediately prior to dissection, blood was collected via retro‐orbital sinus with heparinized capillary tubes, placed on ice, and centrifuged (10 000 × g for 10 min at 4°C). The supernatant was removed and stored for plasma IL‐6 analysis. Plasma and tissue samples were stored at −80°C until analysis.
Western blotting
Western blot analysis was performed as previously described.36 Briefly, frozen TA muscle was homogenized in Mueller buffer, and protein concentration was determined by the Bradford method. Crude TA muscle homogenates were fractionated on 7–15% sodium dodecyl sulphate‐polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were stained with Ponceau red to verify equal loading and transfer. Membranes were then blocked at room temperature for 1–2 h in 5% non‐fat milk Tris‐buffered saline with 0.1% Tween‐20 (TBST). Primary antibodies for puromycin (Millipore, cat#MABE343, 1:2000), phospho P70S6K (T389) (cat#9205, 1:1000), total P70S6K (cat#2708, 1:1000), RPS6 (S240/244) (cat#2215, 1:500), total RPS6 (cat#2708, 1:1000), phospho Akt (S473) (cat#4060, 1:1000), total Akt (cat#9272, 1:2000), phospho NFκB (S563) (cat#3033, 1:500), total NFκB (cat#4764, 1:2000), phospho STAT3 (Y750) (cat# 9145, 1:1000), total STAT3 (cat#4904, 1:2000), phospho AMPK (T172) (cat#, 1:2000), total AMPK (cat#2603, 1:1000), phospho Acetyl‐CoA carboxylase (ACC) (S59) (cat#3661, 1:1000), total ACC (cat#3662), phospho ERK1/2 (T202/Y204) (cat#4370,1:1000), total ERK1/2 (cat#4695, 1:1000), PGC‐1α (Abcam, cat#ab54481, 1:1000), COXIV (cat#4844, 1:1000), Glyceraldehyde‐3‐Phosphate Dehydrogenase (GAPDH) (cat#2118, 1:10000), MuRF1 (ECM Biosciences, cat#MP3401, 1:2000), Atrogin‐1 (ECM Biosciences, cat#AP2041, 1:5000), and ubiquitin (cat#3933, 1:2000) were incubated overnight in 5% TBST milk. We have previously validated the specificity of this PGC‐1α antibody in tibialis anterior skeletal muscle through somatic gene transfer of empty or PGC‐1α overexpression plasmid (data not shown). Membranes were then incubated in 5% milk‐TBST containing anti‐rabbit (cat#7074, 1:5000) or anti‐mouse (cat#7076, 1:5000) IgG horseradish‐peroxidase conjugated secondary antibodies for 1 h at room temperature. Exceptions to the aforementioned procedures were that for puromycin incorporation 1% BSA‐TBST was used for primary antibody and horseradish‐peroxidase conjugated rabbit anti‐mouse IgG2a antibody (LifeTechnologies, cat#610220, 1:5000) in 5% milk‐TBST was used for secondary antibody. All antibodies were from Cell Signalling Technology unless otherwise stated. Tibialis anterior protein extracts from a mouse that did not receive puromycin at sacrifice were included on all puromycin gels as a negative control. Enhanced chemiluminescence (GE Healthcare Life Sciences) was used to visualize the antibody‐antigen interactions. Immunoblot images were collected using a digital imager (G:BOX Chemi XX6, Syngene, Frederick, MD, USA) and quantified by densitometry using imaging software (Image J; NIH). Each gel contained samples from all groups, and data were normalized to the respective control group (e.g. C57BL/6 control).
Plasma interleukin‐6 concentration
Plasma IL‐6 concentrations were determined as previously described.37 A commercially available IL‐6 enzyme‐linked immunosorbent assay kit was obtained from BD Biosciences, and the manufacturer's protocol was followed. Briefly, clear 96‐well plates were coated and incubated overnight with an IL‐6 capture antibody. The next morning, the plate was blocked with assay diluent buffer and washed, and equal volumes of standards and plasma samples were added in duplicate. After a 2 h incubation, the plate was washed, and sAV‐HRP reagent was added to each well. After several washes, TMB substrate was added, and the reaction was developed for 20 min. The reaction was stopped with sulfuric acid, and absorbance was read at 450 nm using an iMark microplate absorbance reader (Bio‐Rad, Hercules, CA, USA).
Statistical analysis
Results are reported as the means ± standard error. A repeated‐measures two‐way analysis of variance was performed to determine differences between cancer cachexia and eccentric contractions in C57BL/6 and Apc Min/+ mice. Post‐hoc analyses were performed with Student‐Newman‐Keuls methods when appropriate. Student's t‐test was used to determined differences between two groups when appropriate. The accepted level of significance was set at P < 0.05 for all analysis. Statistical analysis and figure generation were performed using Prism 5 for Mac OS X (GraphPad Software Inc, La Jolla, CA, USA).
Results
Systemic and muscle cachectic phenotype
In order to determine if wasting skeletal muscle could respond to a novel ECC bout, we first established the cachectic phenotype in two separate cohorts of male Apc Min/+ mice. Apc Min/+ mice displayed several key features of severe cachexia that included body weight loss, muscle atrophy, adipose tissue depletion, high tumour burden, elevated plasma IL‐6 levels, and hypogonadal features (levator ani‐bulbocavernosus and seminal vesicle atrophy) (Table 1). Body weights at sacrifice were lower when compared with C57BL/6 mice, and significant body weight loss from peak measurement was observed within Apc Min/+ mice. Body weight loss was accompanied by reduced TA muscle mass and epididymal fat loss in Apc Min/+ mice. Cachexia increased spleen weight and plasma IL‐6 compared with C57BL/6 mice (Table 1). There were no differences in tibia length between C57BL/6 and Apc Min/+ mice. To further characterize the cachectic phenotype, protein expression related to protein turnover, inflammation, and metabolism was examined in the non‐contracted control TA muscle (Figure 1). Cachectic muscle demonstrated perturbed protein turnover regulation by disrupted mTORC1 signalling (increased Akt and decreased P70S6K), E3 ligase expression (Atrogin‐1 and MuRF1), and total ubiquitin (Figure 1A). Moreover, cachexia activated inflammatory/mechanical related proteins (STAT3, NFκB, ERK1/2, and P38) (Figure 1B). Lastly, cachexia altered the expression of proteins related to mitochondrial content (PGC‐1α and COXIV) and metabolic regulation (AMPK and ACC), respectively (Figure 1C). Overall, these findings demonstrate that severe cachexia in this cohort of mice was associated with high circulating IL‐6, enhanced muscle inflammatory signalling, and disrupted anabolic and metabolic regulation.
Mechanical signalling response to eccentric contractions
Having established the severe cachectic phenotype in this cohort of male Apc Min/+ mice, we then examined mechano‐sensitive pathways immediately following a single ECC bout (Figure 2A). First, we examined two MAPKs that have been shown to be activated immediately post‐ECC.18, 21 While cachexia increased basal P38 and ERK1/2 phosphorylation in the non‐contracted control TA muscle, ECC induced their activation irrespective of cachexia (Figure 2B). While absolute P38 and ERK1/2 phosphorylation were greater following ECC during cachexia, there were no differences in the degree of activation from the non‐contracted TA muscle. Given that ECCs are a potent stimulator of Akt/mTORC1 signalling,18, 21, 38 we next determined Akt/mTORC1 activation through phosphorylation of Akt and the downstream mTORC1 target P70S6K. While cachexia increased basal Akt phosphorylation in the non‐contracted control TA muscle, ECC induced its activation irrespective of cachexia (Figure 2C). Interestingly, while cachexia decreased basal P70S6K phosphorylation in the non‐contracted control TA muscle, ECC induced its activation irrespective of cachexia (Figure 2C). While absolute P70S6K phosphorylation was decreased following ECC during cachexia, there were no differences in the degree of activation from the non‐contracted control TA muscle. We further validated P70S6K activation through the phosphorylation of the direct P70S6K target RPS6. Similarly, cachexia decreased basal RPS6 phosphorylation in the non‐contracted control TA muscle, while ECC induced its activation irrespective of cachexia (Figure 2C). Collectively, these findings demonstrate that mechano‐sensitive pathways were induced immediately post‐ECC despite disrupted basal Akt/mTORC1 signalling in cachectic Apc Min/+ mice.
Figure 2.
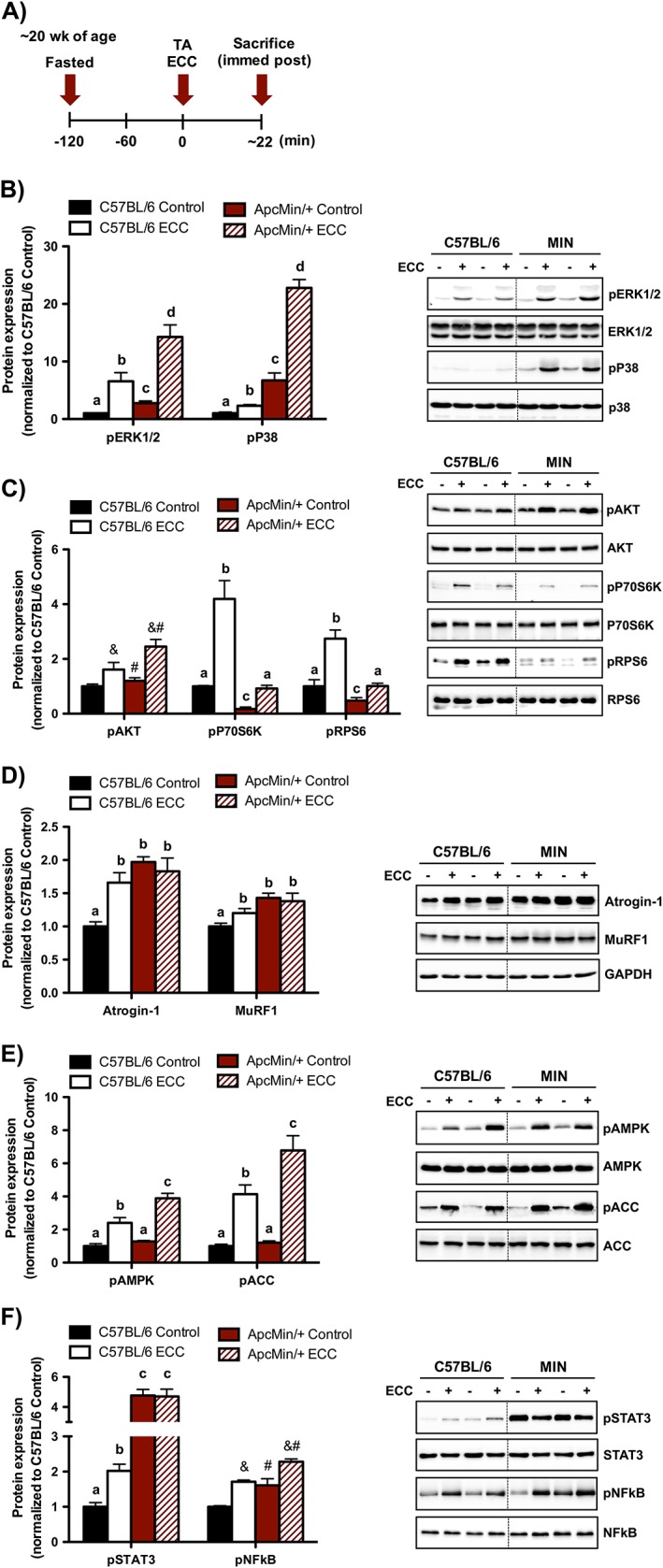
Muscle mechano‐sensitive signalling immediately post‐eccentric contractions (ECC). (A) Experimental design. C57BL/6 and Apc Min/+ mice were sacrificed immediately post‐ECC. Mice were fasted 2 h prior to contraction. (B) Muscle mitogen‐activated protein kinase signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. (C) Muscle Akt/mechanistic target of rapamycin complex 1 signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. (D) Muscle proteolytic regulation by ECC in C57BL/6 and Apc Min/+ mice. (E) Muscle metabolic signalling regulation in C57BL/6 and Apc Min/+ mice. (F) Muscle inflammatory signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. The activation of signalling molecules was determined by the phosphorylated and total ratio when appropriate. For protein expression, values were corrected for equal protein loading using GAPDH. All samples were run on the same gel and normalized to C57BL/6 Control values. Dotted lines indicate that images were cropped for representative purposes. Data are means ± standard error; n = 6; C57BL/6, n = 7; Apc Min/+. A two‐way repeated measures analysis of variance was used to determine differences between treatment groups. Post‐hoc analyses were performed with Student‐Knewman‐Keuls methods when appropriate. Statistical significance was set at P < 0.05. Different letters are statistically different. &, Main effect of ECC; #, main effect of Apc Min/+; ERK1/2, extracellular signal‐regulated kinases 1 and 2; TA, tibialis anterior.
Proteolytic, metabolic, and inflammatory signalling response to eccentric contractions
Given that the ubiquitin‐proteasome pathway has been implicated in the proteolytic response to acute resistance exercise,39 we examined the expression of two E3 ligases immediately following a single ECC bout. While ECC induced Atrogin‐1 and MuRF1 protein expression in C57BL/6 mice, this was not observed in Apc Min/+ mice (Figure 2D). Given the potential role of metabolic stress to inhibit anabolic processes, we next examined AMPK and its direct downstream target ACC in response to ECC. Interestingly, while the absolute AMPK and ACC phosphorylation were greater immediately post‐ECC during cachexia, there were no differences in the degree of activation from the non‐contracted TA muscle (Figure 2E). Lastly, we examined muscle inflammatory signalling pathways that have been implicated in cachexia and muscle contraction. Interestingly, STAT3 and NFκB were activated immediately post‐ECC in C57BL/6 (Figure 2F). While basal STAT3 and NFκB were induced by cachexia, only NFκB was further induced immediately post‐ECC in Apc Min/+ mice (Figure 2F). Collectively, these findings demonstrate that mechano‐sensitive pathways remained intact and was not associated with an exacerbated metabolic and proteolytic response immediately post‐ECC in cachectic Apc Min/+ mice.
Cachectic muscle anabolic signalling response to eccentric contractions
Having established the cachectic muscle's mechano‐sensitive response to a single ECC bout, we then determined if cachexia disrupted ECC‐induced protein synthesis and mTORC1 signalling. Therefore, anabolic signalling was examined 3 h after a single ECC bout (Figure 3A). While cachexia suppressed protein synthesis, ECC activated protein synthesis irrespective of cachexia. Although the relative induction by ECC was not altered by cachexia, the absolute protein synthesis rate remained suppressed relative to C57BL/6 mice (Figure 3B). We then examined several upstream regulators and downstream targets implicated in the mechanical activation of mTORC1 signalling. Interestingly, while cachexia increased basal P38 and ERK1/2 phosphorylation, these mechano‐sensitive signalling molecules were not altered 3 h post‐ECC irrespective of cachexia (Figure 3C). We then examined Akt/mTORC1 activation in response to ECC. While Akt was not altered by ECC regardless of cachexia, there was a strong trend (P = 0.07) for ECC to decrease Akt phosphorylation in C57BL/6 mice (Figure 3D). While cachexia decreased the phosphorylation of P70S6K, ECC induced its activation irrespective of cachexia (Figure 3D). ECC also increased RPS6 phosphorylation irrespective of cachexia (Figure 3D). Collectively, these findings demonstrate that protein synthesis and mTORC1 signalling was induced 3 h post‐ECC in severely cachectic Apc Min/+ mice.
Figure 3.
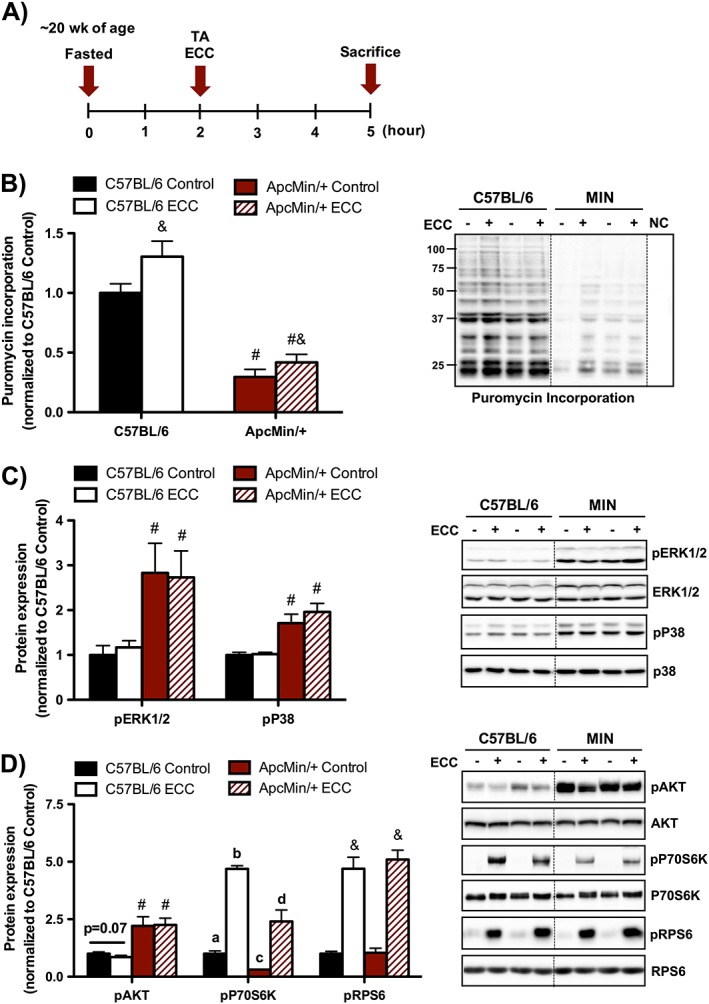
Muscle protein synthesis and mechanistic target of rapamycin complex 1 signalling 3 h post‐eccentric contractions (ECC). (A) Experimental design. C57BL/6 and Apc Min/+ mice were sacrificed 3 h post a single bout of eccentric contractions. Mice were fasted 2 h prior to contraction and remained fasted during the 3 h recovery until sacrifice. Mice were injected with puromycin 30 min prior to sacrifice. (B) Muscle protein synthesis regulation by ECC in C57BL/6 and Apc Min/+ mice. (C) Muscle mitogen‐activated protein kinase signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. (D) Muscle Akt/mechanistic target of rapamycin complex 1 signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. The activation of signalling molecules was determined by the phosphorylated and total ratio when appropriate. For protein expression, values were corrected for equal protein loading using GAPDH. All samples were run on the same gel and normalized to C57BL/6 Control values. Dotted lines indicate that images were cropped for representative purposes. Data are means ± standard error; n = 9; C57BL/6, n = 10; Apc Min/+. A two‐way repeated measures analysis of variance was used to determine differences between treatment groups. Post‐hoc analyses were performed with Student‐Knewman‐Keuls methods when appropriate. Statistical significance was set at P < 0.05. Different letters are statistically different. &, Main effect of ECC; #, main effect of Apc Min/+; ERK1/2, extracellular signal‐regulated kinases 1 and 2; TA, tibialis anterior.
Cachectic muscle proteolytic, metabolic, and inflammatory response to eccentric contractions
Muscle protein breakdown, metabolic dysfunction, and enhanced inflammatory signalling have established roles during cancer‐induced muscle wasting, and these same signalling pathways are perturbed in response to muscle contraction.40, 41, 42, 43 ECC did not alter the expression of Atrogin‐1 and MuRF1 irrespective of cachexia (Figure 4A). We have previously shown that the sustained activation of AMPK coincided with suppressed mTORC1 signalling 3 h after a single bout of concentric muscle contractions.32 Interestingly, while AMPK and ACC were activated by cachexia, ECC decreased their activation irrespective of cachexia (Figure 4B). Lastly, we found that both STAT3 and NFκB phosphorylation were induced by cachexia and were further increased by ECC irrespective of cachexia (Figure 4C). Collectively, these data demonstrate that muscle metabolic and inflammatory signalling molecules were sensitive to both cachexia and ECC.
Figure 4.
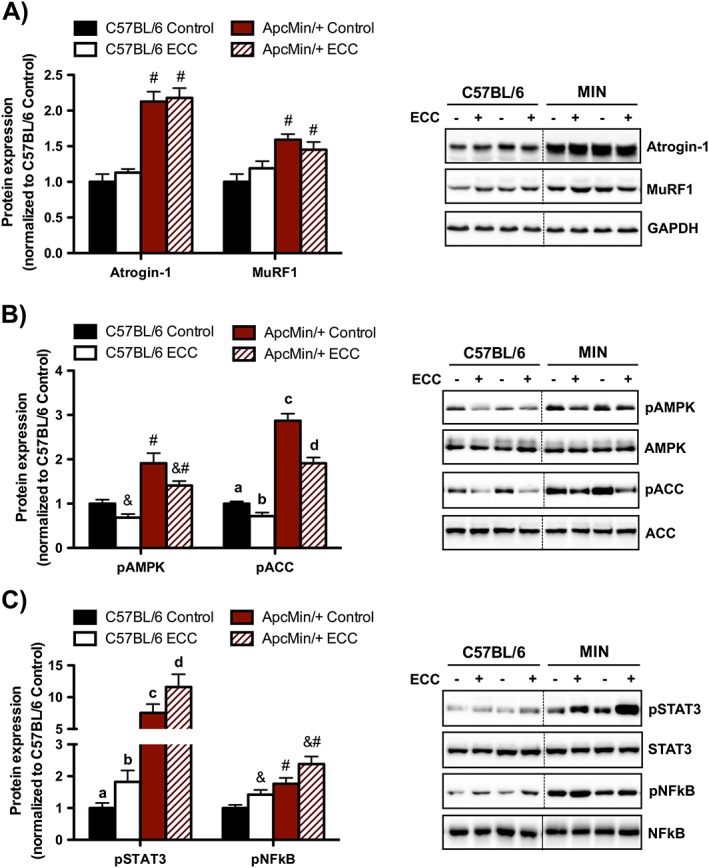
Muscle proteolytic, metabolic, and inflammatory signalling 3 h post‐eccentric contractions (ECC). (A) Muscle proteolytic regulation by ECC in C57BL/6 and Apc Min/+ mice. (B) Muscle metabolic signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. (C) Muscle inflammatory signalling regulation by ECC in C57BL/6 and Apc Min/+ mice. The activation of signalling molecules was determined by the phosphorylated and total ratio when appropriate. For protein expression, values were corrected for equal protein loading using GAPDH. All samples were run on the same gel and normalized to C57BL/6 Control values. Dotted lines indicate that images were cropped for representative purposes. Data are means ± standard error; n = 9; C57BL/6, n = 10; Apc Min/+. A two‐way repeated measures analysis of variance was used to determine differences between treatment groups. Post‐hoc analyses were performed with Student‐Knewman‐Keuls methods when appropriate. Statistical significance was set at P < 0.05. Different letters are statistically different. &, Main effect of ECC; #, main effect of Apc Min/+; ACC, Acetyl‐CoA carboxylase; AMPK, adenosine monophosphate‐activated protein kinase; NFκB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3.
Muscle inflammatory signalling regulation of basal and eccentric contractions‐induced protein synthesis
Given that muscle STAT3 and NFκB signalling was induced by cachexia and ECC, we next examined its involvement in the regulation of basal and ECC‐induced protein synthesis in cachectic Apc Min/+ mice. To accomplish this, we used an established pharmacological approach to lower basal muscle inflammatory signalling prior to ECC.29, 32 This experimental paradigm has previously been used by our laboratory to lower muscle STAT3/NFκB signalling prior to a single bout of low‐frequency electrical stimulation.32 Therefore, we administered PDTC 24 h prior to a single ECC bout in cachectic Apc Min/+ mice (Figure 5A). As expected, PDTC decreased basal STAT3 and NFκB phosphorylation in the non‐stimulated control TA muscle (Figure 5B). There was no effect of PDTC on basal P38, ERK1/2, or Akt in cachectic skeletal muscle (data not shown). Interestingly, PDTC induced basal protein synthesis in cachectic muscle and was further increased 3 h post‐ECC (Figure 5C). While there was a trend (P = 0.08) for PDTC to increase basal P70S6K phosphorylation, there was a robust induction 3 h post‐ECC. Similarly, PDTC increased basal RPS6 phosphorylation and was further increased 3 h post‐ECC in cachectic skeletal muscle (Figure 5D). As previously observed, the activation of mechano‐sensitive pathways (P38, ERK1/2, and Akt) were not altered 3 h post‐ECC (data not shown). Collectively, these data demonstrate that acute muscle STAT3 and NFκB inhibition improved basal and ECC‐induced protein synthesis in cachectic Apc Min/+ mice.
Figure 5.
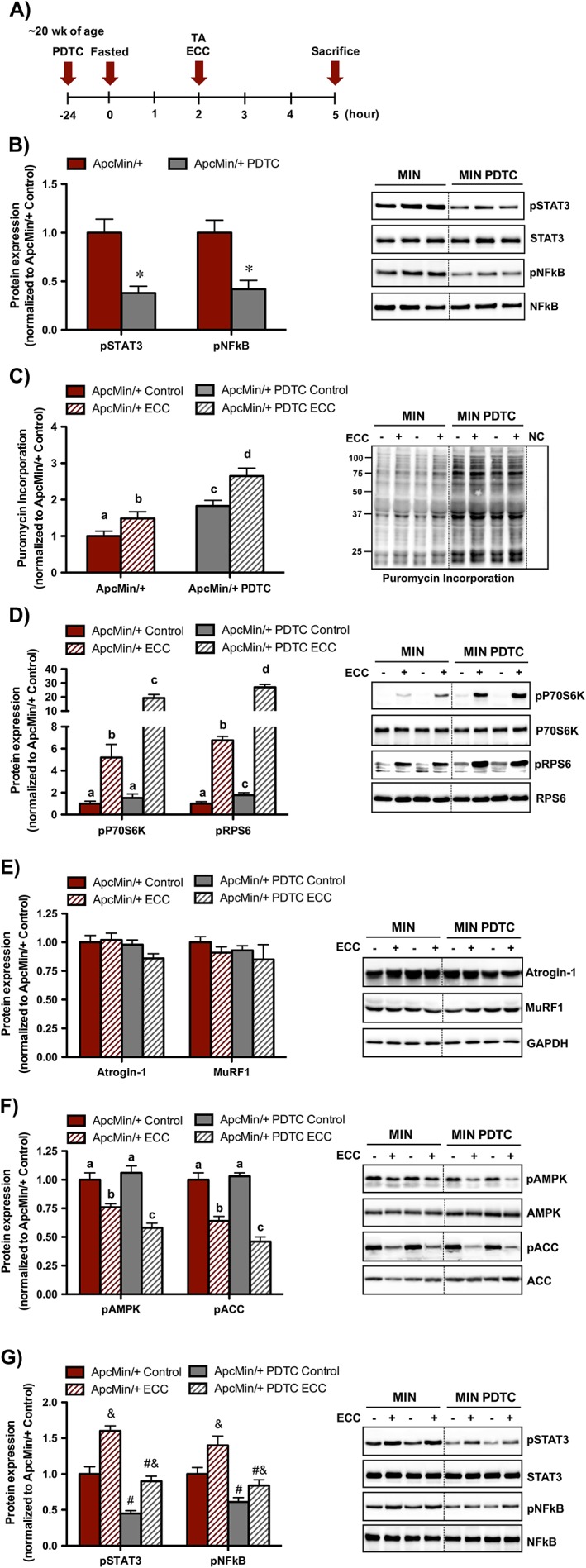
Muscle inflammatory signalling regulation of eccentric contractions (ECC)‐induced protein synthesis in Apc Min/+ mice. (A) Experimental design. Apc Min/+ mice were sacrificed 3 h post‐ECC. A cohort of Apc Min/+ mice was given a single pyrrolidine dithiocarbamate (PDTC) treatment (10 mg/kg body weight) 24 h prior to ECC. Mice were fasted 2 h prior to ECC and remained fasted during the 3 h recovery until sacrifice. Mice were injected with puromycin 30 min prior to sacrifice. (B) Muscle signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB) signalling in the non‐contracted, control muscle following a single PDTC treatment. (C) Muscle protein synthesis regulation by PDTC and ECC in Apc Min/+ mice. (D) Muscle mTORC1 signalling regulation by PDTC and ECC in Apc Min/+ mice. (E) Muscle proteolytic regulation by PDTC and ECC in Apc Min/+ mice. (F) Muscle metabolic signalling regulation by PDTC and ECC in Apc Min/+ mice. (G) Muscle inflammatory signalling regulation by PDTC and ECC in Apc Min/+ mice. The activation of signalling molecules was determined by the phosphorylated and total ratio when appropriate. For protein expression, values were corrected for equal protein loading using GAPDH. All samples were run on the same gel and normalized to Apc Min/+ Control values. Dotted lines indicate that images were cropped for representative purposes. Data are means ± standard error; n = 10; Apc Min/+, n = 10; Apc Min/+ PDTC. A two‐way repeated measures analysis of variance was used to determine differences between treatment groups. Post‐hoc analyses were performed with Student‐Knewman‐Keuls methods when appropriate. Statistical significance was set at P < 0.05. Different letters are statistically different. &, Main effect of ECC; #, main effect of PDTC; TA, tibialis anterior.
Proteolytic, metabolic, and inflammatory signalling response to pyrrolidine dithiocarbamate and eccentric contractions
Lastly, we examined the proteolytic, metabolic, and inflammatory signalling response to PDTC and ECC. Neither PDTC or ECC altered the expression of Atrogin‐1 and MuRF1 in Apc Min/+ mice (Figure 5E). While PDTC did not alter basal AMPK and ACC activation in cachectic skeletal muscle, these molecules were further suppressed 3 h post‐ECC (Figure 5F). Interestingly, while PDTC suppressed basal muscle STAT3 and NFκB signalling, it did not block the induction 3 h post‐ECC (Figure 5G). Altogether, these data demonstrate that improved ECC‐induced protein synthesis by PDTC corresponded to suppressed metabolic signalling but was independent to altered proteolytic E3 ligase expression in cachectic Apc Min/+ mice.
Discussion
Healthy skeletal muscle stimulates protein synthesis in response to anabolic stimuli associated with daily living, which can include physical activity and feeding. While basal muscle protein synthesis and mTORC1 signalling is suppressed in tumour‐bearing mice and some human cancer patients,7, 44 the capacity for cachectic muscle to respond to an anabolic stimulus is not well understood. This knowledge could have significant ramifications for the treatment of the cachectic cancer patient. Resistance exercise consisting of ECC is a potent stimulator of protein synthesis and muscle growth.11, 12, 45 We have previously found that repeated ECC bouts after the initiation of cachexia attenuated myofiber atrophy and was accompanied by suppressed muscle inflammatory signalling. However, the capacity to activate mTORC1 signalling and protein synthesis by ECC has not been investigated. Therefore, we examined if cachectic muscle maintained the ability to induce mechano‐sensitive pathways and protein synthesis in response to a single ECC bout. We report that ECC‐induced mechanical signalling was maintained in cachectic muscle, but the capacity for increased protein synthesis was attenuated. This finding demonstrates an unexpected uncoupling between the activation of known mechano‐sensitive regulators of anabolic signalling and the absolute protein synthesis induction. Therefore, we also examined if the cachectic environment involving muscle inflammatory signalling could regulate ECC‐induced protein synthesis. Interestingly, both cachexia and ECC induced muscle STAT3 and NFκB signalling. However, muscle STAT3/NFκB inhibition by PDTC increased basal and ECC‐induced protein synthesis in cachectic Apc Min/+ mice. These findings demonstrate that mechano‐sensitive signalling is responsive to ECC and highlight muscle inflammatory signalling's role in the altered regulation of both basal and ECC‐induced protein synthesis during cancer cachexia.
While exercise training has been discussed as a potential therapy to mitigate muscle atrophy during cancer cachexia, there is currently a limited understanding of the acute response and training adaptation to exercise. Whole‐body treadmill exercise prevented muscle mass loss in tumour‐bearing mice46, 47, 48 and blocked the disruption of muscle oxidative metabolism regulation at the initiation of cachexia.49, 50 However, the inability of severely cachectic mice to perform voluntary exercise remains a consistent barrier and has limited our understanding of the muscle response to exercise during refractory cachexia. To address this, we have reported that cachexia disrupted the metabolic and anabolic signalling response to a single bout of stimulated low‐frequency concentric contractions,32 which mimics low intensity, endurance type exercise. Given that exercise involves muscle contractions that can vary in overall intensity and metabolic demand, the molecular responses related to growth and metabolism are distinct between contraction types. Furthermore, the response of cachectic muscle to high force contractions is not well established. Therefore, we first examined the mechanical and metabolic response to a single ECC bout. We found that several contraction and mechano‐sensitive kinases (MAPKs, Akt, and P70S6K) were induced by ECC in cachectic muscle. The activation of P38, ERK1/2, and Akt was transient, which extends previous observations in mouse skeletal muscle.18 In addition, P70S6K activation by ECC remained elevated irrespective of cachexia, which is in contrast to our previous observations using concentric muscle contractions.32 Interestingly, we also observed a transient AMPK induction by ECC, which was not associated with mTORC1 signalling inhibition. Collectively, we provide initial evidence that the mechanical and metabolic plasticity of muscle to ECC is maintained despite the presence of a systemic cachectic environment.
There is considerable interest in understanding the mechanisms that serve to repress cachectic muscle anabolic signalling. We previously reported that repeated ECC bouts performed after the initiation of cancer cachexia could attenuate myofiber atrophy24; however, this study did not determine whether these improvements were related to the induction of muscle growth or the attenuation of muscle breakdown. While our current study has extended these findings to demonstrate that mechano‐signalling in cachectic muscle is maintained, the ability to synthesize protein remained dramatically suppressed. These findings further demonstrate that the capacity for either basal or contraction‐induced muscle protein synthesis is suppressed by cachexia. The chronic activation of AMPK in cachectic skeletal muscle from tumour‐bearing mice has been implicated as a potential mechanism for mTORC1 and protein synthesis suppression.7, 28 Interestingly, our current study demonstrates the induction of protein synthesis after ECC coincides with reduced AMPK activation in tumour‐bearing mice. Furthermore, reduced AMPK activation after a single ECC bout has also recently been observed in castrated mice.51 These findings are in contrast to our previous observations using low‐frequency electrical stimulation and point to the differential regulation of muscle metabolic signalling by different types of contraction. We previously found a sustained AMPK activation following a single bout of low‐frequency concentric contractions in cachectic skeletal muscle.32 The specificity of the responses induced by different types of contractions may be related to metabolic stress as cachectic muscle develops mitochondrial dysfunction.52, 53, 54 The cachectic muscle's anabolic and metabolic response to different contraction types will require further investigation. Nonetheless, our findings suggest that ECC may be a potential therapeutic treatment to promote muscle anabolism during cancer cachexia progression.
Muscle signalling related to inflammation, energy status, and proteostasis are disrupted during cachexia progression and have been implicated in the regulation of muscle wasting.55, 56 Interestingly, many of these same pathways are induced by muscle contraction and exercise.57 IL‐6 and muscle STAT3 signalling through the gp130 receptor are activated during the progression of cachexia30, 52, 58 and are associated with mTORC1 and protein suppression in pre‐clinical cachexia models.7, 28 Inhibition of muscle IL‐6 signalling through either direct STAT3 inhibition or gp130 loss can attenuate wasting in mouse models of cachexia.30, 31 We have previously found that short‐term PDTC treatment attenuated the suppression of mTORC1 signalling and protein synthesis while concomitantly reducing muscle STAT3 and NFκB activation in Apc Min/+ mice.29, 32 We extend these findings by demonstrating a single PDTC dose improved basal protein synthesis. It has recently been suggested that intermittent cycles of pathway inhibition/activation may be required to combat muscle wasting during cancer cachexia.59 Indeed, many cytokine‐related signalling pathways have established roles in myogenesis60 and load‐induced muscle growth and remodelling.41, 42, 61 Therefore, we utilized an experimental paradigm that lowered chronic muscle inflammatory signalling but did not block contraction‐induced signalling.32 Importantly, we found that PDTC treatment increased both basal and ECC‐induced protein synthesis in cachectic muscle. These findings demonstrate that cachectic muscle retains the anabolic capacity to increase protein synthesis, and inflammatory signalling contributes to the suppression of these processes. Additional research is warranted to determine the specific mechanisms related to STAT3 and NFκB that serve to diminish the capacity for protein synthesis in cachectic muscle. Moreover, further research is also required to determine the effect of muscle inflammatory signalling on metabolic remodelling in response to repeated contraction bouts, which could dramatically impact health outcomes related to exercise.
Conclusions
In summary, we examined cachectic muscle's ability to induce protein synthesis in response to ECC and determined the role of muscle inflammatory signalling involving STAT3 and NFκB on ECC‐induced anabolic signalling. We found that mechano‐sensitive signalling pathways related to P38, ERK1/2, and Akt were not altered by the cachectic environment in wasting muscle. While cachexia did not attenuate the ECC induction of mTORC1 signalling, the capacity for protein synthesis remained suppressed compared with healthy controls. Interestingly, we found that reducing muscle STAT3/NFκB signalling improved basal and ECC‐induced protein synthesis during severe cachexia. These studies demonstrate that mechano‐sensitive signalling pathways are maintained in skeletal muscle, but STAT3/NFκB signalling serves to attenuate basal and ECC‐induced protein synthesis. Further work is necessary to determine whether intermittent anti‐inflammatory therapies combined with exercise training may be useful to alleviate suppressed muscle protein synthesis during cancer cachexia.
Conflict of interest
Justin P. Hardee, Brittany R. Counts, Song Gao, Brandon N. VanderVeen, Dennis K. Fix, Ho‐Jin Koh,, and James A. Carson declare that they have no conflict of interest.
Acknowledgements
The authors thank Gaye Christmus and Dr Bradley Gordon for editorial review of the manuscript.
This work was supported by National Institutes of Health [grants R01 CA‐121249 (National Cancer Institute) and P20 RR‐017698 (National Institute of General Medical Science)] to J.A.C, SPARC Graduate Research Grant from the Office of the Vice President for Research at the University of South Carolina to J.P.H and an ACSM Foundation Research Grant from the American College of Sports Medicine Foundation to J.P.H. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.62
Hardee, J. P. , Counts, B. R. , Gao, S. , VanderVeen, B. N. , Fix, D. K. , Koh, H.‐J. , and Carson, J. A. (2018) Inflammatory signalling regulates eccentric contraction‐induced protein synthesis in cachectic skeletal muscle. Journal of Cachexia, Sarcopenia and Muscle, 9: 369–383. doi: 10.1002/jcsm.12271.
References
- 1. Fearon KC. The Sir David Cuthbertson Medal Lecture 1991. The mechanisms and treatment of weight loss in cancer. Proc Nutr Soc 1992;51:251–265. [DOI] [PubMed] [Google Scholar]
- 2. Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 2009;89:381–410. [DOI] [PubMed] [Google Scholar]
- 3. Maddocks M, Murton AJ, Wilcock A. Improving muscle mass and function in cachexia: non‐drug approaches. Curr Opin Support Palliat Care 2011;5:361–364. [DOI] [PubMed] [Google Scholar]
- 4. Murton AJ, Greenhaff PL. Physiological control of muscle mass in humans during resistance exercise, disuse and rehabilitation. Curr Opin Clin Nutr Metab Care 2010;13:249–254. [DOI] [PubMed] [Google Scholar]
- 5. Samuels SE, Knowles AL, Tilignac T, Debiton E, Madelmont JC, Attaix D. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am J Physiol Regul Integr Comp Physiol 2001;281:R133–R139. [DOI] [PubMed] [Google Scholar]
- 6. Smith KL, Tisdale MJ. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br J Cancer 1993;67:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One 2011;6: e24650. doi:https://doi.org/10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sandri M. Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 2013;45:2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charette SL, McEvoy L, Pyka G, Snow‐Harter C, Guido D, Wiswell RA, et al. Muscle hypertrophy response to resistance training in older women. J Appl Physiol 1991;70:1912–1916. [DOI] [PubMed] [Google Scholar]
- 11. Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA, Smith K. Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol (1985) 1992;73:1383–1388. [DOI] [PubMed] [Google Scholar]
- 12. Eliasson J, Elfegoun T, Nilsson J, Kohnke R, Ekblom B, Blomstrand E. Maximal lengthening contractions increase p70 S6 kinase phosphorylation in human skeletal muscle in the absence of nutritional supply. Am J Physiol Endocrinol Metab 2006;291:E1197–E1205. [DOI] [PubMed] [Google Scholar]
- 13. Alberga AS, Segal RJ, Reid RD, Scott CG, Sigal RJ, Khandwala F, et al. Age and androgen‐deprivation therapy on exercise outcomes in men with prostate cancer. Support Care Cancer 2012;20:971–981. [DOI] [PubMed] [Google Scholar]
- 14. Sharif S, Thomas JM, Donley DA, Gilleland DL, Bonner DE, McCrory JL, et al. Resistance exercise reduces skeletal muscle cachexia and improves muscle function in rheumatoid arthritis. Case Rep Med 2011;2011:205691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hardee JP, Porter C, Sidossis LS, Borsheim E, Carson JA, Herndon DN, et al. Early rehabilitative exercise training in the recovery from pediatric burn. Med Sci Sports Exerc 2014;46:1710–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baar K, Esser K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am J Phys 1999;276:C120–C127. [DOI] [PubMed] [Google Scholar]
- 17. Chen YW, Nader GA, Baar KR, Fedele MJ, Hoffman EP, Esser KA. Response of rat muscle to acute resistance exercise defined by transcriptional and translational profiling. J Physiol 2002;545:27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nader GA, Esser KA. Intracellular signaling specificity in skeletal muscle in response to different modes of exercise. J Appl Physiol (1985) 2001;90:1936–1942. [DOI] [PubMed] [Google Scholar]
- 19. Witkowski S, Lovering RM, Spangenburg EE. High‐frequency electrically stimulated skeletal muscle contractions increase p70s6k phosphorylation independent of known IGF‐I sensitive signaling pathways. FEBS Lett 2010;584:2891–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacobs BL, You JS, Frey JW, Goodman CA, Gundermann DM, Hornberger TA. Eccentric contractions increase the phosphorylation of tuberous sclerosis complex‐2 (TSC2) and alter the targeting of TSC2 and the mechanistic target of rapamycin to the lysosome. J Physiol 2013;591:4611–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Neil TK, Duffy LR, Frey JW, Hornberger TA. The role of phosphoinositide 3‐kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol 2009;587:3691–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. West DW, Baehr LM, Marcotte GR, Chason CM, Tolento L, Gomes AV, et al. Acute resistance exercise activates rapamycin‐sensitive and ‐insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiol 2016;594:453–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Al–Majid S, McCarthy DO. Resistance exercise training attenuates wasting of the extensor digitorum longus muscle in mice bearing the colon‐26 adenocarcinoma. Biol Res Nurs 2001;2:155–166. [DOI] [PubMed] [Google Scholar]
- 24. Hardee JP, Mangum JE, Gao S, Sato S, Hetzler KL, Puppa MJ, et al. Eccentric contraction‐induced myofiber growth in tumor‐bearing mice. J Appl Physiol (1985) 2016;120:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Norton JA, Lowry SF, Brennan MF. Effect of work‐induced hypertrophy on skeletal muscle of tumor‐ and nontumor‐bearing rats. J Appl Physiol Respir Environ Exerc Physiol 1979;46:654–657. [DOI] [PubMed] [Google Scholar]
- 26. Otis JS, Lees SJ, Williams JH. Functional overload attenuates plantaris atrophy in tumor‐bearing rats. BMC Cancer 2007;7:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao S, Carson JA. Lewis lung carcinoma regulation of mechanical stretch‐induced protein synthesis in cultured myotubes. Am J Physiol Cell Physiol 2016;310:C66–C79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL‐6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 2013;304:E1042–E1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Narsale AA, Puppa MJ, Hardee JP, VanderVeen BN, Enos RT, Murphy EA, et al. Short‐term pyrrolidine dithiocarbamate administration attenuates cachexia‐induced alterations to muscle and liver in ApcMin/+ mice. Oncotarget 2016;https://doi.org/10.18632/oncotarget.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma‐induced cachexia. FASEB J 2014;28:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puppa MJ, Murphy EA, Fayad R, Hand GA, Carson JA. Cachectic skeletal muscle response to a novel bout of low‐frequency stimulation. J Appl Physiol (1985) 2014;116:1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong TS, Booth FW. Skeletal muscle enlargement with weight‐lifting exercise by rats. J Appl Physiol (1985) 1988;65:950–954. [DOI] [PubMed] [Google Scholar]
- 34. Wong TS, Booth FW. Protein metabolism in rat tibialis anterior muscle after stimulated chronic eccentric exercise. J Appl Physiol (1985) 1990;69:1718–1724. [DOI] [PubMed] [Google Scholar]
- 35. Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 2011;25:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hardee JP, Puppa MJ, Fix DK, Gao S, Hetzler KL, Bateman TA, et al. The effect of radiation dose on mouse skeletal muscle remodeling. Radiol Oncol 2014;48:247–256, rado‐48‐03‐247 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hetzler KL, Hardee JP, Puppa MJ, Narsale AA, Sato S, Davis JM, et al. Sex differences in the relationship of IL‐6 signaling to cancer cachexia progression. Biochim Biophys Acta 2015;1852:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomson DM, Fick CA, Gordon SE. AMPK activation attenuates S6K1, 4E‐BP1, and eEF2 signaling responses to high‐frequency electrically stimulated skeletal muscle contractions. J Appl Physiol (1985) 2008;104:625–632. [DOI] [PubMed] [Google Scholar]
- 39. Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, et al. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J Gerontol A Biol Sci Med Sci 2013;68:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Washington TA, White JP, Davis JM, Wilson LB, Lowe LL, Sato S, et al. Skeletal muscle mass recovery from atrophy in IL‐6 knockout mice. Acta Physiol (Oxf) 2011;202:657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. White JP, Reecy JM, Washington TA, Sato S, Le ME, Davis JM, et al. Overload‐induced skeletal muscle extracellular matrix remodelling and myofibre growth in mice lacking IL‐6. Acta Physiol (Oxf) 2009;197:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Serrano AL, Baeza‐Raja B, Perdiguero E, Jardi M, Munoz‐Canoves P. Interleukin‐6 is an essential regulator of satellite cell‐mediated skeletal muscle hypertrophy. Cell Metab 2008;7:33–44. [DOI] [PubMed] [Google Scholar]
- 43. Begue G, Douillard A, Galbes O, Rossano B, Vernus B, Candau R, et al. Early activation of rat skeletal muscle IL‐6/STAT1/STAT3 dependent gene expression in resistance exercise linked to hypertrophy. PLoS One 2013;8: e57141. doi:https://doi.org/10.1371/journal.pone.0057141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emery PW, Edwards RH, Rennie MJ, Souhami RL, Halliday D. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. Br Med J (Clin Res Ed) 1984;289:584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, et al. Aging impairs contraction‐induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle 2011;1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deuster PA, Morrison SD, Ahrens RA. Endurance exercise modifies cachexia of tumor growth in rats. Med Sci Sports Exerc 1985;17:385–392. [PubMed] [Google Scholar]
- 47. Penna F, Busquets S, Pin F, Toledo M, Baccino FM, Lopez‐Soriano FJ, et al. Combined approach to counteract experimental cancer cachexia: eicosapentaenoic acid and training exercise. J Cachexia Sarcopenia Muscle 2011;2:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salomao EM, Toneto AT, Silva GO, Gomes‐Marcondes MC. Physical exercise and a leucine‐rich diet modulate the muscle protein metabolism in Walker tumor‐bearing rats. Nutr Cancer 2010;62:1095–1104. [DOI] [PubMed] [Google Scholar]
- 49. Puppa MJ, White JP, Velazquez KT, Baltgalvis KA, Sato S, Baynes JW, et al. The effect of exercise on IL‐6‐induced cachexia in the Apc ( Min/+) mouse. J Cachexia Sarcopenia Muscle 2012;3:117–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, et al. IL‐6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2012;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steiner JL, Fukuda DH, Rossetti ML, Hoffman JR, Gordon BS. Castration alters protein balance after high‐frequency muscle contraction. J Appl Physiol (1985) 2017;122:264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL‐6‐dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2011;300:R201–R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tzika AA, Fontes‐Oliveira CC, Shestov AA, Constantinou C, Psychogios N, Righi V, et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int J Oncol 2013;43:886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Antunes D, Padrao AI, Maciel E, Santinha D, Oliveira P, Vitorino R, et al. Molecular insights into mitochondrial dysfunction in cancer‐related muscle wasting. Biochim Biophys Acta 2014;1841:896–905. [DOI] [PubMed] [Google Scholar]
- 55. Carson JA, Hardee JP, VanderVeen BN. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer‐induced muscle wasting. Semin Cell Dev Biol 2016;54:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 57. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 2013;17:162–184. [DOI] [PubMed] [Google Scholar]
- 58. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 2011;6: e22538. doi:https://doi.org/10.1371/journal.pone.0022538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Au ED, Desai AP, Koniaris LG, Zimmers TA. The MEK‐inhibitor selumetinib attenuates tumor growth and reduces IL‐6 expression but does not protect against muscle wasting in Lewis lung cancer cachexia. Front Physiol 2016;7:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Costamagna D, Costelli P, Sampaolesi M, Penna F. Role of inflammation in muscle homeostasis and myogenesis. Mediat Inflamm 2015;2015: 805172. doi:https://doi.org/10.1155/2015/805172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spangenburg EE, Booth FW. Leukemia inhibitory factor restores the hypertrophic response to increased loading in the LIF(‐/‐) mouse. Cytokine 2006;34:125–130. [DOI] [PubMed] [Google Scholar]
- 62. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]